

Abstract
Glioblastoma remains as the most common and aggressive primary adult brain tumor to date. Within the last decade, cancer immunotherapy surfaced as a broadly successful therapeutic approach for a variety of cancers. However, due to the neuroanatomical and immunosuppressive nature of malignant gliomas, conventional chemotherapy and radiotherapy treatments garner limited efficacy in patients with these tumors. The intricate structure of the blood brain barrier restricts immune accessibility into the tumor microenvironment, and malignant gliomas can activate various adaptive responses to subvert anticancer immune responses and reinstate an immunosuppressive milieu. Yet, evidence of lymphocyte infiltration within the brain and recent advancements made in cell engineering technologies implicate the vast potential in the future of neuro-oncological immunotherapy. Previous immunotherapy platforms have paved way to improved modalities, which includes but is not limited to personalized vaccines and chimeric antigen receptor T-cell therapy. This review will cover the various neuroanatomical and immunosuppressive features of central nervous system tumors and highlight the innovations made in T-cell based therapies to overcome the challenges presented by the glioblastoma microenvironment.
Keywords: Immunotherapy, immunosuppression, glioblastoma, T-cell, vaccine, CAR
INTRODUCTION
Immunotherapy has been a long-studied approach that has garnered the attention as a promising mode of cancer treatment. The maturation and impact of the field helped certain sets of immunotherapies in achieving status as first-line treatments, and recent advances in cancer immunotherapy within the last decade effectively improved outcomes for several human cancers. [1][2][3][4] FDA-approved nivolumab and ipilimumab treatment demonstrated remarkable clinical efficacy in some patients with melanoma and renal carcinoma [1][2], and early clinical studies of CD19-specific chimeric antigen receptor T-cells showcased a 90% complete response rate in patients with relapsed or refractory B-cell acute lymphoblastic leukemia. [3][4] Despite the many exciting advances made in the field, the complex relationship between brain tumors and the immune system has limited the quality of response of many immunotherapeutic strategies. This review focuses on recent advances made in T-cell-based immunotherapies for malignant gliomas. We discuss the neuroanatomical challenges for immunotherapy and highlight approaches that may facilitate better efficacy in delivering and enabling immunotherapies towards these unique sites.
NEUROANATOMICAL CHALLENGES FOR IMMUNOTHERAPY
The ability of therapeutic immune cells to reach the brain parenchyma is a critical prerequisite for inducing an anti-tumor response. Entry of innate immune cells into the central nervous system (CNS) is tightly regulated by the blood brain barrier (BBB) in order to limit the potential effects of neuroinflammation, such as edema, cytokine-induced toxicity, and neurodegeneration. Traditionally, the brain has been regarded as an immune-privileged site, which was heavily supported by the lack of immunological responses by brain allografts [5] and inoculation of pathogens into the brain parenchyma. [6][7] However, this view has greatly changed, and while the brain is demonstrated to be privileged to a certain degree, consequent studies have proven that the brain does not completely preclude blood-borne immune cells. [8][9][10] The increasing attention towards immunotherapies underscores the importance of understanding the unique anatomical features and immunosuppressive nature of the CNS that impede proper T-cell infiltration into the brain. This segment will focus on outlining the distinct neuroanatomical and immunosuppressive features of the CNS and discuss approaches in addressing these challenges.
Immunological Accessibility of the Brain Parenchyma
The BBB serves as the principal gateway that restricts the entry of ions, large molecules, and naïve immune cells from peripheral blood by the formation of capillary tight junctions created by the interactions of endothelial cells with the luminal and abluminal membranes. [11] This protective barrier between the CNS and the periphery typically retain immune cells in the CSF-drained perivascular, leptomeningeal or ventricular spaces, but during neuroinflammation, immune cells are capable of breaching the glia limitans and entering the CNS parenchyma. [12] Early studies indicated the importance of α4 integrin surface expression in the passage of activated effector T-cells into the brain parenchyma [13], and a murine study demonstrated the selective trafficking of antigen-specific CD8+ T-cells into the brain and its dependency on the luminal expression of major histocompatibility complex (MHC) class I by cerebral endothelium. [14] While this infers the lack of T-cell infiltration in the absence of inflammation, T-cell recruitment to the brain parenchyma can still occur under specific circumstances and mechanisms upon entry to the CNS.
