

Abstract
We report the synthesis of several related 4-anilinoquinazolines as inhibitors of cardiac troponin I–interacting kinase (TNNi3K). These close structural analogs of 3-((6,7-dimethoxyquinazolin-4-yl)amino)-4-(dimethylamino)-N-methylbenzenesulfonamide (GSK114) provide new understanding of structure–activity relationships between the 4-anilinoquinazoline scaffold and TNNi3K inhibition. Through a small focused library of inhibitors, we observed that the N-methylbenzenesulfonamide was driving the potency in addition to the more traditional quinazoline hinge-binding motif. We also identified a compound devoid of TNNi3K kinase activity due to the addition of a methyl group in the hinge binding region. This compound could serve as a negative control in the study of TNNi3K biology. Small molecule crystal structures of several quinazolines have been solved, supporting observations made about overall conformation and TNNi3K inhibition.
Keywords: cardiac troponin I–interacting kinase (TNNi3K), 4-anilino-quinazolines, conformational flexibility, hinge binder, kinase inhibitor design
1. Introduction
Kinases have been successfully targeted with 52 kinase inhibitors approved by the FDA to date. These kinase inhibitor drugs tend to be predominantly multi-targeted tyrosine kinase inhibitors for the treatment of cancer [1,2]. However, with over 500 kinases in the human genome, kinase inhibitors have therapeutic potential that extends beyond cancer [3], and there remains vast untapped potential within the kinome for treating a wide range of human diseases [4].To identify which kinases play key roles beyond oncology, highly potent and selective inhibitors are required [2].
One example of less-studied kinase is cardiac troponin I-interacting kinase (TNNi3K or CARK), a member of the wider tyrosine-like kinase (TLK) family. TNNi3K is selectively expressed in heart tissues and has been linked to cardiac hypertrophy dilated cardiomyopathy, pressure overload-induced heart failure, and ischemia/reperfusion injury in an in-vivo neonatal rat model [5,6,7]. A TNNi3K knockout mouse exhibited reduced ischemic injury suggesting that deletion of TNNi3K is cardioprotective [5,6,7]. These studies suggest that TNNi3K inhibition could serve as a unique strategy for addressing acute ischemic injury and heart failure [5,6,7].
TNNi3K inhibitor development could serve the purpose of developing novel heart failure therapeutics and probing biological function of TNNi3K. Several TNNi3K inhibitor structures have been previously disclosed (1–8), each with some common structural themes (Figure 1) [8,9,10,11]. These include the structural element of a N-methylbenzenesulfonamide and a preference for a flat molecular conformation around the pyrimidine/quinazoline hinge binding moeity [8,9,10,11]. We now describe a small array of new inhibitors of TNNi3K based on GSK114 (7) to gain additional insights into the structural requirements for TNNi3K inhibition.
Figure 1.

Previously reported TNNi3K inhibitors (1–8).
2. Results and Discussion
2.1. Synthesis of Aniline 13
To synthesize analogs of GSK114 (7) we first needed to access the previously reported aniline building block (Scheme 1) [8,9,10]. First 1-fluoro-2-nitrobenzene (9) was treated with neat chlorosulfonic acid at 0 °C and then refluxed for 2 h to afford the sulfonyl chloride derivative (10) in 69% yield (5 g scale). Next, 10 was then treated with methyl amine to afford the N-methylbenzenesulfonamide product (11) in 81% yield (5 g scale) followed by a fluoride displacement using dimethylamine affording the nitro compound (12) in good yield (84%). The nitro group was reduced with hydrogen and Pd/C to furnish the aniline (13) in quantitative yield.
Scheme 1.

Synthetic route to access non-commercial aniline 13.
2.2. Synthesis of 4-Anilinoquinazolines 2, 7, 15–17 & 20
Aniline 13 was then reacted with the corresponding 6-bromo-4-chloroquinoline derivatives (14a–d) to afford 4-anilinoquinazolines (7 and 15–17) via a nucleophilic aromatic substitution reaction in good overall yields (53–67%) (Scheme 2) [12,13,14,15,16,17,18,19]. The two other derivatives (2 and 20) were afforded by reaction of commercially available anilines 18 and 19, respectively, with 4-chloro-6,7-dimethoxyquinazoline (14a) via the same route as 7 and 15–17 to afford 2 in excellent yield (78%) and 20 in good yield (57%).
