

Abstract
Both NF-κB pathway and complement activation appear to be involved in kidney damage induced by trichloroethylene (TCE). However, any relationship between these two systems has not yet been established. The present study aimed to clarify the role of NF-κB in complement activation and renal injury in TCE-sensitized BALB/c mice. Mice were sensitized by an initial subcutaneous injection and repeated focal applications of TCE to dorsal skin at specified timepoints. NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) was injected (intraperitoneal) before the final two focal TCE challenges. In the experiments, mice had their blood and kidneys collected. Kidney function was evaluated via blood urea nitrogen (BUN) and creatinine (Cr) content; renal histology was examined using transmission electron microscopy (TEM). Kidney levels of phospho-p65 were assessed by Western blot and kidney mRNA levels of interleukin (IL)-1β, IL-6, IL-17, tumor necrosis factors (TNF)-α, and p65 by real-time quantitative PCR. Presence of C3 and C5b-9 membrane attack complexes in the kidneys was evaluated via immunohistochemistry. The results showed there was significant swelling, vacuolar degeneration in mitochondria, shrinkage of microvilli, disappearance of brush borders, segmental foot process fusion, and glomerular basement membrane thickening (or disrobing) in kidneys from TCE-sensitized mice. In conjunction with these changes, serum BUN and Cr levels were increased and IL-1β, IL-6, IL-17, and TNFα mRNA levels were elevated. Levels of p65 and phospho-p65 protein were also up-regulated, and there was significant C3 and C5b-9 deposition. PDTC pre-treatment attenuated TCE-induced up-regulation of p65 and its phosphorylation, complement deposition, cytokine release and renal damage. These results provide the first evidence that NF-κB pathway has an important role in TCE-induced renal damage mediated by enhanced complement activation in situ.
Keywords: Trichloroethylene, renal injury, NF-κB, p65
Introduction
Trichloroethylene (TCE) is a solvent used for cleaning electronic and metal components in many industrial applications. Growth in the electronics industry in many countries resulted in increased numbers of TCE-exposed workers (Zhu et al. 2005; Watanabe et al. 2011). TCE can enter the body via the respiratory tract, gastrointestinal tract, and skin. Workplace exposure occurs primarily by inhalation of vapors. However, skin absorption can occur if a worker comes into contact with TCE-contaminated liquids. Accordingly, the latter route is particularly pertinent to the issue of skin sensitization. Based on known health effects and risks for exposure, government agencies have established permissible exposure limits. For example, the Ministry of Health in China defined the Maximum Allowable Concentration (MAC) for TCE in the workplace at 30–50 mg/m3 and a Permissible Exposure Concentration-Time-Weighted Average (PEC-TWA) at 30 mg/m3.
Increasing evidence has demonstrated exposure to TCE may cause organ damage in humans, including trichloroethylene hypersensitivity syndrome (THS), as well as renal and liver injury (Khan et al. 2009; Xu et al. 2011). Allergic hepatitis in a guinea pig maximization test (Tang et al. 2008) and mouse liver injury (Ramdhan et al. 2010) have also been shown to evolve from TCE exposure. Zhang et al. (2016) found THS caused by TCE could lead to immune-mediated kidney injury. When taken together, these TCE-induced multi-system injuries are categorized under the term “occupational dermatitis medicamentosa-like of TCE’’ (ODMLT) (Xia et al. 2006).
Renal damage associated with TCE exposure has become a major problem in workers (Chiu et al.2006; Xu et al. 2011). This type of chronic injury is mainly a result of repeated exposures to TCE, but acute kidney injury may also occur following a large single (dose) exposure. While the pathogenesis of the damage is not fully clear, its pathology is similar to T-lymphocyte-mediated Type IV allergic reactions (Sheman et al. 2004). However, the clinical manifestations of ODMLT cannot be explained solely by Type IV allergic reactions. A previous study reported complement activation played an important role in pathologies in TCE-treated mice (Karami et al. 2012). The three pathways of complement activation are all involved in generating key fragments including C3 and C5b-9 that can contribute to the tissue damage. It has been found that C5b-9 ultimately acts on kidney cells in a manner that leads to overall kidney damage (Wang et al. 2014). Yu et al (2012) showed that the level of deposition of C5b-9 (as well as C3) in renal sites was correlated with the extent of TCE-induced kidney disease, an outcome verified later by Wang et al. (2016).
NF-κB is an important nuclear transcription factor in eukaryotic cells (Lu et al. 2015) and has an important role in the immune response and cell apoptosis (Iwanaszko et al. 2012). The NF-κB family includes p65(RelA), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2). All these proteins share an N-terminus Rel homology domain (RHD) required for dimerization, nuclear targeting, binding to DNA, and interaction with inhibitory IκB proteins (except p50 and p52) (Patel et al. 2017). NF-κB signaling actives mainly through a canonical pathway, and occurs downstream of many stimuli, including pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α or interleukin (IL)-1. Under physiological conditions, p65-p50 dimers are bound by IκB (Johnson et al. 1999; Huang et al. 2000; Carlotti et al. 2000; Birbach et al. 2002). On activation, IκB kinase (IκK) complex (kinase IKKα\IΚKβ plus regulatory IΚKγ subunits) phosphorylates IB leading to its ubiquitination and proteasomal degradation (Figure 1). Loss of IκB enhances NF-κB nuclear accumulation and DNA binding, promoting transcription of target genes such as those for IL-1β, IL-6, IL-17, and TNFα (Ramaglia et al. 2008; Ghosh et al. 2012).
Figure 1.

Pathways involved in NF-κB and the complement system. IκKα = IκB kinase that phosphorylates IκKα; IκB = κB protein inhibitor; p50 and p65 = NF-κB family members; p-p65 = phosphorylated p65; C3 = Complement 3.