One such mechanism by which T-cells are recruited into the brain parenchyma is through post-capillary venules into the perivascular space. The recruitment of any circulating immune cell into a given tissue is mediated by the sequential interaction of various adhesion and signaling molecules expressed on the surface of immune cells and endothelial cells lining the vessel wall. (Figure 1) Activated T-cells express α4/β1-integrins and lymphocyte-associated antigen-1 (LFA-1) which bind to vascular cell adhesion molecule 1 (VCAM1) and intracellular cell adhesion molecule 1 (ICAM1), respectively, on the surface of endothelial cells. The initial tether formed by these interactions elicits rolling along the vascular wall and allows G-protein-coupled receptors (GPCRs) on T-cells to bind chemotactic factors on the endothelial surface. A GPCR inside-out signal to the α4/β1-integrins leads to increased avidity and conformational changes that results in T-cell diapedesis and traversal through the endothelial lining. [12] Passage through the glia limitans requires the secretion of active matrix metalloproteinases MMP-2 and MMP-9 by leptomeningeal macrophages. MMP-2 and MMP-9 cleave the extracellular matrix receptor β-dystroglycan from astrocytes, which allow T-cell diapedesis across the glia limitans into the brain parenchyma. [15] Moreover, astrocytes lining the glia lamitans can facilitate activated T-cell infiltration into the parenchyma by secreting factors such as TNFα, IL-12, and IL-6. [16][17] These intricacies underscore the need for integrating a deeper understanding of the multifaceted integrity of the BBB and the complex mechanisms involving T-cell recruitment in order to develop of effective T-cell therapies for brain tumors, especially malignant gliomas.
Figure 1: Neuroanatomical challenges for T-cell infiltration into the brain parenchyma:
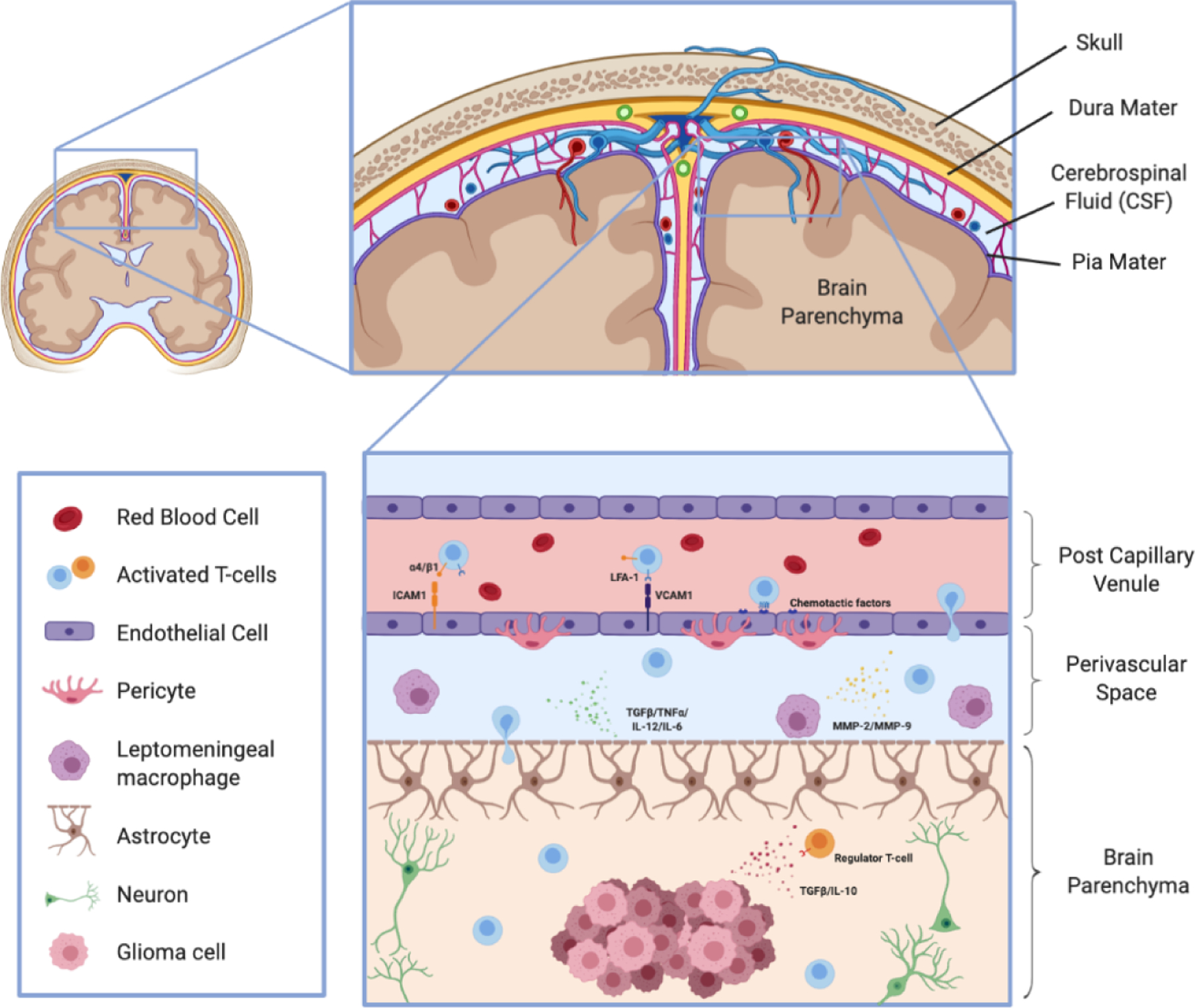