Scheme 2.

Synthetic route to access 4-anilinoquinazolines (2, 7, 15–17 & 20).
2.3. Results of 4-Anilinoquinazolines 2, 7, 15–17 & 20 in TNNi3K Binding Assay
GSK114 (7) provided good baseline activity with an IC50 = 25 nM (Table 1) in an enzyme assay [10]. The incorporation of a methyl group in the hinge-binding region (15) led to complete loss of TNNi3K activity. The removal of the methoxy group in the 7-position (16) led to a decrease in potency by roughly 4-fold. Interestingly, removal of the other methoxy group at the 6-position (17) resulted in a 2-fold increase in potency. The removal of the dimethylamine substitution (2) on the aniline led to a nearly 5-fold loss of activity. However, removal of the N-methylbenzenesulfonamide (20) was more significant with an almost 250-fold drop in potency compared to GSK114 (7).
Table 1.
Results of 4-anilinoquinazolines 2, 7, 15–17 & 20 in the TNNi3K binding assay.
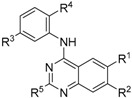
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | TNNi3K (Kd) a (nM) |
| 7 (GSK114) | OMe | OMe | N-SO2NHMe | NMe2 | H | 25 b |
| 15 | OMe | OMe | N-SO2NHMe | NMe2 | Me | 0% Inh. at 1 µM |
| 16 | OMe | H | N-SO2NHMe | NMe2 | H | 90 |
| 17 | H | OMe | N-SO2NHMe | NMe2 | H | 11 |
| 2 | OMe | OMe | N-SO2NHMe | H | H | 120 |
| 20 | OMe | OMe | H | NMe2 | H | 6200 |
a binding assay (n = 1); b previously reported enzyme assay IC50 [10].
2.4. KinomeScan® of GSK114 (7) and 15
GSK114 (7) and 15 were submitted for a KINOMEscan® assay to explore the selectivity profile across >400 human kinases at a compound concentration of 1 µM. The screen (Figure 2 and Figure 3) showed activity on only six kinases for compound 7 (with TNNi3K added from Lawhorn BG et al. [10]: MEK5, STK36, ZAK, GAK, PDFRB, BRAF and TNNi3K [10]) and two kinases (MEK5 and GAK) for 15. GSK114 (7) is a narrow-spectrum TNNi3K inhibitor but has some potent off-targets including GAK which has been previously reported for this chemotype [12,13,14,15]. GAK and MEK5 were also identified in the screening of the hinge-blocked analog (15), albeit significantly weaker activity than in the case of 7. If utilized together this set of compounds could be useful to investigate TNNi3K biology.
Figure 2.

Representation of the human kinome based on sequence identity displaying KINOMEscan® results for compound 7 screened at 1 μM. Red dots represent kinases inhibited by >80%, differentiated by size; wild-type human kinases inhibited >80% at 1 μM MEK5, 100%; STK36, 94%; ZAK, 93%; GAK, 92%; PDFRB, 91%; BRAF 84% and TNNi3K, IC50 = 25 nM [10] (see Supplementary Materials).
Figure 3.

Representation of the human kinome based on sequence identity displaying KINOMEscan® results for compound 15 screened at 1 μM. Red dots represent kinases inhibited by >75%, differentiated by size; kinases inhibited at 1 μM MEK5, 92% and GAK, 78% (see Supplementary Materials).
2.5. Modelling of Inhibitors in TNNi3K
The molecular modelling demonstrates that the N-methylbenzenesulfonamide interaction formed through a water-mediated bridge with the carboxylic acid of E509 and with the alcohol of T539 provides significant binding affinity in addition to the hinge contact (Figure 4). Literature compounds 6 and 8 (Figure 4A,B) show a binding pose mediated by the hinge-binding motif, but also by the N-methyl-benzenesulfonamide as a main interaction. The same binding interactions are also observed in compounds 17, 16, 2 in Figure 4C–E respectively, with the dimethylamine assisting in the conformational preference of the optimal inhibitor. Interestingly, despite the hinge binder being present in compound 20, the potency was significantly weaker, and this was supported by the modelling of 20 showing a loose, non-coordinated interaction of the aniline portion of the molecule in Figure 4F.
