

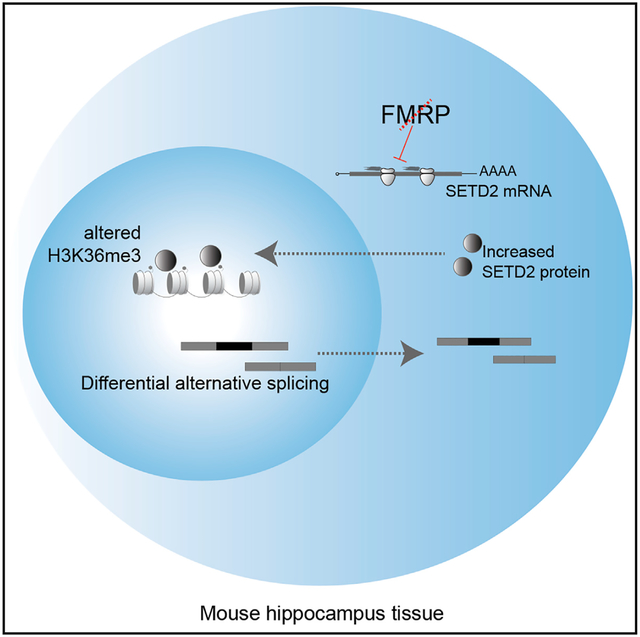
SUMMARY
Silencing of FMR1 and loss of its gene product, FMRP, results in fragile X syndrome (FXS). FMRP binds brain mRNAs and inhibits polypeptide elongation. Using ribosome profiling of the hippocampus, we find that ribosome footprint levels in Fmr1-deficient tissue mostly reflect changes in RNA abundance. Profiling over a time course of ribosome runoff in wild-type tissue reveals a wide range of ribosome translocation rates; on many mRNAs, the ribosomes are stalled. Sucrose gradient ultracentrifugation of hippocampal slices after ribosome runoff reveals that FMRP co-sediments with stalled ribosomes, and its loss results in decline of ribosome stalling on specific mRNAs. One such mRNA encodes SETD2, a lysine methyltransferase that catalyzes H3K36me3. Chromatin immunoprecipitation sequencing (ChIP-seq) demonstrates that loss of FMRP alters the deployment of this histone mark. H3K36me3 is associated with alternative pre-RNA processing, which we find occurs in an FMRP-dependent manner on transcripts linked to neural function and autism spectrum disorders.
Graphical Abstract
In Brief
Shah et al. show that many neuronal mRNAs are associated with FMRP-stalled ribosomes. FMRP-regulated ribosome stalling influences the chromatin landscape. These events in turn mediate alternative processing of pre-mRNAs linked to autism.
INTRODUCTION
Fragile X syndrome (FXS) is characterized by intellectual disability, developmental delays, social impairment, and other maladies. The cause of FXS is an iteration of ~200 or more CGG triplets in FMR1 that induces DNA methylation and transcriptional inactivation. Loss of the FMR1 gene product FMRP results in synaptic dysfunction and aberrant circuit formation, which produces neuro-pathological conditions during development (Santoro et al., 2012). FXS is modeled in Fmr1 knockout (KO) mice, which mimic many facets of the human disease, including learning and memory deficits, repetitive disorders, and susceptibility to seizures. In these animals, neuronal communication is frequently examined at hippocampal Schaffer collateral-CA1 synapses, which exhibit exaggerated metabotropic glutamate-receptor-dependent long-term depression (mGluR-LTD) (Huber et al., 2002). This form of synaptic plasticity normally requires protein synthesis, but in Fmr1-deficient animals, protein synthesis is both unnecessary for mGluR-LTD and excessive in neurons, which contributes to aberrant circuit formation and other hallmarks of the syndrome (Waung and Huber, 2009).
FMRP is an RNA-binding protein present in most cells; in neurons, its localization at postsynaptic sites is thought to control activity-dependent synapse remodeling by regulating mRNA expression (Richter et al., 2015). Considerable effort has been made to identify FMRP target mRNAs, but the most rigorous involves in vivo crosslink and immunoprecipitation (CLIP). CLIP combined with RNA sequencing (RNA-seq) has identified 842 target mRNAs in the post-natal day 11 (P11)–P25 mouse cortex and cerebellum (Darnell et al., 2011); predominantly a single mRNA in cultured cortical neurons (Tabet et al., 2016); 1,610 RNAs in the P13 cortex, hippocampus, and cerebellum (Maurin et al., 2018); and ~6,000 RNAs in HEK cells expressing epitope-tagged FMRP (Ascano et al., 2012). This diversity of FMRP CLIP targets could reflect the different procedures employed or the different brain cell types examined. Surprisingly, the majority of CLIP sites in mRNA are in coding regions (Darnell et al., 2011; Maurin et al., 2018), with a bias to some cis sequences (Anderson et al., 2016; Maurin et al., 2018). FMRP association with mRNA coding regions and co-sedimentation with polyribosomes (Khandjian et al., 1996; Feng et al., 1997; Stefani et al., 2004) suggests it normally inhibits translation by impeding ribosome translocation. Indeed, studies that examined polypeptide elongation (Udagawa et al., 2013) or susceptibility to puromycin release of nascent polypeptides (Darnell et al., 2011) indicate that FMRP regulates ribosome transit. Supporting evidence comes from studies showing that Drosophila FMRP interacts with ribosomal protein L5 (Ishizuka et al., 2002), which may preclude tRNA or elongation factors from engaging the ribosome and causing it to stall (Chen et al., 2014). Although these observations need to be extended to mammalian FMRP, they suggest a molecular mechanism by which FMRP stalls ribosomes. Such observations do not indicate how FMRP could stall ribosomes on specific mRNAs.
Ribosome profiling is a whole-transcriptome method for analyzing the number and positions of ribosomes associated with mRNA; when combined with RNA-seq, it yields heretofore unobtainable information on gene expression at high resolution (Ingolia et al., 2009; Ingolia, 2014; Brar and Weissman, 2015). We used ribosome profiling to explore FMRP regulation of gene expression in mouse adult neural stems cells (aNSCs), which in FXS have a proclivity to differentiate into glia at the expense of neurons (Liu et al., 2018; Luo et al., 2010; Guo et al., 2011). We identified mRNAs whose ribosome occupancy was either up or downregulated in FMRP-deficient cells or had altered steady-state levels reflected by their association with ribosomes. We identified yet additional mRNAs whose ribosome occupancy was “buffered” so that changes in their levels were compensated for by opposite changes in ribosome association (Liu et al., 2018).
Ribosome profiling at steady state cannot distinguish between translocating and stalled ribosomes, suggesting that studies examining FMRP-regulated translation by this method or translating ribosome affinity purification (TRAP) and RNA-seq (TRAP-seq) (Thomson et al., 2017; Liu et al., 2018, 2019; Greenblatt and Spradling, 2018; Das Sharma et al., 2019) may overlook key events leading to FXS. Moreover, if intact brain circuitry is important for FMRP regulation, then cultured neurons might not reveal important aspects of FMRP function. To circumvent these issues, we have developed a hippocampal slice assay to detect mRNA-specific ribosome translocation dynamics. Hippocampal slices maintain the in vivo synaptic connectivity and cellular architecture and express protein synthesis and FMRP-dependent forms of synaptic plasticity (Huber et al., 2002). Profiling over a time course of ribosome runoff in slices demonstrates a wide range of ribosome translocation rates on specific mRNAs, from rapid (rates similar to those in embryonic stem [ES] cells; Ingolia et al., 2011) to near-complete stalling. Many mRNAs retain 4–6 ribosomes after runoff in wild-type (WT), but not in FMRP-deficient, slices. Some RNAs associated with FMRP-stalled ribosomes encode proteins involved in neurologic function and transcriptional regulation. One such RNA encodes SETD2, a methyltransferase for the chromatin mark H3K36me3. We find increased SETD2 protein in Fmr1-deficient hippocampus. Chromatin immunoprecipitation sequencing (ChIP-seq) demonstrates that distribution of this mark varies in a genotype (i.e., WT versus Fmr1-deficient)-dependent manner in hippocampal tissue. In mammals, apart from blocking cryptic transcription initiation and DNA damage response, H3K36me3 is correlated with pre-mRNA processing events such as alternative splicing (Kim et al., 2011; Kolasinska-Zwierz et al., 2009). RNA-seq reveals that splicing events such as exon skipping are altered in FMRP-deficient slices and that these transcripts are linked to neural function and autism spectrum disorders. Aberrant RNA splicing of synapse related genes is prevalent in autism spectrum disorders (Lee et al., 2016; Quesnel-Vallières et al., 2016; Smith and Sadee, 2011). Our study demonstrates that a reduction in ribosome stalling in the Fmr1-deficient brain results in a cascade of epigenetic and RNA processing changes that likely contribute to neurologic disease.
RESULTS
Hippocampal Schafer collateral-CA1 synapses exhibit an exaggerated mGluR-LTD in Fmr1-deficient mice that is aberrantly protein-synthesis independent (Nosyreva and Huber, 2006; Huber et al., 2000). To identify mRNAs whose basal expression is misregulated, we prepared CA1-enriched hippocampal slices (400 μm) (Figure 1A), performed ribosome profiling and RNA-seq (Figure 1B), and calculated translational efficiency (TE), the ratio of ribosome footprints to RNA-seq reads. We identified RNAs with changes in TE (TE up/TE down) or RNAs whose levels were elevated or reduced but without changes in TE (mRNA up/mRNA down) (Figure 1C; Table S1) (nominal p value < 0.01, false discovery rate [FDR] = 0.097 by permutation test). A Gene Ontology (GO) analysis of the biological processes most enriched in each group shows that the mRNA-down group encodes factors that mediate taxis/chemotaxis, while the mRNA-up group encodes factors involved in ribonucleotide metabolism (Figure 1D, S1A, and S1B). The TE-down mRNAs encode factors that control synaptic transmission (Figures 1D and S1C). The mRNA-down group is enriched for cellular components linked to the extracellular matrix, while the mRNA-up group encodes the myelin sheath and spliceosome-interacting factors. The TE-down group is involved in the extracellular matrix and synaptic architecture (Figures 1E and S1C). No GO terms from the TE-up group were statistically significant.
Figure 1. Ribosome Profiling Reveals Diverse Changes of Gene Expression in Fmr1 KO Hippocampus.
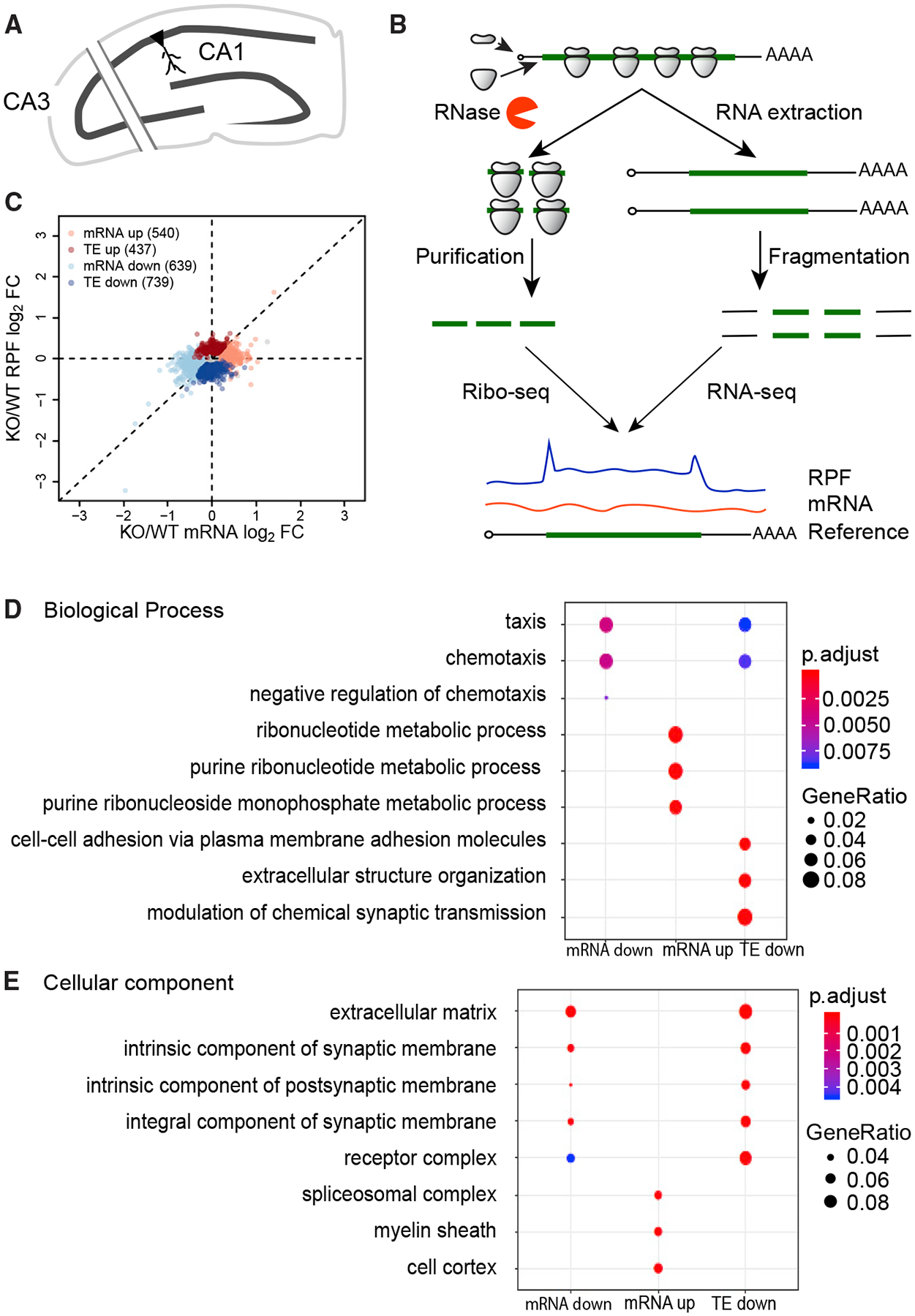
(A) Schematic diagram of the hippocampal slice preparation. To reduce spontaneous electrical activity, region CA3 was excised. 8–10 slices from six to eight mice/genotype were pooled per biological replicate (wild-type [WT], n = 4 and Fmr1 KO, n = 3).
