

Abstract
Endothelial activation by proinflammatory cytokines is closely associated to the pathogenesis of atherosclerosis and other vascular diseases; however, the molecular mechanisms controlling endothelial activation are not fully understood. Here we identify TRIM14 as a new positive regulator of endothelial activation via activating NF-κB signal pathway. TRIM14 is highly expressed in human vascular endothelial cells (ECs) and markedly induced by inflammatory stimuli such as TNF-α, IL-1β, and LPS. Overexpression of TRIM14 significantly increased the expression of adhesion molecules such as VCAM-1, ICAM-1, E-selectin, and cytokines such as CCL2, IL-8, CXCL-1, and TNF-α in activated ECs and by which it facilitated monocyte adhesion to ECs. Conversely, knockdown of TRIM14 has opposite effect on endothelial activation. Upon TNF-α stimulation, TRIM14 is recruited to IKK complex via directly binding to NEMO and promotes the phosphorylation of IκBα and p65, which is dependent on its K63-linked ubiquitination. Meanwhile, p65 can directly bind to the promoter regions of human TRIM14 gene and control its mRNA transcription. Finally, TRIM14 protein level is significantly upregulated in mouse and human atheroma compared to normal arteries. Taken together, these results indicate that TRIM14–NF-κB forms a positive feedback loop to enhance EC activation and TRIM14 may be a potential therapeutic target for vascular inflammatory diseases such as atherosclerosis.
Keywords: TRIM14, NF-κB, endothelial activation, atherosclerosis
Introduction
Vascular endothelium plays a central role in both health and disease throughout the circulation, and endothelial dysfunction is intimately associated with atherosclerosis (Gimbrone and Garcia-Cardena, 2016). Endothelial dysfunction is usually preceded by `activation’, which is defined by the induction of proinflammatory signaling cascades and increased expression of adhesion molecules, such as VCAM-1, ICAM-1, and E-selectin (Liao, 2013). These events stimulate the arterial recruitment of inflammatory cells from circulation and their transendothelial migration into the subendothelial space of large arteries where they differentiate into macrophages and promote atheroma initiation and progression (Weber and Noels, 2011). Endothelial cell (EC) activation is typically induced by proinflammatory cytokines, such as TNF-α and IL-1β. Cytokine-activated ECs can also synthesize and secrete chemokines on their luminal surface, such as IL-1β, CCL2, G-CSF, and IL-8 (Sprague and Khalil, 2009).
Nuclear factor kappa B (NF-κB) signaling plays a critical role in the proinflammatory activation of endothelium in atherogenesis (de Winther et al., 2005). Many of the effector molecules associated with EC activation in atherogenesis (e.g. VCAM-1, ICAM-1, E-selectin, and CCL2) are under the control of NF-κB. In quiescent cells, NF-κB binds to the inhibitory IκBα and forms an inactive complex. Upon stimulation by proinflammatory cytokines such as TNF-α, IκBα was phosphorylated by a large multi-unit complex that contains two catalytic subunits (IKKα and IKKβ), as well as the regulatory subunit IKKγ or NEMO, and then degraded via the ubiquitin pathway. Finally, NF-κB is released and translocated to the nucleus where it activates transcription of its specific targets. EC activation induced by proinflammatory cytokines (TNF-α or IL-1β) may also involve other signaling pathways such as ERK1/2, JNK, and p38 (Pober, 2002; Sethi et al., 2008). The signaling transduction of these pathways has been well studied, but the regulation is not fully understood.
The tripartite motif (TRIM) family proteins contain more than 80 members, which are defined by an evolutionarily conserved domain architecture composed of three zinc-binding regions: RING finger, B-box, and coiled-coil domains (Galkina and Ley, 2009). Most of the TRIM family proteins have been defined as ubiquitin E3 ligases (Jiang et al., 2017). TRIM14 was first discovered to be overexpressed in human immunodeficiency virus (HIV)-infected human and simian immunodeficiency virus (SIV)-infected simian non-Hodgkin’s lymphomas (Pober, 2002; Sethi et al., 2008). Accumulating evidences indicate that TRIM14 is an important mediator of antiviral immunity, which stabilizes not only cGAS upon DNA virus infection but RIG-1 and double-stranded RNA interaction to achieve a robust and optimal range of antiviral activity (Chen et al., 2016; Tan et al., 2017). Importantly, TRIM14 has been reported as a mitochondrial adaptor associated with MAVS to recruit NF-κB essential modulator (NEMO) and WHIP/PPP6C complex to elicit maximal antiviral signaling (Zhou et al., 2014; Tan et al., 2017). Furthermore, several groups reported that TRIM14 plays a role in activation of the NF-κB pathway and tumorigenesis (Su et al., 2016; Hai et al., 2017; Xu et al., 2017). Most recently, we found that the expression of TRIM14 could be induced by different toll-like receptor ligands in THP1-derived macrophages (Jiang et al., 2017). However, the functional role of TRIM14 in EC activation remains unknown.
Here, we report that TRIM14 is a positive regulator in EC activation. Our results indicate that TRIM14 enhances TNF-α-induced expression of adhesion molecules and monocyte adherence to ECs via activating NF-κB signaling pathway in HUVECs. The expression of TRIM14 is significantly induced by TNF-α, IL-1β, and LPS in human ECs and in the aortas of LPS-injected mice via NF-κB. Moreover, TRIM14 protein level is markedly increased in mouse and human atheroma compared to normal arteries. These results suggest that TRIM14–NF-κB may form a positive feedback loop to drive endothelial activation and the progress of atherogenesis in humans.
Results
TRIM14 is highly expressed in ECs and induced by inflammatory stimuli
To study the role of TRIM14 in ECs, we first detected the expression of TRIM14 in a variety of primary ECs including human aortic ECs (HAEC), human coronary artery ECs (HCAEC), human dermal microvascular ECs (HDMEC), human lung microvascular ECs (HLMEC), and human umbilical vein ECs (HUVEC). As shown in Figure 1A, TRIM14 was highly expressed in all human ECs compared to non-ECs. Moreover, the expression of TRIM14 was further induced by TNF-α, IL-1β, and LPS in HUVECs in time- and dose-dependent manners (Figure 1B–G). Consistently, TRIM14 mRNA levels were also induced by TNF-α, IL-1β, and LPS (Figure 1H), suggesting that these inflammatory stimuli can drive the transcription of TRIM14 in ECs. To examine whether the expression of TRIM14 is also induced in vivo during inflammation, aortas from mice injected with LPS (25 mg/kg body weight, intraperitoneal) were harvested at 0, 4, and 8 h after injection and subjected to analysis by western blot. As shown in Figure 1I, TRIM14 protein levels were significantly induced by LPS treatment in aorta. Together, TRIM14 was highly expressed in ECs and induced by inflammatory stimuli both in vitro and in vivo, suggesting that it may involve in the regulation of vascular endothelial activation in response to these stimuli.
Figure 1.