Figure 4.

Modelling of compounds 6 (A), 8 (B), 17 (C), 16 (D), 2 (E) and 20 (F) docked in the TNNi3K ATP binding site (PDB: 4YFF).
One interesting observation of this focused library was the slight improvement in potency of 17 over 7 despite the very small difference with removal of just one methoxy group. In order to investigate this effect and rationalize the improved potency we simulated the bound water networks of 17 and 7 (Figure 5) and found less disruption to the solvent network of 17 compared to 7. In the absence of direct interactions outside of simple Van der Waals contacts this provides a logical rationale for the increased potency observed with 17 [20].
Figure 5.

Docked 17 and 7 in the TNNi3K ATP binding site (PDB: 4YFF). WaterMap of 17 (A) and 7 (B) showing water molecules as spheres and WaterMap of 17 (C) and 7 (D) showing water molecules with hydrogen bonding networks.
2.6. Small Molecule Crystal Structures of 16, 17 and 20
A crystallographic analysis revealed 16, 17 and 20 crystallized as chloride salts (Figure 6) with 16 and 17 crystallizing in the triclinic P-1 space group and 20 crystallizing in the monoclinic I2/a space group. Each of the molecules comprise a planar quinazoline moiety and substituted aniline moiety. The quinazoline moiety, N1-N2 and C1-C8, exhibit root mean square (r.m.s) deviations between 0.011 Å (20) and 0.034 Å (16) with the maximum deviation from the plane being between 0.019 Å (20) and 0.0057 Å (16) for the aniline N-bonded C2 atom in all molecules. The r.m.s. deviation of the aniline moieties (excluding the dimethylamine and N-methylbenzenesulfonamide substituents) is between 0.021 Å (16) and 0.059 Å (20) with the aniline N-atom bonded C9 displaying the maximum deviation of 0.029 Å (16) or 0.031 Å (17) or for 20, the aniline N-atom N3 (0.093 Å). The dihedral angles between the two aforementioned planes are 55.75(3)° (16), 56.42(5)° (17) and 64.57(3)° (20). The C2-N3 bond distances of 1.3423(13) Å (16), 1.344(2) Å (17) and 1.3309(12) Å (20) are indicative of a double bond character in this bond consistent with conjugation in the quinazoline moieties. All other bond distances and angles are within expected limits.
Figure 6.

Molecular structure of 16H+Cl−, 17H+Cl− and 20H+Cl− showing atomic labeling and displacement ellipsoids at a 50% probability level.
The chloride anion is integral to the solid-state structures of each of the compounds (Figure 7). The structures of 16 and 17 are isostructural to a reasonable approximation and in these structures stacks, parallel to the a-axis, of protonated cationic molecules are hydrogen bonded to Cl− via the quinazoline N-H+ (N-H+…Cl− (3.0741(10) Å (16), 3.1185(17) Å (17)), aniline N-H (N-H…Cl− (3.128(1) Å (16), 3.1896(16) Å (17)) and the N-methylbenzenesulfonamide N-H (N-H…Cl− (3.2989(11) Å (16), 3.2747(18) Å (17)). These hydrogen bonds link neighboring molecules in each stack with molecules in adjacent stacks via each chloride anion to form two-dimensional layers which stack via π-π interactions to form the structures. The structure of 20 comprises one-dimensional ‘tapes’ of 20H+ hydrogen bonded to Cl− via the quinazoline N-H+ and aniline N-H respectively (N-H+…Cl−…H-N (2.9732(9) Å, 3.1444(9) Å)) parallel to the b-axis. These tapes form two-dimensional, interleaved layers via π-π interactions parallel to the a-axis which close-pack to form the structure.
Figure 7.

Unit cell contents of 16H+Cl−, 17H+Cl− and 20H+Cl− shown in projection down the a-axis (16, 17) or b-axis (20). Hydrogen bonds are shown as dashed green lines.
