

Abstract
We used the molecular modeling program Rosetta to identify clusters of amino acid substitutions in antibody fragments (scFvs and scAbs) that improve global protein stability and resistance to thermal deactivation. Using this methodology, we increased the melting temperature (Tm) and resistance to heat treatment of an antibody fragment that binds to the Clostridium botulinum hemagglutinin protein (anti-HA33). Two designed antibody fragment variants with two amino acid replacement clusters, designed to stabilize local regions, were shown to have both higher Tm compared to the parental scFv and importantly, to retain full antigen binding activity after 2 hours of incubation at 70 °C. The crystal structure of one thermostabilized scFv variants was solved at 1.6 Å and shown to be in close agreement with the RosettaAntibody model prediction.
Keywords: Biomolecular Engineering, Bioengineering, Biochemicals, Biofuels, Food Rosetta, antibody engineering, thermostable antibodies, scFv, scAb
INTRODUCTION
Antibody fragments, such as single chain variable fragment (scFv) and single chain antibody (scAb), have been utilized for numerous applications1. Antibody fragments can be efficiently expressed in microorganisms2, they offer several advantages for diagnostic applications and also afford greater tissue penetration properties, which is useful for therapeutic purposes especially in cancer3. However, compared to full length antibodies, scFv and scAb fragments have generally lower stability and are more susceptible to thermal stress4,5. Low thermal stability can lead to decreased shelf-life, poor pharmacokinetics, and reduced biodistribution6. Additionally, the formation of partially unfolded structures at elevated temperatures exposes the hydrophobic core that can initiate protein aggregation7,8.
A number of approaches for scFv stabilization have been reported including directed evolution of variants with increased stability using display systems9-13, stabilization of the interface between variables regions of heavy (VH) and light (VL) chains through the introduction of a non-native disulfide bond between VH and VL14-16, and rational design17-19. Recent advances in computational protein modeling also have accelerated stability design and optimization processes of enzymes and biotherapeutics, including antibodies20-24. RosettaAntibody in particular has become an important tool for predicting antibody structures by first using sequence homology to generate a starting model, then refining the conformations of complementarity determining region (CDR) loops as well as VH-VL orientations25,26. Rosetta-based approaches have been used to generate “supercharged” antibody variants27 as well as for the de novo design of antigen binders28-30.
Here, starting with the VH and VL sequences of a scFv molecule that binds to Clostridium botulinum hemagglutinin 33 protein (HA33)31 as a model system, we first produced a structural model using RosettaAntibody32 and then designed in silico clusters of mutations that stabilize local regions that are expected to be most susceptible to unfolding. The engineered variants were expressed, purified and evaluated in terms of thermodynamic stability (Tm) and resistance to inactivation at elevated temperatures. We identified two variants C1+C9 and C9+C14 in which increased local hydrophobic interactions and rigidity confer an increased Tm by ~4.5 °C and >20-fold greater resistant to inactivation at 70 °C for 2 hours. The structure of one of these computationally stabilized variants, C9+C14 scFv, was solved at 1.6 Å to validate the accuracy of the model and the designed stabilizing mutations.
RESULTS
Homology modeling and engineering of anti-HA33 scFv
The RosettaAntibody variable region homology modeling server32 was used to generate a structural model of anti-HA33 scFv based on the amino acid sequence. Antibodies with known structures that have VH and VL framework sequences (from Protein Data Bank [PDB] 1RUR and 1DCF) and H1, H2, L1, L2, and L3 loops (1JO5, 1AJ7, 1A2Y, 1WEJ, and 1WEJ) identical or similar to those of anti-HA33 scFv were identified. H3 loop conformations were modeled de novo, and after several cycles of modeling and refinement, the ten scFv models with the lowest energies based on the Rosetta score function were selected (Supplementary Figure 1).
We sought to redesign small clusters of residues (≤5 residues) to increase local stability. Without any special insight on framework regions likely to be destabilizing, we visually inspected the predicted structures to generate a diverse set of clusters in the orders of hundred that would cover most framework regions. The Rosetta protein structure prediction and design software uses a weighted energy function33, incorporating hydrogen bonding, salt-bridge interactions, van der Waals forces, and solvation energies to assign a Rosetta Score that reflects relative protein stability. We used the software to introduce cluster mutations in framework regions, excluding cysteine residues, in the ten homology models. Ultimately, the top 15 lowest energy-scored cluster variants were selected for further experimental validation for their expression and thermostability (Figure 1). These models showed a tendency to introduce bulkier side-chains and add more hydrophobic contacts. We note that all the mutations identified by computational modeling were proximal to the protein surface as mutations in the hydrophobic core of the protein failed to show improvements based on the energy-score.