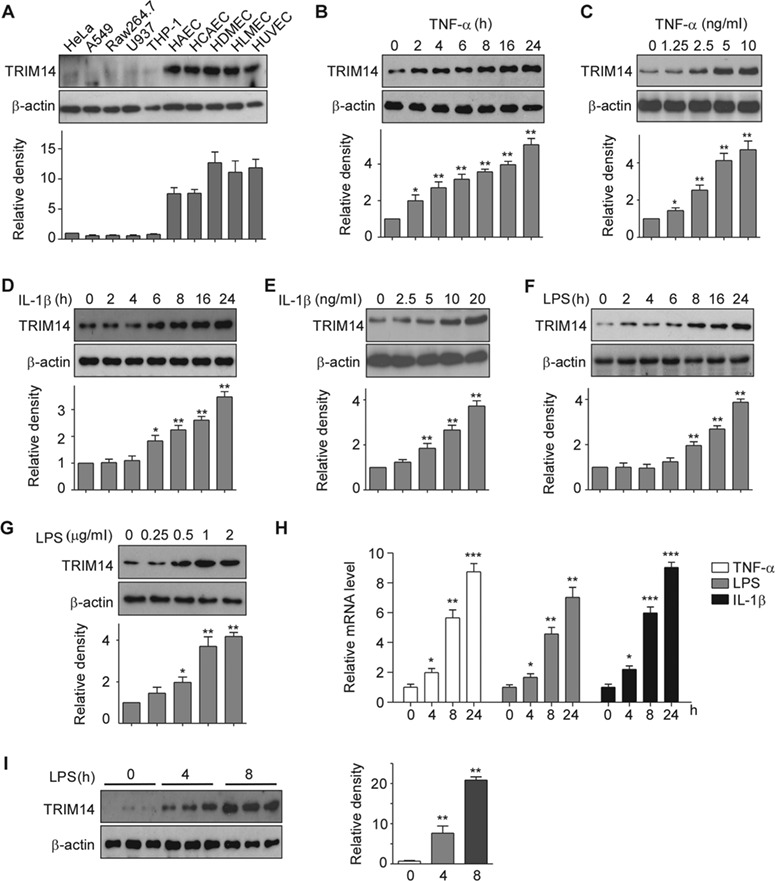
Expression of TRIM14 in human ECs. (A) Whole-cell lysates were isolated from human primary vascular ECs including HAEC, HCAEC, HDMEC, HLMEC, HUVEC, and other human cell lines as indicated and used for western blot assays to detect TRIM14. β-actin served as a loading control. Relative fold changes were determined by densitometry and normalized to β-actin. (B–G) HUVECs were treated with 10 ng/ml TNF-α (B), 10 ng/ml IL-1β (D), or 1 μg/ml LPS (F) for 0, 2, 4, 6, 8, 16, and 24 h, respectively. HUVECs were also treated with TNF-α (C), IL-1β (E), or LPS (G) in different doses as indicated for 24 h. Whole-cell lysates were harvested for detection of TRIM14 protein levels, β-actin served as a loading control. Western blot bands were quantified using Gel-Pro Analyzer software and presented as fold changes under the images. *P < 0.05, **P < 0.01 vs. 0 group by Student’s t-test. (H) HUVECs were stimulated with TNF-α, IL-1β, and LPS for 24 h. Total RNA was extracted, and mRNA levels of TRIM14 were detected by qPCR. Data are presented as mean ± SD (n = 3); *P < 0.05, **P < 0.01, ***P < 0.001 vs. PBS-treated group. (I) TRIM14 protein levels in the aortas of adult C57BL/6 mice treated with LPS for different times as indicated. Fold-change of the protein levels were determined by densitometry and normalized to β-actin. Quantitative data are presented as mean ± SD (n = 3); **P < 0.01 vs. 0 h group by Student’s t-test.
Overexpression of TRIM14 increases the expression of adhesion molecules and cytokines and monocyte adherence to HUVEC
To explore the function of TRIM14 in ECs, TRIM14 plasmids were transfected into HUVECs for 24 h and then the transfected cells were activated by TNF-α treatment. The expression of adhesion molecules and cytokines were examined by western blot and quantitative real-time RT-PCR (qPCR). As shown in Figure 2A and B, TRIM14 overexpression significantly increased the protein levels of adhesive molecules including VCAM-1, ICAM-1, and E-selectin but not the membrane protein VE-cadherin. Consistently, overexpression of TRIM14 also enhanced the mRNA levels of adhesion molecules and cytokines including VCAM-1, ICAM-1, E-selectin TNF-α, CCL2, IL-8, CXCL-1, and CCL-5 in activated HUVECs (Figure 2C). Similarly, TRIM14 overexpression also increased IL-1β-induced expression of VCAM-1, ICAM-1, and E-selectin in HUVECs (Figure 2D and E). As activated ECs can recruit the monocytes from blood to subspace of ECs, next we transfected Flag-TRIM14 or Flag vector into HUVECs and then the transfected cells were incubated with or without TNF-α. The activated HUVECs were co-cultured with fluorescence-labeled THP-1 cells (a human monocyte cell line). After repeated washes, the adherence THP-1 cells were imaged and counted. As shown in Figure 2F, TNF-α treatment dramatically increased THP-1 cell adherence to ECs. TRIM14 overexpression further promoted THP-1 cells adherence to HUVECs. These results suggest that TRIM14 is a positive regulator in cytokine-induced EC activation.
Figure 2.
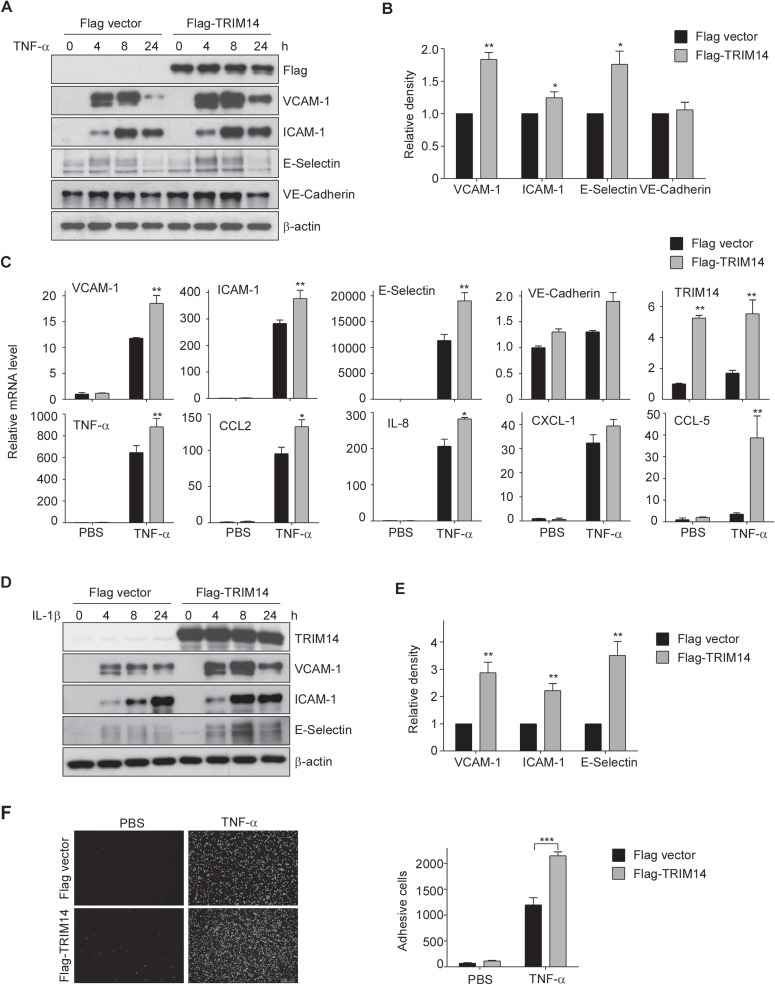
Overexpression of TRIM14 increased the expression of adhesion molecules and cytokines and monocyte adherence to HUVEC. (A) HUVECs were transiently transfected with Flag-TRIM14 or empty vector for 24 h and then treated with 10 ng/ml TNF-α for 0, 4, 8, and 24 h. Cell lysates were extracted and used to detect the protein levels of VCAM-1, ICAM-1, E-selectin, and VE-cadherin. (B) Relative fold changes of proteins after 8 h treatment of TNF-α were determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); *P < 0.05, **P < 0.01 by Student’s t-test. (C) HUVECs were transfected with Flag-TRIM14 or empty vector for 24 h. Transfected cells were then stimulated with 10 ng/ml TNF-α or PBS (as controls) for 4 h. The mRNA levels of adhesive molecules and cytokines were determined by qPCR and normalized to β-actin. (D) Flag-TRIM14/Flag was transfected into HUVECs, and the transfected cells were treated with 10 ng/ml IL-1β for 0, 4, 8, and 24 h. The protein levels were detected by western blot analysis. (E) Relative fold changes of proteins after 8 h treatment of IL-1β were determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); **P < 0.01 by Student’s t-test. (F) Flag-TRIM14 or empty vector were transfected into HUVECs, and the transfected cells were incubated with TNF-α or PBS for 8 h and then co-cultured with fluorescence-labeled THP-1 cells for 1 h. After carefully washing, adhesive cells were visualized by Cytation 3 Cell Imaging Multi-Reader (Biotek Instruments). Attached cells were counted from 5 random pictures in three independent experiments. ***P < 0.001.
