

Abstract
Epigenetic modifiers frequently harbor loss-of-function mutations in lung cancer, but their tumor-suppressive roles are poorly characterized. Histone methyltransferase KMT2D (a COMPASS-like enzyme, also called MLL4) is among the most highly inactivated epigenetic modifiers in lung cancer. Here we show that lung-specific loss of Kmt2d promotes lung tumorigenesis in mice and upregulates pro-tumorigenic programs, including glycolysis. Pharmacological inhibition of glycolysis preferentially impedes growth of human lung cancer cells bearing KMT2D-inactivating mutations. Mechanistically, Kmt2d loss widely impairs epigenomic signals for super-enhancers/enhancers, including the super-enhancer for the circadian rhythm repressor Per2. Loss of Kmt2d decreases expression of PER2, which regulates multiple glycolytic genes. These findings indicate that KMT2D is a lung tumor suppressor and KMT2D deficiency confers a therapeutic vulnerability to glycolytic inhibitors.
Significance
Lung cancer is the leading cause of cancer deaths. The overall survival rate for lung cancer patients remains low despite recent therapeutic advances, and a majority of lung cancer patients lack a druggable target. Therefore, there is a great need for a better understanding of the molecular mechanisms driving lung cancer. Epigenetic modifiers are frequently lost in lung cancer, but how this provokes lung tumorigenesis remains unclear. We show that KMT2D, which is recurrently mutated in lung cancer, is a lung tumor suppressor. Our results uncover a tumor-promoting epigenetic mechanism by which KMT2D-inactivating mutations induce aberrant metabolic reprogramming via super-enhancer impairment in lung cancer. Our findings support a glycolysis-inhibitory approach as a therapeutic intervention strategy against KMT2D-mutant lung cancer.
Keywords: Epigenetic modifier, Lung Cancer, Super-enhancer, Histone methyltransferase, Histone methylation, KMT2D, Glycolysis, Metabolism, Inhibitor, Tumor suppressor
Graphical Abstract

eTOC Blurb
Histone methyltransferase KMT2D is frequently mutated in lung tumors, and Alam et al. identify KMT2D as a lung tumor suppressor. KMT2D deficiency induces aberrant metabolic reprogramming via super-enhancer impairment, conferring sensitivity to glycolytic inhibitors in lung cancer with KMT2D-inactivating mutations.
Introduction
Lung cancer accounts for more cancer deaths than any other types of cancer among both men and women. The overall 5-year survival rate for lung cancer is low (~18.1%). Lung cancer is often characterized by gain-of-function mutations and amplification of oncogenic kinase genes (e.g., KRAS and EGFR) as well as loss-of-function alterations in tumor suppressor genes (e.g., TP53 and LKB1) (Cancer Genome Atlas Research, 2014; Herbst et al., 2008; Imielinski et al., 2012). Much research for lung cancer has focused on kinase signaling pathways, leading to the development of the kinase-targeted therapies, such as the EGFR mutant inhibitors and the ALK inhibitors. However, a vast majority of lung cancer patients who are treated with the kinase-targeted therapies later experience tumor relapse and drug resistance (Herbst et al., 2008; Sharma et al., 2007). Recently, the use of immune checkpoint inhibitors (e.g., anti-PD1 and anti-PD-L1) has provided significant survival benefit for lung cancer patients whose lung tumor expresses a high level of PD-L1. However, the prognosis for many lung cancer patients remains poor (Herbst et al., 2018; Malhotra et al., 2017). Moreover, a majority of lung cancer patients do not have a well-defined drug target (Chan and Hughes, 2015; Herbst et al., 2018). For these reasons, there is a great need for further mechanistic understanding of lung cancer to be used for more effective therapeutic approaches for lung cancer treatment.
Epigenetic alterations, which represent heritable aberrations in gene expression or cellular phenotype without alterations of DNA sequences, have emerged as a major type of cancer-driving events (Dawson and Kouzarides, 2012). Covalent modifications of DNA and histones play a key role in epigenetic regulation of gene expression and include DNA methylation, histone acetylation, and histone methylation. Interestingly, histone methylation, which can occur at lysine and arginine residues in histones, is linked to either gene activation or silencing, depending on the methylation residues within the histones (Barski et al., 2007). Notably, histone methylation modifiers and other epigenetic modifiers are often mutated in lung tumors, including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), which comprise 40% to 50% and 25% to 30% of lung cancer, respectively (Campbell et al., 2016; Cancer Genome Atlas Research, 2012, 2014; Imielinski et al., 2012; Kandoth et al., 2013). In fact, a substantial percentage of such mutations results in loss of function, suggesting that epigenetic modifiers can have tumor-suppressive functions. However, their tumor-suppressive roles in lung cancer remain to be established. In the present study, we sought to identify an epigenetic modifier with tumor-suppressive function in lung cancer.
Results
Lung-specific loss of Kmt2d strongly promotes KRASG12D-induced LUAD in mice
To identify an epigenetic modifier with tumor-suppressive function in lung cancer, we examined which epigenetic modifiers most frequently undergo genomic alterations (i.e., truncations, missense mutations, and insertions/deletions) that are related to loss-of-function mutations in lung tumors. Our analysis of Pan-lung cancer data (LUAD and LUSC) in The Cancer Genome Atlas (TCGA) database showed that the gene encoding the histone methyltransferase KMT2D (a COMPASS-like enzyme, also called MLL4, MLL2, and ALR) (Shilatifard, 2012) harbored such genomic alterations in about 14% of lung tumor samples (Figures 1A and S1A). Of the genomic alterations in KMT2D, 48.7% were truncating mutations, which was the highest percentage of all the epigenetic modifiers examined. A vast majority of the KMT2D truncations cause loss of function because the catalytic SET domain (5397– 5513 aa) is located at the C-terminus of KMT2D. Thus, our analysis indicates that KMT2D is among the most highly inactivated epigenetic modifiers (Figure 1A). Interestingly, KMT2D was mutated in substantial portions of both LUAD (e.g., ~7.8%) and LUSC (e.g., ~22.4%) samples (Figures S1B and S1C). Of note, it has been reported that a majority of KMT2D missense mutations in lymphoma significantly reduce KMT2D enzymatic activity, indicating a negative effect for KMT2D missense mutations in KMT2D activity (Zhang et al., 2015a). Because these findings indicate that KMT2D is a candidate lung tumor suppressor with frequent genomic alterations, we chose to characterize the role of KMT2D in lung cancer in subsequent analyses.
Figure 1. The loss of Kmt2d, whose human homologue is among the most highly mutated epigenetic modifiers in lung cancer, strongly accelerates KRAS-driven LUAD in mice.
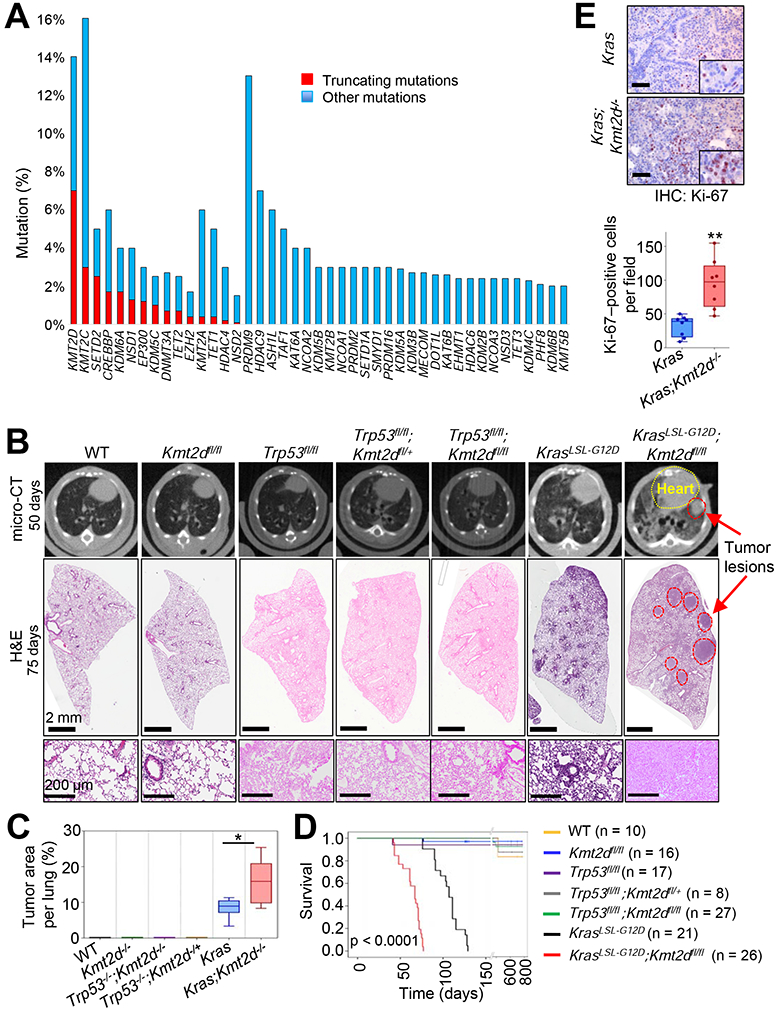
(A) Comparison of gene alterations of epigenetic modifiers (histone acetyltransferases and deacetylases, histone methyltransferases and demethylases, and DNA modifiers) in the TCGA Pan-lung cancer dataset. Other mutations represent missense and inframe mutations. (B) Representative images of micro-CT scans (top panels) and H&E-stained tissues (middle and bottom panels) of wild-type (WT), Kmt2dfl/fl, Trp53fl/fl, Trp53fl/fl;Kmt2dfl/+, Trp53fl/fl;Kmt2dfl/fl, KrasLSL-G12D, and KrasLSL-G12D;Kmt2dfl/fl mice. The lungs of the mice were infected with Adeno5 (Ad5)-CMV-Cre viruses. (C) Comparison of tumor area (%) per mouse in the indicated groups of mice (n = 6). (D) Kaplan-Meier survival analysis of the indicated groups of mice. The statistical analysis was performed using the two-sided log-rank test. (E) immunohistochemistry (IHC) analysis for the cell proliferation marker Ki-67 in Kras and Kras;Kmt2d−/− lung tumors. Ki-67-positive cells in ten random fields of three different tumors each from Kras and Kras;Kmt2d−/− groups were quantified. Black scale bars represent 50 μm. In the boxplots in (C) and (E), the bottom and the top rectangles indicate the first quartile (Q1) and third quartile (Q3), respectively. The horizontal lines in the middle signify the median (Q2), and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively. *, p < 0.05; **, p < 0.01 (two-tailed Student’s t-test). See also Figures S1 and S2.
We analyzed whether KMT2D alterations frequently co-occur with alterations in the two most frequently mutated genes TP53 and KRAS in human lung cancer. About 71% of KMT2D alterations co-occur with TP53 alterations in lung tumors, and about 22% of KMT2D alterations coincide with KRAS alterations in LUAD (Figure S1D). Therefore, we sought to determine whether lung-specific loss of Kmt2d cooperates with Trp53 inactivation or Kras activation for lung tumorigenesis in mice. We first generated several genetically engineered mouse models (GEMMs), including Trp53fl/fl;Kmt2dfl/fl and KrasLSL-G12D;Kmt2dfl/fl (Figures S2A-S2C). We then induced Cre-mediated deletion of loxP sites by infecting the lungs of 6- to 10-week old GEMM mice with Adeno-Cre viruses via the commonly used intra-tracheal intubation (DuPage et al., 2009) and monitored their lungs at 6- to 8-week intervals for up to 125 days post-infection by performing micro-computed tomography (micro-CT) scans. Lung-specific single loss or co-loss of Kmt2d and Trp53 by Adeno-Cre-mediated deletion neither induced any detectable tumor in mouse lungs for up to 16 months post-infection nor changed mouse survival times (Figures 1B-1D and S2D). This may be partly because Trp53 loss alone rarely induces lung tumors in mice although Trp53 loss promotes oncogene-driven lung tumorigenesis (Donehower et al., 1992; DuPage et al., 2009; Lang et al., 2004; Olive et al., 2004). In addition, because KMT2D and p53 in a multi-protein complex may activate a similar set of the DNA damage response pathway genes (Lee et al., 2009), their co-loss may not cooperate for tumorigenesis. For KRAS-induced lung tumorigenesis, we used a KrasLSL-GI2D model (hereinafter referred to as the Kras model) because it is a well-established GEMM for LUAD (DuPage et al., 2009). Kras activation was induced by lung-specific deletion of the LSL (loxP-STOP-loxP) cassette via Adeno-Cre virus infection, as previously described (Jackson et al., 2001). To examine whether Kmt2d loss promotes KRAS-induced lung tumorigenesis, we monitored tumor formation in Kras and Kras;Kmt2dfl/fl lungs, as described above.