Figure 1. Description of the cluster variants designed with Rosetta.
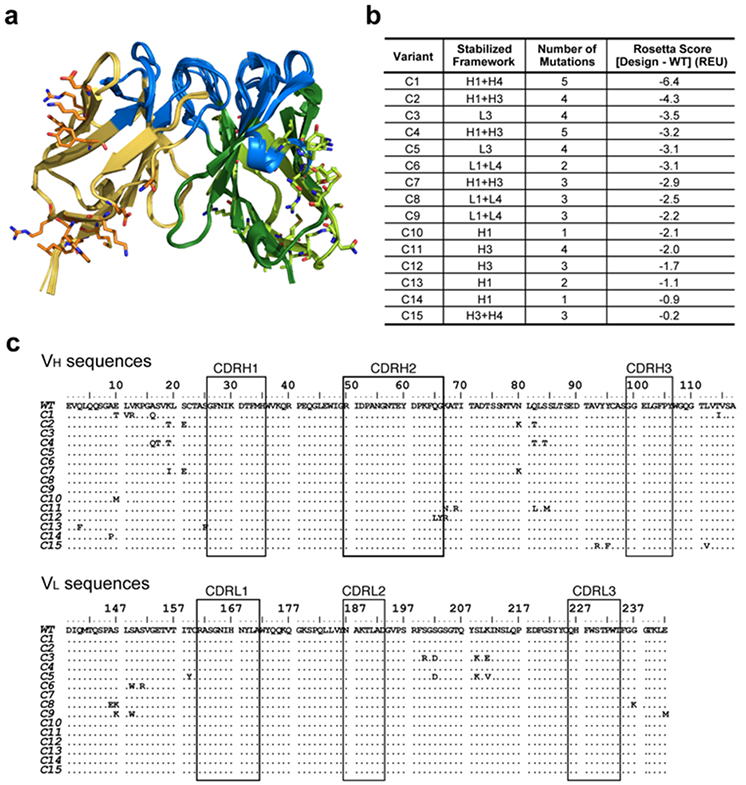
(a) Superimposed modeled structures of the 15 cluster variants. A diverse set of cluster mutations were designed to cover most regions of the anti-HA33 VH and VL regions. The designed mutations are depicted as stick representations. Yellow, VL; green, VH; blue, CDR. (b) Description of the 15 variants. Framework regions of heavy (H) and light (L) with introduced mutations are shown in the second column. WT, wildtype; REU, Rosetta Energy Unit. More negative energies represent more stable folds. (c) Sequence alignments of the 15 cluster variants against wildtype. CDRs are delimited with boxes, and VH and VL regions are linked with a (GGGGS)4 linker.
Characterization of the designed antibody variants
Fusion of the CL chain to scFvs (resulting in scAb fragments) increases the expression yield of scFv antibody fragments in bacteria by assisting with folding efficiency in vivo, but it has been reported not to affect the thermal stability of the protein34. To validate that the thermostability of scAb proteins reflects that of scFvs, we first expressed the anti-HA33 antibody in both scFv and scAb formats in E. coli. Because we had found that the yield of HA33 scFv in E.coli is very low due to aggregation, we produced scFvs by first expressing the corresponding scAbs containing a Factor Xa cleavage site between the VL and CL domains. Following purification from E.coli, scFvs were produced by treatment with Factor Xa and subsequent removal of the CL domain (Supplementary Figure 2). We measured Tm via differential scanning fluorometry (DSF), and scFv produced by this method had a comparable Tm (69.2 ± 0.1 °C) as the anti-HA33 scAb (69.3 ± 0.1 °C) (Figure 2a).
Figure 2. Characterization of the cluster variants.