Activated T-cells can be recruited into the brain parenchyma through post-capillary venules. LFA-1 and α4/β1-integrins expressed on the surface of activated T-cells bind to ICAM1 and VCAM1, respectively, on endothelial cells to allow the T-cells to reduce their velocity and undergo diapedis through the endothelial lining. Secretion of MMP-2 and MMP-9 by leptomeningeal macrophages and other astrocytic secreting factors permit T-cell infiltration through the glia limitans. Upon entering the brain parenchyma and reaching the tumor microenvironment, T-cells must overcome various mechanisms of tumor-mediated immunosuppression to elicit a robust and persistent immune response.
Immunosuppression in the Glioma Microenvironment
T-cell infiltration into the brain parenchyma does not infer sustained adaptive immune responses, and for all intent and purposes, the immunosuppressive nature of the glioma microenvironment inhibits proper effector T-cell proliferation and function. Patients with malignant glioma and murine glioma models have both demonstrated impaired cellular immunity as a result of systemic immunosuppression. [18][19][20] The underlying mechanisms facilitating immunosuppression in brain tumors largely stem from three key features: secreted factors within the microenvironment, tumor cell-intrinsic properties, and the enrichment of immunosuppressive myeloid cells (Table 1). However, preclinical and early-phase clinical studies of antigen-specific immunotherapies have demonstrated antigen-specific anti-tumor effects in CNS tumors [21][22][23][24], providing evidence that certain activated immune cells are capable of bypassing immunosuppression.
Table 1: Methods of immunosuppression and immune evasion employed by the glioblastoma milieu:
The lack of efficacy in conventional immunotherapy approaches in glioblastomas is due largely in part to the multitude of immunosuppressive mechanisms employed by the tumor microenvironment.