Data from micro-CT scans and hematoxylin and eosin (H&E)-stained tumor sections showed that lung-specific loss of Kmt2d promoted KRAS-induced lung tumorigenesis (Figures 1B and 1C). Specifically, our H&E data demonstrated that a higher percentage of the pulmonary parenchyma was effaced by tumors in the Kras;Kmt2d−/− group of mice than in the Kras group of mice (Figures 1B, 1C, S2E, and S2F). Consistent with the enhancement of tumorigenicity by Kmt2d loss, immunohistochemistry (IHC) analysis showed that levels of the cell proliferation marker Ki-67 were increased by Kmt2d loss (Figure 1E). Like Kras lung tumors, Kras;Kmt2d−/− lung tumors were positive for the well-known LUAD marker TTF-1 (alias NKX2.1) and weak for expression of Keratin 5 (an LUSC marker), indicating their LUAD characteristics (Figure S2G). Our survival analysis demonstrated that Kmt2d loss significantly shortened the survival of mice bearing Kras pulmonary tumors (Figure 1D) and that low KMT2D mRNA levels were associated with poor prognosis in LUAD but not LUSC patients (Figures S2H and S2I). These results, along with the above results, suggest that Kmt2d loss, like Trp53 loss, promotes rather than initiates LUAD tumorigenesis.
Glycolysis program upregulated by Kmt2d loss has human LUAD relevance
To understand how lung-specific loss of Kmt2d promotes LUAD, we isolated tumor lesions from Kras and Kras;Kmt2d−/− lungs and compared their gene expression profiles using RNA-seq. Our RNA-seq data confirmed Cre-mediated deletion of loxP in Kmt2d, as RNA peaks corresponding to exons 16–19 of Kmt2d were greatly reduced (Figure S2J). Bioinformatic analyses of RNA-seq data using DAVID tools and Gene Set Enrichment Analysis (GSEA) both showed that genes for glycolysis and oxidative phosphorylation (OXPHOS) pathways were upregulated by Kmt2d loss (Figures 2A-2D and Table S1). In contrast, these two analyses did not share a pathway downregulated by Kmt2d loss, as GSEA analysis did not show any pathway that was significantly downregulated by Kmt2d loss (Figure 2E). In line with our RNA-seq results, analysis of the TCGA lung cancer database showed that glycolytic and OXPHOS genes were enriched in human KMT2D-low/mutant LUAD samples as compared with human KMT2D-high LUAD samples (Figures 2F-2H and Table S2). Robust upregulation of glycolytic genes Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 by Kmt2d loss was confirmed by quantitative RT-PCR (Figure 2I). In contrast, most genes for OXPHOS were weakly upregulated by Kmt2d loss (Figure 2I). These results and other results described below prompted us to delve into the regulation of glycolytic genes by KMT2D.
Figure 2. Kmt2d loss upregulates tumor-promoting programs, such as glycolysis.
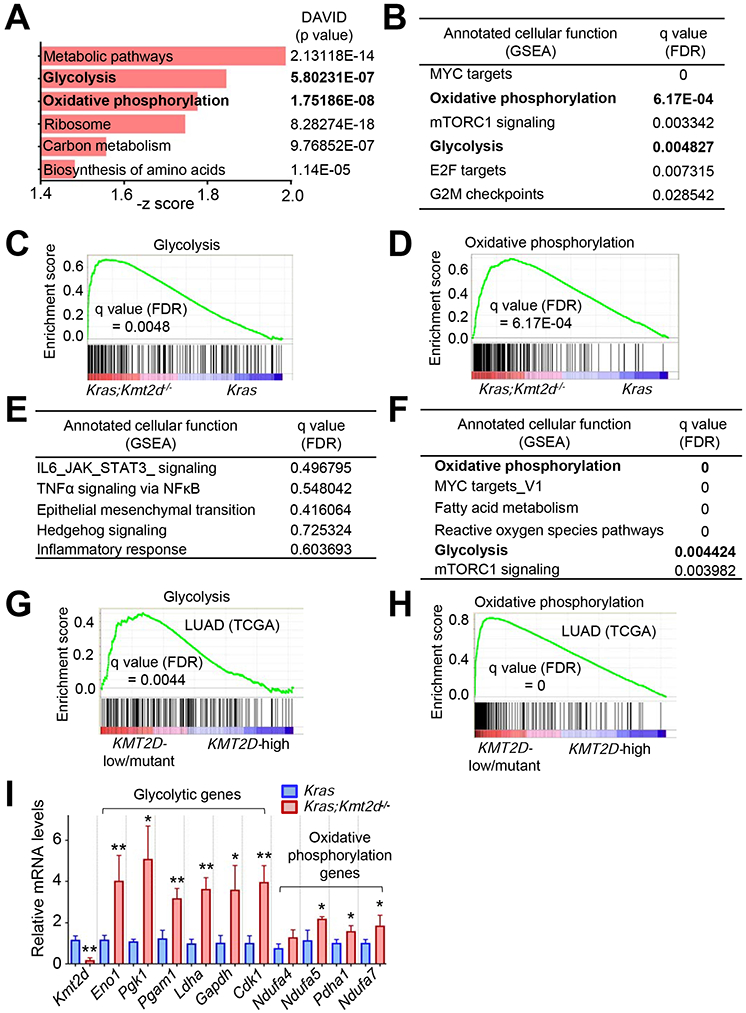
(A) Ontology analysis of genes (n = 1113, p < 0.05) upregulated by Kmt2d loss in KRAS-driven mouse LUAD. Gene expression profiles were compared between Kras tumors and Kras;Kmt2d−/− tumors. The functional annotation tool DAVID was used for the analysis. Metabolic pathways include genes for glycolysis, oxidative phosphorylation (OXPHOS), and TCA cycle. (B) Pathways upregulated by Kmt2d loss in KRAS-driven mouse LUAD. FDR, false discovery rate. (C and D) Enrichment plot of glycolytic (C) and OXPHOS (D) genes in Kras;Kmt2d−/− tumors as compared with Kras tumors. Each of the black bars represents a gene in the pathway. (E) Pathways downregulated by Kmt2d loss in KRAS-driven mouse LUAD. (F) Pathways upregulated in human LUAD samples bearing low KMT2D mRNA expression (n = 20) or KMT2D-inactivating mutations (n = 4) as compared with human LUAD samples (n = 24) bearing high KMT2D mRNA expression. (G and H) Enrichment plot of glycolytic (G) and OXPHOS (H) genes in human LUAD samples bearing low KMT2D mRNA expression or KMT2D inactivation as compared with human LUAD samples bearing high KMT2D mRNA expression For (B–H), the GSEA was performed, and the Benjamini-Hochberg procedure was used for statistical analyses. (I) Analysis of mRNA levels of glycolytic genes and OXPHOS genes in Kras and Kras;Kmt2d−/−lung tumors using quantitative RT-PCR (n = 3). Data are presented as the mean ± SEM (error bars). *, p < 0.05; **, p < 0.01 (two-tailed Student’s t-test). See also Tables S1 and S2.
It has been shown that tumorigenesis is promoted by upregulation of glycolysis by ENO1 (Capello et al., 2011), PGK1 (Li et al., 2016), PGAM1 (Hitosugi et al., 2012), LDHA (Sheng et al., 2012), and GAPDH (Colell et al., 2009). CDK1 is a cell cycle-associated gene (Malumbres and Barbacid, 2009) and increases glycolysis (Wang et al., 2017b). Glycolytic enzymes are considered to be potential therapeutic targets for cancer treatment (Capello et al., 2011; Ganapathy-Kanniappan et al., 2013). In line with the aforementioned mRNA expression data, immunofluorescence (IF) and IHC analysis showed that Kmt2d loss increased ENO1, PGK1, and PGAM1 levels while decreasing KMT2D levels (Figures 3A-3C). Analysis of a human LUAD dataset showed that ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels negatively correlated with KMT2D mRNA levels in human LUAD samples (Figure 3D). Moreover, high mRNA levels of ENO1, PGK1, PGAM1, LDHA, and GAPDH were associated with poor survival in LUAD patients (Figure 3E). These results indicate human relevance of our findings that Kmt2d loss upregulates expression of glycolytic genes in mouse LUAD.
Figure 3. High expression levels of several glycolytic genes negatively correlate with KMT2D mRNA levels in LUADs and are linked to poor prognosis in LUAD patients.
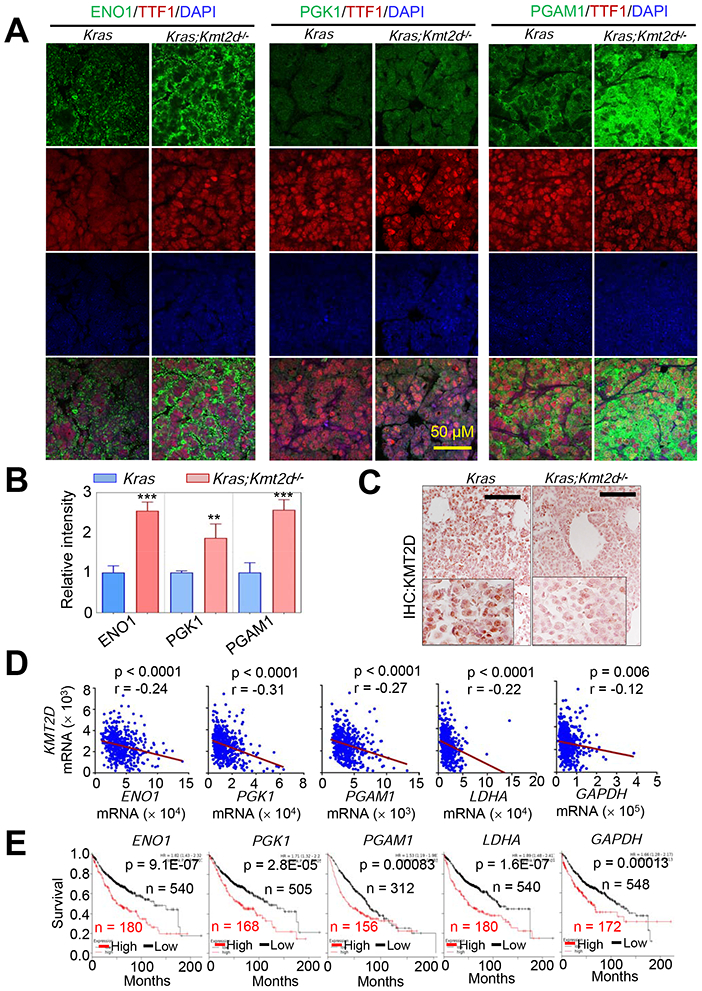
(A and B) Immunofluorescence (IF) analysis for ENO1, PGK1, and PGAM1 in Kras and Kras;Kmt2d−/− lung tumor tissues. Representative IF images are shown (A), and the IF images were quantified (B). TTF1 is an LUAD marker. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. **, p < 0.01; ***, p < 0.001 (two-tailed Student’s t-test). (C) IHC analysis for KMT2D in Kras and Kras;Kmt2d−/− lung tumor tissues. Black scale bars, 50 μm. (D) Inverse correlations of KMT2D mRNA levels with ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels in human LUADs in the TCGA dataset (n = 517). The statistical analysis was performed using two-tailed Student’s t-test. r, Pearson correlation coefficient. (E) Kaplan-Meier survival analysis of human LUAD patients using ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels. Higher quartile (except a best cutoff for PGAM1 data) was used as a cutoff to divide the samples into high (the highest 25%) and low (the remaining 75%) groups in the KM Plotter database (http://kmplot.com/analysis). The statistical analysis was performed using the two-sided log-rank test. KMT2D probe set, 227527_at; ENO1 probe set, 201231_at; PGK1 probe set, 227068_at; PGAM1 probe set, 200886_at; LDHA probe set, 200650_s_at; GAPDH probe set, 212581_at.
Kmt2d loss globally reduces super-enhancer and enhancer signals but has no obvious effect on H3K4me3 and H3K27me3 signals in mouse lung tumors
We and others have shown that KMT2D can catalyze monomethylation, dimethylation, and trimethylation at H3K4 (Dhar et al., 2012; Herz et al., 2012; Lee et al., 2013; Lee et al., 2007b; Zhang et al., 2015b). Monomethyl H3K4 (H3K4me1), together with acetyl H3K27 (H3K27ac), marks enhancers, which spatiotemporally activate gene expression in various locations (Smith and Shilatifard, 2014). KMT2D is required for enhancer formation regulated by the H3K27 acetyltransferases CBP and p300 (Lai et al., 2017; Wang et al., 2017a). We and others have also shown that KMT2D interacts with the H3K27me3 demethylase UTX (Cho et al., 2007; Issaeva et al., 2007; Lee et al., 2007b). For these reasons, we performed chromatin immunoprecipitation (ChIP)-seq for H3K4me1, H3K27ac, H3K4me3, and H3K27me3 to analyze the effect of Kmt2d loss on epigenomic landscapes and chromatin states in Kras tumors and Kras;Kmt2d−/− tumors. In the ChIP-seq experiment, we also included H3K79me2 and H3K9me3, because H3K79me2 is associated with transcriptional elongation and H3K9me3 is a marker for heterochromatin repression (Hawkins et al., 2010; Nguyen and Zhang, 2011). We modeled combinatorial patterns of the six histone modifications using ChromHMM algorithm (Ernst and Kellis, 2012) and categorized the data into 10 different states, as shown in Figure 4A. We noted three prominent transitions in chromatin states from Kras tumors to Kras;Kmt2d−/− tumors: E2 (Active enhancer) to E3 (Weak enhancer), E3 to E9 (Low state = very low signal), and E5 (Active enhancer containing low H3K4me1) to E7 (H3K27ac-lacking transcribed enhancer) (Figures 4B and S3A). Further analysis showed that Kmt2d loss did not have any obvious effect on ChIP-seq signals of H3K4me3 and H3K27me3 marks (Figures S3B-S3G). In agreement with these, IF analyses showed that global levels of H3K4me1 and H3K27ac but not H3K4me3 were downregulated by Kmt2d loss (Figure S3H). These results indicate that Kmt2d loss has a strong negative impact on enhancer states.