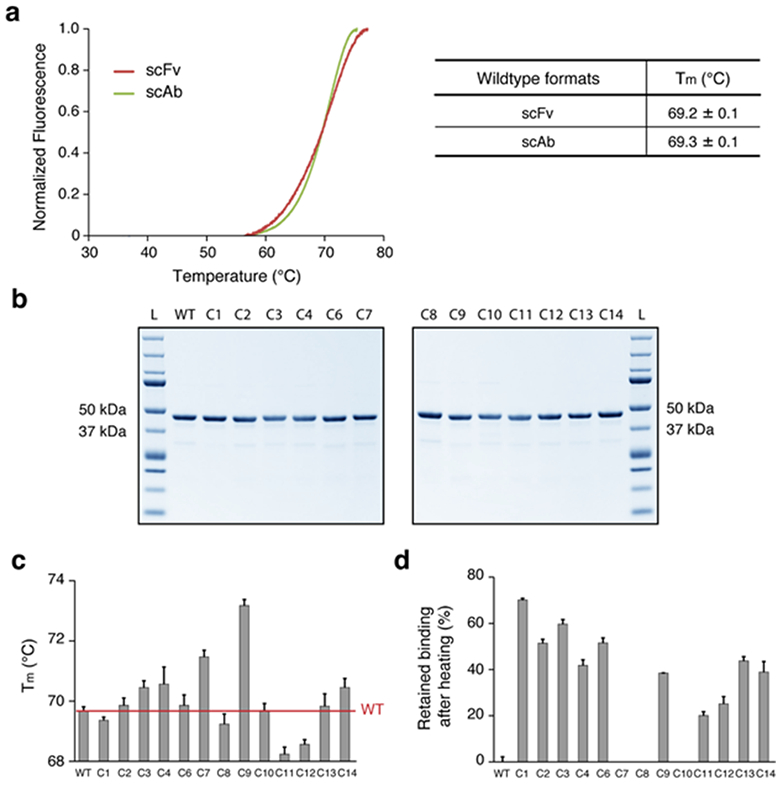
(a) Melting temperatures (Tm) of the anti-HA33 antibody in scFv and scAb formats. scFv were generated using the approach summarized in Supplementary Figure 2. (b) SDS-PAGE of the HA33 and the 13 variants expressed in scAb format (expected size: ~43 kDa). L, protein ladder. (c) Tm measurements of the designed variants expressed as scAbs. (d) Retained binding activity of the designed scAb variants after thermal challenge at 70 °C for 1 hour. The percent binding affinity retained for each variant was calculated by setting the half maximal effective concentration (EC50) of each variant without the heat treatment as 100% and the EC50 of the wildtype after the thermal challenge as 0% (the C7, C8, and C10 variants were worse than the wildtype and set to 0). For (a), (c) and (d), averages are calculated as mean with error indicating s.d., repeated in triplicates.
After confirming that the Tm of the wildtype anti-HA33 antibody in the scAb format reflects that of the scFv form, we expressed all the cluster variants in the scAb formats to examine their thermostability. Overall, 13 out of 15 variants were successfully expressed and purified with >95% purity (Figure 2b), and all showed comparable affinity to HA33 as determined by ELISA (Supplementary Table 1).
For each variant, we measured both the thermodynamic stability by determining the Tm and also the resistance of the proteins to deactivation and loss of binding activity following prolonged incubation at 70 °C. The latter assay reflects the fraction of the protein that irreversibly unfolds at 70 °C35. 5 out of 13 variants showed small but significant increases in Tm when compared to the wildtype scAb (Figure 2c), with the C9 variant showing the highest ΔTm of 3.9 ± 0.1 °C. Next, each variant was incubated at 70 °C for 1 hour, and then the remaining binding activity at room temperature was determined by ELISA. 10 out of 13 variants exhibited dramatic improvement with regards to resistance to inactivation at 70 °C, with the C1 scAb retaining 70.2 ± 0.7 % of its binding affinity (Figure 2d and Supplementary Table 2). We noted that Tm did not correlate with resistance to thermal stress; for example, C1 showed the highest resistance to thermal stress despite having a lower Tm than the wildtype scAb, whereas C7 was more susceptible to thermal challenge than the wildtype despite showing an increase in Tm.
We sought to further improve scAb thermal stability by examining whether mutations that stabilize different regions exert synergistic effects. For this purpose, we combined the mutations in the C9 variant which showed the highest increase in Tm due to amino acid substitutions in the VL with those from variants C1, C2, C4, and C14, which had shown improved thermal stability with the mutations introduced in the VH region (Supplementary Table 3). The two best variants were C1+C9 and C9+C14, which showed ΔTm of 4.5 ± 0.1 °C and 4.6 ± 0.1 °C, respectively (Figure 3a). Significantly, these two variants retained 100% of their binding affinity to HA33 after the thermal challenge at 70 °C of 1 hour as well as after a 2 hour thermal challenge (Figure 3b and c, and Supplementary Figure 3).
Figure 3. Characterization of the best combinatorial variants.
(a) Tm measurements of wildtype (WT), C1+C9 and C9+C14 variants in scAb format. (b) ELISA signal before and after the thermal challenges. Wildtype and each variant underwent thermal challenge of heating at 70 °C for 1 hour or 2 hours before OD450 was measured. (c) Summary of binding affinities (EC50) to HA33 before and after thermal challenges of 1 hour or 2 hours. For (a)-(c), averages are calculated as mean with error indicating s.d., repeated in triplicates.