The observation NF-κB activation occurred in epidermal tissues after repeated combined exposures to TCE and ultraviolet radiation suggested a potential role for NF-κB in TCE-induced toxicities (Farrah and Sarwat 2012). Other evidence has also indicated that NF-κB activation leads to increases in C3 release (Gui et al. 2013). This would be in keeping with a previous study in which it was found that B7-H1 expression on epithelial cells induced by C5b-9 was also partly regulated by NF-κB activation (Chen et al. 2009). Because levels of complement in serum and tissues are elevated in TCE-treated mice, and this may lead to generation of C3a and C5a that bind to corresponding receptors (C3aR/C5aR), it ultimately may be these inter-related effects that give rise to the toxicities induced by TCE in the kidney (Zhang et al. 2013).
Whether NF-κB contributes to complement activation in TCE-induced sensitization and renal damage has not yet been examined. It was hypothesized here that TCE activates NF-κB and up-regulates C3 and C5b-9 expression, each of which then contributes to renal injury. To confirm this, studies a mouse model of TCE sensitization were performed here. In addition, to provide additional support for the hypothesis, studies using pyrrolidine dithiocarbamate (PDTC), an anti-oxidant that specifically inhibits NF-κB activation, was also employed.
Materials and Methods
Animals
BALB/c mice (female, 16-week-old, 18–24 g) were obtained from the Experimental Animal Center of Anhui (Anhui, China). All animals were healthy, housed at five/cage in SPF (specific pathogen-free) laboratory animal rooms maintained at 20°C and a 50% relative humidity with a 12-hr light: dark cycle. All mice had ad libitum access to standard rodent chow and filtered tap water. Mice were acclimatized 1 wk before being used in the experiments. All experimental protocols used were approved by the Anhui Medical University Animal Care and Use Committee.
Reagents
TCE and Freund’s complete adjuvant (FCA) were purchased from Sigma (St. Louis, MO). Olive oil (solvent for TCE) was bought from Shanghai Chemical Reagent Company (Shanghai, China). Pyrrolidine dithiocarbamate (PDTC) was purchased from Santa Cruz Biotechnology (CA, USA). Rabbit anti-mouse C5b-9 and C3 antibodies (polyclonal) were from Abcam (Cambridge, UK), anti p65 from Cell Signaling Technology (Danvers, MA), and anti-rabbit IgG from Beijing Zhong Shan-Golden Bird (Beijing, China). Other materials purchased were: RevertAid First Strand cDNA Synthesis Kits (ThermoScientific, Pittsburgh, PA); Histostain Plus and DAB substrate kits (Beijing Zhong Shan-Golden Bird), and LightCycler 480 SYBR Green I Master kits (Roche, Basel, Switzerland). Spurr resin was purchased from SPI Supplies (West Chester, PA).
Experimental design
In these studies, mice were randomly allocated into a blank control group (N = 5), a solvent control group (N = 5), a TCE-treatment group (N = 15), and PDTC+TCE pre-treatment group (N = 15). The TCE-sensitization model was referred to Wang et al. (2016). This route of skin application mimics the skin exposure in humans that might occur in the workplace and in the environment. As shown in Figure 2, in brief, all mice had an area of 2 cm × 2 cm on dorsal skin shaved with an electric clipper. 24 hr later, mice in the TCE sensitization groups were injected with 100 μl of a mixture of 50% FCA and 50% TCE (TCE: olive oil: acetone [5:2:3]) for the first stimulation (Day 1). On Days 4, 7, and 10, 100 μl of 50% TCE solution was painted on the shaved area for continued sensitization. On Days 17 and 19, the same area of the dorsal skin was then challenged with TCE (100 μl of 30% TCE [TCE: olive oil: acetone=3:2:5] solution). Following painting with TCE, the shaved area on each mouse was covered with filter paper and sealed with non-irritating tape for 24 hr. In PDTC+TCE pre-treatment group, an intraperitoneal injection of PDTC (100 mg/kg) was given to each mouse 24 hr before TCE challenge on Days 16 and 18 (Adams et al. 2011). Blank control mice received no treatments while solvent control mice were treated with the same proportions of olive oil and acetone without TCE on the above-specified days. Mice were weighed on Days 1, 5, 8, 11, 18, 20, and 22 to discern any overt toxicities from each treatment regimen. Weights of mice in all groups were unaffected by treatment (Table 1).
Figure 2.

Protocol for TCE induction of sensitization in mice. After 7 days of adaptive feeding, TCE was applied to the abdomen by intradermal injection. On Days 4, 7, and 10, TCE (100 μl of 30% TCE [TCE:olive oil:acetone, 3:2:5 {v/v/v}] solution) was applied to the dorsal skin. Animals were then re-challenged with TCE on Days 17 and 19. NF-κB inhibitor PDTC was applied to the PDTC pre-treatment group of mice 24 hr before each challenge.
Table 1.
Body weight (g) of mice in the different groups.
| Group | N | 1 d | 5 d | 8 d | 11 d | 18d | 20d | 22d |
|---|---|---|---|---|---|---|---|---|
| Blank control | 5 | 19.30 ± 1.86 | 19.82 ± 1.81 | 20.00 ± 1.91 | 20.24 ± 2.12 | 20.82 ± 2.58 | 20.88 ± 2.37 | 21.36 ± 2.49 |
| Solvent control | 5 | 20.34 ± 1.76 | 21.06 ± 2.03 | 21.04 ± 2.05 | 21.20 ± 1.90 | 21.96 ± 2.06 | 22.12 ± 2.19 | 22.16 ± 2.16 |
| TCE+ | 6 | 19.47 ± 1.98 | 19.58 ± 2.13 | 19.78 ± 2.14 | 20.08 ± 2.50 | 20.50 ± 2.67 | 21.00 ± 2.54 | 21.05 ± 2.60 |
| TCE− | 9 | 18.781 ± 1.22 | 18.77 ± 1.47 | 19.02 ± 1.49 | 19.63 ± 1.81 | 20.28 ± 2.38 | 20.69 ± 2.24 | 20.93 ± 2.30 |
| PDTC+ | 5 | 20.48 ± 1.82 | 21.02 ± 2.04 | 21.08 ± 2.22 | 21.02 ± 2.17 | 21.90 ± 2.31 | 21.94 ± 2.12 | 21.62 ± 2.10 |
| PDTC− | 10 | 19.52 ± 1.78 | 19.80 ± 2.69 | 20.04 ± 2.61 | 19.76 ± 2.16 | 21.37 ± 2.96 | 21.27 ± 2.67 | 21.23 ± 2.54 |
All data shown are means ± SD.