Figure 4. Kmt2d loss reduces epigenomic signals for super-enhancers and to a lesser extent enhancers at the genome-wide level.
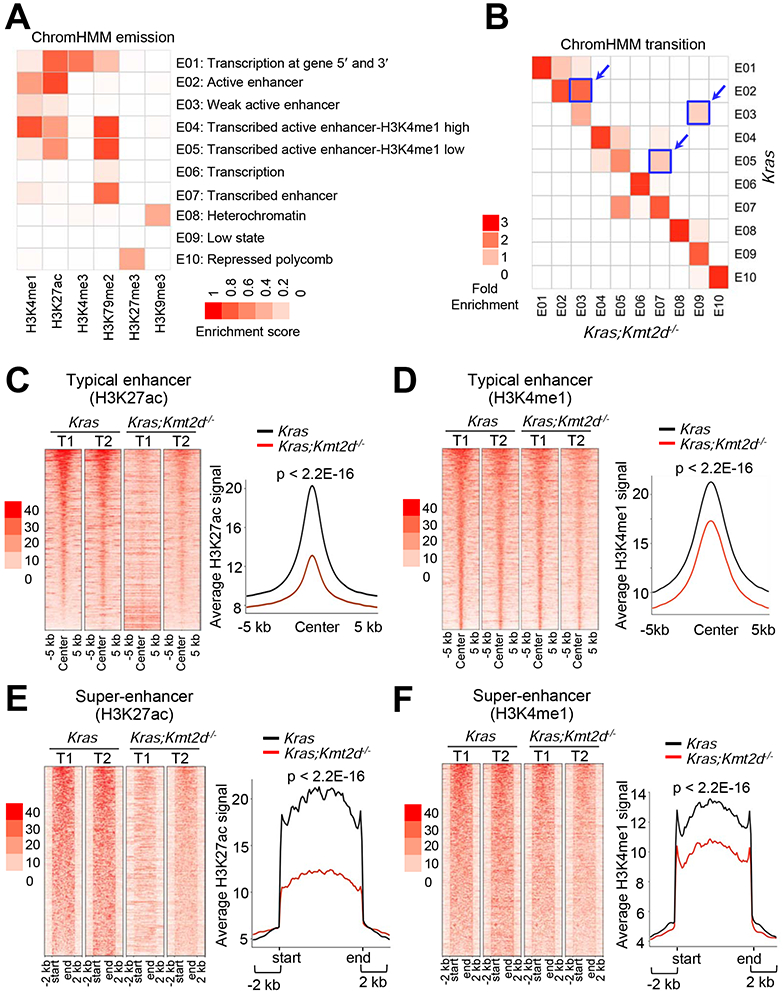
(A) Emission probabilities of the 10-state ChromHMM model calculated from six histone modification profiles in Kras and Kras;Kmt2d−/− lung tumors. Each row represents one chromatin state. The 10 states predicted using ChromHMM represent various enhancer states (E2, E3, E4, E5, and E7), promoter state (E1), transcription states (E1, E6, and E7), polycomb-repressed state (E10), and heterochromatin state (E8). Each column corresponds to a histone modification. The intensity of the color in the scale from 0 (white) to 1 (red) in each cell reflects the frequency of occurrence of each histone mark in the indicated chromatin state. (B) Heat map showing the fold enrichment of transitions of chromatin states from Kras lung tumors to Kras;Kmt2d−/− lung tumors. The color intensities represent the relative fold enrichment. (C and D) Heat maps (left panels) and average intensity curves (right panels) of ChIP-seq reads (RPKM) for H3K27ac (C) and H3K4me1 (D) at typical enhancer regions. Enhancers are shown in a 10-kb window (centered on the middle of the enhancer) in Kras and Kras;Kmt2d−/− lung tumors. (E and F) Heat maps (left panel) and average intensity curves (right panels) of ChIP-seq reads for H3K27ac (E) and H3K4me1 (F) at the super-enhancer regions plus their flanking 2-kb regions in Kras and Kras;Kmt2d−/− lung tumors. Wilcoxon rank sum test was used for statistical analysis of (C–F). T1, tumor 1; T2, tumor 2. See also Figures S3 and S4.
It has been documented that enhancers can be categorized as super-enhancers and typical enhancers (Whyte et al., 2013). Super-enhancers are characterized by clusters of enhancers that are much larger than typical enhancers with a median size of 0.7 kb to 1.3 kb and highly activate gene expression (Loven et al., 2013; Whyte et al., 2013). Interestingly, our enhancer analysis showed that Kmt2d loss strongly decreased average H3K4me1 and H3K27ac levels in super-enhancers and to a lesser extent in typical enhancers (Figures 4C-4F and S4A-S4D). Expression levels of lung-enriched, super-enhancer-associated genes were downregulated by Kmt2d loss (Figures S4E and S4F).
Pharmacological inhibition of glycolysis impedes the tumorigenic growth of LUAD cell lines with KMT2D-inactivating mutations
As described above, Kmt2d loss upregulated glycolysis program but decreased H3K27ac levels. This led us to test whether the inhibition of glycolytic pathways or H3K27 deacetylation would impede the growth of human lung cancer cell lines bearing loss-of-function mutations in KMT2D. To inhibit glycolysis, we used the following inhibitors: 2-deoxy-D-glucose (2-DG), a hexokinase inhibitor and glucose analog in a phase I/II clinical trial (Huang et al., 2015; Pelicano et al., 2006); POMHEX, an enolase inhibitor (Lin et al., 2018); koningic acid (KA), a selective inhibitor of GAPDH (Liberti et al., 2017); Lonidamine, a hexokinase and mitochondrial respiration inhibitor in a phase II clinical trial (Brawer, 2005); and Dinaciclib, an inhibitor of CDK1 and CDK2/5/9 in a phase III clinical trial (Parry et al., 2010; Santo et al., 2010). We also tested the OXPHOS inhibitor IACS-010759 (Molina et al., 2018) because Kmt2d loss somewhat upregulated the OXPHOS program. To increase histone acetylation via inhibition of histone deacetylase (HDAC), we used AR-42 (in phase I clinical trial) and Vorinostat (also known as SAHA), which has been approved for cancer treatment by the U.S. Food and Drug Administration (Bjornsson et al., 2014; Jacob et al., 2012). We compared the inhibitory effects of these agents on cell confluency of human LUAD cell lines with wild-type (WT) KMT2D (A549, H1792, H1437, H23, and H358) and human LUAD cell lines bearing KMT2D-truncating mutations (i.e., H1568, with nonsense at E758; DV-90, with the truncating mutation P2118fs; and CORL-105, with the truncating mutation R2188fs) (Table S3). Of note, glucose uptake, lactate excretion, and glycolysis metabolites (e.g., glucose-6-phosphate, fructose-6-phosphate, glucose-1,6-bisphosphate, and fructose-1,6-bisphosphate) tended to be higher in the KMT2D-mutant cell lines than in the KMT2D-WT cell lines (Figures S5A-S5C). 2-DG (and to a lesser extent POMHEX, KA, and Vorinostat) inhibited the confluency of the KMT2D-mutant cell lines to a greater extent than that of the KMT2D-WT cell lines (Figures 5A and 5B). In contrast, AR-42, Lonidamine, Dinaciclib, and IACS-010759 did not exert an obvious selective inhibition against the confluency of the KMT2D-mutant cell lines (Figures S5D-S5G).
Figure 5. Inhibition of glycolysis suppresses tumorigenic growth of KMT2D-mutant LUAD cell lines.
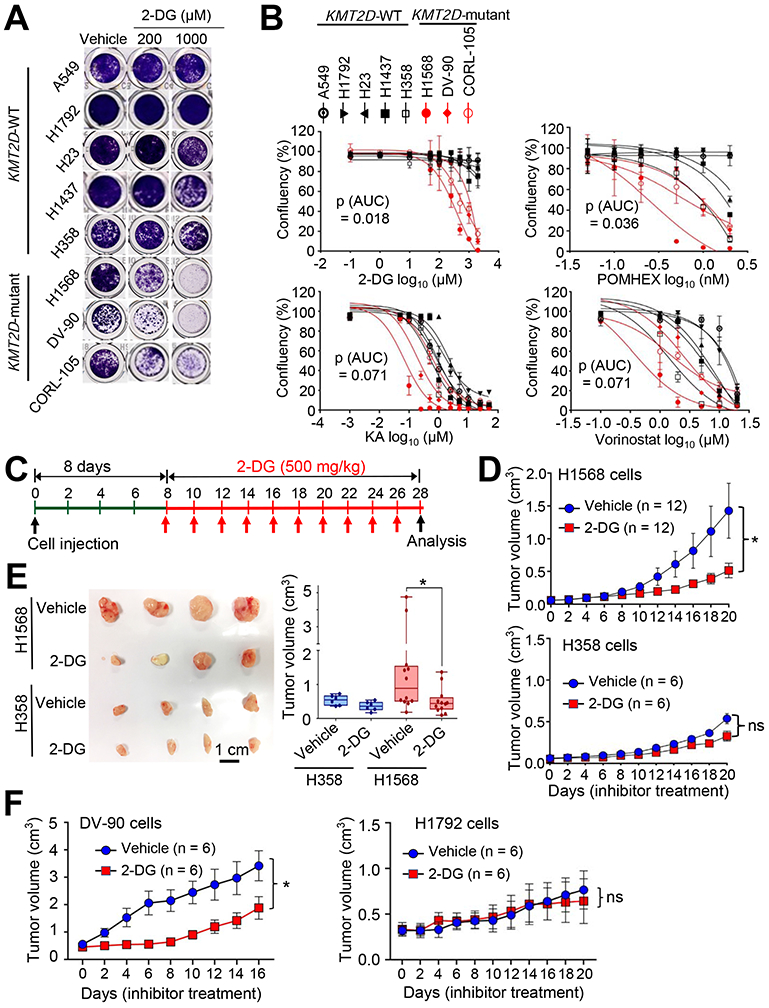
(A and B) The effect of 2-DG on the confluency of human LUAD cell lines bearing WT KMT2D (A549, H1792, H23, H1437, and H358) and LUAD cell lines bearing KMT2D-truncating mutations (H1568, DV-90, and CORL-105) (A) and dose-response curves of several inhibitors on these LUAD cell lines (B). Cells were treated with various concentrations of the hexokinase inhibitor 2-deoxy-D-glucose (2-DG), the enolase inhibitor POMHEX, the GAPDH inhibitor koningic acid (KA), and the HDAC inhibitor SAHA/vorinostat. Wilcoxon rank sum test (n = 3) was used for statistical analysis. AUC, Area under the curve. (C–E) The effect of 2-DG on tumorigenic growth of H1568 cells bearing a KMT2D-truncating mutation and of H358 cells bearing WT KMT2D in a mouse subcutaneous xenograft model. The schedule of treatment of mice with 2-DG is shown (C). The sizes of xenograft tumors after treatment with 2-DG or vehicle control were measured (D). Tumors were dissected from the mice (E). In the boxplots, the bottom and the top rectangles indicate the first quartile (Q1) and third quartile (Q3), respectively. The horizontal lines in the middle signify the median (Q2), and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively. (F) The effect of 2-DG on tumorigenic growth of DV-90 cells bearing a KMT2D-truncating mutation and of H1792 cells bearing WT KMT2D in a mouse subcutaneous xenograft model. Mice were treated with 2-DG (500 mg per kg body weight every other day) or vehicle. ns indicates non-significant. *, p < 0.05 (two-tailed Student’s t-test). See also Figures S5 and S6 and Table S3.