3. Experimental Section
3.1. Chemistry
All reactions were performed using flame-dried round-bottomed flasks or reaction vessels. Where appropriate, reactions were carried out under an inert atmosphere of nitrogen with dry solvents, unless otherwise stated. Yields refer to chromatographically and spectroscopically pure isolated yields. Reagents were purchased at the highest commercial quality and used without further purification. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60F-254) using ultraviolet light as the visualizing agent. NMR spectra were recorded on a Varian Inova 400 spectrometer (Varian, Palo Alto, CA, USA) and were calibrated using residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26, 13C NMR = 77.16). The following abbreviations or combinations thereof have been used to explain the multiplicities observed: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Liquid chromatography (LC) and high-resolution mass spectra (HRMS) were recorded on a ThermoFisher hybrid LTQ FT (ICR 7T) (ThermoFisher, Waltham, MA, USA). The University of Southampton (Southampton, UK) small molecule X-ray facility collected and analyzed all X-ray diffraction data. Compounds 10 and 11 were prepared as previously described [8].
4-(dimethylamino)-N-methyl-3-nitrobenzene-1-sulfonamide (12) was obtained as a yellow solid (465 mg, 1.793 mmol, 84%). MP 132–134 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 2.3 Hz, 1H), 7.75 (dd, J = 9.1, 2.3 Hz, 1H), 7.32 (d, J = 9.1 Hz, 1H), 2.93 (s, 6H), 2.40 (s, 3H). HRMS m/z [M + H]+ calculated for C9H14N3O4S: 260.0705, found 260.0696, LC tR = 4.00 min, >98% purity, consistent with previously reported results [8].
3-amino-4-(dimethylamino)-N-methylbenzene-1-sulfonamide (13) was obtained as a purple solid (442 mg, 1.928 mmol, 100%). MP 66–68 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.15–7.04 (m, 2H), 7.00 (d, J = 8.2 Hz, 1H), 6.93 (dd, J = 8.2, 2.2 Hz, 1H), 5.14 (s, 2H), 2.61 (s, 6H), 2.38 (d, J = 5.0 Hz, 3H). HRMS m/z [M + H]+ calculated for C9H16N3O2S: 230.0963, found 230.0956, LC tR = 2.77 min, >98% purity consistent with previously reported results [8].
3-((6,7-dimethoxyquinazolin-4-yl)amino)-4-(dimethylamino)-N-methylbenzenesulfonamide (7) was obtained as a yellow solid (98.5 mg, 0.236 mmol, 53%). MP 244–246 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 1H), 8.75 (s, 1H), 8.34 (s, 1H), 7.70–7.55 (m, 2H), 7.40 (s, 1H), 7.32 (q, J = 5.0 Hz, 1H), 7.22–7.15 (m, 1H), 3.98 (s, 6H), 2.79 (s, 6H), 2.40 (d, J = 5.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 159.0, 156.4, 151.9, 150.3, 148.8, 135.4, 129.2, 128.6, 126.8, 125.9, 117.7, 107.2, 103.9, 99.7, 56.9, 56.5, 42.2 (2C, s), 28.7. HRMS m/z [M + H]+ calculated for C19H24N5O4S: 418.1549, found 418.1540, LC tR = 3.04 min, >98% purity consistent with previously reported results [10].
3-((6,7-dimethoxy-2-methylquinazolin-4-yl)amino)-4-(dimethylamino)-N-methylbenzenesulfonamide (15) was obtained as a yellow solid (101 mg, 0.235 mmol, 56%). MP 252-254 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.23 (s, 1H), 8.28 (s, 1H), 7.67 (d, J = 2.3 Hz, 1H), 7.63 (dd, J = 8.7, 2.3 Hz, 1H), 7.39 (s, 1H), 7.32 (q, J = 5.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 1H), 3.98 (s, 6H), 2.80 (s, 6H), 2.52 (s, 3H), 2.44 (d, J = 5.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 159.0, 158.4, 156.2, 151.6, 149.7, 136.0, 129.0, 128.5, 126.5, 125.9, 117.8, 105.7, 103.8, 99.1, 56.8, 56.4, 42.0 (2C, s), 28.7, 21.9. HRMS m/z [M + H]+ calculated for C20H26N5O4S: 432.1706, found 432.1700, LC tR = 3.25 min, >98% purity.