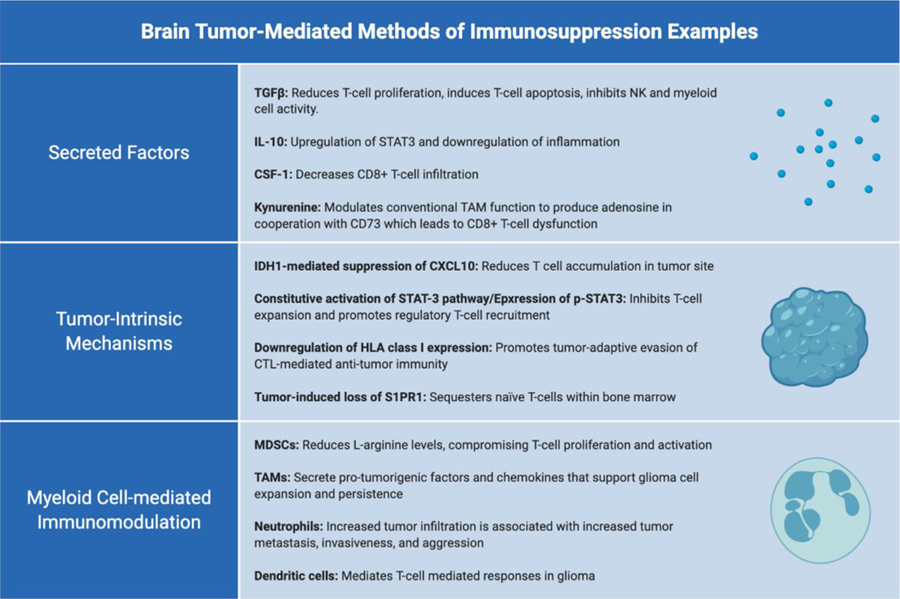 |
Immunosuppression Regulated by Secreted Factors
The CNS tumor stroma has been previously demonstrated to release immunosuppressive cytokines, such as transforming growth factor β (TGFβ) and IL-10, that can downregulate inflammation and attract regulatory T-cells. TGFβ induces immune suppression by reducing T-cell proliferation, inducing T-cell apoptosis, and inhibiting natural killer (NK) cell and myeloid cell activity. [25] Early studies demonstrated elevated levels of serum-TGFβ in mice bearing brain tumors, and the pharmacological inhibition of TGFβ signalling was capable of partially reversing immune suppression. [26] As such, the secretion of TGFβ by microglia was initially thought to be the predominant source of brain tumor-related immune suppression. Additional studies investigating the role of IL-10 in glioblastoma immunosuppression have also inferred its involvement in upregulation of signal transducer and activator of transcription 3 (STAT3) in suppressing immune cell activity. [27] However, recent studies suggested that the recruitment and infiltration of CD8+ T-cells into malignant gliomas can similarly be inhibited by macrophage and microglia-secreted colony-stimulating factor (CSF-1) and kynurenine. [28] CSF-1 receptor blockade by PLX3397 has demonstrated increased CD8+ T-cell infiltration and tumor regression in murine tumor models, [29][30] and these findings are further investigated in a Phase II clinical trial of adult patients with recurrent glioblastoma (NCT01349036). The catabolic production and secretion of kynurenine can activate aryl hydrocarbon receptors (AHRs) on tumor-associated macrophages (TAMs) to drive the expression of CD39. This modulation of conventional TAM functions promotes CD8+ T-cell dysfunction by producing adenosine in cooperation with CD73. [31] Indoleamine-2,3-dioxygenase (IDO) and tryptophan dioxygenase (TDO) catabolizes tryptophan into kynurenine, and IDO- or TDO-blockade have been demonstrated to alleviate tumor-associated immune suppression in preclinical cancer models. [32][33] Furthermore, a Phase I study (INCB024360) treating patients with advanced malignancies, including gliomas, with epacadostat is currently investigating the therapeutic effect of IDO inhibition. [34]
Immunosuppression Mediated by Tumor Cell-Intrinsic Mechanisms
Brain tumor-associated mutations in signature oncogenes and chromosome modifying proteins can have immense repercussions on the immunological and genomic landscape of the tumor microenvironment. 70–80% of WHO grade 2 and 3 low grade glioma (LGG) or secondary glioblastoma cases report single amino-acid mutations in two isocitrate dehydrogenase (IDH) isoforms: R132 in IDH1 and R140 or R172 in IDH2. [35] Our group previously reported mutant IDH-associated suppression of type I immune response genes and a decrease in overall expression and activation of STAT1 and T-cell attracting chemokines in the glioma immune environment. [36] These findings were further supported by the identification of significantly reduced levels of T-cell infiltration in mutant IDH gliomas relative to their wild-type counterparts [37], signifying that genetic alterations intrinsic to glioma cells are capable of promoting immune evasion through modifying the tumor microenvironment cellular composition.
The constitutive activation of the STAT3 pathway in glioblastoma-initiating cells and expression of phosphorylated STAT3 (p-STAT3) in glioblastoma was shown to suppress T-cell expansion and promote regulatory T-cell (T-reg) recruitment. [38][39] In one particular study, the treatment of glioblastoma patient-derived myeloid cells with WP1066, a small molecule inhibitor of p-STAT3, reversed immune cell tolerance by inducing IL-2, IL-4, IL-12, and IL-15 production while simultaneously inducing proliferation of effector T-cells that were otherwise refractory to CD3 stimulation. [39] These findings led to the evaluation of WP1066 in a Phase I clinical trial (NCT01904123) for recurrent glioblastoma patients and melanoma patients with brain metastases.