(B) Schematic diagram of the experimental procedures for ribosome profiling.
(C) Scatterplot of expression changes of mRNA levels and ribosome-protected fragments (RPFs). Dysregulated mRNAs in the absence of FMRP are classified into four regulatory groups. 14,459 genes past filtering are used for the scatterplot. Nominal p < 0.01, FDR = 0.097 by permutation test. FC, fold change; TE, translational efficiency.
(D) Top three GO terms of Biological Process enriched in each regulatory group. The enrichment (gene ratio) is represented by the size of dots. The enrichment significance (adjusted p value) is colorcoded.
(E) Top three GO terms of Cellular Component enriched in each regulatory group.
Heatmaps of the top 20 mRNAs with altered up and down TEs in Fmr1-deficient slices compared to the WT are shown in Figures 2A and 2B. Examples of the change in TEs are depicted in Figures 2C and 2D. Gfpt2 (glutamine-fructose-6-phosphate transaminase 2) mRNA is nearly unchanged in Fmr1 KO but has an increase in ribosome footprints; Figure 2C). Conversely Lhfpl5 (lipoma high-mobility group protein isoform C [HMGIC] fusion partner-like 5) mRNA has reduced numbers of ribosome footprints while RNA levels are mostly unchanged (Figure 2D). Neither Gfpt2 nor Lhfpl5 mRNAs have FMRP CLIP sites. Figure 2E shows boxplots for all 843 FMRP CLIP RNAs (p value < 0.0001, Wilcoxon test) (Darnell et al., 2011). As a group, these RNAs decrease significantly in Fmr1 KO, which is tracked by commensurate decreases in ribosome footprints resulting in no change in TE.
Figure 2. Characterization of Genes with TE Changes in Fmr1 KO Hippocampus.
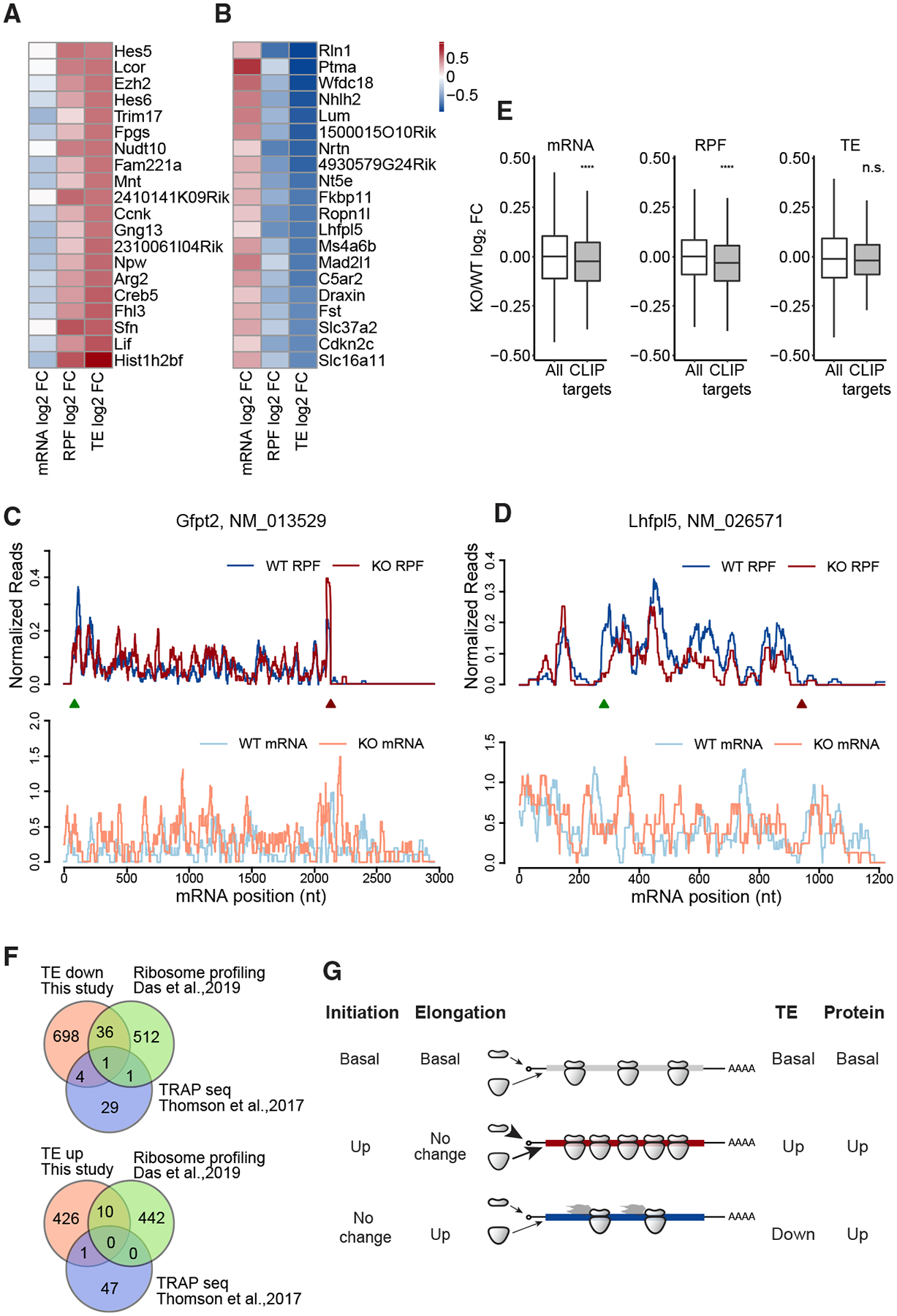
(A) Heatmap of expression changes (log2FC KO/WT) for the top 20 RNAs in the “TE up” group.
(B) Heatmap of expression changes (log2FC KO/WT) for the top 20 RNAs in the “TE down” group.
(C) Read distributions on the Gfpt2 RNA of TE-up group. Normalized RPF reads (top) and mRNA reads (bottom) averaged across all replicates are plotted along the mRNA nucleotide positions with green and red triangles for annotated start and stop codons, respectively. For visualization purposes, the curves were smoothed within a 30-nt window.
(D) Read distributions on the Lhfpl2 RNA of the TE-down group.
(E) Boxplots visualize the medians of expression changes for FMRP CLIP targets. The lower and upper hinges correspond to the first and third quartiles. The whiskers extend from the hinges to the largest and smallest values no further than 1.5-fold of interquartile range. Outliers are not shown. Gene expression changes of CLIP genes were compared to those of all genes used for differential expressed genes (DEG) analysis (ns, not significant; ****p < 0.0001; Wilcoxon test). CLIP genes are the FMRP targets identified in (Darnell et al., 2011). All grouped data are presented as mean ± s.e.m.
(F) Overlap of the TE-down and TE-up genes detected in this study (orange) with those of Das Sharma et al. (2019) (green) and the TRAP-seq data of Thomson et al. (2017) (blue).
(G) Schematic models of ribosome density (TE) changes that reflect increased protein synthesis rates.
See also Table S1.
We compared our TE-down and TE-up groups in Fmr1 KO hippocampus with results from Das Sharma et al. (2019), who performed ribosome profiling on mouse frontal cortex, and Thomson et al. (2017), who performed TRAP-seq from hippocampal CA1 pyramidal neurons (Figure 2F). Only 37 of our TE-down RNAs were detected by Das Sharma et al., 2019 (~7% overlap), and only 5 were detected by Thomson et al. (2017) (~17% overlap). Even fewer of our TE-up RNAs were detected in either of those studies (Figure 2F) (~2% overlap). We surmise that the differences in methods and source material are likely responsible for little overlap among the studies.
Our data show almost all changes in ribosome footprint number in Fmr1-deficient slices can be attributed to altered RNA levels irrespective of whether the transcripts are FMRP CLIP targets. This suggests that the increase in incorporation of labeled amino acids into protein in Fmr1 KO hippocampal slices (e.g., Dölen et al., 2007; Udagawa et al., 2013; Bowling et al., 2019) is mostly due to changes in RNA levels and/or ribosome transit rate. However, TE may not necessarily reflect protein production. For example, increased initiation with no change in elongation would produce increased ribosome footprints and yield an elevated level of protein product (Figure 2G). If there is no change in initiation but elevated elongation, the TE will appear to be reduced but the production of protein will be increased. Consequently, ribosome profiling at steady state, which cannot distinguish between transiting and stalled ribosomes, may inadequately represent translation if there are changes in elongation. To assess whether FMRP regulates ribosome dynamics, we modified our procedure to investigate temporal changes in transcriptome-wide ribosome translocation rates.
Ribosome Runoff Dynamics in Hippocampal-Cortical Slices
We next performed ribosomal profiling and RNA-seq in hippocampal-cortical slices at different time points after blocking initiation with homoharringtonine (HHT). HHT halts ribosomes on initiation codons but allows elongating ribosomes to continue translocating until they run off (Ingolia et al., 2011; Lee et al., 2012). We aimed to determine differences in ribosome translocation rates of all expressed genes in WT tissue slices. Slices were incubated with HHT for 0–60 min and ribosome transit stopped at specific times with cycloheximide; the samples were then used for ribosome profiling (Figure 3A; Table S1). A metagene analysis of the ribosome footprints on 1,401 mRNAs with open reading frames (ORFs) of >3,000 nt aligned with the start and stop codons is shown in Figure 3B, indicating that the ribosomes runoff in a 5′→3′ direction. We chose mRNAs with ORFs >3,000 nt to ensure that ribosome runoff was not complete within 10 min of HHT treatment and read densities could be obtained at each nucleotide position. The read densities were normalized to the average densities of the last 500 nt of the protein coding sequence (CDS) and is based on the P site of the ribosome footprints. A linear regression analysis between the HHT treatment time and ribosome runoff distances was then used to estimate a global elongation rate of 4.2 nt/s at 30°C (Figure 3C). By comparison, the global elongation rate in cultured ES calls is 1.7 nt/s at 37°C (Ingolia et al., 2011).
Figure 3. Runoff Ribosome Profiling of WT Mouse Brain Slices.
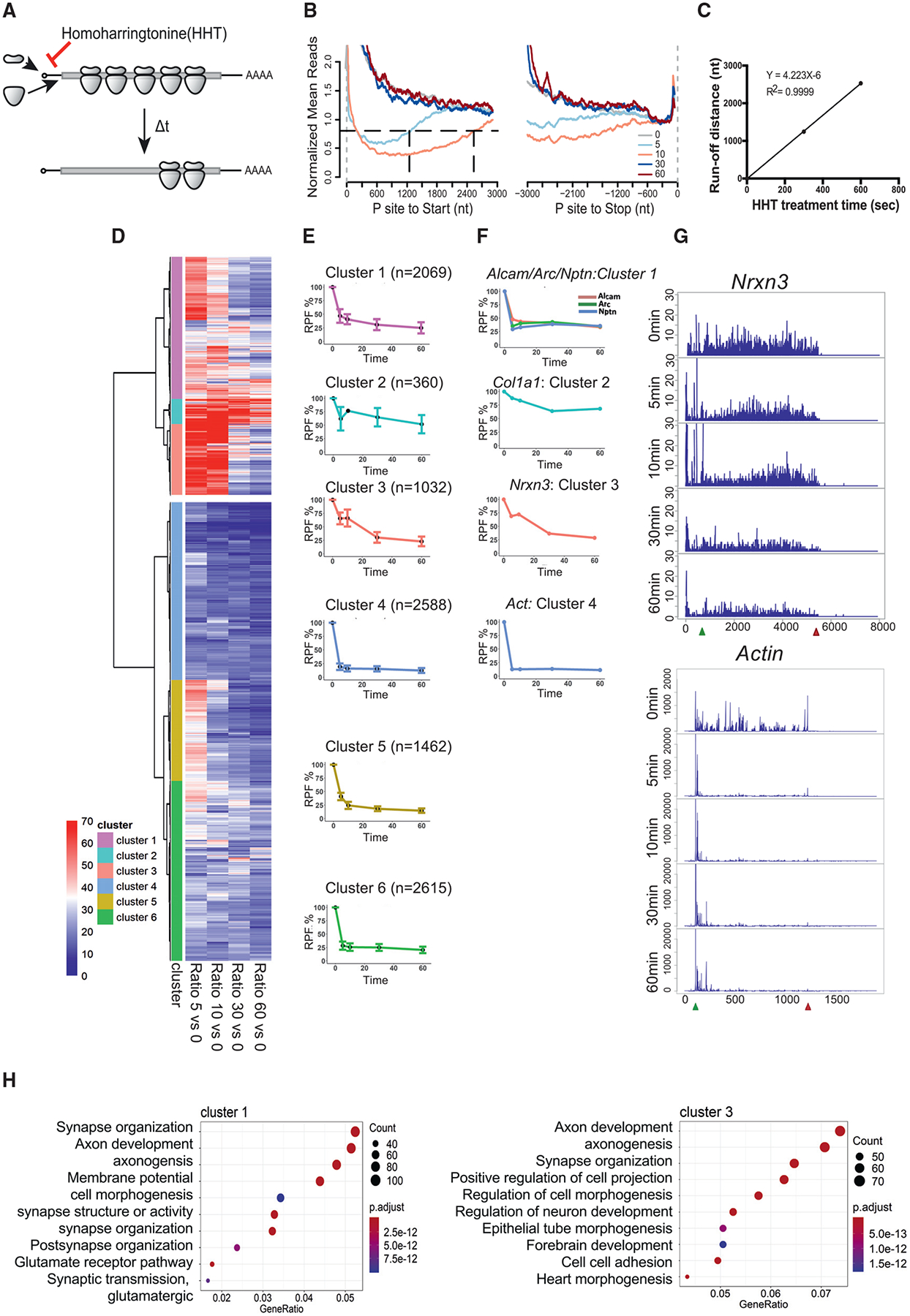
(A) Schematic diagram of homoharringtonine (HHT) runoff ribosome profiling. WT mouse brain slices were treated with 20 mM HHT, an inhibitor of translation initiation, to allow ribosome runoff for 5, 10, 30, and 60 min (t) at 30°C. 10 slices from two or three mice were pooled per time point of HHT treatment.
(B) Metagene plot of RPFs after HHT treatment. Reads are mapped transcripts (n = 1,401) with CDS longer than 3,000 nt and aligned at the annotated start and stop codons (gray vertical dash lines). The read densities at each nucleotide position are normalized to the average density of the last 500 nt of CDS and averaged using the P sites of RPFs. For visualization purposes, the curves were smoothed within a 90-nt window. Black horizontal dash line indicates the arbitrary 0.8 threshold to estimate the relative runoff distances (black vertical dash lines).
