

Abstract
Motivation
Gene lists are routinely produced from various omic studies. Enrichment analysis can link these gene lists with underlying molecular pathways and functional categories such as gene ontology (GO) and other databases.
Results
To complement existing tools, we developed ShinyGO based on a large annotation database derived from Ensembl and STRING-db for 59 plant, 256 animal, 115 archeal and 1678 bacterial species. ShinyGO’s novel features include graphical visualization of enrichment results and gene characteristics, and application program interface access to KEGG and STRING for the retrieval of pathway diagrams and protein–protein interaction networks. ShinyGO is an intuitive, graphical web application that can help researchers gain actionable insights from gene-sets.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
For a set of genes identified in genome-wide studies, enrichment analysis can be done to see if the set is enriched with genes of a certain pathway or functional category, such as those defined by gene ontology (GO) (Ashburner et al., 2000). Dozens of tools have been developed for enrichment analysis (Khatri et al., 2012). A small subset of these tools is listed in Supplementary Table S1. Some tools are designed for biomedical research and thus focus primarily on human and mouse. For example, Enrichr (Kuleshov et al., 2016) includes a gene-sets ranging from GO, co-expression, tissue-specific genes, transcriptional factor (TF) or microRNA (miRNA) target genes, to various pathway databases. Similarly, tools like PlantGSEA are focused on 15 plants species (Yi et al., 2013).
g: Profiler is based on gene annotation in Ensembl (Aken et al., 2017) for over 300 plant and animal species. STRING (Szklarczyk et al., 2015) is a large database of protein–protein interactions (PPI). It also provides functionality for enrichment analysis of GO and protein domains in 5090 species (version 11). On the other extreme is DAVID (Huang da et al., 2009) which is able to include a large database of tens of thousands of organisms because it derives information from many sources including NCBI, UniProt, KEGG, GO, Biocarta, REACTOME, etc. These tools have helped biologists gain insights from gene lists.
Taking advantage of the Shiny framework, which enable access to many powerful R packages for visualization and statistical analyses, we developed a new tool based on the annotation database at Ensembl and pathway databases from many other sources. Unique features of ShinyGO include: (i) display query genes on pathway diagrams and PPI networks based on application program interface (API) access to KEGG and STRING, (ii) visualize the overlaps among enriched pathways using hierarchical clustering and interactive networks and (iii) identify statistically significant differences in gene type, length, GC content, chromosomal distribution between query genes and the background. For several model organisms, we also enhanced the annotation database by incorporating other gene-sets, especially TF and miRNA target genes.
2 Materials and methods
ShinyGO is a Shiny application developed based on several R/Bioconductor packages, and a large annotation and pathway database compiled from many sources. See Supplementary Files S1 and S2 for more details. Source code is available at https://github.com/iDEP-SDSU/idep/tree/master/shinyapps/go61. Current database files are available at https://doi.org/10.5281/zenodo.1451847.
3 Results
We developed ShinyGO for in-depth analysis of gene lists, with graphical visualization of enrichment, pathway, gene characteristics and protein interactions (Fig. 1). It is based on annotation databases for 315 organisms, including 184 at Ensembl (vertebrates, release 96) (Aken et al., 2017), 59 from Ensembl Plants (release 43) (Bolser et al., 2017) and 72 from Ensembl Metazoa (release 43). See Supplementary File S2 for a list. We batch downloaded not only GO functional categorizations, but also gene ID mappings and other quantitative gene characteristics. Query genes are mapped to all gene IDs in the database, for both ID conversion and suggestion of possible organisms.
Fig. 1.