4-(dimethylamino)-3-((6-methoxyquinazolin-4-yl)amino)-N-methylbenzenesulfonamide (16) was obtained as a yellow solid (127 mg, 0.387 mmol, 64%). MP 224–226 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 8.81 (s, 1H), 8.35 (d, J = 2.6 Hz, 1H), 7.94 (d, J = 9.2 Hz, 1H), 7.76 (dd, J = 9.2, 2.6 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.65 (dd, J = 8.6, 2.3 Hz, 1H), 7.33 (q, J = 5.0 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 3.98 (s, 3H), 2.82 (s, 6H), 2.43 (d, J = 4.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 159.9, 159.1, 151.8, 149.1, 133.6, 129.3, 128.5, 127.0, 126.9, 125.7, 121.7, 117.8, 114.6, 104.3, 56.6, 42.2 (2C, s), 28.7. HRMS m/z [M + H]+ calculated for C18H22N5O3S: 388.1443, found 388.1434, LC tR = 3.08 min, >98% purity consistent with previously reported results [10].
4-(dimethylamino)-3-((7-methoxyquinazolin-4-yl)amino)-N-methylbenzenesulfonamide (17) was obtained as a mustard solid (133 mg, 0.344 mmol, 67%). MP 216–219 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.86 (d, J = 9.2 Hz, 1H), 8.82 (s, 1H), 7.73–7.57 (m, 2H), 7.49 (dd, J = 9.3, 2.5 Hz, 1H), 7.42 (d, J = 2.5 Hz, 1H), 7.36 (q, J = 5.1 Hz, 1H), 7.23–7.18 (m, 1H), 3.98 (s, 3H), 2.80 (s, 6H), 2.42 (d, J = 4.5 Hz, 3H).13C NMR (100 MHz, DMSO-d6) δ 164.9, 159.9, 151.8, 150.9, 140.8, 129.3, 128.5, 127.0, 126.9, 125.8, 119.2, 117.9, 107.2, 100.2, 56.4, 42.2 (2C, s), 28.7. HRMS m/z [M + H]+ calculated for C18H22N5O3S: 388.1443, found 388.1436, LC tR = 3.07 min, >98% purity.
3-((6,7-dimethoxyquinazolin-4-yl)amino)-N-methylbenzenesulfonamide (2) was obtained as a beige solid (195 mg, 0.521 mmol, 78%). MP 246–248 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.77 (s, 1H), 8.85 (s, 1H), 8.44 (s, 1H), 8.16 (q, J = 1.4 Hz, 1H), 8.09–8.02 (m, 1H), 7.78–7.63 (m, 2H), 7.60 (q, J = 5.0 Hz, 1H), 7.40 (s, 1H), 4.01 (s, 3H), 3.96 (s, 3H), 2.47 (d, J = 4.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 158.2, 156.4, 150.3, 148.5, 139.8, 137.7, 135.7, 129.6, 128.4, 123.9, 122.7, 107.4, 104.2, 99.6, 57.1, 56.5, 28.8. HRMS m/z [M + H]+ calculated for C17H19N4O4S: 375.1127, found 375.1112, LC tR = 2.91 min, >98% purity consistent with previously reported results [9].
N1-(6,7-dimethoxyquinazolin-4-yl)-N2,N2-dimethylbenzene-1,2-diamine (20) was obtained as a beige solid (124 mg, 0.381 mmol, 57%). MP 224–226 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H), 8.69 (s, 1H), 8.33 (s, 1H), 7.44 (s, 1H), 7.34 (ddd, J = 15.6, 7.7, 1.6 Hz, 2H), 7.22 (d, J = 8.1 Hz, 1H), 7.08 (td, J = 7.5, 1.4 Hz, 1H), 3.99 (d, J = 2.6 Hz, 6H), 2.68 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 159.0, 156.1, 150.1, 148.7, 135.9, 129.1 (2C, s), 129.0 (2C, s), 128.2, 122.0, 118.9, 107.3, 103.9, 100.2, 56.8, 56.4, 43.3 (2C, s). HRMS m/z [M + H]+ calculated for C18H21N4O2: 325.1665, found 325.1657, LC tR = 2.60 min, >98% purity.
3.2. Mass Spectrometry
Samples were prepared as previously described (see Supplementary Materials) [19].