At 24 hr after the final treatment (i.e., on Day 20), cutaneous reactions in the dorsal skin were scored: 0 = no reaction; 1 = scattered mild redness; 2 = moderate and diffuse redness; and 3 = intensive erythema and swelling (Wang et al. 2016). Based on scores, mice were then divided into TCE sensitization-positive (TCE+), TCE sensitization-negative (TCE−), PDTC pre-treatment but TCE+ (PDTC+), and PDTC pre-treatment but TCE− (PDTC−) groups. Thereafter, mice in all groups were euthanized by CO2 asphyxiation and kidneys/blood was collected for investigation. Blood from the eye angular vein was collected into non-coated tubes, centrifuged at 4°C for 5 min at 3000 rpm to generate serum, and the serum was then analyzed for blood urea nitrogen (BUN) and creatinine (Cr) levels using an AU5800 automated biochemistry analyzer (Beckman Coulter, Brea, CA) to provide assessments of kidney function. The kidneys were removed, weighed, trimmed of extraneous tissue/adrenals, and processed for use in the protocols below.
Transmission electron microscopic (TEM) observation
The same portion of the left kidney of each mouse was isolated and then fixed in glutaral-dehyde-paraformaldehyde for 8–10 hr. Each specimen was then washed three times with phosphate buffer (pH 7.0), post-fixed with 1% OsO4 in phosphate buffer for 1 hr, and then washed three times with phosphate buffer. Thereafter, each sample was dehydrated through a graded series of ethanol (30, 50, 70, 80, 90, 95, and 100%) for 15–20 min at each step before being transferred into absolute acetone for 20 min. Specimens were then placed in 1:1 mixture of absolute acetone-Spurr resin for 1 hr at room temperature (RT) and then in a 1:3 mixture of these materials for 3 hr. After overnight incubation in resin mixture, specimens were placed in capsules containing Spurr resin and heated at 70°C for 9 hr. Sections were then stained in uranyl acetate\alkaline lead citrate for 15 min and then examined in a blinded manner by a certified histologist using a Hitachi H-7650 transmission electron microscope (Shanghai-ran Ultra Photoelectric Technology, Shanghai, China).
Immunohistochemical analyses of tissue C5b-9 and C3
Additional renal tissue from each left kidney was fixed as above and then de-paraffinized, re-hydrated, mounted on slides, and then treated for antigen retrieval [microwave]. Sections were then incubated in goat serum (15 min, RT) to block non-specific binding, and then overnight (4°C) in phosphate-buffered saline (PBS, pH 7.4) containing anti-C5b-9 antibody or anti-C3 antibody (each at 1:1000 dilution). The slides were then washed with PBS and incubated for 15 min at RT in a solution of anti-rabbit IgG in PBS (1:1000 dilutions). After rinsing with PBS, slides were processed using Histostain Plus and DAB kits, and then analyzed in a blinded manner using a light microscope. Grading of deposition in tissues\tubules was based on a semi-quantitative assessment of deposition area: 0 = area of positive reaction was ≤ 5% of total area analyzed on slide; 1 = 5–25%; 2 = 26– 50%; 3 = 51–75%; and, 4 = > 75%. Grading of immunoreaction intensity was estimated with four standard grades: 0 = no positive reaction; 1 = light brown; 2 = moderate brown; and, 3 = dark brown. Five areas in each slide (3 slides/mouse) were analyzed, and mean IHC group scores for renal C3 and C5b-9 were calculated.
Real-Time Quantitative PCR
From each mouse, the same portion of each right kidney was taken and its total RNA was extracted using Trizol as per manufacturer directions. Purity of the isolated RNA in each case was verified in a spectrophotometer (OD 260/280) and by examination of 28S and 18S rRNA band levels in agarose gels. After confirming sample purity/integrity, a RevertAid First Strand cDNA Synthesis Kit was used to produce cDNA. This material, in turn, was amplified by PCR using a SYBR Green I Master kit and a Light Cycler 480 system (Roche). Primers used for analyses are listed in Table 2. The amplification protocol used was: initial denaturation at 95°C for 10 min, followed by 45 cycles, consisting of 95°C for 15 sec, 60°C for 15 sec, and 72°C for 20 sec. Relative quantification of gene expression was analyzed using the 2-ΔΔCt method using the values for the blank control group for ‘calibration’. Fold-change values for IL-1β, IL-6, IL-17, TNFα, and p65 mRNA were all calculated based on GAPDH (housekeeping gene) levels.
Table 2.