Because 2-DG most selectively inhibited cell confluency of the KMT2D-mutant LUAD cell lines over several KMT2D-WT cell lines, we used 2-DG for subsequent experiments. We tested the effect of 2-DG on cell death. Consistent with the notion that 2-DG can induce cell death in a cell-type- and dose-dependent manner (Zhang et al., 2014), 2-DG induced more cell death in the three KMT2D-mutant cell lines than in two KMT2D-WT cell lines tested (Figure S5H). In addition, 2-DG tended to inhibit the extracellular acidification rates (ECARs; an indicator for glycolysis) of the KMT2D-mutant cell lines to a greater extent than those of the KMT2D-WT cell lines (Figures S5I and S5J). We also tested the effect of 2-DG on the growth of the lung cancer cell line LKR10, which is derived from a mouse KrasG12D tumor. A three-dimensional (3D) cell culture system was used, because 1) it better mimics an in vivo situation than a 2D culture system does and 2) KMT2D knockdown increased the sizes of the spheroids formed from LKR10 cells in the 3D culture (Figure S6A) — In contrast, KMT2D knockdown decreased the proliferation of several LUAD cell lines in the 2D culture system (data not shown). In line with its inhibitory effect on confluency of human LUAD cells, 2-DG inhibited spheroid growth of KMT2D-depleted LKR-10 cells to a greater extent than that of shLuciferase (shLuc)-treated LKR-10 cells (a control) in the 3D culture system (Figures S6A and S6B).
To examine whether 2-DG selectively inhibits tumorigenic growth of KMT2D-mutant LUAD cells over KMT2D-WT LUAD cells, we used H1568 cells harboring a KMT2D’s truncation mutation and H358 cells harboring WT KMT2D. H1568 cells and H358 cells have similar proliferation rates in 2D cultures (Figure S6C). Notably, 2-DG preferentially inhibited the tumorigenicity of KMT2D-mutant H1568 cells as compared with KMT2D-WT H358 cells in a mouse subcutaneous xenograft model (Figures 5C-5E). However, KA, which less selectively inhibited KMT2D-mutant cell lines than did 2-DG, weakly and insignificantly impeded the tumorigenicity of H1568 cells as compared with H358 cells (Figures S6D and S6E). Using another KMT2D-truncated cell line DV-90 and another KMT2D-WT cell line H1792, we compared the in vivo inhibitory effect of 2-DG on their tumorigenic growth. H1792 cells grew faster than DV-90 cells did in a 2D culture (Figure S6C), but DV-90 cells formed tumors faster than H1792 cells did in a mouse xenograft model (Figure 5F). In agreement with the above results, 2-DG selectively inhibited tumorigenic growth of KMT2D-mutant DV-90 cells over KMT2D-WT H1792 cells (Figure 5F). Finally, to test the effect of 2-DG on the tumorigenicity of KMT2D-WT and KMT2D-mutant LUAD cells in an isogenic cell state, we generated KMT2D-mutant H1568 cells harboring doxycycline (Dox)-inducible WT KMT2D (Figure S6F) and then compared the inhibitory effect of 2-DG on tumorigenic growth of these cells between Dox treatment and Dox untreatment. KMT2D induction decreased ECAR (Figure S6F). In line with the above results, 2-DG tended to preferentially impede tumorigenic growth of KMT2D-inducible H1568 cells in Dox-untreated mice as compared with Dox-treated mice (Figure S6G). These results suggest that increased glycolysis in KMT2D-mutant LUAD tumors can be targeted by the glycolysis inhibitor 2-DG.
KMT2D is required for the activity of the Per2 super-enhancer, and PER2 represses glycolytic genes
Because KMT2D is a transcriptional co-activator, Kmt2d loss may not directly upregulate tumor-promoting glycolysis program. Thus, we hypothesized that the glycolysis program upregulated by Kmt2d loss may be repressed by a tumor-suppressive, transcription-repressive regulator encoded by a KMT2D-activated gene (i.e., a gene downregulated by Kmt2d loss). To identify such a regulator, we first searched the genes that were both downregulated by Kmt2d loss (n = 522) and associated with significant decreases in H3K27ac levels (n = 3751), because Kmt2d loss strongly downregulated the enhancer mark H3K27ac to decrease gene expression (Figures 6A and 6B). To further reduce the number of candidate regulators, we incorporated aspects of human lung cancer into our analysis. Specifically, we examined which genes among those downregulated by Kmt2d loss correlate with KMT2D mRNA levels in more than 0.3 of the correlation coefficient value (r) in human LUAD samples in the TCGA dataset. This analysis resulted in 14 candidate genes (Figures 6C and S7A). Of note, Kmt2d loss in KRAS-driven lung tumors did not affect expression of Dnmt3a (a transcriptional co-repressor gene) and DNMT3A-regulated Ras activator genes (Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1) (Figure S7B), although expression of Dnmt3a was downregulated by Kmt2d loss in other types of tumors (e.g., medulloblastoma) (Dhar et al., 2018). Of these 14 genes, expression levels of five (SHANK2, ACACB, PER2, NFASC, and CLCN6) were lower in LUAD tumors than in adjacent normal tissues, and their low levels correlated with poor prognosis in lung cancer patients (Figures 6D-6F and S7C-S7G).
Figure 6. Integrative analysis of expression, enhancers, and clinical relevance for KMT2D-regulated genes.
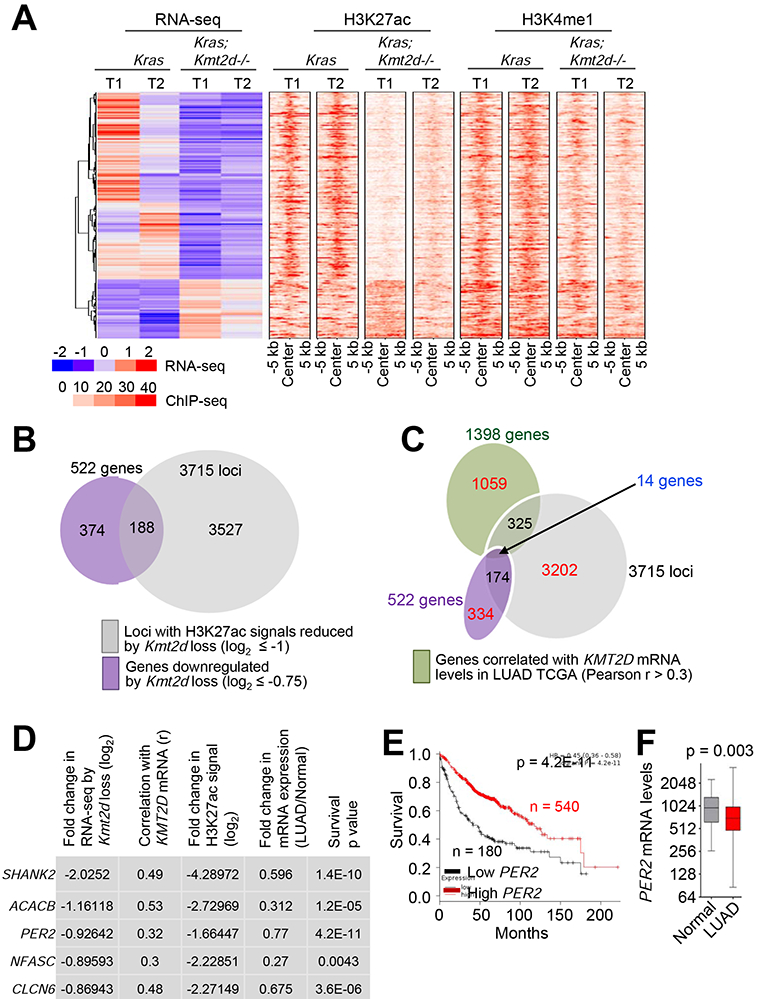
(A) Heat maps for genes differentially expressed between Kras and Kras;Kmt2d−/− lung tumors and for the signals of their closest H3K27ac and H3K4me1 peaks. (B) A Venn diagram showing the overlap between genes downregulated by Kmt2d loss (n = 522) and genes with H3K27ac ChIP-seq signals reduced by Kmt2d loss (n = 3715). (C) A Venn diagram showing the overlap between genes downregulated by Kmt2d loss (n = 522), genes with H3K27ac ChIP-seq signals reduced by Kmt2d loss (n = 3715), and genes correlated with KMT2D expression (n = 1398 with r ≥ 0.3) in LUAD samples (n = 357) in the TCGA database. (D) The top five hits on the basis of the five different parameters indicated. r, Pearson correlation coefficient. (E) The Kaplan-Meier survival analysis showing the association of low PER2 mRNA levels with poor survival in lung cancer patients. The lower quartile was used as a cutoff to divide the samples into low (the lowest 25%) and high (the remaining 75%) KMT2D mRNA groups. PER2 probe set, 205251_at. In (D) and (E), the p values were calculated using using the two-sided log-rank test. (F) Boxplots showing downregulation of PER2 mRNA levels in LUAD samples (n = 517) as compared with their adjacent normal samples (n = 54) in the TCGA dataset. In the boxplots, the bottom and the top rectangles indicate the first quartile (Q1) and third quartile (Q3), respectively. The horizontal lines in the middle signify the median (Q2), and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively. The p value was determined using two-tailed Student’s t-test. See also Figure S7.
Of these five genes, the circadian rhythm repressor gene Per2 was particularly interesting for the following reasons: 1) Per2 loss in mouse lung promoted lung tumorigenesis (Papagiannakopoulos et al., 2016); 2) Per2 loss increased glucose metabolism, implicating PER2 in regulation of glycolysis (Papagiannakopoulos et al., 2016); and 3) disruption of circadian rhythm increased the tumorigenicity (Filipski et al., 2003; Fu et al., 2002). As aforementioned and also shown in Figures 7A and 7B, Kmt2d loss decreased Per2 expression levels, and PER2 mRNA levels significantly correlated with KMT2D mRNA levels. Kmt2d loss also reduced PER2 protein levels (Figure 7C). Interestingly, Per2 was occupied by a large cluster of H3K4me1 and H3K27ac peaks that are indicative of a super-enhancer (Figures 7D and 7E). In enhancer regions, RNAs called enhancer RNAs (eRNAs) are bidirectionally transcribed by RNA Polymerase II, and enhancer activities can be assessed by eRNA levels (Kim and Shiekhattar, 2015). Therefore, we measured the effect of Kmt2d loss on eRNA levels in the Per2–associated super-enhancer. Kmt2d loss decreased eRNA levels in the super-enhancer region, suggesting that Kmt2d loss reduces the activity of the Per2 super-enhancer (Figure 7F).
Figure 7. KMT2D positively regulates Per2 expression, and PER2 occupies glycolytic genes.
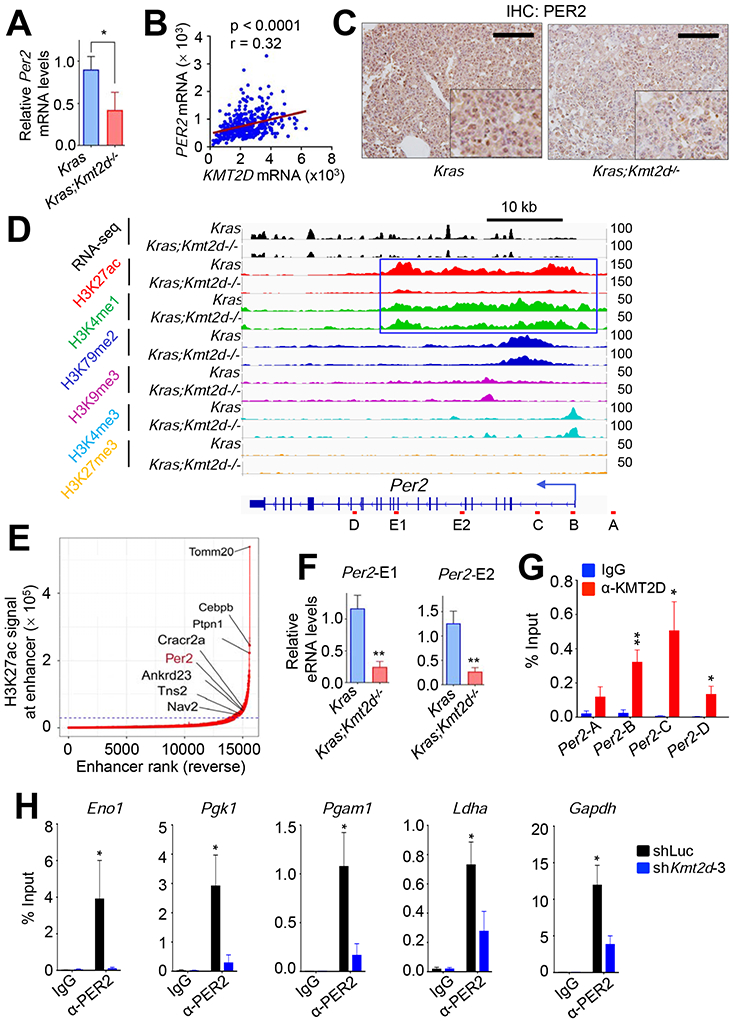
(A) Analysis of relative Per2 mRNA levels in Kras and Kras;Kmt2d−/− lung tumors using quantitative RT-PCR. (B) A scatter plot showing a positive correlation between KMT2D and PER2 mRNA levels in the TCGA LUAD dataset (n = 517). The statistical analysis was performed using Two-tailed Student’s t-test. r, Pearson correlation coefficient. (C) Analysis of PER2 protein levels in Kras and Kras;Kmt2d−/− lung tumors using IHC. Black scale bars, 50 μm. (D) Genome browser view of normalized ChIP-seq signals of six chromatin marks (H3K27ac, H3K4me1, H3K79me2, H3K9me3, H3K4me3, and H3K27me3) at the Per2 locus in Kras and Kras;Kmt2d−/− lung tumors. All the tracks were average of two biological replicates. The Per2-associated super-enhancer is indicated by the blue-outlined box. (E) A plot indicating super-enhancers identified on the basis of H3K27ac signals. The numbers in X-axis are in reverse order. (F) Analysis of eRNA levels for two different regions (E1 and E2) of the Per2 super-enhancer in Kras and Kras;Kmt2d−/− lung tumors using quantitative RT-PCR. (G) Quantitative ChIP analysis of KMT2D in Per2 in LKR10 cells. (H) Quantitative ChIP analysis of PER2 in glycolytic genes in LKR10 cells. ChIP amplicons are indicated in Figure S7H. In (A), (F), (G), and (H), data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. *, p < 0.05; **, p < 0.01 (two-tailed Student’s t-test).