The crystal structure of C9+C14 (in the scFv format) was solved at 1.6 Å resolution (Supplementary Table 4). Overall, the structure was in close agreement with the RosettaAntibody model, with the overall root-mean-square deviation (RMSD) being 0.56 Å, and RMSD values ranging between 0.58 Å to 0.84 Å for individual antibody framework and CDR regions (Figure 4a). In the design model of C9, the hydrophobic groups in the side chains of S147K, S149W and E242M pack together to form a hydrophobic cluster on the surface of the VL domain, demonstrated by the increased local hydrophobicity based on the Eisenberg hydrophobicity scale36 (Figure 4b and Supplementary Figure 4). Similar interactions are observed in the crystal structure, although the chi2 torsion angle on W149 is flipped 180 degrees so that the opposite face of the indole ring is presented to the methylene and methyl groups of K147 and M242. It is interesting that placing hydrophobic residues on the solvent exposed face of a β-sheet can raise protein stability. We observed a similar result in a previous study with a β-sandwich protein that adopts an antibody-like fold37. Although the hydrophobic groups are partially solvent exposed in this scenario, they are still burying more hydrophobic surface area than is buried in the unfolded state. This additional hydrophobic burial occurs when the β-strands partner and pack adjacent side chains against each other. In C9, M242 is brought next to W149 when the full β-sheet forms. Overall, our results are consistent with other studies that show that hydrophobic interactions substantially improving protein stability38-40.
Figure 4. Comparisons between the Rosetta-predicted and experimentally-determined crystal structure of the variant C9+C14.
(a) Superimposed structures of the modeled and experimentally determined structures. Root mean square deviation (RMSD) between the two structures for each region is shown. (b) Predicted interactions of the substituted residues (left) compared to the observed interactions formed by the residues (right) for the mutations included in the variant C9. (c) Predicted interactions of the substituted residues (left) compared to the observed interactions formed by the residues (right) for the mutations included in the variant C14.
The amino acid substitution derived from the C14 variant (A9P) in the VH rendered that loop more rigid (Figure 4c). Proline residue has lower configurational entropy than any other amino acid residue due to its pyrrolidine ring hindrance, and prolines have been shown to be beneficial for increasing thermal stability when they are introduced within loops41-43. Taken together, our data demonstrate that the overall stabilization of anti-HA33 antibody can be explained and rationalized by the interactions and effects introduced by the mutations and this, in turn, validates the accuracy of the design through the Rosetta.
DISCUSSION
Antibody fragments, such as scFvs and scAbs, are widely used for diagnostics; however, their utility can be limited by loss of binding activity upon storage especially at elevated temperatures. Stability at elevated temperatures is particularly relevant for the design of diagnostic kits suitable for desert and tropical environments under conditions where refrigeration is not an option. Here we report the successful stabilization of an antibody fragment against deactivation at elevated temperatures by computational design. We introduced stabilizing amino acid substitutions only in the frameworks to minimize effects on antigen binding affinity and we confirmed experimentally that this is the case (Supplementary Table 1). 5 out of 13 variants showed a significant increase in Tm, and 10 out of 13 variants showed increased resistance to thermal deactivation following incubation at 70 °C (Figure 2c and d). Interestingly, variants that had stabilizing substitutions at the lower part of VH (C1 and C14) and VL (C9) regions showed the highest increase in thermostability. Two variants with combinations of mutations, C1+C9 and C9+C14, showed about ~4.5 °C improvement in Tm and improved resistance to thermal challenge (Figure 3). The crystal structure of the C9+C14 variant was in excellent agreement (<1Å overall RMSD) with the RosettaAntibody model (Figure 4a). We note that in the C9+C14 variant two serine residues (S147K and S149W) had been replaced. Of note, a systematic analysis of proteins from thermophilic organisms has shown that serine is considered a thermolabile amino acid and the serine content is significantly lower in thermostable proteins44. Overall, the three mutations in the VL domain likely increased the stability of the antibody by introducing new hydrophobic contacts between β-strands. The bottom of the VH domain likely is stabilized through the rigidity introduced by the proline substitution.
In conclusion, we used the Rosetta platform for protein design to achieve thermal stabilization of an antibody fragment; our best design was completely resistant to loss of binding activity even after 2 hours at 70 °C. It remains to be seen whether the amino acid substitutions in C9+C14 can be imported into antibody fragments that recognize other antigens but are derived from the same framework (and have different CDRs). Overall, the Rosetta based workflow reported here is likely to be useful as a strategy for engineering enhanced resistance to thermal deactivation in other antibody fragments of practical interest.
Materials and Methods
Antibody Modeling Methods
The RosettaAntibody variable region homology modeling server32 was used to generate a structural model of anti-HA33 scFv based on the amino acid sequence. First, from antibodies with known structures that have VH and VL framework sequences (from Protein Data Bank [PDB] 1RUR and 1DCF) and H1, H2, L1, L2, and L3 loops (1JO5, 1AJ7, 1A2Y, 1WEJ, and 1WEJ) were used for modeling. H3 loop conformations were modeled de novo, while allowing the VH-VL orientation and the conformations of other regions to change. After several cycles of modeling and refinement, the ten scFv models with the lowest energies based on the Rosetta score function were selected.