Real-time quantitative PCR primers sequences.
| Primer | Sequences |
|---|---|
| GAPDH forward | 5’-ACCCCAGCAAGGACACTGAGCAAG-3’ |
| GAPDH reverse | 5’-GGCCCCTCCTGTTATTATGGGGGT-3’ |
| IL-17 forward | 5’-TGTCAATGCGGAGGGAAAGC-3’ |
| IL-17 reverse | 5’-CCCACCAGCATCTTCTCGAC-3’ |
| TNFα forward | 5’-CCCTCCTGGCCAACGGCATG-3’ |
| TNFα reverse | 5’-TCGGGGCAGCCTTGTCCCTT-3’ |
| IL-1 forward | 5’-GCCCATCCTCTGTGACTCAT-3’ |
| IL-1 reverse | 5’-AGGCCACAGGTATTTTGTCG-3’ |
| IL-6 forward | 5’-CCACTTCACAAGTCGGAGGCTTA-3’ |
| IL-6 reverse | 5’-GCAAGTGCATCATCGTTGTTCATAC-3’ |
| p65 forward | 5’-CCTCTGGCGAATGGCTTTACT-3’ |
| p65 reverse | 5’-GCTGGCTCTGAGGGAAAGATG-3’ |
Western blot analysis of p-p65 (phosphorylated p65)
The remaining portions of each right kidney were used for Western blot analyses. In brief, proteins in each sample were extracted in RIPA lysis buffer (1% Triton X-100, 1% deoxycholate, 0.1% SDS) to which protease inhibitor PMSF and phosphatase inhibitors had been added. After determining total protein content in bicinchoninic acid (BCA) assays, aliquots of each sample (10 μg/mouse) were resolved over 10% acrylamide gels and separated components electrotransferred to nitrocellulose membranes. Each membrane was then blocked (1.5 hr, RT) in PBS-5% non-fat milk, and then incubated overnight at 4°C in PBS containing anti-p65 antibody (1:2000 dilution) and anti-LaminB1 (loading control). Each membrane was then washed in PBS (4X) before being incubated (2 hr, RT) in PBS containing goat anti-rabbit IgG (1:1000 dilution). After a final wash, membrane proteins were visualized using a chemiluminescence (ECL) detection kit. Signal density was analyzed using Image Pro-plus software (v.6.0, MediaCybernetics, Rockville, MD).
Statistical analysis
All data were presented as means ± SD. One-way analysis of variance (ANOVA) and least significant difference (LSD) and Dunnett post-hoc tests were used to analyze the differences between the intervention groups and the appropriate control. A chi-square test was used to analyze differences between proportions or percentages (rates). All analyses were performed using SPSS v.11.0 software (SPSS, Chicago, IL). A p-value < 0.05 was considered significant.
Results
Sensitization rates
Sensitization rates were calculated (as number of redness or swelling/number of mice) in each group. Based on cutaneous reaction scores, mice were divided into sensitization-positive (dermatitis [+]) and -negative (dermatitis [-]) groups after 24 hr. The sensitization rate was 42.9% (6/15 positive) in the TCE+ group and 33.3% (5/15 positive) in the PDTC+ pre-treatment group. The difference between these outcomes was not significant (χ2 =0.14, P=0.71).
TCE-induced kidney injury
TEM revealed no significant pathological changes or cell infiltration in kidney tissues from blank or solvent control mice, suggesting healthy kidney in untreated control and no toxic effect of the solvents. There were however indications of significant swelling, vacuolar degeneration in mitochondria, shrinkage of microvilli, disappearance of brush borders, segmental foot process fusion, and basement membrane thickening (or peeling) in the glomerulus in kidneys of the TCE+ mice (Figure 3). Of note, TEM showed foot process fusions and mitochondria vacuolar degeneration were attenuated in PDTC+ pre-treated mice compared with what was seen in the TCE+ hosts.
Figure 3.
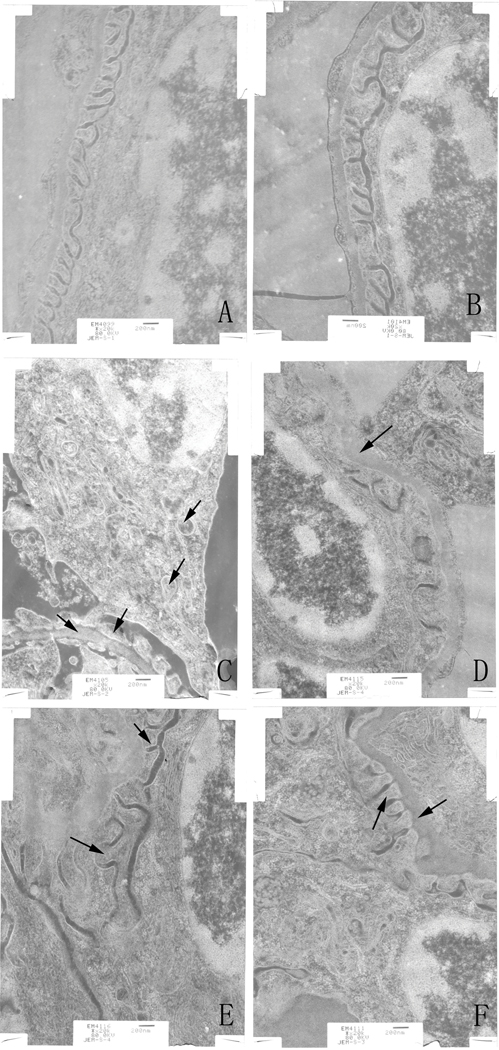
Ultrastructure changes of renal tissue as monitored by transmission electron microscopy. (A) Control. (B) Solvent control. (C) TCE+, with basement membrane thickening, foot process fusions, swelling and vacuolar degeneration of mitochondria. (D) TCE-, with basement membrane thickening. (E) PDTC+, basement membrane thickening, foot process fusions. (F) PDTC-, with basement membrane thickening, foot process fusions. Black arrows indicate podo-cytes and mitochondrial vacuolar degeneration, and glomerular basement membrane thickening. Changes noted in (C) were alleviated in (E). Magnification = 20,000X.