(C) Linear regression between the HHT treatment time and ribosome run-off distances (from B) to estimate the global elongation rate (4.2 nt/s).
(D) Cluster analysis of gene groups with distinct ribosome runoff patterns. The RPFs of each gene at each time point was normalized to time 0. The Euclidean distance matrix was then calculated, followed by hierarchical clustering using Ward’s agglomeration method (Ward, 1963).
(E) Ribosome runoff patterns for each subcluster. The global pattern of each subcluster was summarized using the corresponding median and standard deviation in each time point. The number of RNAs in each subcluster is shown in parentheses.
(F) Representative ribosome runoff profiles that reflect each subcluster. The runoff pattern for Actin is similar to subclusters 4–6.
(G) Ribosome footprints for Nrxn3 and Actin mRNAs during the runoff time period.
(H) GO terms for subclusters 1 and 3. Gene ratio refers to the percentage of total differentially expressed genes in the given GO term.
We performed a cluster analysis of all mRNAs with respect to number of ribosome-protected fragments (RPFs) at each time point relative to time 0. Briefly, Euclidean distance was first computed, followed by hierarchical clustering using Ward’s algorithm (Ward, 1963), and the analysis of group similarities (ANOSIM) test (Clarke, 1993) was then used to assess whether distances between clusters were statistically greater than within clusters (Figure 3D). The mRNAs fell into two broad clusters (ANOSIM R = 0.56, p value < 0.001): those whose ribosome runoff occurred relatively quickly (i.e., within 5–10 min, bottom cluster), and those whose runoff occurred slowly, if at all (top cluster). These two clusters were further grouped into three sub-clusters each for six total subclusters (ANOSIM R = 0.64, p < 0.001). Subclusters 1–3 had slow runoff times, while subclusters 4–6 had relatively fast ribosome runoff times (Figures 3D and 3E). The ribosomes on mRNAs in subcluster 2 had the slowest runoff time. We determined the ribosome runoff patterns for the mRNAs in each subcluster (Figure 3E). For example, mRNAs for Alcam (activated leukocyte cell adhesion molecule), Arc (activity-regulated cytoskeleton), and Nptn (neuroplastin) are present in subcluster 1, Col11 (collectin subfamily 11) in subcluster 2, Nrxn3 (neurexin 3) in subcluster 3, and actin in subcluster 4 (Figure 3F). As examples, Figure 3G shows the ribosome footprints of Nrxn3 and actin over time; Figure S2A shows ribosome footprints for additional mRNAs. Although there was some CDS-length dependency of the ribosome runoff rates, the correlation was not linear (Figure S2B). GO term analysis showed that specific biological activities were associated with each subcluster. Genes in subclusters 1 and 3 (slow runoff times) were associated with neural function such as axon development and synapse organization (Figure 3H). Subcluster 2, which contained ribosomes with the slowest runoff rates, had genes encoding the extracellular matrix; subcluster 4 contained genes involved in metabolic functions; subcluster 5 was associated with mRNA processing and regulation; and subcluster 6 was associated with organelle localization (Figure S2C). These results suggest that clusters of mRNAs with specific functions have differential ribosomal transit rates.
FMRP Controls Ribosome Translocation on Specific mRNAs
To investigate whether FMRP controls ribosome stalling on specific mRNAs, we first determined whether it is present in ribosome-containing complexes (Feng et al., 1997; Khandjian et al., 2004; Stefani et al., 2004; El Fatimy et al., 2016). Extracts from cortices were treated with nonionic detergent (NP40) in the presence or absence of RNase and sedimented through sucrose gradients. Figure S3A shows FMRP sedimented to a “heavy” ribosome-containing region of the gradient (Stefani et al., 2004; Darnell et al., 2011). These data suggest that FMRP is associated with ribosomes in heavy fractions and thus could play a role in ribosome stalling.
We next performed sucrose gradient analysis of slices treated with HHT for 30 min (Figure 4), which allows ribosomes on most genes to run off (cf. Figure 3). Figure 4A shows that most FMRP co-sediments with polysomes as shown previously (e.g., Feng et al., 1997; Stefani et al., 2004), but after HHT treatment, it is more prevalent in “medium” gradient fractions that correspond to four to six ribosomes most likely due to association with stalled ribosomes. Other proteins (e.g., UPF1, MAP2, and eEF2) do not show a similar change in sedimentation (Figure S3B). A small amount of FMRP remains in a heavy (>7 ribosomes) region of the gradient (Figure 4B). The same experiments were performed with Fmr1 KO slices (Figures 4C and 4D). RNA-seq of the heavy fractions showed that 1,574 RNAs and 686 RNAs were reduced or elevated, respectively, in both genotypes, indicating a sensitivity or resistance to HHT treatment (Figure 4E). Actin is one RNA that was sensitive to HHT and was depleted by ribosome runoff (Figure 4F), which is consistent with results from the HHT time course (Figures 3F and 3G). Map1b RNA, on the other hand, was enriched in the heavy polysomes by HHT treatment (Figure 4G). Figure 4H shows GO terms for the RNAs depleted in the heavy fractions after runoff, which include factors involved in RNA metabolism. Figure 4I shows GO terms for RNAs enriched in the heavy polysomes after HHT treatment, which include factors involved in membrane and ion transport.
Figure 4. Substantial FMRP Remains Associated with Polysomes after HHT Treatment.
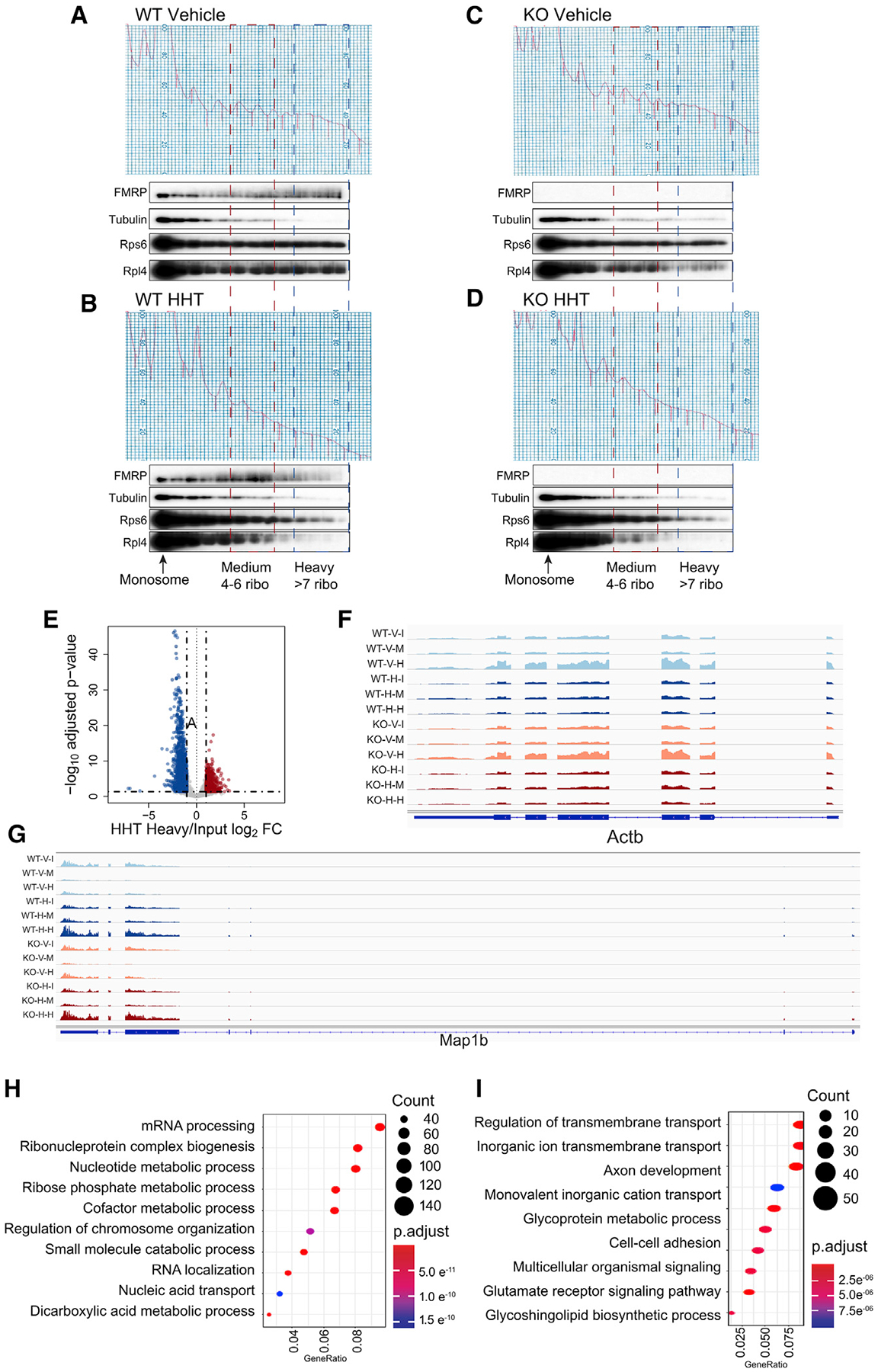
(A–D) WT mouse hippocampal slices were treated with DMSO vehicle (A) or 20 mM HHT (B) for 30 min at 30°C. In parallel, Fmr1 KO mouse hippocampal slices were also treated with DMSO vehicle (C) or 20 mM HHT (D) for 30 min at 30°C. Slices were homogenized and applied to 15%–45% (w/w) sucrose gradients, which were fractionated with continuous monitoring of A260 after ultracentrifugation. Fractions were collected for immunoblotting with indicated antibodies to detect the association of FMRP with polysomes. 10 slices from two or three mice/genotype were pooled per biological replicate (WT, n = 2; Fmr1 KO, n = 2).
(E) RNA sedimenting to heavy fractions containing more than seven ribosomes from WT slices treated with HHT was analyzed relative to input. 1,574 RNAs were reduced and 686 were elevated relative to input after HHT treatment.
(F) Actin mRNA reads in the designated conditions (WT; V, vehicle, I, input; H, heavy fractions; KO, Fmr1 KO).
(G) Map 1b mRNA reads in the designated conditions.
(H) GO terms of RNAs depleted in heavy fractions relative to WT.
(I) GO terms of RNAs enriched in heavy fractions relative to WT.
In contrast to the heavy polysome fractions, we found genotype-specific differences in the RNA-seq of the medium polysome fractions (Table S1). Forty-six RNAs were downregulated in HHT-treated Fmr1 KO slices relative to WT, indicating that they are normally bound by FMRP-stalled ribosomes (Figure 5A). Eight of these mRNAs encode chromatin and transcriptional regulators, and 23 encode proteins involved in neural function (Figure 5B). One is Ankrd12 (ankyrin repeat domain containing 12), which is depleted in the KO slices following HHT treatment (Figure 5C). Figure 5C shows that after HHT treatment, Ankrd12 RNA contains fewer stalled ribosomes in the medium fractions of Fmr1 KO slices, confirming this mRNA is under FMRP regulation. Figure 5D demonstrates that compared to all RNAs, the HHT-sensitive RNAs (Figure 5B) showed significant decreases in RPFs and TEs, but not RNA levels, in Fmr1 KO relative to WT (ribosome profiling data of these RNAs from Figure 2; **p < 0.01; ***p < 0.001, Wilcoxon test) showing the consistency between steady-state ribosome profiling and the dynamic ribosome runoff assay.
Figure 5. FMRP Stalls Ribosomes on Specific mRNAs.
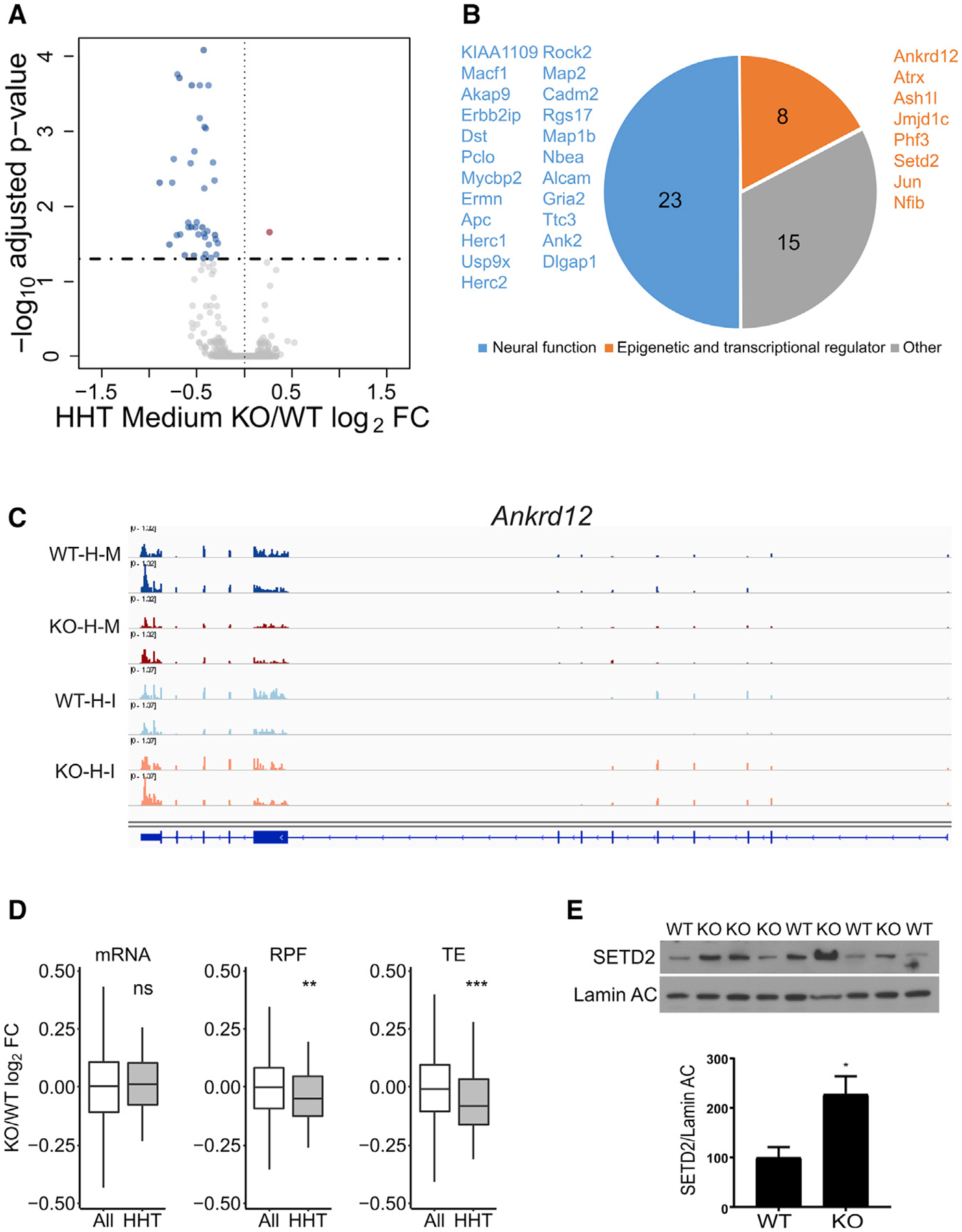
(A) RNA sedimenting to medium polysomes containing four to six ribosomes after HHT treatment of hippocampal slices; 46 RNAs are downregulated and 1 is upregulated in Fmr1 KO relative to WT.