Knockdown of TRIM14 attenuates the expression of adhesion molecules and cytokines in activated ECs
To further confirm its role in endothelial activation, we next performed loss-of-function study on HUVECs using pooled TRIM14 siRNA. Pooled TRIM14 siRNA or control siRNA were transfected into HUVECs for 24 h and then the transfected cells were incubated with TNF-α for 0, 4, 8, and 24 h. The expression of adhesion molecules and inflammatory cytokines as well as TRIM14 was examined by western blot and qPCR. As shown in Figure 3A, TRIM14 protein levels were decreased by >90% in si-TRIM14 group compared to that in si-Control group. As expected, knockdown of TRIM14 significantly attenuated the protein levels of adhesion molecules including VCAM-1, ICAM-1, and E-selectin but not VE-cadherin (Figure 3A and B). In consistent, knockdown of TRIM14 also attenuated the mRNA levels of VCAM-1, ICAM-1, E-selectin, TNF-α, CCL2, IL-8, and CXCL1 in activated HUVECs (Figure 3C). The functional impact of reducing VCAM-1 and ICAM-1 expression in TRIM14 siRNA-transfected HUVECs was demonstrated by THP-1 cell adhesion assay. siRNA-mediated knockdown of TRIM14 expression consistently decreased adhesion of THP-1 cells to HUVECs by over 60% (Figure 3D). These results suggest that silencing of TRIM14 has opposite effect on EC activation.
Figure 3.
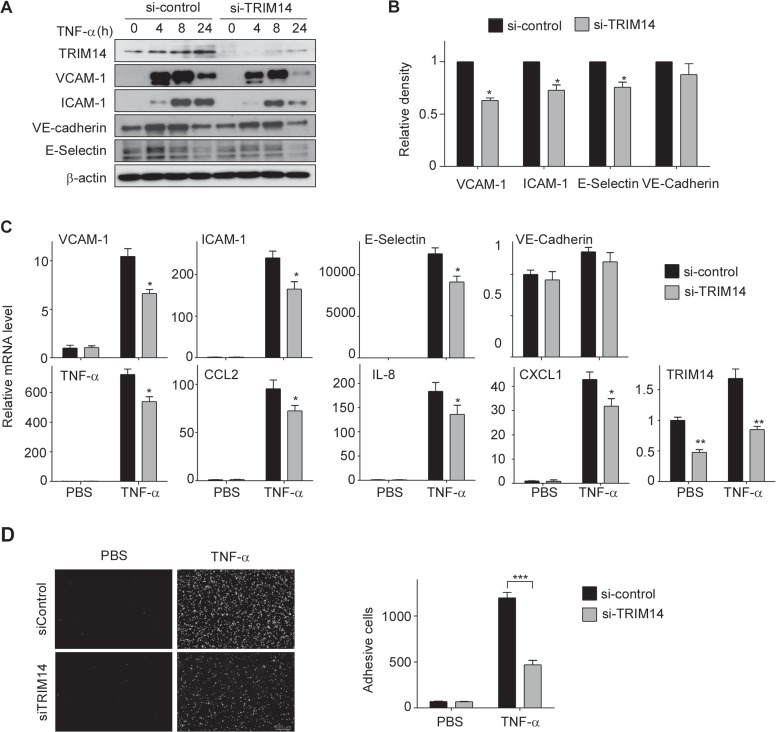
Knockdown of TRIM14 attenuated the expression of adhesion molecules and cytokines and monocyte adherence to HUVEC. (A) HUVECs were transfected with TRIM14 siRNAs or control siRNAs. Twenty-four hours later, the transfected cells were treated with 10 ng/ml TNF-α for 0, 4, 8, and 24 h. Expression of VCAM-1, ICAM-1, E-selectin, and VE-cadherin were determined by western blot analysis. (B) Relative fold changes of proteins after 8 h treatment of TNF-α were determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); *P < 0.01 by Student’s t-test. (C) TRIM14 siRNAs or control siRNAs were transiently transfected into HUVECs. Transfected cells were quiescent for 24 h and then treated with 10 ng/ml TNF-α or PBS (as controls) for 4 h. The mRNA levels of adhesive molecules and cytokines were determined by qPCR and normalized to β-actin. Data are presented as mean ± SD (n = 3); *P < 0.05, **P < 0.01 by Student’s t-test. (D) TRIM14 siRNAs or control siRNAs were transfected into HUVECs, and the transfected cells were incubated with TNF-α or PBS for 8 h and then co-cultured with fluorescence-labeled THP-1 cells for 1 h. After carefully washing, adhesive cells were visualized and counted as described as above. ***P < 0.001.
TRIM14 promotes TNF-α-induced NF-κB activation in ECs
As TNF-α-induced expression of adhesive molecules and cytokines mainly attributes to MAPK and NF-κB signaling pathways, next we investigated the effect of TRIM14 on the activation of MAPK and NF-κB signaling pathways. Flag-TRIM14 or Flag vector (as controls) was transfected into HUVECs for 24 h and then the transfected cells were incubated with 10 ng/ml of TNF-α for different times as indicated (Figure 4A). Whole-cell lysates were collected for analysis by western blot. As shown in Figure 4A and B, overexpression of TRIM14 significantly enhanced TNF-α-induced phosphorylation of p65 and IκBα but not affected the phosphorylation of ERK1/2, JNK, and p38. Conversely, knockdown of TRIM14 using siRNAs attenuated TNF-α-induced phosphorylation of p65 and IκBα (Figure 4C). As phosphorylated p65 translocate into the nucleus and activate the transcription of downstream genes, we next examine whether TRIM14 regulates p65 translocation. As shown in Figure 4D and E, TRIM14 overexpression promoted p65 entering in nucleus, whereas silencing of TRIM14 caused the retention of p65 in the cytoplasm. To examine if the effect of TRIM14 on NF-κB signaling is restricted in ECs, we performed transient transfection experiments in Raw264.7 cells, a murine macrophage cell line. As shown in Supplementary Figure S1, overexpression of TRIM14 did not affect the activation of both NF-κB and MAPK signaling in Raw264.7 cells, suggesting that the effect of TRIM14 on NF-κB activation may depend on cellular context. Taken together, these results suggest that TRIM14 acts as an important regulator in TNF-α-induced NF-κB signaling in ECs.
Figure 4.