Serum BUN and Cr levels did not significantly differ between solvent and blank control mice (Figure 4), with respective values of 2.82 [± 0.22] and 2.81 [± 0.13] for BUN and 54.59 [± 8.56] and 56.68 [± 8.73] for Cr. In contrast, BUN and Cr levels were increased significantly in TCE+ mice (4.75 [± 0.11] [BUN], and 175.81 [± 6.80] [Cr]) compared with respective levels in solvent control mice. Values for PDTC+TCE mice were 3.77 [± 0.13] [BUN], and 133.78 [± 7.47] [Cr], significantly higher than in solvent control mice, but significantly lower than in TCE+ mice.
Figure 4.


Serum BUN and Cr levels in the study mice. (A) BUN. (B) Cr. Value significantly different vs. asolvent control, bcorresponding sensitization-negative group, or c corresponding sensitization-positive group (each p < 0.05).
C3 and C5b-9 deposition in kidneys
Representative immunohistochemical sections shown in Figures 5 and 6 indicate there were no visible immunoreactive depositions in the tissues from the blank and solvent control mice. In contrast, deposition of C3 was evident in samples from the TCE+ mice. The main deposition site for C3 was on the glomerulus. The IHC ratings revealed that C3 and C5b-9 deposition were not significantly different between solvent control and blank control group (Table 3), and C3 deposition in TCE+ was significantly increased compared with solvent control group (p < 0.05). This increase in C3 was dramatically inhibited in the PDTC+ group. Deposition of C5b-9, which was mainly in the renal tubules and abundant along the cortical tubular basement membrane, followed the same trends as C3. These data suggest that TCE stimulated-C3 and C5b-9 mediated complement activation and PDTC suppressed the effect.
Figure 5.

C3 deposition in mouse renal tissue. (A) Control. (B) Solvent control. (C) TCE+. (D) TCE−. (E) PDTC+. (F) PDTC−. Black arrows indicate deposition of C3. Magnification = 200×.
Figure 6.

C5b-9 deposition in mouse renal tissue. (A) Control; (B) solvent control; (C) TCE+; (D) TCE−; (E) PDTC+;(F) PDTC−. Black arrows indicate C5b-9 deposition. Magnification = 200X.
Table 3.
IHC ratings for C5b-9 and C3 in the different groups.
| Group | N | C5b-9 | C3 |
|---|---|---|---|
| Blank control | 5 | 1.11 ± 0.11 | 0.89 ± 0.38 |
| Solvent control | 5 | 0.89 ± 0.22 | 1.33 ± 0.34 |
| TCE+ | 6 | a3.89 ± 0.11 | a3.78 ± 0.19 |
| TCE− | 9 | 2.44 ± 0.29 | 2.56 ± 0.20 |
| PDTC+ | 5 | ab2.67 ± 0.19 | ab2.22 ± 0.19 |
| PDTC− | 10 | 1.78 ± 0.22 | 1.67 ± 0.34 |
Value significantly different from solvent control group, p < 0.05.
Value significantly different from TCE+, p < 0.05.
Role of NF-κB in TCE-induced kidney injury
There was no difference in p65 mRNA expression levels (normalized to GAPDH expression) in tissues from the blank (0.47 [± 0.07]) and solvent control mice (0.44 [± 0.05]) (Figure 7A). However, levels of p65 mRNA were significantly increased in the TCE+ (1.11 [± 0.06]) and PDTC+ (0.87 [± 0.04]) hosts. Figure 7B shows a representative Western blot for assessing expression of phospho-p65 (p-p65) protein in tissues from the solvent control, TCE+, and PDTC+ mice. At equal loading levels, compared to the value for control (1.21 [± 0.07] relative units [Figure 7C]) mice, p-p65 levels were significantly increased in samples from TCE+ (12.02 [± 1.12]) and PDTC+ (5.65 [± 0.65]) mice, with the latter values far lower than those in TCE+ mice (p < 0.05).
Figure 7.



Renal p65 gene and p-p65 protein expression. (A) p65 mRNA. Values significantly different vs. asolvent control or bTCE+ (each p < 0.05). (B) p-p65 expression. (C) Result expressed as ratio of p-p65:GAPDH. Values shown are means ± SD. Significant vs. asolvent control, or bTCE+ 72 hr group (each p < 0.05).
The mRNA levels of IL-1β, IL-6, IL-17 and TNFα in kidney tissues did not differ between the blank and solvent control mice (Figures 8A-8D). Values for these groups (normalized to GAPDH expression) were, 0.45 [± 0.05] (blank) and 0.53 [± 0.11] (solvent) for IL-1β, 0.25 [± 0.03] and 0.21 [± 0.03] for IL-6, 0.21 [± 0.05] and 0.22 [± 0.06] for IL-17, and 0.69 [± 0.02] and 0.56 [± 0.01] for TNFα. By contrast, IL-1β, IL-6, IL-17 and TNFα levels increased significantly in TCE+ mice (1.84 [± 0.10], 1.18 [± 0.03], 2.26 [± 0.02], and 2.87 [± 0.05], respectively; all p < 0.05 vs. controls. These effects however were drastically attenuated in PDTC+ mice (1.04 [± 0.05], 0.50 [± 0.01], 0.90 [± 0.07], and 1.81 [± 0.09], respectively; all p < 0.05 vs. TCE+ levels), even though the values were still significantly greater compared to control values for each gene. In all cases, non-sensitized mice values (TCE−, PDTC−) did not significantly differ from control levels.
Figure 8.




IL-1β, IL-6, IL-17, and TNFα gene expression in kidney. Expression levels were normalized against GAPDH. (A) IL-1β. (B) IL-6 (C) IL-17 (D) TNFα. Data shown are means ± SD. Value significantly different vs. asolvent control, bcorresponding sensitization-negative group, or vs. c corresponding sensitization-positive group (each p < 0.05).