For the protein design simulations, the ten lowest scoring homology models from RosettaAntibody of the starting antibody were used as templates. The homology models output from RosettaAntibody were subjected to two additional rounds of rotamer optimization and gradient-based minimization of side chain and backbone torsion angles before the design simulations were performed. This initial preparation of the antibody model and the subsequent design simulations were performed with the “score12” energy function in Rosetta45. Without relying on any foreknowledge in regards to unstable regions of scFv to avoid biasing our redesigning efforts, visual inspection was used to identify small clusters of amino acid side chains (2-5 residues) that interact with each in the homology models. Overall, we identified a diverse set of clusters that would cover most regions of the scFv. The sequences for these clusters were then optimized using the pack_rotamers protocol in Rosetta, which performs fixed backbone rotamer-based sequence optimization46. Additional sub-rotamers were included by sampling torsion angles near the most preferred values for chi1 and chi2 (ex1 and ex2 options) in Rosetta47. All amino acids except for cysteine were allowed during the sequence optimization. On the order of hundred different residue clusters were submitted to this protocol. Each cluster optimization was performed on all ten of the homology models and the homology model which produced the lowest scoring design for a given cluster was passed forward for consideration. The lowest scoring designs (~40) were visually inspected to select the designs for experimental characterization. The latest up-to-date directions can also be found at: https://www.rosettacommons.org/docs/latest/ application_documentation/antibody/antibody-protocol.
The predicted structures of C9+C14 scFv modeled using the RosettaAntibody 3.0 antibody modeling protocol as described26,48. A total of 2000 candidate structures were generated per sequence, with the ten lowest scoring models, as evaluated by the Rosetta scoring function, being chosen for visual inspection and further analysis. Root-mean square deviation (RMSD) of the top models to the solved structure was calculated over the backbone atoms (C, Cα, N, O) using the McLachlan algorithm49 as implemented in the ProFit software (Martin ACR, Porter CT. Available at: http://www.bioinf.org.uk/soft-ware/profit).
Gene synthesis and cloning
Genes encoding the scFv variants were synthesized as previously described50, and the inserts were cloned into a modified version of pMoPac1651, where Factor Xa protease cut-site was introduced between the scFv and CL domain (PelB-scFv-Strep-Factor Xa-Ck-His-cMyc).
Expression and purification of HA33 and antibody variants from E. coli
For the HA33 expression, Jude-1 cells with pMoPac12 vector containing HA33 sequences with 6x his31 were grown overnight at 37 °C, then diluted 1:1000 into 1 L TB media and grown until OD600 around 0.5 at 30 °C. To induce expression of the proteins, 500 μL of IPTG (Sigma-Aldrich) was added and incubated overnight at 25 °C. Cells were isolated by centrifugation at 120 Hz for 10 min and resuspended in 12.5 mL of 100 mM Tris, 0.75 M sucrose solution at pH 7.5 with EDTA-free protease inhibitor cocktail added (Roche). 1.25 mL of lysozyme at 20 mg/mL in Tris-sucrose solution was added and the culture was placed in a shaking incubator at 4 °C for 20 min. After 25 mL of 1 mM EDTA was added, the culture was placed back in a shaking incubator at 4 °C for 30 min. Cells were pelleted by centrifuging at 280 Hz for 30 min and the supernatant containing the periplasmic fraction was collected. These samples were filtered through 0.22 μm syringe filter. The collected periplasmic fraction was passed through 4 mL Ni-NTA agarose (QIAGEN) affinity column in gravity mode. Flow-through sample was collected and passed through the column three times. The column was washed with 15 mL of 10 mM imidazole 150 nM NaCl, 100 mM Tris-HCl, pH 7.5 buffer prior to eluting with 5 mL of 250 mM imidazole, 150 nM NaCl, 100 mM Tris-HCl, pH 7.5 buffer. Eluates were then buffer-exchanged into PBS using Amicon Ultra-15 (10K) centrifugal spin columns (Millipore).