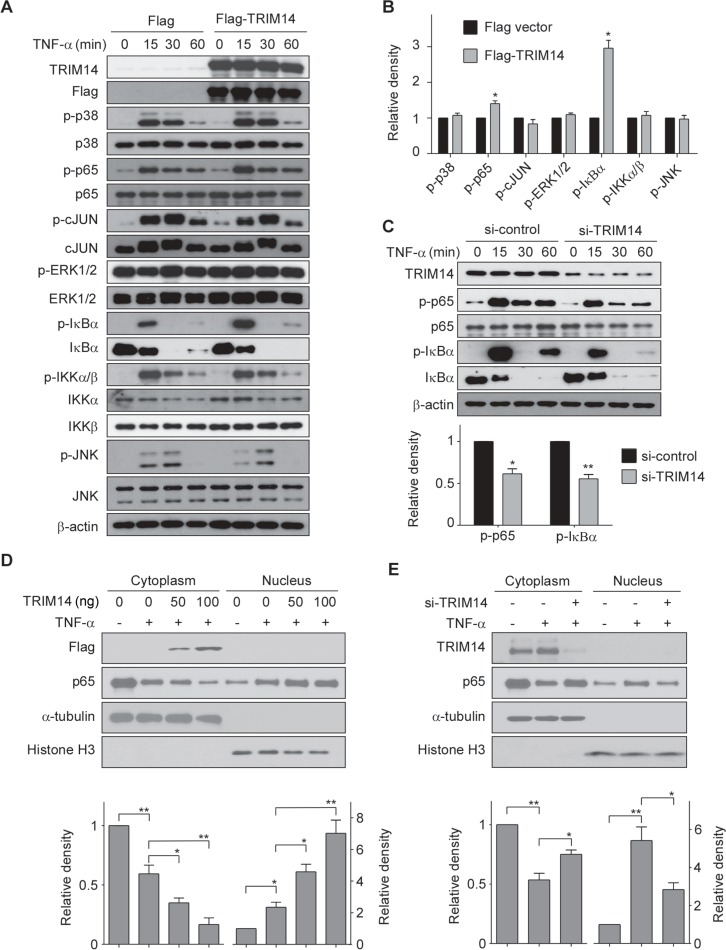
TRIM14 facilitated the activation of NF-κB signaling pathway. (A) HUVECs were transiently transfected with Flag-TRIM14 or Flag vector (control) for 24 h and then stimulated with 10 ng/ml TNF-α for 0, 15, 30, and 60 min. Cell lysates were harvested for detection of the phosphorylated protein levels by western blot. (B) Relative fold changes of phosphorylated protein levels after 15 min treatment of TNF-α were determined by densitometry and normalized to its total protein levels. Phosphorylation of p38, p65, c-JUN, ERK1/2, IκBα, IKKα/β, and JNK were determined and quantified. Data are presented as mean ± SD (n = 3); *P < 0.05 by Student’s t-test. (C) HUVECs were transiently transfected with TRIM14 siRNAs or control siRNAs. 24 h later, cells were stimulated with 10 ng/ml TNF-α for 0, 15, 30, and 60 min. Cell lysates were extracted and western blots were carried out to detect phosphorylation of IκBα and p65 as well as total protein levels. Relative fold changes of phosphorylated proteins after 15 min treatment of TNF-α was determined by densitometry and normalized to total protein levels. Data are presented as mean ± SD (n = 3); *P < 0.05, **P < 0.01 by Student’s t-test. (D and E) HUVECs were transiently transfected with 50 ng or 100 ng Flag-TRIM14 or empty vector for 24 h and then treated with TNF-α for 1 h (D). HUVECs were transiently transfected with TRIM14 siRNAs or control siRNAs for 24 h and then treated with TNF-α for 1 h (E). Cell lysates were harvested and used to extract proteins from cytoplasm and nucleus. Western blots were performed to detect protein expression level of p65 both in the cytoplasm and nucleus. Western blot bands were quantified using Gel-Pro Analyzer software and presented as fold changes (relative level was set as p65 band density/α-tubulin or histone H3 band density) under the images. *P < 0.05, **P < 0.01 by Student’s t-test.
TRIM14 promotes the phosphorylation and degradation of IκBα via interacting with NEMO
In the canonical NF-κB signal pathway, activated IKKβ/IKKα phosphorylates IκBα and leads IκBα degradation, then NF-κB (p65/p50) translocate into nucleus and activate gene transcription. Thus, we investigated if TRIM14 affected the phosphorylation of IκBα. As shown in Figure 5A and B, TRIM14 expression enhanced the phosphorylation and degradation of IκBα in a dose-dependent manner. Moreover, TRIM14 expression increased the degradation rate of IκBα (Figure 5C). Conversely, siRNA-mediated knockdown of TRIM14 obviously stabilized IκBα and prolonged its half-life to ∼8 h (Figure 5D). To further investigate the mechanism by which TRIM14 regulates the phosphorylation and degradation of IκBα, we examined the interaction of TRIM14 with proteins involved in TNF-α stimulated pathway, including TNF receptor-associated factor 2 (TRAF2), TRAF5, NEMO, and TAK1. Upon 15 min treatment of TNF-α, TRIM14 can bind NEMO and TAK1, but not TRAF2 and TRAF5 in HUVECs (Figure 5E). Since Zhou et al. (2014) has proved that TRIM14 interacts with NEMO directly upon viral infection, we next examined if TRIM14 can bind to TAK1 without NEMO. TRIM14 bound much less efficiently to TAK1 when siRNA knockdown of NEMO, which indicated that TRIM14 may not directly interact with TAK1 (Figure 5F). GST pull-down assays showed that recombinant TAK1 did not interact with TRIM14 upon TNF-α treatment (Figure 5G). These results indicated that TRIM14 promotes the phosphorylation and degradation of IκBα via interacting with NEMO.
Figure 5.
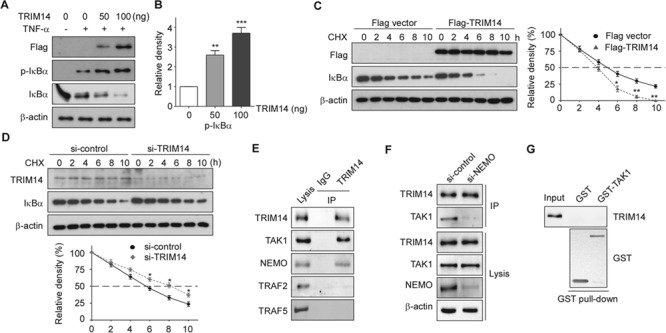
TRIM14 facilitated IκBα phosphorylation and degradation in activated ECs. (A) HUVECs were transiently transfected with 50 ng or 100 ng Flag-TRIM14 or empty vector for 24 h and then treated with TNF-α for 15 min. Cell lysates were collected, and western blots were performed to detect phosphorylation of IκBα and total protein levels of IκBα. (B) Relative fold changes of phosphorylated protein was determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); **P < 0.01, ***P < 0.001. (C) Flag-TRIM14 or empty vector transfected HUVECs were incubated with 50 μg/ml CHX for indicated time intervals. IκBα were detected with IκBα antibody by western blot. Quantitative analysis of IκBα protein levels was shown. Data are presented as mean ± SD (n = 3); *P < 0.05, **P < 0.01. (D) HUVECs were transfected with control siRNAs or TRIM14 siRNAs for 24 h and then treated with 50 μg/ml CHX for indicated time intervals. Cell lysates were collected for western blot. Quantitative analysis of IκBα protein levels was shown. Data are presented as mean ± SD (n = 3); *P < 0.05. (E) HUVECs were treated with TNF-α for 15 min, and whole-cell lysates were immunoprecipitated with TRIM14 antibody (mouse) or mouse IgG as control. The precipitates were immunoblotted with the indicated antibodies. (F) HUVECs were transiently transfected with control siRNAs or NEMO siRNAs for 24 h and then treated with TNF-α for 15 min, and whole-cell lysates were immunoprecipitated with TRIM14 antibody (mouse) or mouse IgG as control. The precipitates were immunoblotted with TAK1 antibody. (G) Purified GST or GST-TAK1 protein were incubated with the cell lysates of TNF-α treated HUVECs. After GST pull-down experiment, the elute protein and cell lysates were analyzed by western blot with indicated antibodies.