Discussion
Results from this study indicated that TCE sensitization caused specific changes in sub-cellular structures and function in the kidneys in complement-associated inflammatory responses. The data also point to an involvement of NF-κB activation in these outcomes, as the NF-κB inhibitor PDTC effectively reduced these changes and inflammatory responses mirrored by consistent changes to NF-κB pathway signaling. To our knowledge, this is the first evidence that the NF-κB signal is involved in TCE-mediated immune responses associated with renal damage.
Exposure to TCE, a significant occupational hazard, causes ODLMT (IARC 1995; Hansen et al. 2001). It has been recognized that ODLMT-associated skin injury is often accompanied by renal damage, including chronic damage to renal tubules, i.e., both proximal and distal (Jollow et al. 2009). However, the mechanisms underlying renal injury caused by TCE are poorly defined and a subject of intense interest in environmental/occupational health research. A previous study showed complement system activation was evident in TCE-induced renal injury (Zhang et al. 2016) and C3 and C5b-9 deposition present in the kidney in TCE-sensitized guinea pigs (Tang et al. 2008). Taken together, these outcomes suggested that complement activation was important in TCE-associated kidney injury. However, it is not clear what factors in the cells/hosts might be being impacted to give rise to these changes in complement expression.
It is of interest to note that Qiang et al. (2012) reported that trichloroethanol, a metabolite of TCE, induced expression of Il-1β, Il-6, and TNFα in HaCaT cells, and this event appeared to be mediated in part by activation of NF-κB. Subsequent work by Woodruff et al. (2015) then found that C3 levels could be impacted by changes in expression of NF-κB transcription factor. When one takes these two separate studies together, this could suggest that the NF-κB pathway may potentially affect complement activation in TCE-mediated immune responses. However, to date, any specific role for NF-κB in TCE-induced renal damage has not yet been documented.
We tentatively hypothesize that activation of complement in TCE-induced immune renal injury is linked to NF-κB activation, and performed the current studies in an established TCE sensitization model to explore if/how NF-κB contributed to the process. To provide support for the hypothesis, this study also utilized PDTC to uncover the role NF-κB in TCE sensitization-induced kidney damage. Many investigations have demonstrated PDTC is a specific inhibitor of NF-κB and thus a powerful tool to evaluate the impact of NF-κB in biological processes (An et al. 2017; Bhat et al. 2017). The current study found that the rate of sensitization in PDTC pre-treated (PDTC+) mice was lower than that in TCE+ mice. This demonstrated to us that PDTC counteracted TCE-induced sensitization and provided evidence that NF-κB was likely to be activated in the process of TCE sensitization.
The present results also showed that TCE sensitization resulted in significant subcellular lesions in the kidneys, such as swelling, vacuolar degeneration of the mitochondria, microvilli shrinking, disappearance of brush border, segmental foot process fusion and glomerular basement membrane thickening or disrobing. PDTC pre-treatment attenuated these TCE-induced damages, implying again an important role for NF-κB in TCE-induced renal injury. Moreover, TCE-induced functional damage (revealed by increases in serum Cr and BUN) was also markedly reduced by PDTC pre-treatment.
Increased nucleoprotein p65and p-p65 expression are hallmarks of NF-κB activation (Shernan et al. 2004). The current investigation shows that levels of the p65 gene and its phosphorylated protein in kidneys were increased by TCE sensitization. The observed antagonistic effect of PDTC on these outcomes lends further credence to the notion that NF-κB activation was critically involved in TCE-induced sensitization/renal damage. Taking all the data together, we surmise that increases in p65 and its phosphorylation are the key steps in TCE-induced renal damage. The present study also investigated if the expression of the key pro-inflammatory cytokines TNFα, IL-1β, IL-6, and IL-17 (all regulated by NF-κB) in renal tissues was affected by TCE sensitization. The data show that the expression of genes for each of these pro-inflammatory cytokines was markedly increased in renal tissues of TCE-sensitized mice. Previous in vitro experiments showed PDTC could inhibit secretion of pro-inflammatory factors secondary to reductions in NF-κB activation (Liu et al. 2015). Confirming a role for NF-κB in the induction of the inflammatory cytokines, the study found that these above-noted increases in gene expression were each significantly reversed by pre-treatment of the mice with PDTC.
Given the new evidence of the relationship between NF-κB activation and the complement system (Cao et al. 2016; Nitkiewicz et al. 2017), the current study also investigated any association between NF-κB and complement in TCE-sensitized mice. By specifically examining deposition of C3 and C5b-9 in the kidneys, the study revealed increased deposition of C3 and C5b-9 in TCE-sensitized mice (which was, in turn, suppressed by PDTC). These effects were mirrored by consistent changes in serum biochemistry, p65 mRNA, and p-p65 protein levels in corresponding renal tissues. As there were no changes in values noted above with the TCE sensitization-negative groups (i.e., with\without PDTC), this demonstrated the changes were TCE sensitization-specific. Of further note, in TCE-sensitized mice, while C5b-9 was mainly deposited in the renal tubules, C3 was mainly deposited in glomeruli.
Evidence suggests that regulation of complement is closely related to inflammatory lesions of the kidney, especially in the glomerulus (Yu et al. 2012). The impact of C3 in renal damage is best exemplified in C3 glomerular nephritis, where defective C3 regulatory proteins [such as C3 nephritis factor] lead to an imbalance in complement and glomerular damage (Pavenatast et al. 2000; Xiao et al. 2013). Similarly, increased C5b-9 deposition in renal tubules contributes to renal tubular interstitial damage (Hori et al. 2013). The present findings that the complement system participates in TCE-sensitization-induced renal injury are in keeping with earlier observations suggesting that C3 and C5b-9 mainly act on, respectively, the glomerulus and renal tubules. The involvement of complement activation and the specific roles of key complement factors in TCE sensitization-mediated tissue damage have also been reported in the liver. These together may suggest that the complement signaling is a common pathway in TCE sensitization-induced multi-organ lesions.