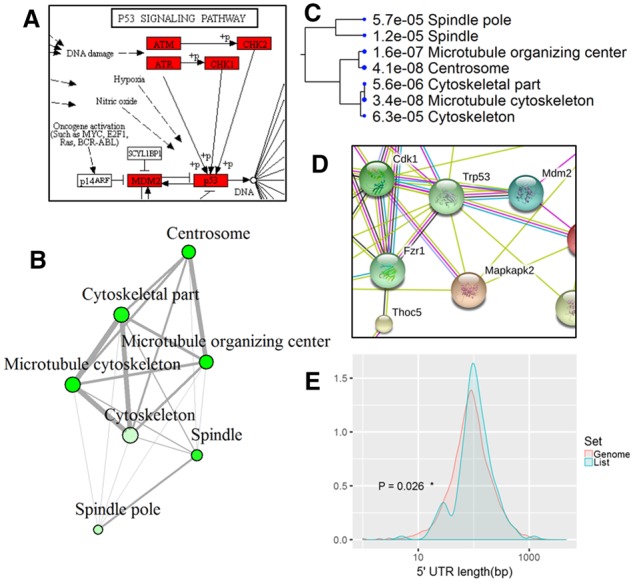
Example outputs of ShinyGO. (A) A partial KEGG pathway diagrams with genes highlighted. Enriched GO molecular component terms visualized as a network (B) and hierarchical clustering tree (C). (D) PPI network. (E) Distribution of the lengths of 5’ UTRs in query genes versus other coding genes in the genome
In addition to GO, pathways were downloaded directly from KEGG (Kanehisa et al., 2017). For human genes, various pathway data are also obtained from MSigDB (Liberzon et al., 2015), GeneSetDB (Araki et al., 2012), Reactome (Fabregat et al., 2016), as well as many sources of verified or predicted miRNA and TF target genes. In total, we compiled 72 394 gene-sets for human (Supplementary Table S2). Similar databases, such as GSKB (Lai, 2016) for mouse and araPath (Lai et al., 2012) for Arabidopsis, are included in ShinyGO.
ShinyGO can retrieve pathways diagrams from KEGG web server via API access using the pathview Bioconductor package (Fig. 1A). To visualize overlapping relationships among enriched gene-sets, we developed a network view (Fig. 1B) and a hierarchical clustering tree (Fig. 1C) of the enriched gene-sets. In a GO cellular component enrichment analysis, Figure 1B and C shows that three terms related to cytoskeleton overlap in many genes.
For species with fully sequenced genomes, ShinyGO plots the chromosomal locations of all the genes in the user’s list and conducts statistical analysis on the genomic features. It detects whether the genes are randomly distributed on the chromosomes using a Chi-squared test, compared with all other background genes in the genome. Similar tests are conducted to see if query genes differ from the rest in terms of the number of exons and transcript isoforms, and the types of genes (coding, non-coding, pseudogenes and so on). We plot the distribution of GC content, and the lengths of coding sequences, transcripts and UTRs (untranslated regions). T-tests are carried out to identify any significant differences between the query genes and all other background genes on the genome. As shown in Figure 1E, the query genes seem to have longer 5’ UTRs than other genes in the genome.
Enrichment analysis can also be conducted through API access to STRING (Szklarczyk et al., 2015), thus expanding the number of covered organisms. PPI networks are retrieved directly from STRING. ShinyGO produces a custom link to an interactive, annotated network on the STRING web site (Fig. 1D) with protein structures and PubMed.
A use case can be found in Supplementary File S1 with many example outputs. Through the analysis of 147 human genes upregulated by radiation, we were able to identify some expected pathways such as p53-mediated DNA damage response, as well as the underlying TFs (p53 and Rela/NF-κB) and even miRNAs (miR-145 and miR-21).
4 Discussion
ShinyGO is an intuitive, graphical tool for enrichment analysis. Even though its species coverage is not a broad as DAVID, ShinyGO has more comprehensive gene-sets regarding TF and miRNA target genes for human, mouse and Arabidopsis. We will continue to compile such information for other organisms and update the annotation database on a yearly basis. To improve reproducibility, older versions of the database will be made available to users.
Supplementary Material
Acknowledgements
The authors thank En Woo Sun, Brian Moore, Chad Julius, Luke Gassman, and Kevin Brandt for technical support, and Jianli Qi for compiling pathway databases.
Funding
This work was partially supported by National Institutes of Health [GM083226]; National Science Foundation/EPSCoR [IIA-1355423]; and by the State of South Dakota.
Conflict of Interest: none declared.
References
- Aken B.L. et al. (2017) Ensembl 2017. Nucleic Acids Res., 45, D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki H. et al. (2012) GeneSetDB: a comprehensive meta-database, statistical and visualisation framework for gene set analysis. FEBS Open Biol., 2, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolser D.M. et al. (2017) Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomic data. Methods Mol. Biol., 1533, 1–31. [DOI] [PubMed] [Google Scholar]
- Fabregat A. et al. (2016) The reactome pathway knowledgebase. Nucleic Acids Res., 44, D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W. et al. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. et al. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P. et al. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol., 8, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V. et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E.A. (2016) GSKB: a gene set database for pathway analysis in mouse. bioRxiv, 0802511. doi: 10.1101/082511. [Google Scholar]
- Lai L. et al. (2012) AraPath: a knowledgebase for pathway analysis in Arabidopsis. Bioinformatics, 28, 2291–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A. et al. (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst., 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. et al. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res., 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X. et al. (2013) PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res., 41, W98–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.