Downregulation of human leukocyte antigen (HLA) class I expression is another immunosuppression mechanism employed by malignant brain tumor cells. [40] While HLA class I expression can be restored by IFN-γ treatment in certain cases, mutation-associated loss of heterozygosity (LOH) of HLA class I and β2-microglobulin regions can result in irreversible downregulation of HLA class I. A previous study conducted by our group demonstrated HLA class I LOH in 41% of informative cases, which were associated with decreased survival in newly diagnosed glioblastoma patients. [41] Tapasin has previously been shown to facilitate peptide binding to HLA class I molecules in the endoplasmic reticulum, and one particular study showed a rapid loss of HLA class I complexes from the surface of tapasin-deficient cells. [42] Admittedly, a recent finding demonstrated a strong correlation between tapasin levels with HLA class I expression and patient survival time. [43]
Yet another tumor-adaptive mode of T-cell dysfunction employed by malignant gliomas is their ability to sequester naïve T-cells in the bone marrow. Treatment-naïve subjects and mice with glioblastoma have severely compromised levels of T-cells in the blood and lymphoid organs, and a recent study by Chongsathidkiet et al. revealed the ‘missing’ naïve T-cells to be sequestered in large numbers within the bone marrow. Sequestration was accompanied by tumor-imposed loss of sphingosine-1-phosphate receptor 1 (S1PR1) from the T-cell surface, and the preclusion of S1P1R internalization led to sequestration reversal. This phenomenom was shown to be characteristic of not only malignant gliomas, but also tumors introduced intracranially. [44] Altogether, these results demonstrate that while brain tumors are capable of employing various methods of initiating T-cell dysfunction, targeted-reversal of specific tumor-intrinsic immunosuppression mechanisms can potentially enable previously ineffective T-cell-activating therapies.
Myeloid Cell-mediated Immunomodulation
Myeloid cells represent the largest immune subset within glioblastomas and largely consists of tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), neutrophils, and dendritic cells (DCs). [45][46] TAMs represent the majority of non-neoplastic cells within gliomas, and support the expansion and persistence of tumor cells by secreting pro-tumorigenic factors and chemokines. [31][47] Neutrophils are recruited to the tumor microenvironment by tumor-derived cytokines, and depending on their maturation and activation state, they can exert either anti- or pro-tumor effects. (Reviewed in [48]) Recruitment accompanied by an overproduction of G-CSF in the tumor microenvironment leads to high neutrophil lymphocyte ratios (NLRs), which is associated with poor prognosis. [49][50] Increased expression of S100A4 within glioma cells by neutrophil infiltration in glioma patients is associated with increased incidences of tumor metastasis, invasiveness, and aggressiveness [51], and inhibition of CSF1R+ myeloid cells by GW2580 decreased molecular signatures of cytokines involved in inflammation and angiogenesis. [52] Conversely, DCs have been suggested in playing a vital role in orchestrating T-cell mediated responses in glioma, and preclinical models utilizing CD103+ DC-deficient mice demonstrated their inability to reject transplantable immunogenic tumors. [53] As such, ongoing Phase II clinical trials aim to validate the efficacy of vaccines with dendritic cells pulsed with glioma-derived antigens, and study results showcased this approach as both safe and feasible. [54]
In a recent study, high levels of MDSCs in patients with recurrent glioblastomas were shown to be associated with poorer overall survival. [55] MDSCs are generally considered as a heterogeneous population of immature myeloid cells, and our group previously demonstrated a novel granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced immunosuppression mechanism employed by glioma-infiltrating MDSCs. In our study, we revealed an upregulation of IL4Rα on glioma-infiltrating MDSCs specifically with no change in expression in myeloid cells derived from peripheral blood or healthy brain tissue. [56] IL4Rα mediates IL-13-induced production of arginase which can reduce L-arginine levels, creating an immunosuppressive environment that compromises cytotoxic lymphocyte proliferation and activation. As such, a plethora of approaches have been developed to reduce and inhibit MDSC recruitment at the tumor site. We previously identified that the inhibition of prostaglandin E2 production by cyclooxygenase-2 (COX-2) inhibitors, celecoxib and acetylsalicyclic acid (ASA), leads to the reduction of MDSC accumulation at the tumor site. [57] ASA-treated mice demonstrated a remarkable enhancement of C-X-C motif chemokine 10 (CXCL10) and infiltration of cytotoxic T-cells, illustrating the ability of CXCL10 in limiting gliomagenesis. Further studies by our group illustrated that administration of cyclic diguanylate monophosphate (c-di-CMP), a STING agonist, increased OVA-specific cytotoxicity and survival of infiltrating lymphocytes while simultaneously reversing the immunosuppressive effects of MDSCs. [58] Overall, these findings showcase the multitude of strategies available in modulating myeloid cell-mediated immunosuppression prior to facilitate more effective immunotherapy outcomes.