(B) Downregulated RNAs in HHT-treated Fmr1 KO slices primarily encode epigenetic and transcriptional regulators and proteins involved in neural function.
(C) Example of Ankrd12 RNA, which has reduced reads in Fmr1 KO slices relative to WT after HHT (H) treatment. Input (I) reads are similar in both genotypes. M refers to medium fraction.
(D) Boxplot showing the fold change of Fmr1 KO versus WT of all RNAs (white) compared to those identified in (A) and (B) (gray) with respect to steady-state RNA levels, RPFs, and TE (ns, not significant; **p < 0.01; ***p < 0.001; Wilcoxon test). All grouped data are presented as mean ± s.e.m.
(E) Western blot analysis of SETD2 and lamin AC in hippocampus from four WT and five Fmr1 KO mice. When quantified and made relative to lamin AC, SETD2 was significantly increased in the KO (p = 0.0245, two-tailed t test). All grouped data are presented as mean ± s.e.m.
See also Table S1 and Figure S5E.
Finally, Figure 5E demonstrates that one of the mRNAs decreased in the medium fraction in the Fmr1 KO slices, SETD2, shows an increase in its protein level in Fmr1 KO hippocampus relative to WT (p = 0.0245, two-tailed t test), which is expected if FMRP stalls ribosome translocation on this RNA. We find no statistically significant increase in SETD2 mRNA in RNA-seq data from the FMRP KO hippocampal slices (from Figure 1). We tested SETD2 protein in immortalized mouse embryonic fibroblasts (MEFs) derived from WT and Fmr1 KO mice and found no change relative to genotype (Figure S5E). This result might reflect a possible brain-specific function of FMRP or that rapidly dividing MEF cell lines do not recapitulate FMRP-dependent ribosome stalling, at least not on SETD2 mRNA. SETD2 is the main histone lysine methyltransferase that trimethylates the histone H3 tail on the lysine36(K36).
FMRP-Mediated Regulation of H3K36me3
To investigate whether altered SETD2 levels influence gene-specific changes in H3K36me3, we performed ChIP-seq from WT and Fmr1 KO hippocampus in duplicate, combining hippocampal tissue from two animals per biologic replicate (Figure 6A). All genotypic replicates were highly correlated with each other(Figure S4A). H3K36me3 is a broad histone mark that is enriched primarily in the body of the gene. To measure regions with enrichment of the mark, we used the SICER v1.1 package (Xu et al., 2014) that annotates ChIP signal over a stretch of DNA as “islands” (an island is defined as a cluster of non-overlapping consecutive DNA regions (200-bp windows) separated by at least a 600-bpgap region of DNA with no ChIP-enriched signal detected above threshold). Annotation of the H3K36me3 islands shows that they were primarily present within the gene body compared to promoter proximal regions, as expected (Krogan et al., 2003; Tiedemann et al., 2016) (Figure 6B). Metagene analysis showed there to be no genotype-specific difference in the distribution of marks overall along the gene body and no effect of gene length (Figures S4B–S4D). We further compared the differential enrichment of the H3K36me3 islands within the replicates by a negative binomial test performed using edgeR (Robinson et al., 2010) (fold change >2 and p < 0.05). We found 363 islands with decreased and 2,223 islands with increased H3K36me3 in the Fmr1KO compared to WT hippocampus ChIP-seq in both replicates (Table S2). Examples of the changes of these marks that are genotype-dependent are shown in Figure 6C where H3K36me3 on Reep4 (receptor expression-enhancing protein 4) is reduced but elevated on Tprkb (TP53-related protein kinase-binding protein). The decreased islands were fewer and mostly found in intragenic regions, whereas the increased islands were enriched and present in both the intragenic and intergenic regions (Figure 6D), demonstrating a redistribution of H3K36me3 in FXS model mice. Many of the islands, especially those that increase in Fmr1 KO animals, are found on genes linked to autism spectrum disorders as defined by Simons Foundation Autism Research Initiative (SFARI) (Figure 6E). A subset of genes (37) showed both increased and decreased islands, but at different locations on the gene body (Figure 6E). GO term enrichment analysis of the genes that increased H3K36me3 islands is shown in Figure 6F. The genes with increased H3K36me3 islands are remarkably enriched for those involved in neural function such as synapse assembly and animal behavior. No significant GO terms were enriched in the group of genes with decreased H3K36me3 islands. We find no correlation between steady-state RNA abundance and H3K36me3 levels on islands in the genes (Figure S4F), which could be attributed to compensatory post-transcriptional mechanisms affecting steady-state RNA. Furthermore, the methylated islands also undergo genotype-specific length changes; they are significantly longer in the WT compared to Fmr1 KO (p < 0.001, Kolmogorov-Smirnov [K-S] test) (Figure S4E). Our data thus show that an increase in the levels of SETD2 in the Fmr1 KO resulting primarily in increased H3K36me3 chromatin marks at a subset of genes.
Figure 6. H3K36me3 Localization Is Altered in Fmr1 KO Hippocampus.
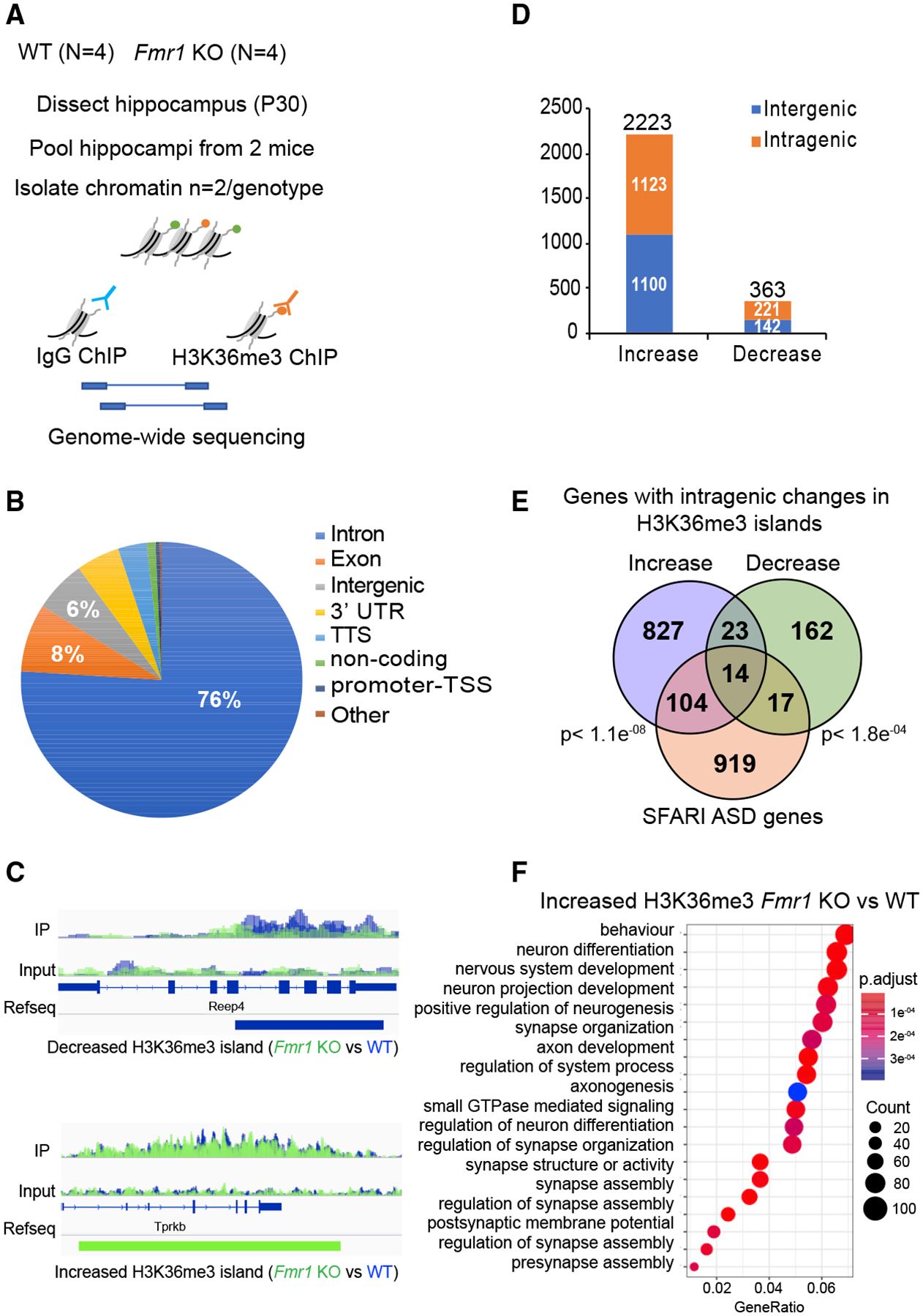
(A) Experimental design for in vivo ChIP-seq of H3K36me3 in hippocampus from adult WT (n = 4) and Fmr1 KO (n = 4) mice.
(B) Pie chart representing the genomic annotation of the total H3K36me3 islands identified in the WT and Fmr1 KO ChIP-seq.
(C) H3K36me3 ChIP-seq gene tracks for WT and Fmr1 KO hippocampal tissue. The two sequencing tracks from each biological replicate (n = 2 [pooled hippocampi from two mice/biological replicate]) of WT and Fmr1 KO were merged and overlaid. WT ChIP-seq tracks are in blue, and KO tracks are in green. The tracks for immunoprecipitation (IP) and input are displayed. The islands with significantly decreased (blue) or increased (green) tracks are shown below the RefSeq gene annotation (FDR < 0.0001; p < 0.01) as identified using the SICER package. Reep4 shows decreased H3K36me3 islands in Fmr1 KO, and Tprkb shows increased islands in Fmr1 KO.
(D) Distribution of significantly increased or decreased H3K36me3 islands in intragenic and intergenic regions of the genome using negative binomial test performed using edgeR (Robinson et al., 2010) (fold change >2 and p <0.05). The total number of increased or decreased islands is indicated above the respective bars in the graph in Fmr1 KO versus WT ChIP-seq.
(E) Venn diagram for significant overlap of Fmr1 KO misregulated H3K36me3 genes (increased islands in blue and decreased islands in green) with the ASD-linked genes from the SFARI database. A subset of genes showed both increased and decreased islands along the length of the gene body. The p value (hypergeometric test) is indicated next to the respective comparisons.
(F) GO term enrichment of H3K36me3 differentially enriched genes in Fmr1 KO versus WT (p adjust value < 0.05). Gene ratio refers to the percentage of total differentially expressed genes in the given GO term.
FMRP Regulates Alternative RNA Processing
H3K36me3 in mammals has been correlated with alternative pre-mRNA splicing (Kim et al., 2011). We analyzed our RNA-seq data from WT and Fmr1 KO hippocampal slices using the rMATS alternative splicing package (Shen et al., 2014) and detected many alternative pre-mRNA processing events including skipped exon (SE), mutually excluded exon (MXE), and alternative 5′ and 3′ splice site (A5SS and A3SS) (Figure 7A; Table S3; FDR < 5% and p < 0.05). The number of exon inclusion events are shown (differences in exon inclusion levels is calculated as percent spliced in [PSI], and a cutoff of |deltaPSI| ≥5% is used). Several of the exon-skipping events were validated by qRT-PCR, with ~20%–40% reduction in exon skipping in the Fmr1 KO relative to WT (Figure 7B). These skipped exons have previously been found to be associated with defects in brain development and disease pathology (An and Grabowski, 2007; Fugier et al., 2011; Rockenstein et al., 1995; Wang et al., 2018). In particular, an increase in microexon 4 skipping in Cpeb4 mRNA was found in postmortem cortical tissue from autism patients. Consequently, a mouse model of Cpeb4 microexon 4 deletion recapitulated the molecular and behavioral phenotypes found in autism patients (Irimia et al., 2014; Parras et al., 2018; Quesnel-Vallières et al., 2016). Figure 7C shows all the RNA processing events that undergo changes in Fmr1-deficient hippocampus relative to WT (p < 0.05, |deltaPSI| ≥5%). Skipped exons are by far the most prevalent category, followed by alternative 3′ splice sites. The genes whose RNA processing events are altered in Fmr1 KO are strongly linked to neural function such as vesicle transport to synapses and vesicle recycling (Figure 7D). Moreover, many of the genes with alternative splicing events (Alt. Spl.) in Fmr1 KO hippocampus (Alt. Spl. Fmr1 KO) are linked to autism (i.e., SFARI autism spectrum disorder database [ASD]) (Figure 7E). In addition, the Alt. Spl. genes in Fmr1 KO significantly overlap with genes previously found to display alternative splicing in patients with autism (Alt. Spl. ASD) (Parikshak et al., 2016) (Figure 7E). We also find an overlap of genes with changes in H3K36me3 islands with those found to harbor aberrant splicing in Fmr1 KO tissue (Figure 7E). We found no correlation between amount of exon skipping and RNA abundance, RPF levels or presence of FMRP CLIP tags at the gene level (Figures S5A–S5C). We next assessed the H3K36me3 levels at the skipped exons in the SE category and found a significant decrease in the H3K36me3 marks at the 5′ splice site (±50 nt) of these exons that are skipped in the Fmr1 KO hippocampus compared to the WT (Figure 7F; p = 0.02 using the Wilcoxon test). Interestingly, when we increased the analysis over a region of ± 150 nt (~2 nucleosomes) we no longer found a decrease in H3K36me3 at the skipped exons in the FMRP KO animal (Figure S5D). Although at the island level, which encompasses mainly regions >1 kb, we see mainly an increase in H3K36me3 levels, but at the 5′ splice site of genes (±50 nt) susceptible to alternative splicing in the FMRP KO, we see a decrease in H3K36me3 levels. Further studies are needed to understand how H3K36me3 modifications and/or other factors might play a role in regulating differential alternative splicing in the FMRP KO background. Thus, as with other autism spectrum disorders, dysregulated alternative splicing is prevalent in a mouse model of FXS and might be linked to changes in H3K36me3 levels at the splice site.