To further assess the role of the KMT2D-PER2 pathway in repressing glycolytic genes that are upregulated by Kmt2d loss, we used the mouse Kras LUAD cell line LKR10. ChIP results showed that KMT2D occupied the Per2 gene in LKR10 cells (Figure 7G). ChIP results also showed that PER2 occupied several glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) and that KMT2D knockdown reduced PER2 occupancy in these genes (Figures 7H and S7H). Similar to the effect of Kmt2d loss on glycolytic genes in Kras tumors, our RNA-seq and RT-PCR data showed that KMT2D knockdown in LKR10 cells robustly upregulated expression levels of the glycolytic genes while downregulating Per2 expression (Figures 8A and S8A). In line with this, KMT2D knockdown increased glucose uptake, lactate excretion, several glycolysis metabolites (e.g., glucose-6-phosphate, fructose-6-phosphate, glucose-1,6-bisphosphate and fructose-1,6-bisphosphate), and ECAR in LKR10 cells (Figures 8B, S8B, and S8C). Of note, KMT2D knockdown increased expression of OXPHOS genes and oxygen consumption rate to a lesser extent than expression of glycolytic genes and ECARs (Figures S8C and S8D). To verify the effect of KMT2D knockdown on glycolytic genes, we ectopically expressed in KMT2D-depleted cells a functional but smaller KMT2D (herein called mini-KMT2D), which rescued the differentiation defect of KMT2D-depleted cells in our previous study (Dhar et al., 2012) and upregulated gene expression via enhancer activation in other study (Wang et al., 2017a). This rescue experiment demonstrated that mini-KMT2D expression restored Per2 expression as well as eRNA and H3K27ac levels for the Per2 super-enhancer while repressing expression of Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 (Figures 8C and 8D). Consistent with this, Dox-induced expression of KMT2D in human H1568 cells increased PER2 expression while decreasing expression of the same glycolytic genes, glucose uptake, lactate excretion, and the spheroid growth (Figures 8E, 8F and S8E). These results suggest that KMT2D directly upregulates Per2 expression by activating the Per2 super-enhancer and that KMT2D-upregulated PER2 represses glycolytic genes.
Figure 8. KMT2D-activated Per2 expression represses glycolytic genes.
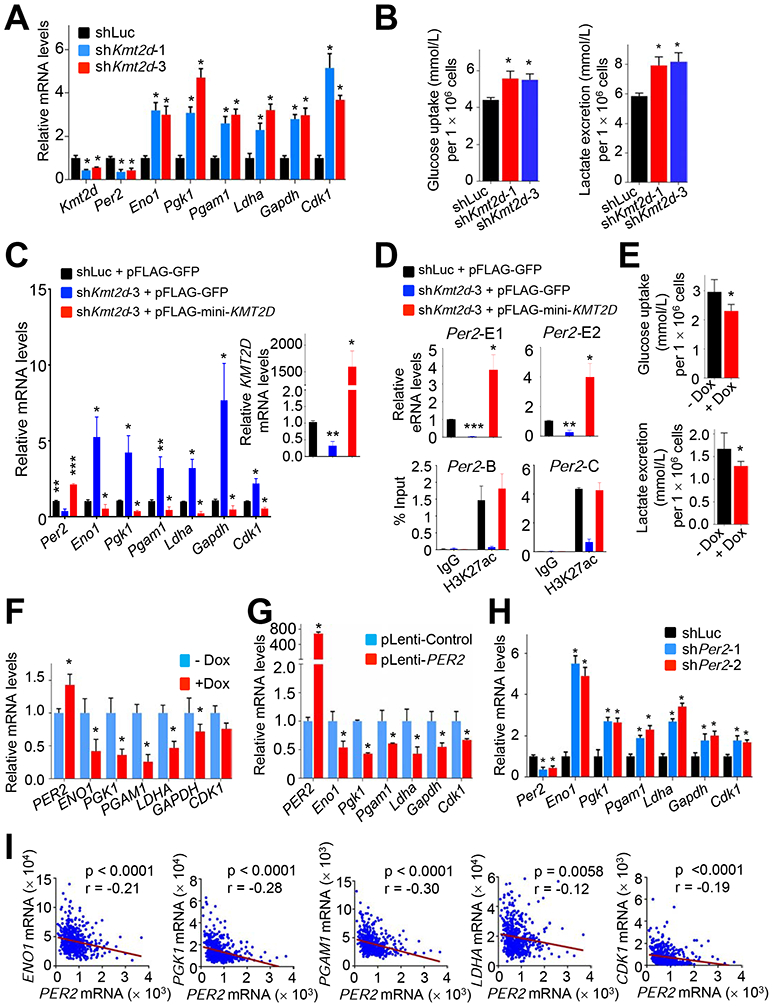
(A) Analysis of the effect of KMT2D knockdown on Per2, Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in mouse LKR-10 LUAD cells bearing KRASG12D using quantitative RT-PCR. (B) The effect of KMT2D knockdown on glucose uptake (Left panel) and lactate excretion (Right panel) in LKR10 cells. (C and D) Rescue experiments by ectopic expression of a functional but smaller KMT2D protein in LKR10 cells. KMT2D-depleted LKR10 cells were transfected with pFLAG-CMV2 expression plasmids encoding mini-KMT2D. Expression of glycolytic genes (C) as well as eRNA levels for two different regions (E1 and E2) of Per2 (D, Top panels) were analyzed using quantitative RT-PCR. H3K27ac levels were analyzed by quantitative ChIP (D, Bottom panels). (E and F) The effect of Dox-induced KMT2D on glucose uptake (E, Top panel) and lactate excretion (E, Bottom panel) in H1568 cells and on expression of glycolytic genes (F). H1568 cells bearing Dox-inducible KMT2D were treated with 10 μg/ml Dox. (G) Analysis of the effect of exogenous PER2 expression on Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in LKR-10 cells using quantitative RT-PCR. (H) Analysis of the effect of PER2 knockdown on Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in LKR-10 cells using quantitative RT-PCR. (I) Scatter plots showing inverse correlations of PER2 mRNA levels with ENO1, PGK1, PGAM1, LDHA or CDK1 mRNA levels in human LUADs in the TCGA dataset (n = 517). r, Pearson correlation coefficient. In (A–H), data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (two-tailed Student’s t-test). See also Figure S8.
To convincingly confirm the regulation of glycolytic genes by PER2, we examined the effect of PER2 overexpression and PER2 knockdown on expression of glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) in LKR10 cells. PER2 overexpression decreased expression of these genes (Figure 8G). PER2 knockdown increased their expression levels and ECAR while slightly increasing expression of several OXPHOS genes (Figures 8H, S8F, and S8G). In agreement with gene expression data, enzyme assays showed that KMT2D or PER2 knockdown increased ENO1 and GAPDH activities whereas KMT2D or PER2 overexpression decreased their activities (Figure S8H). PER2 expression levels negatively correlated with expression of several glycolytic genes (ENO1, PGK1, PGAM1, LDHA, and CDK1) in a TCGA LUAD dataset (Figure 8I). Finally, we examined the effect of ectopic expression of PER2 on the spheroid growth of KMT2D-depleted LKR10 cells. PER2 expression decreased the spheroid growth of these cells in the 3D culture system (Figure S8I). These results indicate that tumor-suppressive function of KMT2D is dependent, at least in part, on the downregulation of glycolytic genes by PER2.
Discussion
In the present study, our results indicate that KMT2D acts as an epigenetic LUAD suppressor by positively regulating super-enhancers (e.g., Per2 super-enhancer) and thereby increasing expression of the tumor suppressor gene Per2. In addition, our findings indicate that KMT2D-mediated upregulation of Per2 represses tumor-promoting glycolytic genes and that KMT2D defectiveness or deficiency decreases Per2 expression to upregulate glycolytic genes. Our in vitro and in vivo experiments showed that increased glycolysis in KMT2D-defective lung cancer cells could be targeted by pharmacological inhibition. Interestingly, glycolysis pathway is enriched not only in our Kras;Kmt2d−/− tumor model over Kras model but also in human LUAD tumors with low/mutant KMT2D over those with high WT KMT2D. In this aspect, Kras;Kmt2dfl/fl mouse model represents an epigenetic LUAD mouse model and may be useful for future studies for human LUAD.
It has been reported that KMT2D is required for the formation of acute myeloid leukemia by the MLL-AF9 oncogene (Santos et al., 2014) and for HOXA9/MEIS1-mediated leukemogenesis (Sun et al., 2018). In contrast, results reported here showed that lung-specific deletion of Kmt2d significantly promoted KRAS–driven lung tumorigenesis in mice and shortened the survival of mice bearing KRAS-driven tumors, suggesting that Kmt2d loss cooperates with other oncogenic aberrations (e.g., Kras activation) to increase LUAD tumorigenicity. The lung tumor-suppressive function of KMT2D is further supported by our following additional results: 1) Kmt2d loss upregulated expression of tumor-promoting glycolytic genes, such as Eno1, Pgk1, Pgam1, Ldha and Gapdh; 2) Kmt2d loss downregulated tumor-suppressive genes, such as Per2; and 3) KMT2D depletion increased sizes of spheroids formed by lung cancer cells in the 3D culture system. In line with the tumor-suppressive function of KMT2D, we have recently shown that brain-specific Kmt2d loss alone induces spontaneous medulloblastoma (cerebellar tumor) in brain (Dhar et al., 2018). Our and other recent studies indicate that KMT2D acts as a tumor suppressor in melanoma (Maitituoheti et al., 2018) and pancreatic cancer cells (Koutsioumpa et al., 2019). Furthermore, other studies have demonstrated that genetic ablation of Kmt2d in B cells enhanced B cell lymphoma genesis, also indicating KMT2D’s tumor-suppressive function (Ortega-Molina et al., 2015; Zhang et al., 2015a). Thus, the anti- or pro-tumorigenicity of KMT2D may be cell-type-dependent, although many studies suggest that KMT2D has a tumor-suppressive function in a majority of tissues.
We have recently shown that in medulloblastoma occurred in brain-specific Kmt2d knockout mice, Kmt2d loss induces Ras signaling pathways by highly increasing expression of several Ras activator genes (e.g., Rasgrp1, Rasgrf1, Rasgrf2, Rapgef5, and Rgl1) (Dhar et al., 2018). In the same study, we have also demonstrated that KMT2D activates expression of the DNA methyltransferase 3A (Dnmt3a) and that DNMT3A represses expression of these Ras activator genes. Similar to this, KMT2D knockdown decreased expression of DNMT3A expression in human lymphoma cells (Ortega-Molina et al., 2015). Contrast to these studies, the present study showed that Kmt2d loss in lung tumorigenesis induced neither expression of Ras activator genes nor decreased Dnmt3a expression. Instead, Kmt2d loss increased expression of glycolytic genes in lung tumorigenesis. Therefore, it is likely that the tissue variation of tumor-suppressive activity of KMT2D would result from KMT2D-mediated regulation of different gene sets in a tissue-specific manner. How does KMT2D-regulated gene expression occur in a tissue-specific manner? Interestingly, it has been shown that the KMT2D complex interacts with tissue-specific DNA-binding transcription factors. For example, the KMT2D complex co-localizes with MyoD during myocyte differentiation while interacting with PPARγ and C/EBP during adipocyte differentiation (Lee et al., 2013). Thus, a tissue-specific factor may direct the KMT2D complex to a unique set of genes to instruct KMT2D-mediated gene activation in a tissue-dependent manner.