Conclusions
This study evaluated the role of NF-κB and the relevant mechanisms associated with generation of inflammatory cytokines and activation of the complement system to give rise to renal damage in a BALB/c mouse model of TCE sensitization. Here, hosts underwent skin challenge with TCE with or without pretreatment with PDTC. Kidney histopathology and functional tests certified that there was renal structural/functional damage after TCE sensitization. The changes were found to be associated with up-regulated pro-inflammatory cytokine expression and increased C3 and C5b-9 renal deposition. PDTC pretreatment attenuated each of these changes. These data show that NF-κB appears to be involved in TCE sensitization-induced kidney damage in part by helping up-regulate inflammatory cytokine expression and the complement system. The data also suggest that C3 mainly causes glomerular lesion and C5b-9 mainly results in renal tubular damage.
Acknowledgments
This project was supported by grants from National Natural Science Foundation of China (81673141, 81502791, 81371730). CW gratefully acknowledges support from Biotechnology and Biological Sciences Research Council (BBSRC) (BB/P004695/1) and National Institute of Aging (NIA, 1R01AG049321–01A1).
Footnotes
Declaration of interest
The authors declare no conflicts of interest. The authors alone are responsible for the content of this manuscript.
References
- Adams G, LaRusch G, Stavrou E, Zhou Y, Nieman M, Jacobs G, Cui Y, Lu Y, Jain M, Mahdi F, et al. 2011. Murine prolyl-carboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood 117:3929–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F, and Sultana S. 2012. Repeated short-term stress synergizes the ROS signaling through up-regulation of NF-κB and INOS expression induced due to combined exposure of trichloroethylene and UVB rays. Mol. Cell. Biochem 360:133–145. [DOI] [PubMed] [Google Scholar]
- An X, Zhang L, Yao Q, Li L, Wang B, Zhang J, He M, Zhang J 2017. The receptor for advanced glycation end-products mediates podocyte heparanase expression through NF-κB signaling pathway. Mol. Cell. Endocrinol May3. pii: S0303–7207(17)30252–6. [DOI] [PubMed] [Google Scholar]
- Bhat O, Uday K, Harishankar N, Harishankar N, Ravichandaran L, Bhatia A, Dhawan V. 2017. IL-18-induced cell adhesion molecule expression is associated with feedback regulation by PPARγ and NF-κB in Apo E−/− mice. Mol. Cell. Biochem 428:119–128. [DOI] [PubMed] [Google Scholar]
- Birbach A, Gold P, Binder B, Hofer E, Schmid J. 2002. Signaling molecules of the NF-κB pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem 277:10842–10851. [DOI] [PubMed] [Google Scholar]
- Cao S, Wang J, Gao J, Wong M, To E, White V, Cui J, Matsubara J. 2016. CFH Y402H polymerphism and complement activation product C5a: Effects on NF-κB activation and inflammasome gene regulation. Br. J. Ophthalmol 100:713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlotti F, Dower S, Qwarnstrom E. 2000. Dynamic shuttling of NF-κB between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J. Biol. Chem 275:41028–41034. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang J, Guo G, Ruan Z, Jiang M, Wu S, Guo S, Fei L, Tang Y, Yang C, et al. 2009. Induced B7-H1 expression on human renal tubular epithelial cells by sub-lytic terminal complement complex C5b-9. Mol. Immunol 46:375–383. [DOI] [PubMed] [Google Scholar]
- Chiu W, Caldwell J, Keshava N, Scott C. 2006. Key scientific issues in the health risk assessment of trichloroethylene. Environ. Health Perspect 114:1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrah A, and Sarwat S. 2012. Repeated short-term stress synergizes ROS signaling through up-regulation of NF-κB and iNOS expression induced due to combined exposure of trichloroethylene and UV-B rays. J. Mol. Cell. Biochem 360:133–145. [DOI] [PubMed] [Google Scholar]
- Ghosh S, and Hayden M. 2012. Celebrating 25 years of NF-κB research. Immunol. Rev 246:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui D, Huang J, Guo Y, Chen J, Chen Y, Xiao W, Liu X, Wang N. 2013. Astragaloside IV ameliorates renal injury in streptozotocin-induced diabetic rats through inhibiting NF-κB-mediated inflammatory genes expression. Cytokine 61:970–977. [DOI] [PubMed] [Google Scholar]
- Hansen J, Raaschou-Nielsen O, Christensen J, Johansen I, McLaughlin J, Lipworth L, Blot W, Olsen J. 2001. Cancer incidence among Danish workers exposed to trichloroethylene. J. Occup. Environ Med 43:133–139. [DOI] [PubMed] [Google Scholar]
- Hori Y, Yamada K, Hanafusa N, Okuda T, Okada N, Miyata T, Couser W, Kurokawa K, Fujita T, Nangaku M. 1999. Crry, a complement regulatory protein, modulates renal interstitial disease induced by proteinuria. Kidney Intl. 56:2096–2106 [DOI] [PubMed] [Google Scholar]
- Huang T, Kudo N, Yoshida M, Miyamoto S. 2000. A nuclear export signal in the N-terminal regulatory domain of IκBα controls cytoplasmic localization of inactive NF-κB/IκBα complexes. Proc. Natl. Acad. Sci. USA 97:1014–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer). 1995. Dry cleaning, some chlorinated solvents and other industrial chemicals. IARC Monographs on the Evaluation of Carcinogenic Risks for Humans Vol. 63 Lyon, France, pp. 33–477. [PMC free article] [PubMed] [Google Scholar]