TRIM14 is subjected to ubiquitination during endothelial activation, which is required for its regulation of NF-κB signaling
It was previously reported that TRIM14 underwent K63 ubiquitination at K365 and recruited NEMO to activate NF-κB signaling pathway in response to viral infection (Zhou et al., 2014). Therefore, we tested if TRIM14 regulates NF-κB activation in dependent on its ubiquitination status. First, we observed that TNF-α stimulation can induce K63-type polyubiquitination of TRIM14 in HUVECs (Figure 6A). Then, we examined whether the K63-linked ubiquitination of TRIM14 is required for its binding to NEMO. HUVECs were transfected with Flag-TRIM14, Flag-TRIM14 mutant (K365R), or Flag vector for 24 h; the transfected cells were treated with TNF-α for 15 min. Co-immunoprecipitation (IP) assays were performed with whole-cell lysates. As shown in Figure 6B, Flag-TRIM14 mutant lost the ability to bind with NEMO and TAK1. Next, we tested if the ubiquitinated status of TRIM14 plays a key role in the regulation of NF-κB signaling. After 8 h treatment of TNF-α, cell lysates of transfected HUVECs were collected for western blot analysis. As shown in Figure 6C, TRIM14, but not TRIM14 mutant, promoted the phosphorylation of IκBα. Consistently, TRIM14, but not TRIM14 mutant, enhanced the expression of VCAM-1, ICAM-1, and E-selectin in activated HUVECs (Figure 6D). Furthermore, unlike TRIM14, its mutant (K365R) had no effect on THP-1 cell adhesion (Figure 6E). These results suggest that TRIM14 was subjected to ubiquitination during endothelial activation, which is required for its regulatory role in the NF-κB signaling.
Figure 6.

The K63-linked ubiquitination of TRIM14 is required for its function. (A) HUVECs were stimulated with 10 ng/ml TNF-α for 0, 5, and 15 min. Cell lysates then were immunoprecipitated using TRIM14 antibody, followed by western blot analysis using the indicated antibodies. (B and C) HUVECs were transiently transfected with TRIM14, TRIM14 mutant or empty vector for 24 h and then treated with TNF-α for 15 min. Whole-cell lysates were immunoprecipitated with Flag antibody and the precipitates were immunoblotted with TAK1, NEMO, TRAF2, TRAF5, and Ub antibodies (B). Cell lysates were collected, and western blots were performed to detect phosphorylation of IκBα and total protein levels of IκBα (C). Relative fold changes of phosphorylated protein was determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); **P < 0.01. (D) HUVECs were transiently transfected with TRIM14, TRIM14 mutant, or empty vector for 24 h and then treated with TNF-α for 8 h. Cell lysates were detected with VCAM-1, ICAM-1, and E-selectin antibodies. Relative fold changes of proteins were determined by densitometry and normalized to β-actin. Data are presented as mean ± SD (n = 3); **P < 0.01. (E) TRIM14 or TRIM14 mutant was transfected into HUVECs, and the transfected cells were incubated with TNF-α or PBS for 8 h and then co-cultured with fluorescence-labeled THP-1 cells for 1 h. After carefully washing, adhesive cells were visualized and counted as described as above. ***P < 0.001.
TRIM14 is a target gene of NF-κB
As three inflammatory signals (LPS, TNF-α, and IL-1β) induce TRIM14 expression via its transcription level and they are all potent activators of NF-κB signaling (Liu et al., 2017), we next examine if NF-κB involves in the regulation of TRIM14 transcription. To elucidate the upstream regulation of TRIM14 and its relationship with NF-κB signaling, we used the JASPAR database for in silico analysis of putative NF-κB binding sites, which were presented within the −2000~+1 region of the human TRIM14 promoter. Two possible binding sites were found in the promoter region of TRIM14 (Figure 7A). Indeed, transfection of p65 can activate the luciferase reporter contained TRIM14 promoter in a dose-dependent manner (Figure 7B). We next constructed a set of luciferase reporter vectors driven by difference promoter regions consist of each putative p65 binding site of TRIM14 promoter (Figure 7C). The reporters were co-transfected with p65 or control vector in HEK293 cells as indicated. As shown in Figure 7C, promoter fragments A and B but not C were significantly activated by p65 expression. To verify that NF-κB binds these regions in vivo, chromatin IP (ChIP) assays were performed in HUVECs treated by TNF-α for 15 min. As shown in Figure 7D, p65 displayed a strong binding enrichment to the specific region −1622~−1613 but not region −1744~−1735 of the TRIM14 promoter. Moreover, the luciferase activity of the promoter mutant that diminished its binding to p65 was significantly attenuated compared with wild-type promoter (Figure 7E). Together, these results suggest that TRIM14 is a target gene of NF-κB.
Figure 7.
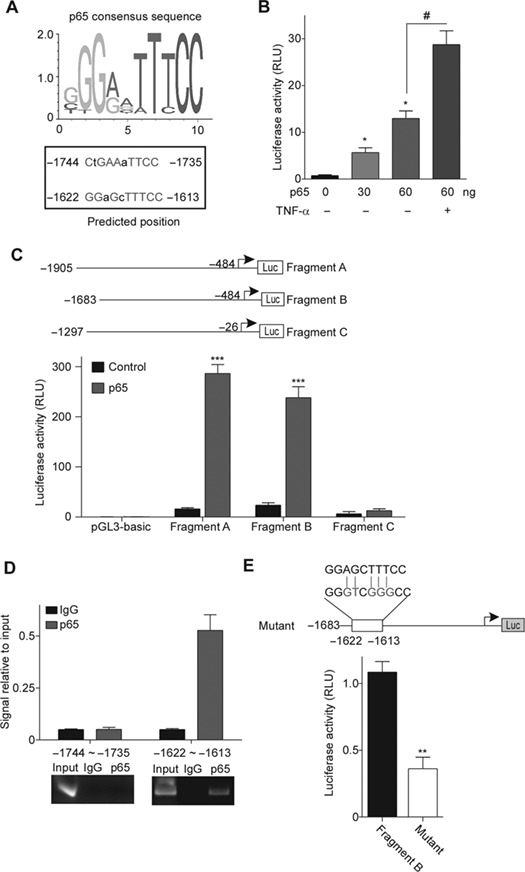
TRIM14 is a target gene of transcription factor p65. (A) Prediction of p65 binding sites in human TRIM14 promoter region using JASPAR database. p65 consensus sequence is shown on the left, and the predicted binding sites are on the right. Nucleotide in red, consensus index value >60. Capitals, core sequence, highest conserved, consecutive positions of the matrix. (B) HUVECs were co-transfected with TRIM14 promoter-reporter vector and different amounts of p65 expression plasmid (0, 30, and 60 ng), and the transfected cells were treated with or without 10 ng/ml TNF-α for 15 min. Relative luciferase activity values are presented as the means ± SEM of three independent experiments. *P < 0.05 vs. 0 ng p65 group; #P < 0.05. (C) The promoter fragments cloned upstream of the luciferase reporter gene are indicated in the left and were transfected into HEK293 cells with or without p65 plasmid. TK-Renilla construct was introduced to normalize firefly luciferase activity. Relative luciferase activity values are presented as the means ± SEM of three independent experiments. ***P < 0.001 vs. control group. (D) ChIP performed with an antibody against p65/NF-κB on chromatin isolated from HUVECs treated by 10 ng/ml TNF-α. The quantity of immunoprecipitated DNA was assessed by qPCR for the two predicted p65 binding sites present in the TRIM14 promoter. The net quantities of enriched DNA were corrected with their corresponding input DNA. Data are expressed as mean ± SEM (n = 3 per group). qPCR products then conduct electrophoresis in 2% agarose gel stained with ethidium bromide and visualized under UV light. (E) The reporter for mutant TRIM14 promoter was constructed. The substituted nucleotides were marked as red. HEK293 cells were co-transfected with p65 with WT and mutant reporters. TK-Renilla was introduced to normalize firefly luciferase activity. All experiments were performed in triplicate. Error bars represent the standard deviation of three independent experiments. **P < 0.01.