For each scAb construct, vector-containing BL21 cells were grown overnight at 37 °C, then diluted 1:1000 into 1 L TB media and grown until OD600 around 0.5 at 30 °C. To induce expression of the proteins, 500 μL of IPTG (Sigma-Aldrich) was added and incubated for 6 hours at 30 °C. Cells were then isolated by centrifugation at 120 Hz for 10 min and resuspended in 12.5 mL of 100 mM Tris, 0.75 M sucrose solution at pH 7.5 with EDTA-free protease inhibitor cocktail added (Roche). 1.25 mL of lysozyme at 20 mg/mL in Tris-sucrose solution was added and the culture was placed in a shaking incubator at 4 °C for 20 min. After 25 mL of 1 mM EDTA was added, the culture was placed back in a shaking incubator at 4 °C for 30 min. Cells were pelleted by centrifuging at 280 Hz for 30 min and the supernatant containing the periplasmic fraction was collected. These samples were filtered through 0.22 μm syringe filter. The collected periplasmic fraction was passed through 4 mL Strep-tactin superflow resins (IBA) affinity column in gravity mode. Flow-through sample was collected and passed through the column three times. The column was washed with 15 mL of 100 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 8.0 prior to elution with 5 mL of 100 mM Tris, 150 mM NaCl, 1 mM EDTA, 2.5 mM desthiobiotin (Sigma), pH 8.0. Each eluted scAb sample was buffer-exchanged into 50 mM Tris, 100 mM NaCl, 5 mM CaCl2, pH 8.0 using Amicon Ultra-15 (10K) centrifugal spin columns (Millipore).
To generate scFv from scAb, we incubated scAb with Factor Xa protease (QIAGEN) for 4 hours at room temperature, and after the completion of the reaction, we removed Factor Xa protease using Factor Xa removal resin (QIAGEN). Subsequently, cleaved CL and uncleaved scAb using 4 mL Strep-tactin superflow resins (IBA).
Size exclusion chromatography (SEC)
Each purified scFv and scAb sample were further purified via SEC performed on Agilent 1100 high performance liquid chromatography system using a Superdex TM 200 10/300GC, (GE Healthcare), with a mobile phase of pH 7.4 PBS at a flow rate of 0.75 mL/min. Fractions corresponding to monomeric proteins were collected for the subsequent analyses.
Differential scanning fluorimetry
Differential scanning fluorimetry (DSF) was performed with SYPRO Orange dye (Life Technologies) to determine the melting temperature of each variant by monitoring the protein unfolding. For each sample, 15 μL of scFv or scAb antibodies varying from 0.1 to 25 μM was mixed with 5 μL of dye, diluted from 5000x to 20x for use. Each experiment included a no protein control containing 100 mM HEPES buffer at pH 7. DSF assay was performed on an Applied Biosystems ViiA 7 Real-Time PCR System using a temperature gradient from 25 °C to 99 °C at a temperature ramp rate of 0.03 °C/s. The fluorescence emissions and melting temperature measurements were analyzed using ViiA 7 Software from Life Technologies.
Thermal challenge
Each scFv and scAb variant was subjected to the heat treatment at 70 °C at the concentration of 1 mg/mL for 1 hour, unless noted otherwise, then cooled down and maintained at 4 °C for 20 min in a thermocycler (Applied Biosystems). Retaining binding affinity after the heat treatment for each sample were subsequently assayed by ELISA.
Enzyme-linked immunosorbent assay (ELISA)
The half maximal effective concentration (EC50) values based on ELISA were used as the apparent affinity values for comparisons between different variants. Costar 96 well ELISA plates (Corning) were coated with 50 μL of 4 μg/mL HA33 antigen in PBS buffer at 4 °C overnight. Wells were then decanted and blocked with 2% skim milk in PBS buffer for 2 hours at RT. After blocking, serially diluted antibodies bound to the plate for 1 hour, followed by 1:5000 diluted mouse anti-NWSHPQFEK-tag HRP-conjugated secondary antibody (GenScript) for 1 hour. For detection, 50 μL TMB substrate (Thermo Scientific) was added and quenched with 50 μL 1 M H2SO4. Absorbance was measured at 450 nm using a Tecan M200 plate reader, and data were analyzed and fitted using a 4-parameter logistic nonlinear regression model in the GraphPad Prism software. All ELISA assays were performed in triplicate.
Structural analysis
Crystals of C9+C14 were grown in hanging drops over a reservoir of 0.1 M Tris-HCl, pH 8.5 buffer with 15% (w/v) polyethylene glycol 2000. X-ray diffraction data were collected from synchrotron radiation at beamline 5.0.3 of the Advanced Light Source (Berkeley, CA). Data were then processed and scaled using the HKL2000 software suite52. The structures of C9+C14 were determined using molecular replacement by Phaser-MR from the PHENIX suite53 with 3UMT as a search model. Structures were refined with phenix.refine53 along with iterative model building in COOT54. In the refinement, 5% of the test set was excluded for Rfree cross-validation55. The final structures were evaluated by MolProbity56. The data collection and refinement statistics are summarized in Supplementary Table 4.