3.3. Molecular Modelling
Prior to modelling studies, the structure of the TNNi3K X-ray structure (PDB: 4YFF) [8] was pre-processed by stepwise manner using the protein preparation wizard tool of Schrödinger Suite 2019-3 (Protein Preparation Wizard uses modules: Epik; Impact and Prime, Schrödinger, LLC, New York, NY, 2019). Preliminary structural inspections of the protein structures, including visualization of electronic density maps of the protein ligand complexes, were done online at the PDB site (https://www.ebi.ac.uk/pdbe/entry/pdb/4yfi). Consequently, the conserved hinge-binding bridged water at the site of back pocket was left in a place, as suggested by original publication [8]. Structures of small molecule ligands were parametrized and minimized using LigPrep module (LigPrep, Schrödinger, LLC, New York, NY, 2019). Molecular docking studies were computed using Glide module of Schrödinger employing XP-setting with softened Van der Waals potential (0.5 in place of default 0.8). A hydrogen bond constraint was added to hinge residue ILE542 to improve convergence of docking poses.
A hydration site analysis was calculated using WaterMap functionality of Schrödinger suite 2019-4. Analysis was computed for empty protein (PDB: 4YFF) and for IFD docking pose of compound 7 and 17 retained in ATP-binding site.
3.4. Crystallography
For each compound a suitable crystal was selected and mounted on a MITIGEN holder (MiTeGen, Ithaca, NY, USA) in perfluoroether oil on a Rigaku FRE+ diffractometer equipped with VHF Varimax confocal mirrors and an AFC12 goniometer and HyPix 6000 detector. The crystal was kept at a steady T = 100(2) K during data collection. The structure was resolved by the ShelXT [21] structure solution program using the using dual methods solution method and using Olex2 [22] as the graphical interface. The model was refined with version 2018/3 of ShelXL [23], using full matrix least squares minimization on F2 minimization. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model, except for those bonded to N-atoms which were located in the difference map and refined with a riding model. The data for 17H+Cl− were processed as a 2-component non-merohedral twin (rotation (UB1, UB2) = 179.7789° around 0.66 0.74 0.13 (rec.) 0.93 0.37 −0.00 (dir) lattice vectors).
Crystal data for 16H+Cl−, Mr = 423.91; triclinic; P-1 (No. 2); a = 6.23720(10)Å; b = 9.54880(10)Å; c = 17.2769(3)Å; α = 91.5310(10)°, β = 92.682(2)°, γ = 99.235(2)°, V = 1013.91(3) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(Mo Kα) = 0.321 mm−1, 27,000 reflections measured, 5645 unique (Rint = 0.0286) which were used in all calculations. The final wR2 was 0.0860 (all data) and R1 was 0.0325 (I > 2(I)).
Crystal data for 17H+Cl−, Mr = 423.91, triclinic, P-1 (No. 2), a = 6.1936(2)Å, b = 9.6619(3)Å, c = 17.1723(5)Å, α = 85.698(2)°, β = 87.900(2)°, γ = 79.989(2)°, V = 1008.84(5)Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(Mo Kα) = 0.322 mm−1, 9229 reflections measured, 9229 unique, which were used in all calculations. The final wR2 was 0.1356 (all data) and R1 was 0.0466 (I > 2(I)).
Crystal data for 20H+Cl−, Mr = 360.84, monoclinic, I2/a (No. 15), a = 13.3145(2)Å, b = 9.38030(10)Å, c = 29.4816(5)Å, α = 97.253(2)°, β = γ = 90°, V = 3652.61(9)Å3, T = 100(2) K, Z = 8, Z’ = 1, μ(Mo Kα) = 0.228 mm−1, 47,137 reflections measured, 5099 unique (Rint = 0.0264) which were used in all calculations. The final wR2 was 0.0915 (all data) and R1 was 0.0330 (I > 2(I)).
3.5. KinomeScan® Assay
KinomeScan® Assay on 7 and 15 was performed as previously described by EuroFins DiscoverX [24].