The expression of TRIM14 is upregulated in mouse and human atheromatous tissues
As endothelial activation is closely associated with the development of atherosclerosis, we first detected the expression of TRIM14, VCAM-1, ICAM-1, and E-selectin in mouse aorta collected from ApoE−/− mice fed a standard chow (SC) or a high-fat (HF)/high-cholesterol diet for 12 weeks. As showed in Figure 8A and B, both protein and mRNA of TRIM14, VCAM-1, ICAM-1, and E-selectin were significantly upregulated in ApoE−/− mouse aorta after feeding an HF diet. Interestingly, the phosphorylation of p65 and IκBα was also upregulated, which is consistent with in vitro results from cultured ECs (Figure 8A). Next, we examined the mRNA levels of TRIM14 in human atheromatous tissues. Both atheromatous tissue and adjacent normal tissue from 11 patients were collected during their carotid endarterectomy operations. Total RNAs were extracted and analyzed by qPCR. Compared to normal tissues, TRIM14 mRNA levels were significantly higher in atheromatous tissues in all 11 samples (Figure 8C). The average level of TRIM14 mRNA in atheroma was increased by 7.1 folds compared to that in normal artery tissues (Figure 8D). These results suggest that TRIM14 may involve in the pathogenesis of atherosclerosis via promoting EC activation.
Figure 8.
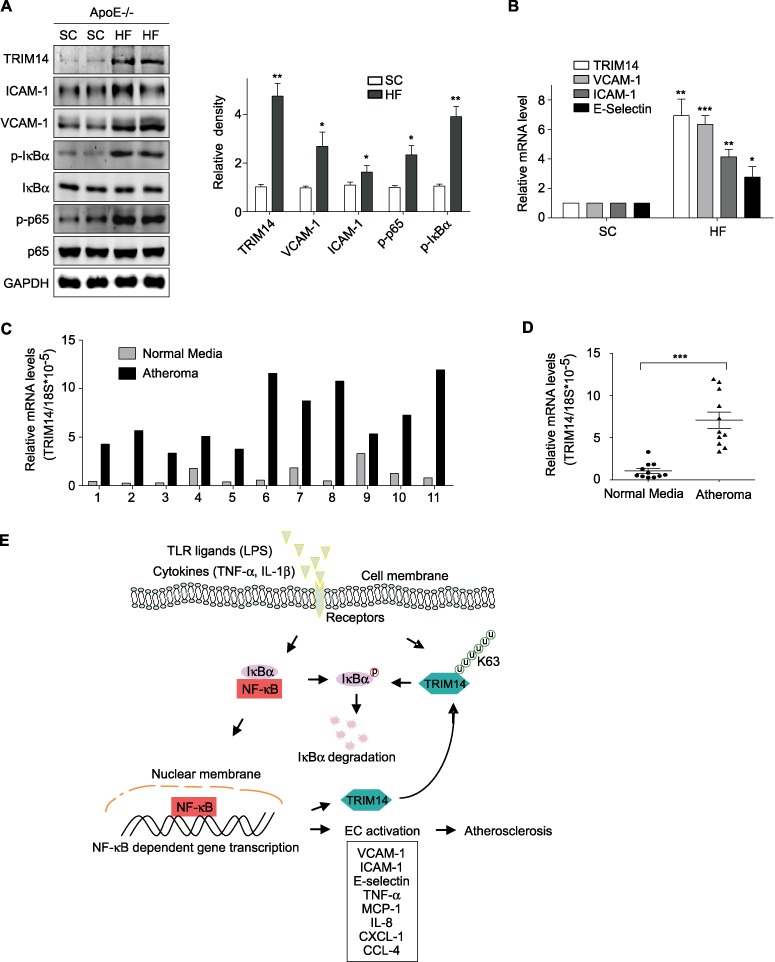
The expression of TRIM14 is elevated in mouse and human atherosclerotic lesions. (A and B) Aortas were collected from ApoE−/− mice with SC diet or HF diet. Protein and RNA were isolated and analyzed by western blot (A) and qPCR (B). Data are representative of three independent experiments. (C and D) Normal artery and atheromatous tissues were collected from 11 patients who received carotid endarterectomy. Total RNAs were extracted and analyzed by qPCR. Data are presented as relative levels to 18S mRNA (C). The average level of TRIM14 in atheroma from 11 patients was 7.1-fold compared to that in normal carotid artery tissues and ***P < 0.001 (D). (E) Working model of TRIM14–NF-κB positive feedback loop in EC activation.
Discussion
Endothelial activation characterized by the expression of multiple chemokines and adhesive molecules is a critical initial step of vascular inflammation, which results in recruitment of leukocytes into the sub-endothelial layer of the vascular wall and triggers vascular inflammatory diseases such as atherosclerosis. Although inhibiting endothelial inflammation has been well recognized as a therapeutic strategy in vascular inflammatory diseases, the therapeutic targets are still elusive. In the present study, we identified TRIM14 as a positive regulator of endothelial activation via activating NF-κB signal pathway. TRIM14 is unexpectedly highly expressed in human vascular ECs and markedly induced by inflammatory stimuli both in vitro and in vivo. Gain-of-function and loss-of-function studies on cultured ECs demonstrated that TRIM14 acts as an adaptor protein to facilitate the interaction of NEMO and TAK, by which it promotes cytokine-induced NF-κB signaling and endothelial activation. Our data further defined that TRIM14 is a target gene of NF-κB, suggesting that TRIM14–NF-κB axis amplifies the inflammatory signal in activated ECs. As TRIM14 expression was significantly upregulated in atherosclerotic artery, TRIM14 may play important roles in the pathogenesis of atherosclerosis and serve as a potential therapeutic target for human atherosclerotic diseases.
The expression of TRIM14 is not specific in ECs. It was previously reported that TRIM14 mRNA was highly expressed in human thymus and spleen and moderate expressed in lung, liver, colon, and bone marrow (Zhou et al., 2014). Several reports also showed that TRIM14 protein was upregulated in various cancer tissues and cell lines including colorectal cancer, hepatocellular carcinoma, glioma, gastric cancer, and breast cancers (Jin et al., 2018; Wang et al., 2018; Hu et al., 2019; Dong and Zhang, 2018). The effect of TRIM14 on the NF-κB signal pathway seems dependent on the cellular and signal context. As shown in Supplementary Figure S1, overexpression of TRIM14 did not affect LPS-induced NF-κB activation in Raw264.7 cells. In current study, we observed that TRIM14 potently promoted TNF-α- and IL-1β-induced NF-κB activation in human ECs. At least two other reports showed that TRIM14 promoted NF-κB activation in A549 human lung epithelial cells (Zhou et al, 2014) and tongue squamous cell carcinoma cells (Su et al., 2016). In addition, several reports demonstrated that TRIM14 also promotes the activation of STAT, AKT, and Wnt/β-catenin signaling though the underlying mechanisms are not well clear (Xu et al., 2017; Jin et al., 2018; Tan et al., 2018; Wang et al., 2018). Nevertheless, the molecular mechanisms of TRIM14 in the regulation of different signal pathways and in different cellular context need to be further investigated.
How does TRIM14 regulate NF-κB signal pathway? In our study, we observed that overexpression of TRIM14 promoted TNF-α-induced IκBα phosphorylation and degradation. Conversely, knockdown of TRIM14 decreased TNF-α-induced IκBα phosphorylation and degradation. Furthermore, TRIM14 directly interacted with NEMO, a component of IKK complex. As reported by Tan et al. (2018) and Zhou et al. (2014), TRIM14 can serve as an adaptor protein to interact with MAVS and recruit NEMO or WHIP/PPP6C to mitochondria, by which it promotes RIG-I-mediated activation of IRF and NF-κB signaling. Though we did not test if TRIM14 is also localized on mitochondria in ECs, we believed that interaction with MAVS would be required for TRIM14-mediated activation of IKK complex and NF-κB signaling.
In current study, we observed that TRIM14 was subject to K63-linked polyubiquitinated during TNF-α-induced endothelial activation, which is required for its interaction with NEMO and TAK1 and regulation of NF-κB activation and endothelial inflammation. Though most TRIM proteins contain an N-terminal RING domain and acts as ubiquitin E3 ligase (Ozato et al., 2008), TRIM14 lacks N-terminal RING domain and does not acts as E3 ubiquitin ligase. The E3 ligase that drives the ubiquitination of TRIM14 at the context of TNF-α-activated ECs remains unclear. As TAK1 complex also recruits TRAF3 E3 ligase (Liu et al., 2013), we postulated that the E3 ligase containing in the signalosome may promote K63-linked polyubiquitination of TRIM14.