Data Availability
Coordinates and diffraction data have been deposited at PDB, accession number 6P79.
Supplementary Material
Acknowledgments
This work was supported by DARPA-09-69-ATP-FP-005 (G.G., A.E., B.K. and J.G.), the Clayton Foundation (G.G.), NIH R01-GM078221 (J.X. and J.G.), NIH R35-GM131923 (B.K.), NIH R01-125882 (Y.Z.), and Welch Foundation F-1654 (A.E.), and Welch Foundation F-1778 (Y.Z.).
Footnotes
Declaration of Interests
JJG is an unpaid board member of the Rosetta Commons. Under institutional participation agreements between the University of Washington, acting on behalf of the Rosetta Commons, Johns Hopkins University may be entitled to a portion of revenue received on licensing Rosetta software including programs described here. As a member of the Scientific Advisory Board of Cyrus Biotechnology, JJG is granted stock options. Cyrus Biotechnology distributes the Rosetta software, which may include methods described in this article.
References
- 1.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotech. 2005;23(9):1126–1136. [DOI] [PubMed] [Google Scholar]
- 2.Power BE, Hudson PJ. Synthesis of high avidity antibody fragments (scFv multimers) for cancer imaging. J Immunol Methods. 2000;242(1-2):193–204. [DOI] [PubMed] [Google Scholar]
- 3.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and Development of Therapies using Chimeric Antigen Receptor-Expressing T cells. Immunological Reviews. 2014;257(1):107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Di De. 2008;11(5):675–687. [PubMed] [Google Scholar]
- 5.Jager M, Pluckthun A. Domain interactions in antibody Fv and scFv fragments: effects on unfolding kinetics and equilibria. FEBS Lett. 1999;462(3):307–312. [DOI] [PubMed] [Google Scholar]
- 6.Honegger A Engineering antibodies for stability and efficient folding. Handb Exp Pharmacol. 2008(181):47–68. [DOI] [PubMed] [Google Scholar]
- 7.Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annu Rev Chem Biomol Eng. 2012;3:263–286. [DOI] [PubMed] [Google Scholar]
- 8.Jefferis R Aggregation, immune complexes and immunogenicity. MAbs. 2011;3(6):503–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22(9):1161–1165. [DOI] [PubMed] [Google Scholar]
- 10.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376(4):926–931. [DOI] [PubMed] [Google Scholar]
- 11.Dudgeon K, Rouet R, Kokmeijer I, et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012;109(27):10879–10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graff CP, Chester K, Begent R, Wittrup KD. Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37 degrees C. Protein Eng Des Sel. 2004;17(4):293–304. [DOI] [PubMed] [Google Scholar]
- 13.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1(2):755–768. [DOI] [PubMed] [Google Scholar]
- 14.Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci U S A. 1993;90(16):7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trivedi MV, Laurence JS, Siahaan TJ. The role of thiols and disulfides on protein stability. Curr Protein Pept Sci. 2009;10(6):614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao JX, Yang L, Gu ZN, et al. Stabilization of the single-chain fragment variable by an interdomain disulfide bond and its effect on antibody affinity. Int J Mol Sci. 2010;12(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ewert S, Honegger A, Pluckthun A. Structure-based improvement of the biophysical properties of immunoglobulin VH domains with a generalizable approach. Biochemistry. 2003;42(6):1517–1528. [DOI] [PubMed] [Google Scholar]
- 18.Ewert S, Honegger A, Pluckthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. 2004;34(2):184–199. [DOI] [PubMed] [Google Scholar]
- 19.Jordan JL, Arndt JW, Hanf K, et al. Structural understanding of stabilization patterns in engineered bispecific Ig-like antibody molecules. Proteins. 2009;77(4):832–841. [DOI] [PubMed] [Google Scholar]
- 20.Borgo B, Havranek JJ. Automated selection of stabilizing mutations in designed and natural proteins. Proc Natl Acad Sci U S A. 2012;109(5):1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sevy AM, Wu NC, Gilchuk IM, et al. Multistate design of influenza antibodies improves affinity and breadth against seasonal viruses. Proc Natl Acad Sci U S A. 2019;116(5):1597–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proceedings of the National Academy of Sciences. 2009;106(29):11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldenzweig A, Fleishman SJ. Principles of Protein Stability and Their Application in Computational Design. Annual Review of Biochemistry. 2018;87(1):105–129. [DOI] [PubMed] [Google Scholar]