Figure 7. Global Analysis of FMRP-Mediated Alternative Splicing.
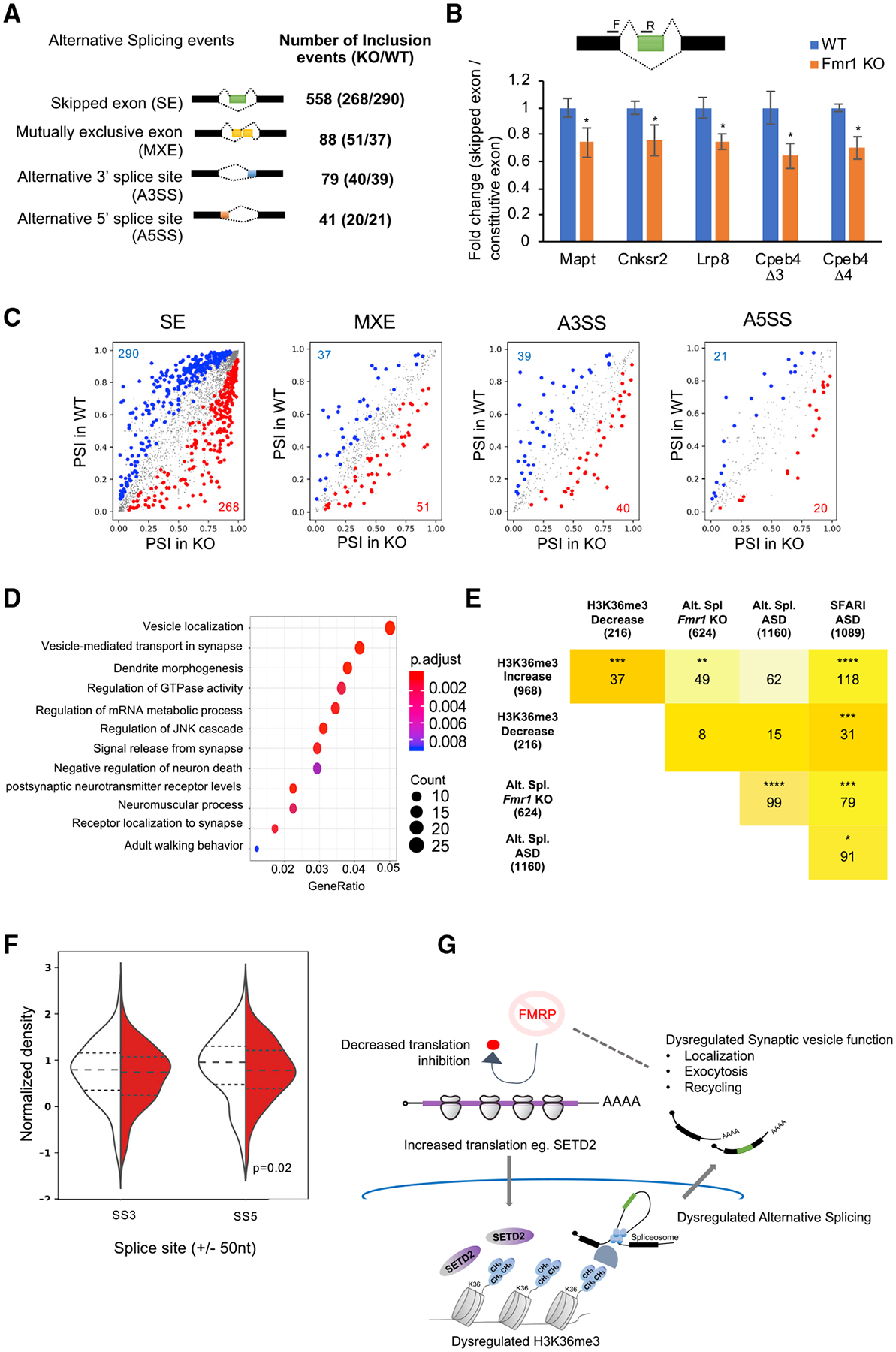
(A) Summary table of total splicing events in Fmr1 KO and WT based on RNA-seq from hippocampal slices. Splicing events detected by rMATS at a FDR <5% and a difference in the exon inclusion levels between the genotypes (deltaPSI) ≥5% are depicted.
(B) Alternative splicing events validated using qRT-PCR are shown for several RNAs (hippocampus tissue, WT, n = 6; Fmr1 KO, n = 6). The illustration depicts an example of exon skipping (green box) in the Fmr1 KO. Primer positions are depicted with black bars. All grouped data are presented as mean ± s.e.m.
(C) Alternative splicing events detected in Fmr1 KO and WT hippocampus. Inclusion events that were significantly (p < 0.05) increased (red), decreased (blue), or unchanged (gray) are indicated. The numbers of events in the up or down category are shown in red and blue, respectively. PSI, percent splice in/exon inclusion levels.
(D) GO term enrichment for all alternative splicing events are shown (p adjust value < 0.05). The total number of genes identified in the RNA-seq from (Figure 1) was used as background. Gene ratio refers to the percentage of total differentially expressed genes in the given GO term.
(E) Table depicting the overlap of the alternative splicing events (Alt. Spl.) in genes in this study with the SFARI autism spectrum disorder database, the Alt. Spl. genes identified in samples from autism patients (Parikshak et al., 2016), and genes with increased or decreased H3K36me3 islands from Figure 6. The intensity of the color represents the increasing number of overlapping genes between the gene sets. Asterisks indicate statistical significance (*p < 0.05; ***p < 0.001; ****p < 0.0001, hypergeometric test).
(F) Violin plot for the H3K36me3 ChIP signal at ±50 nt of the 5′ (SS5) and 3′ (SS3) splice sites of the alternatively skipped exons in WT (white) and Fmr1 KO (red) hippocampus tissue (p < 0.05, K-S test for significance). See also Figures S5E and S5F.
(G) Model for FMRP-mediated alterations in H3K36me3 marks on the chromatin and alternative splicing of transcripts in the hippocampus.
See also Table S3.
DISCUSSION
We used several approaches to investigate FMRP regulation of mRNA expression in the mouse brain, focusing first on steady state ribosome profiling in hippocampal slices. Fmr1 deficiency resulted in ribosome footprint levels that mostly tracked altered levels of RNA, which is consistent with other studies (Liu et al., 2018, 2019; Das Sharma et al., 2019). Because ribosome profiling at steady state cannot distinguish between static and translocating ribosomes, we modified our profiling procedure (Ingolia et al., 2011) to identify the dynamic range of ribosome translocation rates genome-wide in hippocampal slices. These studies in turn resulted in four main findings. First, they showed that mRNAs cluster into defined functional biology groups based on the transit rates of their associated ribosomes. The ribosomes on most mRNAs (>5,000) transit rapidly, whereas on some mRNAs (~3,000), ribosome transit is slow or nearly not at all (~350). There is some CDS length dependency as well. Second, using a combination of ribosome runoff and sucrose gradient centrifugation, we identified ~50 mRNAs associated with FMRP-stalled ribosomes, which strongly imply translational regulation. One of these mRNAs encodes a histone lysine methyltransferase, SETD2, which is elevated in Fmr1 KO hippocampus. Third, the SETD2-dependent histone mark, H3K36me3, is rearranged in both intragenic and intergenic regions upon Fmr1 deficiency; primarily increases in H3K36me3 are observed in Fmr1 KO. Fourth, because H3K36me3 has been correlated with alternative RNA processing in mammals (Kim et al., 2011), we analyzed our RNA-seq data and found widespread aberrant splicing in the Fmr1 KO hippocampus, most prominently exon skipping/inclusion. These altered processing events often correlated with changes in the histone mark, occurred on genes that regulate pre-synaptic functions, such as synaptic vesicle recycling, and were strongly linked to autism spectrum disorders (Figure 7E).
Earlier studies have inferred that ribosome stalling in the brain is linked to RNA transport granules in dendrites (Krichevsky and Kosik 2001; Mallardo et al., 2003; Kanai et al., 2004; Khandjian et al., 2004; Graber et al., 2013). It is possible that at least some of the ribosome stalling we observe may be the result from such compartmentalization in neurons or perhaps is a more general feature of the brain. In contrast, rapidly dividing ES cells do not appear to have stalled ribosomes, at least not of the magnitude we observe here (Ingolia et al., 2011). Irrespective of whether ribosome stalling is a dendritic or neuronal phenomenon, how it occurs mechanistically on specific mRNAs is unclear, but it could be related to secondary structure or association with RNA binding or other proteins. One clue, however, could be the accumulating evidence that strongly implicates phosphorylation of eEF2 as a coordinator of translation and neural activity and thus is likely to be one mediator of ribosome translocation rates (Scheetz et al., 2000; Sutton et al., 2007; Park et al., 2008; McCamphill et al., 2015; Heise et al., 2017). Even so, the involvement of phospho-eEF2, a general translation factor, would unlikely be responsible for mRNA-specific stalling. We also noted that some mRNAs, such as those in subcluster 2 (Figures 3D and 3E), encode proteins involved in the extracellular matrix (Figure S2C). A number of these proteins contain proline-rich stretches, whose geometry in the polypeptide exit tunnel can lead to ribosome stalling (Schuller et al., 2017; Huter et al., 2017).
We have identified 48 mRNAs whose ribosomes are stalled specifically by FMRP. These RNAs include those with (40 mRNAs) and without FMRP CLIP (8 mRNAs) sites in coding regions (Darnell et al., 2011; Van Driesche et al., 2019), so it would appear that a simple FMRP “roadblock” model would not explain ribosome stalling. Alternatively, Ishizuka et al. (2002) showed that Drosophila FMRP binds ribosomal proteins, and in a cryoelectron microscopy (cryo-EM) study, Chen et al. (2014) found that Drosophila FMRP reconstituted with mammalian ribosomes binds a region that would preclude tRNA and elongation factors from engaging in translation. However attractive this model is for FMRP-mediated ribosome stalling, the fact that mRNA or translation factors were not included in the reconstitution assays brings into question whether it reflects the physiological basis for ribosome stalling.
Our studies revealed that FMRP stalls ribosomes on a number of mRNAs that encode epigenetic regulators and that the misregulation of one of them in Fmr1-deficient hippocampus, SETD2, leads to a widespread increase in H3K36me3 chromatin marks (islands). These H3K36me3 islands are both enriched for and depleted from autism-risk genes (Figure 6E) as well as those with synaptic functions (Figure 6F). This suggests that SETD2, the main enzyme that catalyzes this chromatin mark, might also be an autism-risk gene, which has been previously reported (Lumish et al., 2015). Mutations in several chromatin-remodeling genes have been linked to changes in neural function, autism, and other intellectual disabilities (van Bokhoven 2011; Goodman and Azad, 2019). In most cases, however, mutations in epigenetic modifiers result in up or downregulation of transcription. Indeed, previously identified epigenetic changes in FXS model mice by Korb et al. (2017), particularly H3K4me3, H4K8ac, and H4K16ac, were responsible for widespread changes in transcription in cortical neurons and the cerebellum. Importantly, these investigators could rescue several FXS pathophysiologies, many of which resemble those in autism, by targeting the epigenetic reader protein Brd4. H3K36me3 does not mark promoter regions but instead is found in gene bodies and increases as RNA polymerase II proceeds 3′ during catalysis (Neri et al., 2017). In mammals, one of the functions H3K36me3 is correlated with, if not causative for, is alternative pre-mRNA processing (Kim et al., 2011). In the Fmr1-deficient hippocampus, we detected extensive changes in RNA processing, mostly exon skipping. Surprisingly, most of the skipped exons occurred in genes that have pre-synaptic functions such as synaptic vesicle recycling, transport, and exocytosis, which could at least partially explain alterations in synaptic vesicle dynamics and neurotransmitter release in FXS model mice (Deng et al., 2013; Ferron et al., 2014; Broek et al., 2016). Furthermore, a number of skipped exons in Fmr1 KO have been linked to neurological disorders such as autism (skipped microexon 4 in Cpeb4; Irimia et al., 2014; Parras et al., 2018; Quesnel-Vallières et al., 2016), Alzheimer’s disease (skipped exon 7 in APP; Fragkouli et al., 2017; Rockenstein et al., 1995), Parkinson’s disease (skipped exon 3 in Mapt; Lai et al., 2017; Wobst et al., 2017), and intellectual disabilities (skipped exon 12 in Cnksr2; Houge et al., 2012).
Alternative pre-mRNA processing is a common feature in autism (Raj and Blencowe, 2015; Quesnel-Vallières et al., 2019), so it is perhaps not surprising that this would occur in fragile X. However, because FMRP is an RNA-binding protein that regulates translation, a reasonable assumption would be that mRNAs encoding splicing factors are improperly expressed in the disorder and that this leads directly to mis-splicing events. However, our data indicate that this relationship might not be linear. Instead, we find that FMRP controls ribosome dynamics on specific mRNAs, which leads to elevated translation (e.g., SETD2), which leads to chromatin modifications (e.g., H3K36me3), which in turn might lead to alternative splicing of neuronal mRNAs (Figure 7G). These observations do not preclude the possible, if not likely, direct involvement of FMRP in some of these events. Even so, inferences that fragile X is a disorder of improper translation, while not incorrect, are vastly over-simplified. However, the complexity of gene expression changes in fragile X might offer opportunities for therapeutic intervention at multiple steps.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joel Richter (joel.richter@umassmed.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern and University of Massachusetts Medical School and conducted in accordance with the National Institutes of Health Principles of Laboratory Animal Care. Fmr1 KO mice were obtained from Dr. Stephen T. Warren and backcrossed for more than 10 generations on a C57BL/6 background. Food and water were provided ad libitum, and mice were kept on a 12 h light/dark cycle (7am–7pm light period). Pups were kept with their dams until weaning at postnatal day 21. After weaning, mice were group housed (maximum of 5 per cage) by sex. Cages were maintained in ventilated racks in temperature (20–21°C) controlled rooms. P28–35 wild-type or Fmr1 KO male mice littermates were used in this study.