- Iwanaszko M, Brasier A, Kimmel M. 2012. The dependence of expression of NF-κB-dependent genes: Statistics and evolutionary conservation of control sequences in the promoter and in the 3’ UTR. BMC Genomics 13:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, van Antwerp D, Hope T. 1999. An N-terminal nuclear export signal is required for the nucleo-cytoplasmic shuttling of IκBα. EMBO J. 18:6682–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jollow D, Bruckner J, McMillan D, Fisher J, Hoel D, Mohr L. 2009. Trichloroethylene risk assessment: A review and commentary. Crit. Rev. Toxicol 39:782–797. [DOI] [PubMed] [Google Scholar]
- Karami S, Lan Q, Rothman N, Stewart P, Lee K, Vermeulen R, Moore L. 2012. Occupational trichloroethylene exposure and kidney cancer risk: A meta-analysis. J. Occup. Environ. Med 69:858–867. [DOI] [PubMed] [Google Scholar]
- Khan S, Priyamvada S, Khan S, Khan W, Farooq N, Khan F, Yusufi A. 2009. Effect of TCE toxicity on the enzymes of carbohydrate metabolism, brush border membrane and oxidative stress in kidney and other rat tissues. Food Chem. Toxicol 47:1562–1568. [DOI] [PubMed] [Google Scholar]
- Liu W, Fan Z, Han Y, Xu L, Wang M, Zhang D, Mao Y, Li J, Wang H. 2015. Activation of NF-κB signaling pathway in HSV-1-induced mouse facial palsy: Possible relation to therapeutic effect of glucocorticoids. Neuron 289:251–261. [DOI] [PubMed] [Google Scholar]
- Lu T, and Stark G. 2015. NF-κB: Regulation by methylation. Cancer Res. 75:3692–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitkiewicz J, Borjabad A, Morgello S, Murray J, Chao W, Emdad L, Fisher P, Potash M, Volsky D. 2017. HIV induces expression of complement component C3 in astrocytes by NF-κB-dependent activation of IL-6 synthesis. J. Neuroinflam 14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Zaghloul N, Lin K, Liu S, Miller E, Ahmed M. 2017. Hypoxia-induced activation of specific members of the NF-Κb family and its relevance to pulmonary vascular remodeling. Intl. J. Biochem. Cell Biol 92:141–147. [DOI] [PubMed] [Google Scholar]
- Pavenstast H 2000. Roles of the podocyte in glomerular function. Am. J. Physiol 278:F173–179. [DOI] [PubMed] [Google Scholar]
- Ramaglia V, Wolterman R, de Kok M, Vigar M, Wagenaar-Bos I, King R, Morgan B, Baas F. 2008. Soluble complement receptor 1 protects the peripheral nerve from early axon loss after injury. Am. J. Pathol 172:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdhan D, Kamijima M, Wang D, Ito Y, Naito H, Yanagiba Y, Hayashi Y, Tanaka N, Aoyama T, Gonzalez F, et al. 2010. Differential response to trichloroethylene-induced hepato-steatosis in wild-type and PPARα-humanized mice. Environ, Health Perspect. 118:1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenbaugh A, and Baldwin A. 2016. The NF-κB pathway and cancer stem cells. Cell 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shernan S, Fitch J, Nussmeier N, Chen J, Rollins S, Mojcik C, Malloy K, Todaro T, Filloon T, Boyce S. 2004. Impact of pexelizumab, an anti-C5 complement antibody, on total mortality and adverse cardiovascular outcomes in cardiac surgical patients undergoing cardio-pulmonary bypass. Ann. Thorac. Surg 77:942–950. [DOI] [PubMed] [Google Scholar]
- Tang X, Que B, Song X. 2008. Characterization of liver injury associated with hypersensitive skin reaction induced by trichloroethylene in the guinea pig maximization test. J. Occup. Health 50:114–121. [DOI] [PubMed] [Google Scholar]
- Wang F, Zha W, Zhang J, Li S, Wang H, Ye L, Shen T, Wu C, Zhu Q. 2014. Complement C3a binding to its receptor as a negative modulator of TH2 response in liver injury in TCE-sensitized mice. Toxicol. Lett 229:229–239. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang J, Ye L, Li S, Wang F, Zha W, Shen T, Wu C, Zhu Q. 2016. Plasma Kallikrein-Kinin system mediates immune-mediated renal injury in TCE-sensitized mice. J. Immuno-toxicol 30:1–13. [DOI] [PubMed] [Google Scholar]
- Watanabe H 2011. Hypersensitivity syndrome due to trichloroethylene exposure: A severe generalized skin reaction resembling drug-induced hypersensitivity syndrome. J. Dermatol 38:229–235. [DOI] [PubMed] [Google Scholar]
- Woodruff T, and Tenner A. 2015. Commentary on: “NF-κB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s Disease”. A cautionary note regarding C3aR. Front. Immunol 7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Qiu C, Li L. 2006. Editorial explanation for diagnostic criteria of occupational medica-mentosa-like dermatitis (OMLD) induced by trichloroethylene. Chin. Occup. Med 34:383–386. [Google Scholar]
- Xiao H, and He R. 2013. Complement system regulation and C3 glomerulopathy. J. Peking Univ 45:323–326. [PubMed] [Google Scholar]
- Xu H, Leng J, Shen T. 2011. The progress of China’s occupation of DMLT research. J. Chin. Ind. Med 24:196–199. [Google Scholar]
- Yu J, Leng J, Shen T, Zhou C, Xu H, Jiang T, Xu S, Zhu Q. 2012. Possible role of complement activation in renal impairment in TCE-sensitized guinea pigs. Toxicology 302:172–178. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zha W, Ye L, Wang F, Wang H, Shen T, Wu C, Zhu Q. 2016. Complement C5a-C5aR interaction enhances MAPK signaling pathway activities to mediate renal injury in TCE-sensitized BALB/c mice. J. Appl. Toxicol 36:271–284. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Shen T, Ding R, Liang Z, Zhang X. 2005. Cytotoxicity of trichloroethylene and perchloroethylene on normal human epidermal keratinocytes and protective role of Vitamin E. Toxicology 209:55–67. [DOI] [PubMed] [Google Scholar]