- 24.Baran D, Pszolla MG, Lapidoth GD, et al. Principles for computational design of binding antibodies. Proceedings of the National Academy of Sciences. 2017;114(41):10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel. 2012;25(10):507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ. Toward high-resolution homology modeling of antibody Fv regions and application to antibody–antigen docking. Proteins: Structure, Function, and Bioinformatics. 2009;74(2):497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miklos AE, Kluwe C, Der BS, et al. Structure-based design of supercharged, highly thermoresistant antibodies. Chem Biol. 2012;19(4):449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevalier A, Silva D-A, Rocklin GJ, et al. Massively parallel de novo protein design for targeted therapeutics. Nature. 2017;550(7674):74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strauch E-M, Bernard SM, La D, et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nature biotechnology. 2017;35(7):667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, et al. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLOS Computational Biology. 2018;14(4):e1006112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arndt JW, Gu J, Jaroszewski L, et al. The structure of the neurotoxin-associated protein HA33/A from Clostridium botulinum suggests a reoccurring beta-trefoil fold in the progenitor toxin complex. J Mol Biol. 2005;346(4):1083–1093. [DOI] [PubMed] [Google Scholar]
- 32.Sircar A, Kim ET, Gray JJ. RosettaAntibody: antibody variable region homology modeling server. Nucleic Acids Research. 2009;37(Web Server issue):W474–W479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alford RF, Leaver-Fay A, Jeliazkov JR, et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. Journal of Chemical Theory and Computation. 2017;13(6):3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Röthlisberger D, Honegger A, Plückthun A. Domain Interactions in the Fab Fragment: A Comparative Evaluation of the Single-chain Fv and Fab Format Engineered with Variable Domains of Different Stability. Journal of Molecular Biology. 2005;347(4):773–789. [DOI] [PubMed] [Google Scholar]
- 35.Nagano T, Hirotsuka M, Mori H, Kohyama K, Nishinari K. Dynamic viscoelastic study on the gelation of 7 S globulin from soybeans. Journal of Agricultural and Food Chemistry. 1992;40(6):941–944. [Google Scholar]
- 36.Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. Journal of Molecular Biology. 1984;179(1):125–142. [DOI] [PubMed] [Google Scholar]
- 37.Kim DN, Jacobs TM, Kuhlman B. Boosting protein stability with the computational design of β-sheet surfaces. Protein Science. 2016;25(3):702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pucci F, Rooman M. Physical and molecular bases of protein thermal stability and cold adaptation. Curr Opin Struct Biol. 2017;42:117–128. [DOI] [PubMed] [Google Scholar]
- 39.Camilloni C, Bonetti D, Morrone A, et al. Towards a structural biology of the hydrophobic effect in protein folding. Scientific Reports. 2016;6:28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gromiha MM, Pathak MC, Saraboji K, Ortlund EA, Gaucher EA. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins: Structure, Function, and Bioinformatics. 2013;81(4):715–721. [DOI] [PubMed] [Google Scholar]
- 41.Allen MJ, Coutinho PM, Ford CF. Stabilization of Aspergillus awamori glucoamylase by proline substitution and combining stabilizing mutations. Protein engineering. 1998;11(9):783–788. [DOI] [PubMed] [Google Scholar]
- 42.MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. Journal of Molecular Biology. 1991;218(2):397–412. [DOI] [PubMed] [Google Scholar]
- 43.Prajapati RS, Das M, Sreeramulu S, et al. Thermodynamic effects of proline introduction on protein stability. Proteins: Structure, Function, and Bioinformatics. 2007;66(2):480–491. [DOI] [PubMed] [Google Scholar]
- 44.Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein engineering. 2000;13(3):179–191. [DOI] [PubMed] [Google Scholar]
- 45.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science. 2003;302(5649):1364. [DOI] [PubMed] [Google Scholar]
- 46.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proceedings of the National Academy of Sciences. 2000;97(19):10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weitzner BD, Jeliazkov JR, Lyskov S, et al. Modeling and docking of antibody structures with Rosetta. Nature Protocols. 2017;12(2):401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins. 2014;82(8):1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLachlan AD. Rapid comparison of protein structures. Acta Crystallographica Section A. 1982;38(6):871–873. [Google Scholar]
- 50.Miklos AE, Hughes RA, Ellington AD. Design and Assembly of Large Synthetic DNA Constructs. Current Protocols in Molecular Biology. 2012;99(1):3.23. [DOI] [PubMed] [Google Scholar]
- 51.Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. Journal of immunological methods. 2003;276(1-2):185–196. [DOI] [PubMed] [Google Scholar]
- 52.Otwinowski Z. MW. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol 276 New York: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, Grosse-Kunstleve RW, Hung LW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(11):1948–1954. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(12):2126–2132. [DOI] [PubMed] [Google Scholar]
- 55.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355(6359):472–475. [DOI] [PubMed] [Google Scholar]
- 56.Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and diffraction data have been deposited at PDB, accession number 6P79.