Cell culture
Immortalized Mouse Embryonic Fibroblasts (MEFs) were derived from C57BL/6N wild-type or Fmr1 KO mice as described earlier (Groisman et al., 2006. Immortalized MEF cells were cultured in DMEM (Dulbecco’s Modified Eagle’s Medium, GIBCO) with 10% fetal bovine serum using the 3T3 culture protocol and cells derived from Passage number 31 onward were used for western blot analysis.
METHOD DETAILS
Mouse acute brain slice preparation
Transverse hippocampal or hippocampal-cortical brain slices were acutely prepared from P28–35 C57BL/6N wild-type or Fmr1 KO male mice littermates as previously described (Guo et al., 2016). Briefly, mice were anesthetized with ketamine (125 mg/kg)/xylazine (25 mg/kg) and transcardially perfused with chilled (4°C) sucrose dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 mM sucrose, and 10 dextrose aerated with 95% O2/5% CO2. Transverse hippocampal slices (400μm) were obtained on a Leica VT1200S slicer. For ribosomal profiling experiments (Figure 1), CA3 was cut off to enrich for CA1 and for each biological replicate (n), 8–10 slices from 6–8 mice/genotype were pooled. Slices were recovered and maintained at 30°C for 3–4 h in artificial cerebrospinal fluid (ACSF) which contained the following (in mM): 119 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1 NaH2PO4 and 11 D-glucose aerated with 95%/O2/5% CO2 to pH 7.4. Slices were then treated with cycloheximide (100 μg/ml) in ACSF, 4°C, snap-frozen on dry ice/EtOH bath, and stored at 80°C. For homoharringtonine (HHT) experiments (Figures 3, 4, and 5), cortical-hippocampal slices were recovered 3–4 hours in ACSF and then treated with HHT (20 μM) (Tocris) or 0.05% DMSO (vehicle) for different times (0, 5, 10, 30, 60 min). At the end of the incubation time, slices were snap/frozen on dry ice/EtOH bath and stored at −80 C. Ten slices from 2–3 mice/genotype were pooled for each biological replicate.
Sucrose gradients of hippocampal slices for steady-state ribosome profiling
Frozen, isolated CA1 hippocampal slices were thawed in ice-cold homogenization buffer (20mM Tris-HCl pH7.4, 5mM MgCl2, 100mM KCl, 1mM DTT, 100 μg/ml CHX (cycloheximide), 25U/ml Turbo DNaseI (Ambion, #AM2238), 1X EDTA-free protease inhibitor (Roche), avoiding detergent in nuclease-free water) on ice for 5min. Wide orifice tips were used to transfer slices to a pre-chilled detergent-free Dounce homogenizer. Tissues were slowly homogenized by hand (20 strokes of loose pestle A, and 20 strokes of tight pestle B). Homogenates were carefully transferred to clean 1.5ml tubes with clean glass Pasteur pipets and bulbs. 1% NP-40 was added to the homogenates and incubated on ice for 10min. Homogenates were clarified by centrifugation at 2,000 g 4°C for 10min. The supernatants were collected and clarified again by centrifugation at 20,000 g 4°C for 10min. The supernatants were collected, and the amounts of nucleic acid were measured by Nanodrop (A260 units). For each sample, cytoplasmic RNA for RNA-seq was purified from one-fourth of the lysate with TRIzol LS reagent (Invitrogen, #10296028). The other three-fourths of the lysate was digested with 100ng RNase A (Sigma, # R4875) and 60U RNase T1 (Thermo Fisher Scientific, #EN0542) per A260 at 25°C for 30 min and stopped by chilling on ice and adding 50U SUPERase In RNase inhibitor (Ambion, #AM2694). Digested lysates were applied to 10%–50% (w/v) sucrose gradients prepared in 1X polysome buffer (20mM Tris-HCl pH7.4, 5mM MgCl2, 100mM KCl, 1mM DTT, 100 μg/ml CHX in nuclease-free water). After the ultracentrifugation in a SW41Ti rotor (Beckman Coulter) at 35,000 rpm (avg 151,263 g) 4°C for 2.5 h, gradients were fractionated at 1.5 ml/min and 12 s collection intervals through a fractionation system (Brandel) that continually monitored A260 values. Monosome fractions were identified, pooled, and extracted with TRIzol LS.
Sucrose gradients of cortical-hippocampal slices for run-off ribosome profiling
Frozen cortical-hippocampal slices were thawed in ice-cold lysis buffer (20mM Tris-HCl pH7.4, 5mM MgCl2, 100mM KCl, 1mM DTT, 100 μg/ml CHX, 25U/ml Turbo DNaseI, 1X EDTA-free protease inhibitor, 1% NP-40, in nuclease-free water) on ice for 5min. Tissues were dissociated by pipetting and further trituration through 25G needle for 10 times. Lysates were incubated on ice for 10min and then clarified by centrifugation at 2,000 g 4°C for 10min. The supernatants were collected and clarified again by centrifugation at 20,000 g 4°C for 10min. The supernatants were collected, and the amounts of nucleic acid were measured with Qubit HS RNA assays. Lysates containing ~40μg RNA were digested with 600ng RNase A (Ambion, #AM2270) + 75U RNase T1 (Thermo Fisher Scientific, #EN0542)/μg RNA in 0.3ml at 25°C for 30min and stopped by chilling on ice and adding 50U SUPERase In RNase inhibitor. Digested lysates were applied to 10%–50% (w/v) sucrose gradients similarly as above.
Sucrose gradients of cortical-hippocampal slices for polysome profiling
Frozen cortical-hippocampal slices were thawed in ice-cold homogenization buffer on ice for 5min. Wide orifice tips were used to transfer slices to a pre-chilled detergent-free Dounce homogenizer. Tissues were gently homogenized by hand (3 strokes of loose pestle A, and 7 strokes of tight pestle B). Homogenates were carefully transferred to clean 1.5ml tubes with clean glass Pasteur pipets and bulbs. Homogenates were clarified by centrifugation at 2,000 g 4°C for 10min. The supernatants were collected and clarified again by centrifugation at 10,000 g 4°C for 10min. The supernatants were collected, and the amounts of nucleic acid were measured with Nanodrop (A260 units). 1% NP-40 was added to the homogenates and incubated on ice for 10min. ~1A260 unit of lysate was saved for the input RNA. ~5 A260 units of lysate were applied to 15%–45% (w/w) sucrose gradients prepared in 1X polysome buffer and with the lower block mark and long cap. After the ultracentrifugation in a SW41Ti rotor (Beckman Coulter) at 35,000 rpm (avg 151,263 g) 4°C for 2 h, gradients were fractionated at 1.5 ml/min and 20 s collection intervals through a fractionation system (Brandel) that continually monitored A260 values. Some gradients were treated with RNase A prior to centrifugation.
Ribosome profiling
Ribosome profiling libraries were prepared following the published protocols (Heyer et al., 2015). Briefly, rRNA was depleted from the purified monosomal RNA with RiboZero (Illumina, #MRZG12324). Remaining RNA samples were separated on a 15% TBU gel (National Diagnostics, #EC-833) and the ribosome footprints were size-selected between the 26 and 34nt markers. RNA was eluted from the crushed gel pieces in RNA elution buffer (300mM NaOAc pH5.5, 1mM EDTA, 0.25% SDS) at room temperature overnight, filtered with Spin-X Centrifuge Tube Filters (Corning, #8162) and precipitated with equal volume of isopropanol. Recovered RNA was dephosphorylated with T4 Polynucleotide Kinase (NEB, #M0201S) and ligated with preadenylated adaptor in miRCat®−33 Conversion Oligos Pack (IDT) using T4RNL2Tr.K227Q ligase (NEB, #M0351L). Reverse transcription (RT) was performed with primers containing 5nt-barcodes and 8nt-unique molecular identifiers (UMIs) and SuperScript III (Invitrogen, #18080–044) in 1X first-strand buffer without MgCl2 (50 mM Tris-HCl, pH 8.3, 75 mM KCl). RT products were separated on a 10% TBU gel and the 130–140nt region was selected. cDNA was eluted in DNA elution buffer (10mM Tris pH 8.0, 300mM NaCl, 1mM EDTA) at room temperature overnight, filtered, and precipitated with isopropanol. Purified cDNA was circularized with CircLigase (Epicenter, #CL4115K). Except for the RNase titration samples, cDNA derived from remaining rRNA was hybridized to biotin-labeled antisense probes (IDT) and further depleted with Dynabeads MyOne Streptavidin C1 (Invitrogen, #65001). Optimal PCR cycle number was determined empirically by test PCR reactions with titrated cycle numbers. Final PCR amplification was performed with KAPA Library Amplification Kit (Kapa Biosystems, #KK2611) and 180–190bp products were size-selected on an 8% TBE gel. DNA was eluted in DNA elution buffer, filtered, and precipitated with isopropanol. The final library DNA was purified with AMPure XP beads (Beckman Coulter, #A63880). Oligos used for the library preparation are listed in Table S2.
In parallel, input RNA samples were processed similarly as the ribosome footprints except the following steps. After rRNA depletion, input RNA mixed with an equal volume of 2x alkaline fragmentation solution (2 mM EDTA, 10 mM Na2CO3, 90 mM NaHCO3, pH ~9.3) and heated at 95°C for 15min to achieve an average fragment length of ~140nt. Fragmented RNA samples were separated on a 10% TBU gel and size-selected between the 100 and 150nt markers. RT products were separated on a 10% TBU gel and the 200–250nt region was selected. Antisense probe depletion was omitted for input RNA samples. 260–300bp final PCR products were size-selected on an 8% TBE gel.
The size distributions of final libraries were measured by Fragment Analyzer (Advanced Analytical, performed by Molecular Biology Core Labs at UMMS). The concentrations were quantified with KAPA Library Quantification Kit (Kapa Biosystems, #KK4835). Libraries were pooled with equal molar ratios, denatured, diluted, and sequenced with NextSeq 500/550 High Output Kit v2 (Illumina, 75bp single-end runs, #FC-404–2005) on a Nextseq500 sequencer (Illumina).
RNA-seq for polysome fractions
Based on the FMRP signals in the immunoblotting of gradient fractions, medium and heavy fractions were identified, pooled, and extracted with TRIzol LS together with saved input lysates. RNA samples were quantified with Qubit HS RNA assay kit and the integrity was examined with Fragment Analyzer. 200ng RNA was used for BIOO Rapid directional qRNA-seq library preparation following the manufacturer’s instructions. 12 cycles were used for the final PCR amplification. The libraries were quantified with KAPA Library Quantification Kit and the quality was examined with Fragment Analyzer. Libraries were pooled with equal molar ratios, denatured, diluted, and sequenced with NextSeq 500/550 High Output Kit v2 (Illumina, 80bp pair-end runs for RNA-seq, #FC-404–2002) on a Nextseq500 sequencer (Illumina).
Western blotting
Hippocampi were homogenized at 4°C in RIPA buffer. Protein complexes were released by sonication at 4°C and the extract was centrifuged at 13,200 rpm for 10 min at 4°C and the supernatant collected. Protein concentration was determined by BCA reagent. Proteins (10 μg) were diluted in SDS-bromophenol blue reducing buffer with 40 mM DTT and analyzed using western blotting with the following antibodies: SETD2 (ABclonal, A11271) and Lamin AC (Thermo Fisher, 14-9688-80). After incubation in primary antibody, immunoblots were incubated with HRP-conjugated secondary antibodies (Jackson Immunoresearch) and developed by Clarity ECL substrate (Biorad). For western blots for SETD2 detection, Hippocampi or MEF cells were homogenized in Triton Extraction Buffer (TEB: PBS containing 0.5% Triton X 100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3), centrifuged at 2000 rpm. The pellet containing the nuclei was resuspended in RIPA Buffer and Protein complexes were released by sonication at 4°C. Protein concentration was determined by BCA reagent. Proteins (30 μg) were diluted in SDS-bromophenol blue reducing buffer with 40 mM DTT and analyzed using western blotting with the following antibodies: SETD2 (ABclonal, A11271) and Lamin AC (Thermo Fisher, 14-9688-80) or Lamin B1 (Abcam, ab16048). After incubation in primary antibody, immunoblots were incubated with HRP-25 conjugated secondary antibodies (Jackson Immunoresearch) and developed by Clarity ECL. For sucrose gradient fractions, 60μl samples were mixed with 20μl 4X SDS loading dye (240mM Tris-HCl pH 6.8, 5% beta-mercaptoethanol, 8% SDS, 40% glycerol, 0.04% bromophenol blue) and boiled at 95°C for 10min. Samples were briefly heated at 95°C again for 30sec immediately before loading on 10% SDS-PAGE gel (35 ml/sample). Separated proteins were transferred to PVDF membranes at constant 90 mA 4°C for 16 h. Membranes were blocked with 5% non-fat milk in 1X TBST at room temperature for 1hour and incubated with primary antibody at 4°C overnight. FMRP (Abcam, 1: 2000), Tubulin (Sigma, 1:5000), Rpl4 (Proteintech, 1:5000), UPF1 (Abcam, 1: 5000), MAP2 (Millipore, 1:2000), eEF2 (Cell signaling, 1:2000), GAPDH (Cell signaling, 1:2000), MRPS18B (Proteintech, 1:2000) and Rps6 (Cell signaling, 1:4000) were diluted in 1X TBST with 5% non-fat milk. Membranes were washed three times for 10min with 1XTBST and incubated with anti-rabbit or anti-mouse secondary antibodies (Jackson, 1:10000) at room temperature for 1hour. Membranes were washed three times for 10min with 1XTBST, developed with ECL-Plus (Piece), and scanned with GE Amersham Imager
Chromatin immunoprecipitation Sequencing (ChIP-Seq)
ChIP was performed as previously described (Cotney and Noonan, 2015). Briefly, hippocampal tissue was isolated from 4 adult mice (P35) per genotype and minced into tissue < 0.5mm3 in 250ul of ice-cold PBS with protease inhibitors. Tissue was cross-linked with 1% formaldehyde and rotated at room temperature for 15 min at 50 rpm and quenched with 150mm glycine for 10 min in a total volume of 1ml PBS. The supernatant was discarded after centrifugation at 2000 g for 10 min at 4°C and the pellet was resuspended in 300ul of chilled cell lysis buffer for 20 min on ice. Swollen pellets were homogenized using a glass Dounce homogenizer (2ml) with 40 strokes of a tight pestle on ice. Nuclei were harvested after centrifugation at 2000 g for 5min at 4°C and resuspended in 300ul ice-cold nuclear lysis buffer for 20min on ice. SDS was added to make a final concentration of 0.5%. Samples were sonicated on a Bioruptor® sonicator at high power settings for 9 cycles (sonication: 30 s on, 90 s off) of 15min each at 4°C at high power. Supernatants were collected after centrifugation at 16,000 g for 10 min at 4°C. The samples were diluted to bring the SDS concentration < 0.1%; 10% of each sample was reserved as input. The rest of the samples were divided into two and incubated with Protein G dynabeads coupled overnight at 4°C with either H3K36me3 (Abcam ab9050, 5 mg per ChIP) or IgG (Sigma 12–371, 5 mg per ChIP). After IP, beads were washed, and chromatin eluted in elution buffer for 20 min shaking at 65°C. IP and Input samples were de-crosslinked overnight at 65°C. RNase digestion for 1 hr. at 37°C and proteinase K treatment for 30 min at 55°C was performed. DNA was then purified with the QIAGEN PCR purification kit in 50ul elution buffer. For library preparation, purified input and IP DNA was end repaired using T4 DNA polymerase, Klenow polymerase and T4 Polynucleotide kinase from NEB at 20°C for 30 min. DNA was extracted using 35ul of the Agencourt Ampure XP beads and ‘A’ bases were added to the 3′ end using Klenow exonuclease (3′ to 5′ exo minus) from NEB for 30min at 37°C. DNA was purified using 60ul of the Agencourt Ampure XP beads and Illumina adaptor sequences were ligated to the DNA fragments using the Quick Ligase (NEB) for 15min at 20°C. The library was size-selected using 50ul of Agencourt Ampure XP beads. Using the multiplexing barcoded primers, the library was PCR amplified and purified using 50ul of the Agencourt Ampure XP beads and analyzed using a Fragment Analyzer (Advanced Analytical, performed by Molecular Biology Core Labs at UMMS). Libraries were pooled with equal molar ratios, denatured, diluted, and sequenced with NextSeq 500/550 High Output Kit v2.5 (Illumina, 75bp paired-end runs,) on a Nextseq500 sequencer (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
All grouped data are presented as mean ± s.e.m. All tests used to compare the samples are mentioned in the respective figure legends and corresponding text. When exact p values are not indicated, they are represented as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p value < 0.0001; n.s., p > 0.05.