Interestingly, the stimuli that activate ECs also significantly induced TRIM14 transcription via NF-κB signaling. Our study further showed that TRIM14 promoter is directly controlled by NF-κB. These results suggest that TRIM14–NF-κB axis is a driving force to amplify the inflammatory signaling in activated ECs (Figure 8E). As the expression of TRIM14 is increased in human atherosclerotic lesions, TRIM14 may involve in the development of human atherosclerosis and serve as a therapeutic target for the treatment of atherosclerotic diseases.
Materials and methods
Reagents
Human recombinant TNF-α and IL-1β as well as LPS were purchased from Sigma. VCAM-1 (sc-13160), ICAM-1 (sc-1511-R), E-selectin (sc-14011), and β-actin (sc-1616) antibodies were from Santa Cruz Biotechnology. TRIM14 antibodies (ARP34737 and SAB1410027) were purchased from Aviva Systems Biology and Sigma. Phospho-p65 (3033), p65 (8242), IκBα(4812), phospho-IκBα(2859), phospho-IKKα/β (2078), IKKα (11930), IKKβ (8943), phospho-JNK (4668), JNK (9252), phospho-ERK1/2 (4370), ERK1/2 (4695), phospho-p38 (4511), p38 (8690), phospho-C-Jun (2361), C-Jun (9165), Flag (8146), α-tubulin (2125), and histone H3 (4499) antibodies were purchased from Cell Signaling Technology. Ubiquitin (ab7780), ubiquitin (K48, ab140601) and ubiquitin (K63, ab179434) antibodies were purchased from Abcam. TAK1 (12330-2-AP), NEMO (18474-1-AP), TRAF2 (26846-1-AP), and TRAF5 (27065-1-AP) antibodies were purchased from Proteintech. SiRNAs targeting TRIM14 and NEMO were purchased from Santa Cruz Biotechnology. qPCR primers were from Integrated DNA Technologies, Inc.
Cell culture and transfection
All human primary vascular ECs (HAEC, HCAEC, HDMEC, HLMEC, and HUVEC) were purchased from Lonza Walkersville Inc., cultured in EGM or EGM2 medium according to the manufacturer instruction, and used for experiment in less than five passages. The human acute monocytic leukemia cell line THP-1 was obtained from American Type Culture Collection (ATCC) and maintained in RPMI 1640 medium (Corning) containing 10% FBS (Sigma-Aldrich). HeLa, A549, RAW264.7, and U937 were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS. Transient transfection of TRIM14 vector and siRNA into HUVECs was performed by electroporation using Nucleofector device (Lonza) and Nucleofector kits for HUVEC (Lonza) following the manufacturer’s instruction. After electroporation, cells were plated into 35 mm dishes and incubated for 24 h at 37°C, 5% CO2. Then, cells were treated with 10 ng/ml TNF-α or IL-1β for indicated times, and proteins from those cells were extracted and detected by western blot.
Western blot and immunoprecipitation
Cells or mouse tissues were harvested and lysed in the lysis buffer containing 0.5% Brij® L23 solution (Sigma-Aldrich), 50 mM KCl, 2 mM CaCl2, 20% glycerol, 50 mM Tris-HCl, and proteases inhibitors and phosphatases, pH 7.4. After centrifuge at 12000 rpm for 10 min, the protein in the supernatant was quantified using a standard BCA protein assay kit (Pierce). Equal amounts of total protein (20–50 μg) samples were separated on SDS-PAGE and electro-transferred onto a nitrocellulose membrane (Corning). First, the membranes were blocked with 5% non-fat milk dissolved in PBST (PBS, pH 7.4, containing 0.1% Tween-20) for 20 min. Then, they were incubated with primary antibody overnight at 4°C with gentle shaking. After washing with PBST for three times, the membranes were incubated with secondary antibody conjugated to horseradish peroxidase for at least 1 h at room temperature. Finally, the membranes were incubated with HRP chemiluminescence kit (Pierce) and exposed to X-ray film. For immunoprecipitation, HUVECs were collected with lysis buffer and subjected to immunoprecipitation with TRIM14 antibodies and protein-G beads at 4°C overnight with gentle rolling. Bound proteins were analyzed by immunoblotting.
qPCR
Total RNA from HUVECs was extracted using TRIzol® Reagent (Life Technologies™) according to the instructions. Then 2 μg RNA was reverse-transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative PCR was carried out using SYBR Green master mix (ABI) on ABI StepOne Plus real-time PCR system instrument. The results were normalized to β-actin transcript as an internal control. All data were expressed in terms of fold-change relative to the control samples. The primers were validated for their amplification efficiency and specificity. Sequences of the primers are listed in Supplementary Table S1.
Isolation of nuclear and cytoplasm extracts
The nuclear and cytoplasm were separated using NE-PER Nuclear Cytoplasmic Extraction Reagents (Pierce). Briefly, HUVECs were harvested with trypsin-EDTA and washed with cold PBS and then centrifuged at 500× g for 3 min. A total of 200 μl cytoplasmic extraction reagent I was added to the cell pellet to fully suspend it by vigorously vortex for 15 sec. The suspension was kept on ice for 10 min followed by the addition of 11 μl cytoplasmic extraction reagent II, vortexed for 5 sec, incubated on ice for 1 min and centrifuged for 5 min at 16000× g. The cytoplasm in the supernatant fraction was transferred to a pre-chilled tube and stored on ice. The pellet fraction was resuspended in 100 μl nuclear extraction reagent by vortex 15 sec and incubated on ice for 10 min (repeat the vortexing every 10 min and incubate it on ice for 40 min), then centrifuged for 10 min at 16000× g. The resulting supernatant including the nuclear extract was transferred to a new tube for the subsequent experiments.
Monocyte adhesion assay
The adhesion assay was performed as previously described (Nie et al., 2016). After 24 h of transfection, HUVECs grown on 6-well plate were treated with 10 ng/ml TNF-α for 8 h and then washed twice with PBS. THP-1 cells were labeled with fluorescein isothiocyanate using a PKH67 fluorescent staining kit (Zynaxis, Inc.) according to the manufacturer’s instructions. There were 5 × 105 fluorescence dye-labeled THP-1 cells added into each well and allowed to co-cultured with HUVECs for 1 h at 37°C. Non-adherent cells were removed by gently washing with cold PBS. The images of adherent THP-1 cells and the number were determined under Cytation 3 Cell Imaging Multi-mode Reader (Biotek Instruments).
LPS challenge in mice
Adult C57BL/6 mice from Jackson Laboratory were challenged by the intraperitoneal injection of LPS (O127:B8; Sigma-Aldrich) at a dose of 25 mg/kg in 200 μl sterile saline for 0, 4, and 8 h. Mice were sacrificed and the arteries were collected for western blot. All experiments were approved by the Institutional Animal Care and Use Committee of University of Missouri Kansas City. After injection, the mice were closely monitored for general condition and survival.