The circadian rhythm repressor PER2 plays an important role in tumor suppression (Filipski et al., 2003; Fu et al., 2002). Results reported here indicate that KMT2D upregulates Per2 expression and indirectly represses tumor-promoting glycolysis via Per2 activation. Moreover, our results define several oncogenic glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) as target genes of PER2. Therefore, KMT2D-mediated Per2 activation represents a previously unknown tumor-suppressive mechanism that links an epigenetic tumor suppressor to a circadian rhythm regulator. PER2 moves into the nucleus during the evening and downregulates gene expression by antagonizing the circadian rhythm heterodimeric activator CLOCK:BMAL1. During the night, PER2 is gradually phosphorylated and targeted for ubiquitination that leads to proteasomal degradation (Bass and Takahashi, 2010; Koike et al., 2012). Distinct from this type of PER2 regulation, our results uncover a Per2-regulatory mechanism in which KMT2D upregulates Per2 expression by activating the Per2 super-enhancer, providing molecular insights into how Per2 is epigenetically regulated. Super-enhancer formation has been linked to oncogene activation (Sur and Taipale, 2016). However, our results indicate that the Per2 super-enhancer is associated with tumor-suppressive function in LUAD, consistent with our recent finding that super-enhancer diminution downregulates expression of tumor suppressor genes against medulloblastoma genesis (Dhar et al., 2018). Taken together, our findings reveal a tumor-suppressive mechanism in which KMT2D indirectly downregulates glycolytic genes by enhancing Per2 expression via super-enhancer activation and thereby suppresses LUAD.
Increased aerobic glycolysis, known as the Warburg effect, is a major characteristic of cancer cells, providing them with a proliferation advantage by producing ATP as well as glucose-derived metabolites for biosynthesis of nucleotides, lipids, and proteins (Gatenby and Gillies, 2004). Therefore, targeting glycolytic pathways to arrest tumorigenic growth of cancer cells remains an attractive therapeutic intervention. Interestingly, the present study showed that Kmt2d loss increased expression of glycolytic genes. Our other study also showed that Kmt2d loss increased glycolysis via IGFBP5-regulated IGF signaling while promoting melanoma tumorigenesis (Maitituoheti et al., 2018). In line with these findings, we demonstrated that the glycolysis inhibitor 2-DG impeded the tumorigenicity of LUAD cell lines bearing KMT2D-inactivating mutations in a mouse xenograft model. Because KMT2D is mutated in several other types of cancer (Suva et al., 2013) and 2-DG has re-entered clinical trials, our findings may rationalize glycolysis inhibition as an anticancer treatment strategy against other cancer types bearing KMT2D-inactivating mutations besides human KMT2D-inactivated LUAD.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for further information and reagents should be directed to and will be fulfilled by the Lead Contact, Min Gyu Lee (mglee@mdanderson.org). Any plasmid and cell line generated in this study would be available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines.
All lung cancer cell lines (A549, H1792, H23, H1437, H358, H1568, DV-90, CORL-105, and LKR-10) that were used in this study are described in the Key Resources Table, and they were cultured within 10 to 15 passages. Cell culture reagents and other chemicals were purchased from Thermo Fisher Scientific (Gibco & Hyclone), Corning, Sigma-Aldrich, and Fisher Bioreagents.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Abcam | Cat# ab4729, RRID: AB_2118291 |
| H3K27me3 | Abcam | Cat# ab6002, RRID:AB_305237 |
| H3K4me1 | Cell Signaling Technology | Cat# 5326, RRID:AB_10695148 |
| H3K4me3 | Abcam | Cat# ab8580, RRID:AB_306649 |
| H3 | Abcam | Cat# ab1791, RRID:AB_302613 |
| TTF-1 | Seven Hills Bioreagents (WRAB-1231) | Cat# WRAB-1231 RRID: AB_451727 |
| Ki-67 | Cell Signaling Technology | Cat# 9027, RRID:AB_2636984 |
| ENO1 | Abcam (ab227978) | Cat# ab227978 RRID: |
| PGK1 | Abcam | Cat# ab38007, RRID:AB_2161220 |
| PGAM1 | Novus | Cat# NBP1-49532, RRID:AB_10011581 |
| TTF-1 | Abcam | Cat# ab72876, RRID:AB_1271363 |
| KMT2D | Santa Cruz Biotechnology | Cat# sc-68675, RRID:AB_2145784 |
| Actin | Sigma-Aldrich | Cat# A5441, RRID:AB_476744 |
| HRP-conjugated anti-mouse-IgG | Santa Cruz Biotechnology | Cat# sc-2055, RRID:AB_631738 |
| HRP-conjugated anti-rabbit-IgG | Santa Cruz Biotechnology (sc-2004) | Cat# sc-2004, RRID:AB_631746 |
| Alexa 488-conjugated anti-mouse IgG | Life technologies (A11029) | Cat# A-11029, RRID:AB_138404 |
| Alexa 488-conjugated anti-rabbit IgG | Life technologies (A11037) | Cat# A-11037, RRID:AB_2534095 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-Deoxy-D-glucose (2-DG) | Selleck Chemicals | Cat# S4701; CAS: 1032350-13-2 |
| Vorinostat (SAHA, MK0683) | Selleck Chemicals | Cat# S1047; CAS: 149647-78-9 |
| AR-42 (HDAC-42) | Selleck Chemicals | Cat# S2244; CAS: 935881-37-1 |
| Lonidamine | Selleck Chemicals | Cat# S2610; CAS: 50264-69-2 |
| Dinaciclib | Selleck Chemicals | Cat# S2768; CAS: 779353-01-4 |
| Koningic acid (KA) | Focus Biomolecule | Cat# 10-3326; CAS: 74310-84-2 |
| IACS-010759 | Gift from Dr. Joseph R. Marszalek (Molina et al., 2018) | N/A |
| Critical Commercial Assays | ||
| Seahorse XF Glycolysis stress test kit | Agilent | Cat# 103020-100 |
| Seahorse XF Cell Mito stress test kit | Agilent | Cat# 103015-100 |
| Glyceraldehyde 3 Phosphate Dehydrogenase Activity Assay Kit | Abcam | Cat# ab204732 |
| Enolase Assays Kit | Abcam | Cat# ab241024 |
| Deposited Data | ||
| RNA-seq for KrasLSL-G12D and KrasLSL-G12D;Kmt2dfl/fl lung tumors | This paper | GEO: GSE116658 |
| ChIP-seq for KrasLSL-G12D and KrasLSL-G12D;Kmt2dfl/fl lung tumors | This paper | GEO: GSE116659 |
| RNA-seq for LKR10 lung cancer cell line | This paper | GEO: GSE143498 |
| Experimental Models: Cell Lines | ||
| Human: NCI-A549 | ATCC | CCL-185 |
| Human: NCI-H1792 | ATCC | CRL-5895 |
| Human: NCI-H23 | ATCC | CRL-5800 |
| Human: NCI-H1437 | ATCC | CRL-5872 |
| Human: NCI-H358 | ATCC | CRL-5807 |
| Human: NCI-H1568 | ATCC | CRL-5876 |
| Human: DV-90 | DSMZ | ACC 307 |
| Human: CORL-105 | ECACC | 92031918 |
| Mouse: LKR-10 | Gift from Julien Sage | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129-Krastm4Tyj/Nci, | NCI Mouse Repository | Strain: 01XJ6 |
| Mouse: B6.129P2-Trp53tm1Brn/J | The Jackson Laboratory | Strain: 008462 |
| Mouse: Kmt2dfl/fl (also called Mll4fl/fl) | (Dhar et al., 2018) | N/A |
| Oligonucleotides | ||
| Primers for quantitative RT-PCR (mouse): See Table S4 | N/A | N/A |
| Primers for quantitative RT-PCR for enhancer RNAs: See Table S4 | N/A | N/A |
| Primers for genotyping: See Table S4 | N/A | N/A |
| Primers for quantitative ChIP-PCR (mouse): See Table S4 | N/A | N/A |
| Primers for cloning: See Table S4 | N/A | N/A |
| Recombinant DNA | ||
| pInducer20-KMT2D | This paper | N/A |
| shKmt2d-01 (mouse) | Sigma-Aldrich | Cat# TRCN0000239234 |
| shKmt2d-03 (mouse) | Sigma-Aldrich | Cat# TRCN0000239233 |
| shPer2-01 (mouse) | Sigma-Aldrich | Cat# TRCN0000284505 |
| shPer2-02(mouse) | Sigma-Aldrich | Cat# TRCN0000096663 |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| cBio cancer genomics portal | (Cerami et al., 2012; Gao et al., 2013) | http://www.cbioportal.org |
| IBM SPSS Statistics 23 | IBM | https://www.ibm.com/support/pages/downloading-ibm-spss-statistics-23 |
| Pyflow-ChIPseq | (Terranova et al., 2018) | https://github.com/crazyhottommy/pyflow-ChIPseq |
| Bowtie1 | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| samtools | (Li et al., 2009) | http://www.htslib.org/ |
| Deeptools | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/ |
| MACS14 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| ROSE | (Whyte et al., 2013) | http://younglab.wi.mit.edu/super_enhancer_code.html |
| chromHMM | (Ernst and Kellis, 2012) | http://compbio.mit.edu/ChromHMM/ |
| Epilogos | (Marx, 2015) | https://github.com/Altius/epilogos |
| EnrichedHeatmap | (Gu et al., 2018) | https://bioconductor.org/packages/release/bioc/html/EnrichedHeatmap.html |
| ComplexHeatmap | (Gu et al., 2016) | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| featureCounts | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| ChIPseeker | (Yu et al., 2015) | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| DESeq2 | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| TCGAbiolinks | (Colaprico et al., 2016) | https://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html |
| KaryoploteR | (Gel and Serra, 2017) | http://bioconductor.org/packages/release/bioc/html/karyoploteR.html |
| GSEA | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/ |
| Snakemake | (Koster and Rahmann, 2012) | https://snakemake.readthedocs.io/en/stable/ |
| Pyflow-RNAseq | This paper | https://github.com/crazyhottommy/pyflow-RNAseq |
| IGV | (Robinson et al., 2011) | https://software.broadinstitute.org/software/igv/ |
| ggplot2 | (Wickham, 2006) | https://ggplot2.tidyverse.org/ |
Mouse strains and genetically engineered lung cancer mouse models.
The KrasLSL-G12D (strain number 01XJ6) and Trp53fl/fl (strain name, B6.129P2-Trp53tm1Brn/J; stock number, 008462) mice were obtained from the NCI Mouse Repository and Jackson Laboratory, respectively. The Kmt2dfl/fl mice were previously described (Dhar et al., 2018). Kmt2dfl/fl mice were crossed with KrasLSL-G12D or Trp53fl/fl mice to get the desired mouse genotypes of the study. To generate KrasLSL-G12D;Kmt2dfl/fl mice, Kmt2dfl/fl mice were first crossed with KrasLSL-G12D mice and the resulting KrasLSL-G12D;Kmt2dfl/+ mice were then crossed with Kmt2dfl/fl mice. To generate Trp53fl/fl;Kmt2dfl/fl mice, Kmt2dfl/fl were first crossed with Trp53fl/fl, and the resulting Trp53fl/+;Kmt2dfl/+ mice were then crossed with Trp53fl/+;Kmt2dfl/+ to get Trp53fl/fl;Kmt2dfl/fl mice. The genotypes of these mice were confirmed by a standard PCR-based protocol. The primers used for the genotyping are listed in Table S4.
In vivo lung tumorigenesis study.
The protocol for induction and monitoring of lung tumorigenesis was described previously (Alam et al., 2018). In brief, to induce tumors in the mouse lung, 6- to 8-week old mice were infected with about 2.5 × 107 adeno 5 (Ad5)-CMV-Cre virus per mouse by the intratracheal intubation method (DuPage et al., 2009). The tumor progression and survival of mouse groups were compared. For chromatin immunoprecipitation (ChIP) and RNA analyses, the distinct tumors were dissected from the lungs, washed with ice-cold phosphate-buffered saline solution (PBS) and snap frozen. For histology, immunohistochemistry (IHC), and immunofluorescence (IF) analyses, tumor-bearing lungs were isolated, fixed and processed as previously described (Wagner et al., 2013). Hematoxylin and eosin (H&E)-stained sections of tumor-bearing lungs were evaluated microscopically, and tumors were scored 0, I, II, III, and IV on the basis of percentage of pulmonary parenchyma affected by lung adenocarcinoma(s): 0, no tumor present; I, <10% of examined lung affected; II, 10%–20%; III, 21%–50%; IV, >50%. Tumor areas were quantified using ImageScope software.
Study approval.
The care and use of all mice were approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center.
METHOD DETAILS
Micro-computed tomography.