Ribosome profiling analysis
Individual samples were separated from the raw fastq files based on the barcode sequences. Adaptor sequences (TGGAATTCTCGGGTGCCAAGGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG) were removed with cutadapt (1.7.1). Trimmed fastq files were uploaded to the Dolphin platform (https://www.umassmed.edu/biocore/introducing-dolphin/) at the UMMS Bioinformatics Core for the mapping steps. Trimmed reads were quality filtered with Trimmomatic (0.32) and mapped to the mouse rRNA and then tRNA references with Bowtie2 (2.1.0). Unmapped reads were next mapped to the mm10 mouse genome with Tophat2 (2.0.9). Reads mapped to > 1 loci of the genome were classified as “multimapped” reads and discarded. PCR duplicates were marked based on the UMI sequences with custom scripts and only uniquely mapped reads without duplicates were retained with Samtools (0.0.19) for the downstream analyses. RPF length distribution, P-site offsets, and frame preference were calculated with plastid (0.4.8). Counts at each nucleotide position were extracted using P-sites of RPFs and 5′ end of mRNA reads with +11 offset, normalized to the library size, averaged across replicates, and plotted along mRNA positions with custom scripts.
Steady-state differential translation analysis
Cleaned bam files were converted to fastq files with bedtools. For both ribosome profiling and RNA-seq, gene expression was quantified with RSEM (1.2.11) using the cleaned fastq files and Refseq (V69) mouse CDS without the first and last 30nt to avoid the translation initiation and termination peaks. Genes were filtered with minimum 10 reads across all replicates and then the read counts were batch-corrected with the Combat function in sva (3.24.4) using a full model matrix. Batch-corrected counts were normalized with trimmed mean of M values (TMM) method and used to identify differential expressed genes (DEGs) with anota2seq (1.0.0). Instead of the default setting, the priority of TE groups were set to be higher than that of mRNA only groups. A permutation test was performed to estimate the false discovery rate (FDR) with nominal p value < 0.01, which was 0.097. GO analysis was performed and plotted with clusterProfiler (3.10.1; Yu et al., 2012) using all genes past filtering in the dataset as the background.
Hierarchical clustering of ribosome run-off and gene ontology (GO) enrichment of each cluster
RPFs were mapped as described above and the most abundant isoform estimated by RSEM for each gene was selected as the representative transcript. For the metagene analysis, transcripts with CDS longer than 3000nt (n = 1401) were selected to ensure incomplete ribosome run-off after 10min HTT treatment. The read densities at each nucleotide position were normalized to the average density of the last 500nt of CDS and averaged using the P sites of RPFs. For visualization purposes, the curves were smoothed within a 90nt window. An arbitrary 0.8 threshold was used to estimate the relative run-off distances at 5 and 10min. A linear regression analysis between the HHT treatment time and ribosome run-off distances was performed to estimate the global elongation rate. To visualize the run-off patterns for individual transcripts, RPFs were normalized to the abundance of mitochondrial RPFs and plotted along the mRNA nucleotide positions. The same y-scale was used across the time-course, so the initiation peaks were truncated to reveal the patterns over CDS. Stalled ribosomes were quantified by RSEM by mapping RPFs to Refseq (V69) mouse CDS without the first and last 300nt to avoid the translation initiation and termination peaks after prolonged HTT treatment. Counts of RPFs were normalized to the length of CDS minus 600nt and the abundance of mitochondrial RPFs in that sample to calculate the RPK (Reads Per Kilobase of transcript) value for each transcript.
To categorize and group genes with distinct ribosome run-off patterns, the RPF of each gene at each time point was first normalized to time point 0. The Euclidean distance matrix was then calculated, followed by hierarchical clustering using Ward’s agglomeration method (Ward, 1963). The clustering process was performed using the hclust (Galili, 2015), and the clustering heatmap was displayed using pheatmap (Kolde, 2015) in R. To test the reliability of clustering, analysis of group similarities (ANOSIM) test (Clarke, 1993) was performed using vegan (Dixon, 2003) in R with 1000 permutation times on both overall clusters and sub-clusters. The global pattern of each cluster was summarized using the corresponding median and standard deviation in each time point.
GO enrichment analysis for each sub-cluster were performed through clusterProfiler (Yu et al., 2012) using genes covered by run-off ribosome profiling as background. The statistical significance was adjusted using FDR. To remove redundancy in reporting, each reported GO term was required to have at least 25% of genes that were not associated to another term with a more significant p value.
Polysome fraction RNA-seq data analysis
Fastq files were uploaded to the Dolphin platform at the UMMS Bioinformatics Core for mapping and quantification. 9nt molecular labels were trimmed from both 5′ ends of the pair-end reads and quality-filtered with Trimmomatic (0.32). Reads mapped to mouse rRNA by Bowtie2 (2.1.0) were filtered out. Cleaned reads were next mapped to the Refseq (V69) mouse transcriptome and quantified by RSEM (1.2.11).
Estimated counts on each gene were used for the differential gene expression analysis by DESeq2 (1.16.1). After the normalization by median of ratios method, only the genes with minimal 5 counts average across all samples were kept for the DEG analysis. Data were fit to a model of treatment+fraction+treatment:fraction+treatment:genotype:fraction and the treatmenthht.fractionheavy result was used to identify mRNA enriched or depleted in the heavy fractions after HTT treatment. p < 0.05 and 2-fold change were used as the cut-offs.
For the medium fractions after HHT treatment, Fmr1 KO samples was directly compared to the WT samples and p < 0.05 was used as the cut-off for identifying DEGs. Steady-state ribosome profiling data of the decreased DEGs were compared to all the other genes to independently confirm whether these mRNAs indeed had fewer stalled ribosomes in Fmr1 KO.
ChIP-Seq analysis
For ChIP-sequencing analysis, alignments were performed with Bowtie2 (2.1.0) using the mm10 genome, duplicates were removed with Picard and TDF files for IGV viewing were generated using a ChIP-seq pipeline from DolphinNext (Yukselen et al., 2019). The broad peaks for H3K36me3 ChIP-Seq were called using the SICER v1.1 package (Xu et al., 2014). H3K36me3 enriched islands were identified using the parameters set to a window size of 200bp, gap size 600bp and FDR cutoff of 1 × 10−3, and the default value for the redundant rate cutoff. To analyze the H3K36me3 difference level between WT and FMRP KO, the reads count on each island of each replicate was first normalized to total mapped reads and then normalized to its corresponding input sample. The H3K36me3 difference level of each island is then calculated using both replicates in FMRP KO and WT groups; a negative binomial test is performed using edgeR (Robinson et al., 2010) to consider variability between replicates and differences between FMRP KO and WT groups. The islands with significantly different H3K36me3 levels are defined using a cut-off for fold change >2 and p value <0.05. Gene Ontology (GO) enrichment analysis was performed using the clusterProfiler package (Yu et al., 2012) to obtain GO terms related to Biological processes in the genes with differentially enriched islands in the Fmr1 KO versus Wild-type. deepTools (Ramírez et al., 2016) was used to plot heatmaps and profiles for genic distribution of H3K36me3 ChIP signal. Samtools (0.1.19) was used for sorting and converting Bam files. RPGC (per bin) = number of reads per bin / scaling factor for 1× average coverage was used for normalization where the scaling factor was determined using sequencing depth: (total number of mapped reads × fragment length) / effective genome size. IGV tools (2.3.67) was used for visualizing TDF files and all tracks shown are normalized for total read coverage.
To quantify the H3K36me3 level at the 5′ and 3′ splicing sites of the SE, the read density at a region of ± 50nt at each splice site (total 100nt) and ± 150nt at each splice site (total 300nt) was normalized to the read density in the entire gene body, which controls for any fluctuations in total H3K36me3 at the respective genes between both genotypes (p value calculated using the Wilcox test for significance
Alternative splicing analysis
RNA-seq from hippocampal slices was used to analyze alternative splicing (AS) using the rMATS package v3.2.5 (Shen et al., 2014) with default parameters and reads were trimmed to 50bp. The Percent Spliced In (PSI) levels or the exon inclusion levels calculated by rMATS using a hierarchical framework. To calculate the difference in PSI between genotypes a likelihood-ratio test was used. AS events with an FDR < 5% and |deltaPSI| ≥ 5% as identified using rMATS were used for further analysis. We included genes that had at least one read at the differential splice junction for both genotypes. GO enrichment analysis was done by the clusterProfiler package (Yu et al., 2012) to obtain GO term enrichment for the AS events. The genes with significant skipped exons were used for validation using RT-qPCR analysis. One ug of RNA from hippocampal tissue was used to generate cDNA using the Quantitech two-tep cDNA synthesis kit. Primers were designed to overlap skipped/inclusion exon junctions and qPCR was performed using the Bio-Rad SYBR reagent on a Quantstudio3 instrument.
DATA AND CODE AVAILABILITY
Codes and scripts used for quantification analysis were written in Python or R and will be provided upon request to the Lead Contact.. The Gene Expression Omnibus (GEO) accession number for the data reported in this paper is GSE143333.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SETD2 Rabbit pAb-Polyclonal Antibodies | ABclonal | Cat# A11271; RRID: AB_2758486 |
| Lamin A/C Mouse Monoclonal Antibody (4A7) | Thermo Fisher | Cat# 14-9688-80; RRID: AB_2572956 |
| HRP conjugated antimouse secondary antibody | Jackson Immunoresearch | Cat# 115-035-174; RRID: AB_2338512 |
| Rabbit polyclonal to Lamin B1 | Abcam | Cat# ab16048; RRID: AB_443298 |
| Anti-FMRP Rabbit polyclonal antibody | Abcam | Cat# ab17722; RRID: AB_2278530 |
| HRP conjugated mouse anti-rabbit secondary antibody | Jackson Immunoresearch | Cat# 211-032-171; RRID: AB_2339149 |
| Anti-α-Tubulin mouse Monoclonal antibody | Sigma | Cat# T5168; RRID: AB_477579 |
| Anti-Rpl4 mouse monoclonal antibody | Proteintech | Cat# 67028–1-Ig |
| Recombinant Anti-RENT1/hUPF1 antibody [EP4682] | Abcam | Cat# 133564 |
| Anti-MAP2 mouse monoclonal Antibody, clone AP20 | Millipore | Cat# AB5543; RRID: AB_571049 |
| Anti-eEF2 polyclonal rabbit Antibody | Cell Signaling Technology | Cat# 2332; RRID: AB_10693546 |
| Anti- GAPDH (14C10) Rabbit monocloanal Antibody | Cell Signaling Technology | Cat# 2118; RRID: AB_561053 |
| Anti-MRPS18B Rabbit Polyclonal Antibody | Proteintech | Cat# 16139–1-AP; RRID: AB_2146368 |
| Anti-S6 Ribosomal Protein (5G10) Rabbit monoclonal antibody | Cell Signaling Technology | Cat# 2217; RRID: AB_331355 |
| Anti-IgG mouse polyclonal antibody | Sigma | Cat# 12–371; RRID: AB_145840 |
| Anti-Histone H3 (tri methyl K36) Rabbit polyclonal antibody | Abcam | Cat# ab9050; RRID: AB_306966 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide solution | Sigma | Cat#C4859 |
| homoHarringtonine | LKT laboratories | Cat#H0169–5 |
| Turbo DNaseI | Ambion | Cat#AM2238 |
| RNase A | Sigma | Cat#R4875 |
| RNase T1 | Thermo Fisher Scientific | Cat#EN0542 |
| SUPERase In RNase inhibitor | Ambion | Cat#AM2694 |
| RiboZero | Illumina | Cat#MRZG12324 |
| 5 nm miRCat®−33 Conversion Oligos Pack15nm | IDT | Cat#51-01-13-10 |
| SuperScript III | Invitrogen | Cat#18080–044 |
| CircLigase | Epicenter | Cat#CL4115K |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat#65001 |
| KAPA Library Amplification Kit | Kapa Biosystems | Cat#KK2611 |
| AMPure XP beads | Beckman Coulter | Cat#A63880 |
| NextSeq 500/550 High Output Kit v2 (75bp single-end runs) | Illumina | Cat#FC-404–2005 |
| NextSeq 500/550 High Output Kit v2 (80bp pair-end runs) | Illumina | Cat#FC-404–2002 |
| NEXTflex® Rapid Directional qRNA-Seq Kit (24 barcodes) - Set A/B | biooscientific | Cat#NOVA-5130–03D |
| Critical Commercial Assays | ||
| Qubit RNA HS Assay Kit | Fisher scientific | Cat#Q32852 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE143333 |
| Mouse reference genome NCBI build 38,mm10 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/mouse/ |
| Experimental Models: Cell Lines | ||
| Immortalized Mouse Embryonic Fibroblasts (MEFs) | C57BL/6N wild-type or Fmr1 KO mice | NA |
| Experimental Models: Organisms/Strains | ||
| Mouse C57BL/6 wild-type | Jackson lab | Cat# JAX:000664; RRID: IMSR_JAX:000664 |
| Fmr1tm1Cgr/Fmr1tm1Cgr Mus musculus, backcrossed for more than 10 generations on a C57BL/6 background | Dr. Stephen T. Warren | Cat# 2665400; RRID: MGI:2665400 |
| Oligonucleotides | ||
| See Table S1, sheet 5 | NA | NA |
| Software and Algorithms | ||
| DolphinNext | Yukselen et al., 2019 | NA |
| clusterProfiler (3.10.1) | Yu et al., 2012 | NA |
| hclust | Galili, 2015 | NA |
| pheatmap | Kolde, 2015 | NA |
| anota2seq (1.0.0) | Oertlin et al., 2019 | NA |
| SICER v1.1 | Xu et al., 2014 | NA |
| rMATS package v3.2.5 | Shen et al., 2014 | NA |
| deepTools | Ramírez et al., 2016 | NA |
Highlights.