4. Conclusions
We have demonstrated a robust way to access the 4-anilinoquinazoline scaffold in good yield supported by previous literature [12,13,14,15,16,17,18,19]. We report the synthesis of several related 4-anilinoquinazolines as inhibitors of cardiac troponin I–interacting kinase (TNNi3K). These close structural analogs of GSK114 (7) provide new understanding of structure–activity relationships between the 4-anilinoquinazoline scaffold and TNNi3K inhibition. Further, an interesting lesson in kinase design was found: having the hinge binder present does not always guarantee a potent compound, as in the case of 2 vs 20, where 20 without the N-methyl-benzenesulfonamide peripheral interaction is significantly weaker in TNNi3K inhibition. However, both interactions are required for a potent TNNi3K inhibitor. We also show that a simple hinge block with a methyl group (15) is enough to create a scaffold/negative control compound. We hope that the insights provide the medicinal chemist with an enhanced toolbox for kinase inhibitor design.
Acknowledgments
We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data of 16, 17 and 20. We also thank Dr. Brandie Ehrmann for LC-MS/HRMS support provided by the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill.
Supplementary Materials
The following are available online: crystallographic data for compound 16H+Cl−, 17H+Cl− and 20H+Cl− in crystallographic information file (CIF) format, at https://www.mdpi.com/1420-3049/25/7/1697/s1. The data were submitted to the Cambridge Structural Database and given the following CCDC deposition numbers 1980942–1980944. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
Conceptualization and writing the original draft, C.R.M.A.; validation, resources and data curation, C.R.M.A., T.L., C.I.W. and G.J.T.; editing, C.R.M.A., T.L., C.I.W., G.J.T. and W.J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. We also thank CSC—IT Center for Science Ltd., Finland for software management and computational resources and the Biocenter Finland/DDCB for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 2, 7, 15–17, 20 are available from the authors.
References
- 1.Ferguson F.M., Gray N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P., Alessi D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Knapp S., Arruda P., Blagg J., Burley S., Drewry D.H., Edwards A., Fabbro D., Gillespie P., Gray N.S., Kuster B., et al. A public-private partnership to unlock the untargeted kinome. Nat. Chem. Biol. 2013;9:3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 5.Vagnozzi R.J., Gatto G.J., Jr., Kallander L.S., Hoffman N.E., Mallilankaraman K., Ballard V.L.T., Lawhorn B.G., Stoy P., Philp J., Graves A.P., et al. Inhibition of the cardiomyocyte-specific TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart. Sci. Transl. Med. 2013;5:207ra141. doi: 10.1126/scitranslmed.3006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., Wang H., Ye J., Xu R.X., Song L., Shi N., Zhang Y.W., Chen X., Meng X.M. Adenovirus-mediated overexpression of cardiac troponin I-interacting kinase promotes cardiomyocyte hypertrophy. Clin. Exp. Pharmacol. Physiol. 2011;38:278–284. doi: 10.1111/j.1440-1681.2011.05499.x. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler F.C., Tang H., Marks O.A., Hadnott T.N., Chu P.L., Mao L., Rockman H.A., Marchuk D.A. Tnni3k modifies disease progression in murine models of cardiomyopathy. PLoS Genet. 2009;5:e1000647. doi: 10.1371/journal.pgen.1000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawhorn B.G., Philp J., Zhao Y., Louer C., Hammond M., Cheung M., Fries H., Graves A.P., Shewchuk L., Wang L., et al. Identification of Purines and 7-Deazapurines as Potent and Selective Type I Inhibitors of Troponin I-Interacting Kinase (TNNI3K) J. Med. Chem. 2015;58:7431–7448. doi: 10.1021/acs.jmedchem.5b00931. [DOI] [PubMed] [Google Scholar]