Corticosteroid-Induced Immunosuppression
Corticosteroids are often necessary in managing peritumoral edema, but recent studies have shown that steroids can also promote an immunosuppressive milieu. Baseline use of ≥ 10 mg of prednisone (equivalent to ≥ 4 mg dexamethasone) was associated with poorer outcome of non-small-cell lung cancer patients treated with PD-L1 blockade. [59] However, bevacizumab has demonstrated itself as a potential steroid substitute [60], and studies in patients with recurrent glioblastoma illustrated the safety of bevacizumab in treating peritumoral edema without the immunosuppressive effects of steroids. [61] Although preliminary results show no immediate improvements in survival, bevacizumab provides an alternative approach against steroid-induced immunosuppression.
IMMUNOTHERAPY STRATEGIES FOR BRAIN TUMORS
The strength in immunotherapeutic approaches lie in their ability to facilitate the eradication of cancer and prevent recurrence by enhancing the systemic and selective immune response against tumor cells. Recent novel advances in immunotherapy include achievements made in immune checkpoint blockades, adoptive cell therapies (ACT), and vaccine therapy [28], but the aforementioned neuroanatomical challenges limit their efficacy in the brain. Nonetheless, evidence of T-cell infiltration into the parenchyma brought focus towards T-lymphocyte immunotherapy in the brain, and in this section, we will summarize the overall impact of T-cell-based approaches (Figure 2) and their significant breakthroughs in treating brain tumors.
Figure 2: T-cell based immunotherapy modalities:
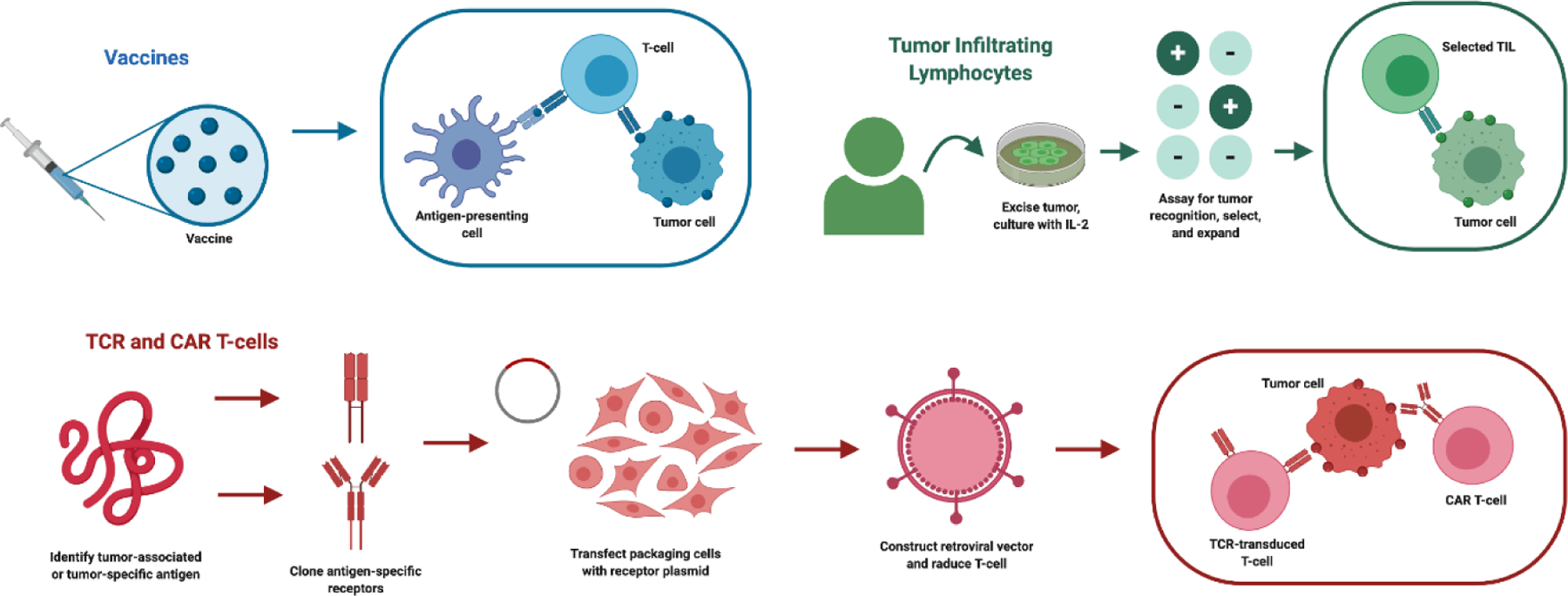
Ongoing T-cell based cancer immunotherapy studies encompass both vaccine therapy and adoptive cell transfer (ACT). Vaccines (upper-left) focus on eliciting a cytotoxic immune response by introducing tumor-associated or tumor-specific antigens for APC presentation. Tumor-infiltrating lymphocyte (TIL) therapy (upper-right) selects for and expands tumor-specific lymphocytes ex vivo for reinfusion into the patient. TCR and chimeric-antigen receptor (CAR) T-cell therapy (bottom) incorporates the design and incorporation of an antigen-specific receptor into a T-cell.
Tumor Vaccination Approaches