Loss of FMRP reduces ribosome stalling on specific mRNAs, including Setd2
Increased SETD2 protein alters H3K36me3 marks in FMRP-deficient hippocampus
Alternative splicing is dysregulated in FMRP-deficient hippocampus
Altered H3K36me3 and splicing occur on genes and transcripts linked to autism
ACKNOWLEDGMENTS
We thank Lori Lorenz, Craig Peterson, Huan Shu, and Danesh Moazed for valuable discussions. J.D.R., K.M.H., B.L., and S.S. conceived various aspects of the project. We thank Maria Ivshina and Suna Jung for providing reagents. This work was supported by grants from the National Institutes of Health (NIH) (grants U54HD82013 to J.D.R. and U54HD082008 to K.M.H.), the Simons Foundation for Autism Research Initiative (grant 400998 to J.D.R. and K.M.H.), and the Charles H. Hood Foundation (to J.D.R.). S.S. was supported by a postdoctoral fellowship from the FRAXA Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.076.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An P, and Grabowski PJ (2007). Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol. 5, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BR, Chopra P, Suhl JA, Warren ST, and Bassell GJ (2016). Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res. 44, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling H, Bhattacharya A, Zhang G, Alam D, Lebowitz JZ, Bohm-Levine N, Lin D, Singha P, Mamcarz M, Puckett R, et al. (2019). Altered steady state and activity-dependent de novo protein expression in fragile X syndrome. Nat. Commun 10, 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, and Weissman JS (2015). Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol 16, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broek JAC, Lin Z, de Gruiter HM, van ‘t Spijker H, Haasdijk ED, Cox D, Ozcan S, van Cappellen GWA, Houtsmuller AB, Willemsen R, et al. (2016). Synaptic vesicle dynamic changes in a model of fragile X. Mol. Autism 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Sharma MR, Shi X, Agrawal RK, and Joseph S (2014). Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 54, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke KR (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol 18, 117–143. [Google Scholar]
- Cotney JL, and Noonan JP (2015). Chromatin immunoprecipitation with fixed animal tissues and preparation for high-throughput sequencing. Cold Spring Harb. Protoc 2015, 191–199. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Sharma S, Metz JB, Li H, Hobson BD, Hornstein N, Sulzer D, Tang G, and Sims PA (2019). Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep. 26, 3313–3322.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, and Klyachko VA (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci 14, 927–930. [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Fatimy R, Davidovic L, Tremblay S, Jaglin X, Dury A, Robert C, De Koninck P, and Khandjian EW (2016). Tracking the fragile X mental retardation protein in a highly ordered neuronal ribonucleoparticles population: a link between stalled polyribosomes and RNA granules. PLoS Genet. 12, e1006192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, and Warren ST (1997). FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol. Cell 1, 109–118. [DOI] [PubMed] [Google Scholar]
- Ferron L, Nieto-Rostro M, Cassidy JS, and Dolphin AC (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat. Commun 5, 3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkouli A, Koukouraki P, Vlachos IS, Paraskevopoulou MD, Hatzigeorgiou AG, and Doxakis E (2017). Neuronal ELAVL proteins utilize AUF-1 as a co-partner to induce neuron-specific alternative splicing of APP. Sci. Rep 7, 44507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, et al. (2011). Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med 17, 720–725. [DOI] [PubMed] [Google Scholar]
- Galili T (2015). dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31, 3718–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman V, and Azad Bonni. (2019). Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Current Opinion in Neurobiology 59, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TE, Hébert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille JC, and Sossin WS (2013). Reactivation of stalled polyribosomes in synaptic plasticity. Proc. Natl. Acad. Sci. USA 110, 16205–16210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, and Spradling AC (2018). Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman I, Ivshina M, Marin V, Kennedy NJ, Davis RJ, and Richter JD (2006). Control of cellular senescence by CPEB. Genes Dev. 20, 2701–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Molinaro G, Collins KA, Hays SA, Paylor R, Worley PF, Szumlinski KK, and Huber KM (2016). Selective disruption of metabotropic glutamate receptor 5-homer interactions mimics phenotypes of fragile X syndrome in mice. J. Neurosci 36, 2131–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zhang L, Christopher DM, Teng ZQ, Fausett SR, Liu C, George OL, Klingensmith J, Jin P, and Zhao X (2011). RNA-binding protein FXR2 regulates adult hippocampal neurogenesis by reducing Noggin expression. Neuron 70, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise C, Taha E, Murru L, Ponzoni L, Cattaneo A, Guarnieri FC, Montani C, Mossa A, Vezzoli E, Ippolito G, et al. (2017). eEF2K/eEF2 pathway controls the excitation/inhibition balance and susceptibility to epileptic seizures. Cereb. Cortex 27, 2226–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer EE, Ozadam H, Ricci EP, Cenik C, and Moore MJ (2015). An optimized kit-free method for making strand-specific deep sequencing libraries from RNA fragments. Nucleic Acids Res. 43, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houge G, Rasmussen IH, and Hovland R (2012). Loss-of-function CNKSR2 mutation is a likely cause of non-syndromic X-linked intellectual disability. Mol. Syndromol 2, 60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, and Bear MF (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. [DOI] [PubMed] [Google Scholar]
- Huter P, Arenz S, Bock LV, Graf M, Frister JO, Heuer A, Peil L, Starosta AL, Wohlgemuth I, Peske F, et al. (2017). Structural basis for polyproline-mediated ribosome stalling and rescue by the translation elongation factor EF-P. Mol. Cell 68, 515–527.e6. [DOI] [PubMed] [Google Scholar]
- Ingolia NT (2014). Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet 15, 205–213. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallières M, Tapial J, Raj B, O’Hanlon D, et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, and Siomi H (2002). A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 16, 2497–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, and Hirokawa N (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. [DOI] [PubMed] [Google Scholar]
- Khandjian EW, Corbin F, Woerly S, and Rousseau F (1996). The fragile X mental retardation protein is associated with ribosomes. Nat. Genet 12, 91–93. [DOI] [PubMed] [Google Scholar]
- Khandjian EW, Huot ME, Tremblay S, Davidovic L, Mazroui R, and Bardoni B (2004). Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc. Natl. Acad. Sci. USA 101, 13357–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Fong N, Erickson B, and Bentley DL (2011). Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. USA 108, 13564–13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, and Ahringer J (2009). Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet 41, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2015). Pheatmap: pretty heatmaps. R Package Version 1.0.10. (CRAN), https://CRAN.R-project.org/package=pheatmap.
- Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, and Darnell RB (2017). Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170, 1209–1223.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky AM, and Kosik KS (2001). Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32, 683–696. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. (2003). Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol 23, 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Bechy AL, Denk F, Collins E, Gavriliouk M, Zaugg JB, Ryan BJ, Wade-Martins R, and Caffrey TM (2017). Haplotype-specific MAPT exon 3 expression regulated by common intronic polymorphisms associated with Parkinsonian disorders. Mol. Neurodegener 12, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, and Martin KC (2016). Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang SX, Shen B, and Qian SB (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U S A 109, E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, and Richter JD (2018). Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. USA 115, E11397–E11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Molinaro G, Shu H, Stackpole EE, Huber KM, and Richter JD (2019). Optimization of ribosome profiling using low-input brain tissue from fragile X syndrome model mice. Nucleic Acids Res. 47, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumish HS, Wynn J, Devinsky O, and Chung WK (2015). Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J. Autism Dev. Disord 45, 3764–3770. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, et al. (2010). Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6, e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallardo M, Deitinghoff A, Müller J, Goetze B, Macchi P, Peters C, and Kiebler MA (2003). Isolation and characterization of Staufen-containing ribonucleoprotein particles from rat brain. Proc. Natl. Acad. Sci. USA 100, 2100–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, Grossi M, Rage F, and Bardoni B (2018). HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 46, 6344–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCamphill PK, Farah CA, Anadolu MN, Hoque S, and Sossin WS (2015). Bidirectional regulation of eEF2 phosphorylation controls synaptic plasticity by decoding neuronal activity patterns. J. Neurosci 35, 4403–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, and Oliviero S (2017). Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, and Huber KM (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol 95, 3291–3295. [DOI] [PubMed] [Google Scholar]
- Oertlin C, Lorent J, Murie C, Furic L, Topisirovic I, and Larsson O (2019). Generally applicable transcriptome-wide analysis of translation using anota2se. Nucleic Acids Research 47, e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, and Worley PF (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras A, Anta H, Santos-Galindo M, Swarup V, Elorza A, Nieto-González JL, Picó S, Hernández IH, Díaz-Hernández JI, Belloc E, et al. (2018). Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallières M, Dargaei Z, Irimia M, Gonatopoulos-Pournatzis T, Ip JY, Wu M, Sterne-Weiler T, Nakagawa S, Woodin MA, Blencowe BJ, and Cordes SP (2016). Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell 64, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Quesnel-Vallières M, Weatheritt RJ, Cordes SP, and Blencowe BJ (2019). Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet 20, 51–63. [DOI] [PubMed] [Google Scholar]
- Raj B, and Blencowe BJ (2015). Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87, 14–27. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, and Klann E (2015). Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, and Mucke L (1995). Levels and alternative splicing of amyloid b protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J. Biol. Chem 270, 28257–28267. [DOI] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, and Warren ST (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol 7, 219–245. [DOI] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, and Constantine-Paton M (2000). NMDA receptor-mediated control of protein synthesis at developing synapses. Nat. Neurosci 3, 211–216. [DOI] [PubMed] [Google Scholar]
- Schuller AP, Wu CC, Dever TE, Buskirk AR, and Green R (2017). eIF5A functions globally in translation elongation and termination. Mol. Cell 66, 194–205.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RM, and Sadee W (2011). Synaptic signaling and aberrant RNA splicing in autism spectrum disorders. Front. Synaptic Neurosci 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Fraser CE, Darnell JC, and Darnell RB (2004). Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J. Neurosci 24, 7272–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, and Schuman EM (2007). Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55, 648–661. [DOI] [PubMed] [Google Scholar]
- Tabet R, Moutin E, Becker JA, Heintz D, Fouillen L, Flatter E, Krężel W, Alunni V, Koebel P, Dembélé D, et al. (2016). Fragile X Mental Retardation Protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc. Natl. Acad. Sci. USA 113, E3619–E3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, and Osterweil EK (2017). Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95, 550–563.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann RL, Hlady RA, Hanavan PD, Lake DF, Tibes R, Lee JH, Choi JH, Ho TH, and Robertson KD (2016). Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget 7, 1927–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. (2013). Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med 19, 1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet 45, 81–104. [DOI] [PubMed] [Google Scholar]
- Van Driesche SJ, Sawicka K, Zhang C, Hung SKY, Park CY, Fak JJ, Yang C, Darnell RB, and Darnell JC (2019). FMRP binding to a ranked subset of long genes is revealed by coupled CLIP and TRAP in specific neuronal cell types. bioRxiv. 10.1101/762500. [DOI] [Google Scholar]
- Wang CX, Cui GS, Liu X, Xu K, Wang M, Zhang XX, Jiang LY, Li A, Yang Y, Lai WY, et al. (2018). METTL3-mediated m 6 A modification is required for cerebellar development. PLoS Biol. 16, e2004880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JH Jr. (1963). Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc 58, 236–244. [Google Scholar]
- Waung MW, and Huber KM (2009). Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol 19, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobst HJ, Denk F, Oliver PL, Livieratos A, Taylor TN, Knudsen MH, Bengoa-Vergniory N, Bannerman D, and Wade-Martins R (2017). Increased 4R tau expression and behavioural changes in a novel MAPT- 296H genomic mouse model of tauopathy. Sci. Rep 7, 43198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Grullon S, Ge K, and Peng W (2014). Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol 1150, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukselen O, Turkyilmaz O, Ozturk A, Garber M, and Kucukural A (2019). DolphinNext: a distributed data processing platform for high throughput genomics. bioRxiv. 10.1101/689539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Codes and scripts used for quantification analysis were written in Python or R and will be provided upon request to the Lead Contact.. The Gene Expression Omnibus (GEO) accession number for the data reported in this paper is GSE143333.