In silico promoter analysis and luciferase reporter assay
To identify p65 binding sites in the TRIM14 promoter, we searched for transcription factor motifs using the JASPAR database at http://jaspar.genereg.net/ (Khan et al., 2018). We found two consensus binding sites (−1744~−1735 and −1622~−1613) in the upstream region of TRIM14 transcription start site. The promoter region contained p65 binding sites of TRIM14 as well as its mutants were cloned into pGL3 basic vector using the primers: fragment A forward primer: 5′GCGCGGTACCATGAGCTTTGGCAGGTATCCA3′, reverse primer: 5′GCGCCTCGAGCGGAGTACTCAGGGAGGCAAA3′; fragment B forward primer: 5′GCGCGGTAC CGTGGAATATGCAATGGAA3′, reverse primer: 5′GCGCCTCGAGCGGAGTACTCAGGGAGGCAAA3′; fragment C-forward primer: 5′GCGCGGTACCGGCGGAGACCAAGGATCTTA3′, reverse primer: 5′GCGCCTCGAGGAGGTGGTCCCACCAGAAAT3′. HEK293 cells were seeded in triplicate in 12-well plates and allowed to settle for 12 h. A total of 300 ng of the above luciferase reporter plasmids and the internal control plasmid pRL-TK (10 ng/well) were co-transfected with p65 expression plasmid into HEK293 cells. The luciferase of both `firefly’ and Renilla signals were measured 24 h after transfection using Dual Luciferase Reporter assay kit (Promega). Each reporter gene assay was performed in triplicate.
ChIP assay
The ChIP assay was performed as previously described with slight modifications (Zhou et al., 2017). Briefly, ~5 × 107 cells were fixed using 4% paraformaldehyde, and chromatin fragments were sonicated into a range of 150–900 bp. The extracts were pre-cleared in BSA-blocked protein A/G beads and incubated with p65 antibodies or IgG control overnight. Beads were washed several times. DNA-protein cross-linking was reversed overnight at 65°C, and DNA was purified and amplified by qPCR. A list of primers used for ChIP-qPCR analysis is provided in Supplementary Table S1. qPCR was performed with three technical and biological replicates.
Human carotid artery sample collection
Collection of human carotid endarterectomy specimens was approved by the institutional review board at Greenville Health System, and all patients gave written informed consent. All procedures were performed in accordance with the National Institutes of Health (NIH) guidelines and regulations on human subjects. Patients enrolled in the study had >50% stenosis in the carotid artery and a prior stroke or TIA or >70% carotid artery stenosis. Carotid endarterectomy specimens were obtained from 11 patients at the time of surgery at Greenville Memorial Hospital. The excised specimens were immediately immersed in Belzer UW Cold Storage Solution (Bridge to Life Ltd.). Intact specimens, ranging from ~2–5 cm in length, were nominally centered around the carotid artery bifurcation and usually contained portions of the common carotid artery, the internal carotid artery, and the external carotid artery. Each specimen was sliced into a series of transverse segments nominally 5-mm thick using a custom slicing device. The slices were dissected by a trained investigator into samples consisting of fibrous cap, atheroma, diseased media, and `normal’ tissue, defined as that part of the common carotid artery devoid of any visible atherosclerotic lesion.
Mice
ApoE−/− mice on C57BL/6J background were purchased from The Jackson Laboratory. All mice were fed a chow diet in a temperature-controlled facility maintained at 23°C ± 1°C and 60% humidity with a 12 h light: 12 h dark cycle. All of the animal breeding and other procedures were approved by the Institutional Animal Care and Use Committee of University of Missouri Kansas City and followed the Institutional and US NIH guidelines. At 8–10 weeks of age, ApoE−/− mice were either continued on SC or switched to an HF/high-cholesterol diet (containing, wt/wt, 20% protein, 50% carbohydrate, 21% fat, and 0.21% cholesterol) for 12 weeks.
In vitro pull-down assay
Recombinant GST-TAK1 protein was purified in E. coli (BL21) as reported. Whole-cell lysates from HUVECs treated with TNF-α were incubated with GST or GST-TAK1 protein in a buffer containing 50 mM Tris-HCI, pH 7.5, 150 mM NaCl, and 1% Triton buffer overnight at 4°C. Protein complexes were precipitated with Glutathione Sepharose 4B (GE healthcare) and subjected to immunoblotting with indicated antibodies.
Statistical analysis
Statistical analysis was performed using SPSS Statistics for Windows (Version 18.0). Comparisons between two groups were performed by paired t-test. All the data were expressed as the mean ± SD. A P-value of <0.05 was considered significant. Experiments were performed at least three times.
Supplementary Material
Funding
This work was supported by The National Natural Science Foundation of China (81660390, 31760329, and 81301964). M.F. was supported by American Heart Association (17AIREA33660073) and NIH Grant (AI103618).
Conflict of interest: none declared.
References
- Chen M., Meng Q., Qin Y., et al. (2016). TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119. [DOI] [PubMed] [Google Scholar]
- Dong B., and Zhang W. (2018). High levels of TRIM14 are associated with poor prognosis in hepatocellular carcinoma. Oncol. Res. Treat. 41, 129–134. [DOI] [PubMed] [Google Scholar]
- Galkina E., and Ley K. (2009). Immune and inflammatory mechanisms of atherosclerosis (*). Ann. Rev. Immunol. 27, 165–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone M.A. Jr, and Garcia-Cardena G. (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118, 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai J., Zhu C.Q., Wang T., et al. (2017). TRIM14 is a putative tumor suppressor and regulator of innate immune response in non-small cell lung cancer. Sci. Rep. 7, 39692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Pen W., and Wang M. (2019). TRIM14 promotes breast cancer cell proliferation by inhibiting apoptosis. Oncol. Res. 27, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M.X., Hong X., Liao B.B., et al. (2017). Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci. Rep. 7, 42781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Li H., Hong X., et al. (2018). TRIM14 promotes colorectal cancer cell migration and invasion through the SPHK1/STAT3 pathway. Cancer Cell Int. 18, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Fornes O., Stigliani A., et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J.K. (2013). Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 123, 540–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Chen J., Cai X., et al. (2013). MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2, e00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Zhang L., Joo D., et al. (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, pii: 17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie C., Zhang Z., Zheng J., et al. (2016). Genome-wide association study revealed genomic regions related to white/red earlobe color trait in the Rhode Island Red chickens. BMC Genet. 17, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozato K., Shin D.M., Chang T.H., et al. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober J.S. (2002). Endothelial activation: intracellular signaling pathways. Arthritis Res. 4(Suppl 3), S109–S116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi G., Sung B., and Aggarwal B.B. (2008). TNF: a master switch for inflammation to cancer. Front. Biosci. 13, 5094–5107. [DOI] [PubMed] [Google Scholar]
- Sprague A.H., and Khalil R.A. (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 78, 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X., Wang J., Chen W., et al. (2016). Overexpression of TRIM14 promotes tongue squamous cell carcinoma aggressiveness by activating the NF-κB signaling pathway. Oncotarget 7, 9939–9950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P., He L., Cui J., et al. (2017). Assembly of the WHIP–TRIM14–PPP6C mitochondrial complex promotes RIG-I-mediated antiviral signaling. Mol. Cell 68, 293–307.e5. [DOI] [PubMed] [Google Scholar]
- Tan Z., Song L., Wu W., et al. (2018). TRIM14 promotes chemoresistance in gliomas by activating Wnt/β-catenin signaling via stabilizing Dvl2. Oncogene 37, 5403–5415. [DOI] [PubMed] [Google Scholar]
- Wang F., Ruan L., Yang J., et al. (2018). TRIM14 promotes the migration and invasion of gastric cancer by regulating epithelial-to-mesenchymal transition via activation of AKT signaling regulated by miR-195-5p. Oncol. Rep. 40, 3273–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C., and Noels H. (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422. [DOI] [PubMed] [Google Scholar]
- de Winther M.P., Kanters E., Kraal G., et al. (2005). Nuclear factor κB signaling in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 25, 904–914. [DOI] [PubMed] [Google Scholar]
- Xu G., Guo Y., Xu D., et al. (2017). TRIM14 regulates cell proliferation and invasion in osteosarcoma via promotion of the AKT signaling pathway. Sci. Rep. 7, 42411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Huang D., Li Y., et al. (2017). Nek2A/SuFu feedback loop regulates Gli-mediated Hedgehog signaling pathway. Int. J. Oncol. 50, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Jia X., Xue Q., et al. (2014). TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl Acad. Sci. USA 111, E245–E254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.