Tumor growth in mice was monitored by using micro-CT as previously described (Alam et al., 2018). Briefly, the mice were anesthetized with 5% isoflurane and maintained at 2% isoflurane. The mice were intubated using a 20 gauge x 1-inch catheter and were transferred onto the bed of X-Rad 225Cx (Precision X-Ray Corporation). The mice were mechanically ventilated in a small animal ventilator, and micro-CT images were captured at 60 KvP, 4 mA, and 3 rpm. Each animal’s breathing was held at 20cmH20 during the 20-second acquisition. Three to five mice (30 days post-infection) per group were subjected to micro-CT.
Histologic, immunohistochemical, and immunofluorescence analyses.
The tumor-bearing lungs were isolated and fixed with 10% formalin buffer. The fixed lung tissues were embedded in paraffin and were cut into 8 μm sections. For histologic examination, a standard H&E staining was performed. IHC and IF experiments were performed as described previously (Alam et al., 2018). Briefly, sections were subjected to antigen retrieval in a solution from Vector Laboratories (Burlingame, CA) and then blocked in 10% horse serum for 1 hr at RT. For IF, anti-mouse IgG antibodies conjugated to Alexa Fluor 488 and anti-rabbit IgG secondary antibodies conjugated to Alexa 568 were used for detection, and images of tumor regions were captured using a laser confocal microscope. For the quantification of IF staining, signal intensities of glycolytic enzymes in TTF1-positive tumor cells were measured using ImageJ software. The primary antibodies used for IHC are listed in the Key Resources Table.
Survival analysis and gene expression correlation analysis.
The LUAD datasets of TCGA database were used for expression and survival analysis. Oncoprint and correlation data were obtained by analysis using the cBioportal for cancer genomics (http://www.cbioportal.org) (Cancer Genome Atlas Research, 2014; Cerami et al., 2012; Gao et al., 2013). For the survival analysis, a publicly available LUAD transcriptomic dataset (http://kmplot.com/analysis/index.php?p=service&cancer=lung) (Gyorffy et al., 2013) was also used.
RNA isolation from lung tumor cells.
The distinct tumor tissues were dissected and cut into < 1-mm pieces. The dissected tumor tissues were then digested in collagenase type 1 and DNAase I for 45 min and treated with 0.25% trypsin for 10 min. To remove red blood cells, the digested single cell suspension was treated with red blood cell lysis buffer for 2–3 min. To further enrich tumor cells, CD45-positive cells were removed using MagniSort™ Mouse CD45 Depletion Kit (ThermoFisher Scientific). The depletion of CD45-positive cells was confirmed by flow cytometry analysis. Total RNA was isolated using Trizol reagent (Life Technologies).
RNA-seq analysis.
The RNAs isolated from CD45-depleted tumor tissue samples were sequenced using the Illumina HiSeq 2000. The RNAs for shLuc-treated and KMT2D-depleted LKR10 lung cancer cells were sequenced using NextSeq 500. Both RNAseq data sets were processed by pyflow-RNAseq (https://github.com/crazyhottommy/pyflow-RNAseq), a Snakemake-based RNAseq pipeline. Raw reads were mapped by STAR (Dobin et al., 2013), RPKM normalized bigwigs were generated by deeptools (Ramirez et al., 2016), and gene counts were obtained by featureCounts (Liao et al., 2014). Differential expression analysis was carried out using DESeq2 (Love et al., 2014). DAVID (version 6.8) was used for gene ontology analysis. Gene Set Enrichment Analysis was carried out using the GSEA tool from the Broad Institute (Subramanian et al., 2005). The pre-rank mode was used. The signed fold change *−log10 (p value) metric was used for pre-ranking the genes.
TCGA RNA-seq data analysis.
RNA-seq raw counts for TCGA lung adenocarcinoma dataset were downloaded using TCGAbiolinks (Colaprico et al., 2016). The mutation MAF files were downloaded with TCGAbiolinks as well, and mutation status was inferred from the MAF files. Twenty four LUAD samples with high WT KMT2D expression were compared with twenty LUAD samples with low WT KMT2D expression and four LUAD samples with KMT2D nonsense mutations using DESeq2 (see Table S2 for sample IDs). The signed fold change *−log10 (p value) metric was used to pre-rank the gene list in GSEA pre-rank analysis.
Quantitative RT-PCR.
Quantitative reverse transcription (RT)-PCR analyses were performed as described earlier (Alam et al., 2018). In brief, iQ SYBR Green Supermix (BioRad) was used for PCR amplification, and signals were acquired using CFX384 Touch™ real-time PCR detection system (BioRad). β-Actin mRNA or 18s rRNA levels were used for data normalization. Each experiment was performed in triplicate. The primers used for quantitative RT-PCR are listed in the Table S4.
ChIP-seq assays.
ChIP for lung tumor tissues was performed with minor modifications of a previous procedure (Terranova et al., 2018). Briefly, distinct lung tumor tissues (3 mg per antibody) were dissected from the lungs, cut into 1-mm pieces, homogenized using a MACS dissociator, and cross-linked using 1% paraformaldehyde for 10 min at 37 °C. Crosslinking was then stopped by adding 0.125 M glycine for 5 min, and tissues were washed with PBS and stored at −80 °C. Later, tissues were thawed on ice and lysed with ChIP harvest buffer (12 mM Tris-HCl, 0.1 × PBS, 6 mM EDTA, 0.5% sodium dodecyl sulfate [SDS]) for 10 min on ice. Sonication conditions were optimized for lung tumor tissues using bioruptor sonicator to achieve a shear length of 250–500 bp. Antibody-dynabead mixtures were incubated for 1 hr at 4 °C and tissue extracts were then incubated overnight with these mixtures. After overnight incubation, immunecomplexes were washed as follows: five times with RIPA buffer, twice with RIPA-500 (RIPA with 500 mM NaCl) and twice with LiCl wash buffer (10 mM Tris-HCl pH8.0, 1 mM EDTA pH8.0, 250 mM LiCl, 0.5% NP-40, 0.1% deoxycholate). For reverse-crosslinking and elution, immunecomplexes were incubated overnight at 65 °C in elution buffer (10 mM Tris-HCl pH8.0, 5 mM EDTA, 300 mM NaCl, 0.5% SDS). Eluted DNA was then treated with proteinase K (20 mg/ml) and RNaseA and DNA clean-up was done using SPRI beads (Beckman-Coulter). Libraries were prepared as previously described (Terranova et al., 2018) using adaptors from New England Biolabs. Libraries were multiplexed together and sequenced in HiSeq2000 or HiSeq4000 (Illumina).
ChIP-seq analysis.
ChIP-seq data that were generated using Kras and Kras;Kmt2d−/− tumors were quality-controlled and processed by pyflow-ChIPseq (Terranova et al., 2018), a Snakemake-based ChIP-seq pipeline (Koster and Rahmann, 2012). Briefly, raw reads were mapped by Bowtie1 (Langmead et al., 2009) and duplicate reads were removed. Only uniquely mapped reads were retained. RPKM-normalized bigwigs were generated by deepTools (Ramirez et al., 2016) and the tracks were visualized with Integrative Genomics Viewer (Broad Institute) (Robinson et al., 2011). Peaks were called using MACS1.4 (Zhang et al., 2008) with a p value of 1.0 × E-8. Chromatin state was called using ChromHMM (Ernst and Kellis, 2012) and the emission profiles were plotted by ComplexHeatmap (Gu et al., 2016). The heatmaps were generated by R package EnrichedHeatmap. ChIP-seq peaks were annotated with the nearest genes using ChIPseeker (Yu et al., 2015). Super-enhancers were identified using ROSE (Loven et al., 2013) based on H3K27ac ChIP-seq signals.
Publicly available PER2 ChIP-seq data for mouse (GSE3997) were reanalyzed by using six samples (GSM982733, GSM982734, GSM982735, GSM982736, GSM982737 and GSM982738). Another data GSM982739 was excluded because it was from a Per2 knock-out mouse. The raw FASTQ files were aligned to mm9 genome separately for each sample using the same Snakemake pipeline. The read duplication rate was high, and the bam files after alignment were merged together. A bigwig file normalized to RPKM was derived from the merged bam file for visualization. The bigwig tracks were plotted using karyoploteR (Gel and Serra, 2017).
ChromHMM transition.
ChromHMM profiles of two Kras;Kmt2d−/− and two Kras samples were consolidated using Epilogos (https://github.com/Altius/epilogos). A pipeline was made to automate the calculation, and the scripts used to re-code the ChromHMM states can be found https://github.com/crazyhottommy/pyflow-chromForest/tree/vsurf_merge. With the Epilogos output, the chromatin state for each bin was chosen for the state that contained the greatest weights. A helper script can be also found at the above link. The output for each group was analyzed by java -mx12000M -jar ChromHMM.jar OverlapEnrichment. The matrix output from OverlapEnrichment was scaled by columns and plotted using ComplexHeatmap (Gu et al., 2016).
Glucose uptake and lactate excretion assay.
Cells were seeded in triplicate in a 12-well plate. Wells containing medium but no cells were used as baseline readings. On the second day, the medium in each well was changed with 1 ml fresh medium (including the wells without cells). After 48 hr, 600 μl medium was collected from each well. Each sample of medium was centrifuged at 3000 rpm for 3–5 min at 4 °C. From each sample, 200 μl were transferred into a 96-well plate, and glucose and lactate levels were measured using an YSI analyzer according to the manufacturer’s protocol. Each level was normalized to the cell number.
Metabolomics experiments.
All the reagents and chemicals for metabolomics experiments were described earlier (Wangler et al., 2017). For sample preparation for mass spectrometry and metabolomics analysis, metabolites were extracted from cell pellets, and mouse liver pool was used for quality control according to the previously described extraction procedures (Putluri et al., 2014; Putluri et al., 2011). Briefly, equal number of cells from each group were used for the metabolic extraction. The metabolites were extracted with polar and non-polar solvents, and the protein content in the extracts was removed using 3 kDa Amicon ultra filters. The filtrate was dried under vacuum (Genevac EZ-2plus; Gardiner, Stone Ridge, NY). The dried extracts were re-suspended in methanol-water (50/50, vol/vol) and were subjected to liquid chromatography-mass spectrometry. Quantitative analysis using Agilent MassHunter Workstation Software was performed for review of chromatograms. Peak-area integration was accessed on the basis of the retention time. After data acquisition, the normalization of each metabolite was performed using an isotopic labeled internal standard, and the data were log2-transformed. For differential analysis, a two sample t-test was conducted. Differential metabolites were identified by adjusting the p values for multiple testing at a false discovery rate (FDR) threshold of < 0.25. The analysis was performed by the R package.
Inhibitor experiments.
Cells were seeded at a density of 1.5 × 103 cells per well in four replicates in 96-well plates. Plated cells were then treated with inhibitors at different concentrations. Human KMT2D-WT LUAD cell lines (A549, H1792, H23, H1437, and H358) and KMT2D-mutant LUAD cell lines (H1568, DV-90, and CORL-105) cells were treated with a range of concentrations of 2-Deoxy-D-Glucose (2-DG; 1 to 1000 μM), POMHEX (0.05 to 2 μM), SAHA (0.1 to 20 μM) and AR-42 (1 to 500 nM) for one week. The cells were replenished with inhibitor-containing medium on alternate days. After 7 days, the cells were stained with crystal violet and quantified using Celigo. Cells treated with dimethyl sulfoxide were used as a control.
Extracellular flux assays using Seahorse analyzer.
The extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR) in lung cancer cell lines were measured using the Seahorse XF96e Extracellular Flux Analyzer (Agilent, USA). Cells (1.6–2 × 104 per well) were seeded in Seahorse XF96 cell culture microplates, and the plates were incubated at 37 °C under 5% CO2. After 24-hr incubation, the media were changed to the assay medium without glucose. The ECAR (e.g., basal glycolysis and glycolytic capacity) was measured by the sequential additions of glucose (at a final concentration of 10 mM), oligomycin (1.0 μM), and 2-DG (50 mM) in the Seahorse Analyzer using the Glycolysis Stress Test Kit (Agilent, USA). The glycolysis and glycolytic capacity were calculated according to manufacturer’s protocol. The OCR was assessed by the sequential additions of oligomycin (1.5 μM), Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 1.0–2.0 μM), and antimycin A (0.5 μM)/rotenone (1 μM) in the Seahorse Analyzer using the Cell Mito Stress Test Kit (Agilent, USA). Both ECAR and OCR were measured over a 3-min period and normalized to the protein concentrations that were determined by the Bradford assay.
Three-dimensional cell culture.
The three-dimensional (3D) cell culture methods were adapted from the previously described procedures (Lee et al., 2007a). In brief, a 96-well plate was coated with 6 μl of Engelbreth-Holm-Swarm tumor matrix (Matrigel, BD Biosciences) and kept on room temperature for 30 min. Cells were trypsinized and suspended in Dulbecco modified essential medium (DMEM) containing 50% Matrigel. Cells (2 × 105 per well) were seeded in the plates. Every third day, the cells were replenished with fresh DMEM containing 10% fetal bovine serum. The cells were cultured for 10–14 days.