- 9.Lawhorn B.G., Philp J., Graves A.P., Holt D.A., Gatto G.J., Jr., Kallander L.S. Substituent Effects on Drug-Receptor H-bond Interactions: Correlations Useful for the Design of Kinase Inhibitors. J. Med. Chem. 2016;59:10629–10641. doi: 10.1021/acs.jmedchem.6b01342. [DOI] [PubMed] [Google Scholar]
- 10.Lawhorn B.G., Philp J., Graves A.P., Shewchuk L., Holt D.A., Gatto G.J., Jr., Kallander L.S. GSK114: A selective inhibitor for elucidating the biological role of TNNI3K. Bioorg Med. Chem Lett. 2016;26:3355–3358. doi: 10.1016/j.bmcl.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 11.Philp J., Lawhorn B.G., Graves A.P., Shewchuk L., Rivera K.L., Jolivette L.J., Holt D.A., Gatto G.J., Jr., Kallander L.S. 4,6-Diaminopyrimidines as Highly Preferred Troponin I-Interacting Kinase (TNNI3K) Inhibitors. J. Med. Chem. 2018;61:3076–3088. doi: 10.1021/acs.jmedchem.8b00125. [DOI] [PubMed] [Google Scholar]
- 12.Asquith C.R.M., Laitinen T., Bennett J.M., Godoi P.H., East M.P., Tizzard G.J., Graves L.M., Johnson G.L., Dornsife R.E., Wells C.I., et al. Identification and Optimization of 4-Anilinoquinolines as Inhibitors of Cyclin G Associated Kinase. Chem. Med. Chem. 2018;13:48–66. doi: 10.1002/cmdc.201700663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asquith C.R.M., Berger B.T., Wan J., Bennett J.M., Capuzzi S.J., Crona D.J., Drewry D.H., East M.P., Elkins J.M., Fedorov O., et al. SGC-GAK-1: A Chemical Probe for Cyclin G Associated Kinase (GAK) J. Med. Chem. 2019;62:2830–2836. doi: 10.1021/acs.jmedchem.8b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asquith C.R.M., Treiber D.K., Zuercher W.J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G associated kinase (GAK) Bioorg. Med. Chem. Lett. 2019;29:1727–1731. doi: 10.1016/j.bmcl.2019.05.025. [DOI] [PubMed] [Google Scholar]
- 15.Asquith C.R.M., Naegeli N., East M.P., Laitinen T., Havener T.M., Wells C.I., Johnson G.L., Drewry D.H., Zuercher W.J., Morris D.C. Design of a cyclin G associated kinase (GAK)/epidermal growth factor receptor (EGFR) inhibitor set to interrogate the relationship of EGFR and GAK in chordoma. J. Med. Chem. 2019;62:4772–4778. doi: 10.1021/acs.jmedchem.9b00350. [DOI] [PubMed] [Google Scholar]
- 16.Asquith C.R.M., Fleck N., Torrice C.D., Crona D.J., Grundner C., Zuercher W.J. Anti-tubercular activity of novel 4-anilinoquinolines and 4-anilinoquinazolines. Bioorg. Med. Chem. Lett. 2019;18:2695–2699. doi: 10.1016/j.bmcl.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asquith C.R.M., Maffuid K.A., Laitinen T., Torrice C.D., Tizzard G.J., Crona D.J., Zuercher W.J. Targeting an EGFR water network using novel 4-anilinoquin(az)olines inhibitors for chordoma. ChemMedChem. 2019;14:1693–1700. doi: 10.1002/cmdc.201900428. [DOI] [PubMed] [Google Scholar]
- 18.Asquith C.R.M., Bennett J.M., Su L., Laitinen T., Elkins J.M., Pickett J.E., Wells C.I., Li Z., Willson T.M., Zuercher W.J. Development of SGC-GAK-1 as an orally active in vivo probe for cyclin G associated kinase through cytochrome P450 inhibition. Molecules. 2019;24:4016. doi: 10.3390/molecules24224016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asquith C.R.M., Tizzard G.J. 6-Bromo-N-(2-methyl-2H-benzo[d][1,2,3]triazol-5-yl)quinolin-4-amine. Molbank. 2019;2019:M1087. doi: 10.3390/M1087. [DOI] [Google Scholar]
- 20.Krimmer S.G., Betz M., Heine A., Klebe G. Methyl, ethyl, propyl, butyl: Futile but not for water, as the correlation of structure and thermodynamic signature shows in a congeneric series of thermolysin inhibitors. Chem. Med. Chem. 2014;9:833–846. doi: 10.1002/cmdc.201400013. [DOI] [PubMed] [Google Scholar]
- 21.Sheldrick G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015;A71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [Google Scholar]
- 23.Sheldrick G.M. Crystal structure refinement with ShelXL. Acta Crystallogr. 2015;C27:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabian M.A., Biggs W.H., Treiber D.K., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M., et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.