In vivo inhibitor experiments.
Cells (5 × 106) in 100 μl Matrigel were subcutaneously injected in both flanks of 6- to 8-week-old athymic nu/nu mice. Eight days later, mice that had developed palpable tumors were randomly separated into two groups. One group of mice was treated with 2-DG (500 mg/kg body weight) or KA (1mg/kg body weight) via intraperitoneal injections on alternate days for 16–20 days. Because the drug was prepared in sterile water, the other group was injected with sterile water as a vehicle control. Tumors were measured on alternate days by caliper. Tumor volumes were calculated using the ellipsoid volume formula (1/2 × ∣ × w × h) as previously described (Wagner et al., 2013). After 16–20 days of treatment, the mice were euthanized and tumors were collected for histologic analysis.
Stable knockdown, overexpression, and rescue experiments.
For knockdown experiments, lentivirus-based, puromycin-resistant shRNAs were purchased from Sigma Chemicals (Key Resources Table). The shRNA-infected cells were selected in puromycin-containing medium (1 μg/ml). shLuc-infected cells were used as a control. For ectopic expression experiments, human PER2 cDNA was cloned into the lentivirus vector pLenti6.3/V5-DEST (Thermo Fisher Scientific) using standard cloning methodology. Cells infected with pLenti-PER2 were selected in blasticidin-containing medium (2 μg/ml). pLenti-GFP-infected-cells were used as controls. For doxycycline (Dox)-inducible KMT2D expression, KMT2D cDNA in pDNR221 (a kind gift from Dr. Laura Pasqualucci) was cloned into pInducer20. The expression plasmid was transfected into H1568 cells using Lipofectamine 3000. The cells were then selected in 1 mg/ml G418 for 2 weeks. KMT2D expression was induced by the medium containing 10 μg/ml Dox.
For rescue experiments, KMT2D-depleted LKR10 cells were seeded and 24 hr later transfected with pFLAG-CMV2 expression plasmids encoding mini-KMT2D or GFP. Of note, the mini-KMT2D construct was previously described as MLL4fusion construct (1358-1572aa/4507-5537aa) (Dhar et al., 2012). As a control, shLuc-treated LKR10 cells were transfected with pFLAG-CMV2 expression plasmid encoding GFP. Two days later, these cells were harvested. Total RNAs were isolated for qRT-PCR analysis. Quantitative ChIP analysis was performed as previously described (Dhar et al., 2012).
Measurement of GAPDH and Enolase activities.
GAPDH and Enolase assays were performed according to the manufacturer’s protocols (Abcam, USA). Briefly, cells (1 × 106 per well) were lysed in GAPDH and Enolase assay buffers. GAPDH and enolase assays were performed using 4–5 μg and 2 μg of lysates, respectively. Protein quantification of lysates was performed using standard BCA Kit (Pierce, USA). The absorbance of 450 nm at a 60 min reaction and 570 nm at a 30 min reaction was measured to calculate GAPDH and Enolase activities, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
In all the boxplots, the bottom and the top rectangles indicate the first quartile (Q1) and third quartile (Q3), respectively. The horizontal lines in the middle signify the median (Q2), and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively. The two-tailed Student’s t-test was used to determine statistical difference between two boxplots as well as between certain two groups (where indicated). Survival differences were compared by the Kaplan-Meier method using the two-sided log-rank test (IBM SPSS Statistics 23). Data are presented as means ± standard error of the mean (SEM; error bars) of at least three independent experiments or three biological replicates. p values less than 0.05 were considered statistically significant. * (p <0.05), ** (p <0.01) and *** (p <0.001) indicate statistically significant differences between two groups.
DATA AND CODE AVAILABILTY
RNA-seq and ChIP-seq data that support the findings of this study have been deposited in the GEO database with the accession numbers GSE116658, GSE116659, and GSE143498.
Supplementary Material
Highlights.
Lung-specific Kmt2d loss in mice promotes lung tumorigenesis
Kmt2d loss impairs enhancers, including a super-enhancer for the tumor suppressor Per2
KMT2D activates Per2 expression and thereby represses glycolytic genes
Glycolysis inhibition impedes the growth of KMT2D-mutant lung cancer
Acknowledgments
We are grateful to Julien Sage, Joseph R. Marszalek, and Laura Pasqualucci for providing their reagents and thank Haoqiang Ying for insightful discussion. We are also thankful to Kathryn Hale (Scientific Publications Services, Research Medical Library, The University of Texas MD Anderson Cancer Center) for the editorial assistance and to Zhenbo Han, Su Zhang, and Charles Kingsley for their technical assistance. This work was supported by grants to MG Lee from the National Institutes of Health (NIH; R01 CA157919, R01 CA207109, and R01 CA207098) and the Center for Cancer Epigenetics (Solexa allowance) at the MD Anderson Cancer Center, by a grant to K Rai from the NIH (CA160578), by a grant to FL Muller from American Cancer Society (RSG-15-145-01-CDD), by a grant to FJ DeMayo from the Intramural Research Program of the National Institute of Environmental Health Sciences (Z1AES103311-10), by a grant to ER Flores from the NIH (R35CA197452), by grants to N Putluri from the CPRIT Proteomics and Metabolomics Core Facility (RP170005), the American Cancer Society (127430-RSG-15-105-01-CNE), and the NIH (R01CA216426 and R01CA220297), by a fellowship to M Kumar from the Center for Cancer Epigenetics at the MD Anderson Cancer Center, and by a fellowship to H Alam from the Odyssey program at the MD Anderson Cancer Center. The animal imagining and histology work were performed at the Small Animal Imaging Facility and Histopathology Core Lab, respectively, at the MD Anderson Cancer Center supported by the NIH National Cancer Institute (P30CA016672).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no potential conflicts of interest.
References
- Alam H, Li N, Dhar SS, Wu SJ, Lv J, Chen K, Flores ER, Baseler L, and Lee MG (2018). HP1gamma Promotes Lung Adenocarcinoma by Downregulating the Transcription-Repressive Regulators NCOR2 and ZBTB7. A. Cancer Res 78, 3834–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Bass J, and Takahashi JS (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, Vaurio RG, Potter MC, Hansen KD, and Dietz HC (2014). Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med 6, 256ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawer MK (2005). Lonidamine: basic science and rationale for treatment of prostatic proliferative disorders. Rev Urol 7 Suppl 7, S21–26. [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capello M, Ferri-Borgogno S, Cappello P, and Novelli F (2011). alpha-Enolase: a promising therapeutic and diagnostic tumor target. FEBS J 278, 1064–1074. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BA, and Hughes BG (2015). Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res 4, 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, et al. (2007). PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. The Journal of biological chemistry 282, 20395–20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colell A, Green DR, and Ricci JE (2009). Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ 16, 1573–1581. [DOI] [PubMed] [Google Scholar]
- Dawson MA, and Kouzarides T (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, and Lee MG (2012). Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev 26, 2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell 70, 825–841 e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, and Bradley A (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. [DOI] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipski E, King VM, Li X, Granda TG, Mormont MC, Claustrat B, Hastings MH, and Levi F (2003). Disruption of circadian coordination accelerates malignant growth in mice. Pathol Biol (Paris) 51, 216–219. [DOI] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, and Lee C (2002). The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50. [DOI] [PubMed] [Google Scholar]
- Ganapathy-Kanniappan S, Kunjithapatham R, and Geschwind JF (2013). Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res 33, 13–20. [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, and Gillies RJ (2004). Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4, 891–899. [DOI] [PubMed] [Google Scholar]
- Gel B, and Serra E (2017). karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M, and Ishaque N (2018). EnrichedHeatmap: an R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics 19, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, and Lanczky A (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. (2010). Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, and Lippman SM (2008). Lung cancer. N Engl J Med 359, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, and Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, and Shilatifard A (2012). Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26, 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, et al. (2012). Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Wang SY, Lin LL, Wang PW, Chen TY, Hsu WM, Lin TK, Liou CW, and Chuang JH (2015). Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis Model Mech 8, 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, and Canaani E (2007). Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol 27, 1889–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Oblinger J, Bush ML, Brendel V, Santarelli G, Chaudhury AR, Kulp S, La Perle KM, Chen CS, Chang LS, et al. (2012). Preclinical validation of AR42, a novel histone deacetylase inhibitor, as treatment for vestibular schwannomas. Laryngoscope 122, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, and Shiekhattar R (2015). Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J, and Rahmann S (2012). Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Koutsioumpa M, Hatziapostolou M, Polytarchou C, Tolosa EJ, Almada LL, Mahurkar-Joshi S, Williams J, Tirado-Rodriguez AB, Huerta-Yepez S, Karavias D, et al. (2019). Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut 68, 1271–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Lee JE, Jang Y, Wang L, Peng W, and Ge K (2017). MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res 45, 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DnA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GY, Kenny PA, Lee EH, and Bissell MJ (2007a). Three-dimensional culture models of normal and malignant breast epithelial cells. Nature methods 4, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, and Lee JW (2009). A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLl4. Proc Natl Acad Sci U S A 106, 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2, e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, and Shiekhattar R (2007b). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, He J, Hunter T, et al. (2016). Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol Cell 61, 705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab 26, 648–659 e648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Satani N, Hammoudi N, Ackroyd JJ, Khadka S, Yan VC, Georgiou DK, Sun Y, Zielinski R, Tran T, et al. (2018). Eradication of ENO1-deleted Glioblastoma through Collateral Lethality. bioRxiv 331538, 10.1101/331538. [DOI] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitituoheti M, Keung EZ, Tang M, Yan L, Alam H, Han G, Raman AT, Terranova CJ, Sarkar S, Orouji E, et al. (2018). Enhancer Reprogramming Confers Dependence on Glycolysis and IGF signaling in KMT2D Mutant Melanoma. bioRxiv 507327, doi: 10.1101/507327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J, Jabbour SK, and Aisner J (2017). Current state of immunotherapy for non-small cell lung cancer. Transl Lung Cancer Res 6, 196–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Marx V (2015). Visualizing epigenomic data. Nat Methods 12, 499–502. [DOI] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, and Zhang Y (2011). The diverse functions of Dot1 and H3K79 methylation. Genes Dev 25, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, and Jacks T (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, and Jacks T (2016). Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab 24, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, et al. (2010). Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 9, 2344–2353. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, and Huang P (2006). Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646. [DOI] [PubMed] [Google Scholar]
- Putluri N, Maity S, Kommagani R, Creighton CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T, et al. (2014). Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia 16, 390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putluri N, Shojaie A, Vasu VT, Nalluri S, Vareed SK, Putluri V, Vivekanandan-Giri A, Byun J, Pennathur S, Sana TR, et al. (2011). Metabolomic profiling reveals a role for androgen in activating amino acid metabolism and methylation in prostate cancer cells. PLoS One 6, e21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santo L, Vallet S, Hideshima T, Cirstea D, Ikeda H, Pozzi S, Patel K, Okawa Y, Gorgun G, Perrone G, et al. (2010). AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 29, 2325–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, et al. (2014). DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 514, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, and Haber DA (2007). Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7, 169–181. [DOI] [PubMed] [Google Scholar]
- Sheng SL, Liu JJ, Dai YH, Sun XG, Xiong XP, and Huang G (2012). Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J 279, 3898–3910. [DOI] [PubMed] [Google Scholar]
- Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, and Shilatifard A (2014). Enhancer biology and enhanceropathies. Nat Struct Mol Biol 21, 210–219. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhou B, Mao F, Xu J, Miao H, Zou Z, Phuc Khoa LT, Jang Y, Cai S, Witkin M, et al. (2018). HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell 34, 643–658 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nat Rev Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Suva ML, Riggi N, and Bernstein BE (2013). Epigenetic reprogramming in cancer. Science 339, 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova C, Tang M, Orouji E, Maitituoheti M, Raman A, Amin S, Liu Z, and Rai K (2018). An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KW, Alam H, Dhar SS, Giri U, Li N, Wei Y, Giri D, Cascone T, Kim JH, Ye Y, et al. (2013). KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest 123, 5231–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, et al. (2017a). A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol Cell 67, 308–321 e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Lo CM, Chen L, Ngan ES, Xu A, and Poon RY (2017b). CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ 24, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangler MF, Chao YH, Bayat V, Giagtzoglou N, Shinde AB, Putluri N, Coarfa C, Donti T, Graham BH, Faust JE, et al. (2017). Peroxisomal biogenesis is genetically and biochemically linked to carbohydrate metabolism in Drosophila and mouse. PLoS Genet 13, e1006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2006). An implementation of the grammar of graphics in R: ggplot. Paper presented at: Book of Abstracts. [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang D, Li J, Wang F, Hu J, Wang S, and Sun Y (2014). 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett 355, 176–183. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015a). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mittal A, Reid J, Reich S, Gamblin SJ, and Wilson JR (2015b). Evolving Catalytic Properties of the MLL Family SET Domain. Structure 23, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.