Vaccination served as the primary approach in redirecting endogenous T-cells against tumor antigens and remained as one of the earliest developed immunotherapeutic modalities. [62] The objective of therapeutic vaccines is to elicit a cytotoxic response in an immune system that has otherwise been induced into tolerance, and overall, cancer vaccines can be broadly classified into multiple categories: peptide vaccines, heat shock protein vaccines, RNA vaccines, and cell-based vaccines. This section will focus primarily on the application of peptide, heat shock protein, RNA vaccines in the recruitment and activation of T-cells.
Peptide Vaccines
Until recently, neuro-oncology vaccines have primarily focused on tumor-associated antigens (TAAs), which are not unique for gliomas. Many of these potential antigen targets are derived primarily from overexpressed normal proteins that are still present in normal tissue, and targeting such shared antigens holds the risk of eliciting collateral autoimmunity. In contrast, antigens found solely in tumor cells and absent from normal cells are called tumor-specific antigens (TSAs), and due to the low mutational burden of gliomas [63], only a minority of mutations are processed into T-cell responsive neoepitopes. As such, little glioma-specific antigens are reported to date, and certain HLA type-restricted antigens further limit the patient population for which vaccines may be considered. The scarcity of tumor-specific and highly expressed antigenic epitopes in the glioma microenvironment serves as the underlying limitation for the development of novel cancer vaccine approaches.
The most widely studied TSA, to date, is a constitutively active mutant form of epidermal growth factor (EGFR), EGFRvIII, which is expressed exclusively on glioblastoma cells in approximately 20 % of glioma patients. [64] Phase I and II clinical trials testing various forms of EGFRvIII vaccines demonstrated promising results, and this led to the subsequent development of rindopepimut (CDX-110) designed to mimic EGFRvIII in order to foster immune recognition of the mutated sequence. Preclinical models utilizing rindopepimut demonstrated impressive efficacy [65], and promising median overall survival (mOS) in Phase I and 2 clinical trials [66] sparked the initiation of a multicenter, double-arm Phase III clinical trial (ACT IV) testing rindopepimut against a control, keyhole limpet haemocyanin (KLH), both concurrent with standard of care temozolomide. The study revealed no significant improvement in mOS of patients treated with rindopepimut when compared to the control arm; interestingly, the antigen loss was seen in both treatment and control arms. [67] This latter point highlights heterogeneous EGFRvIII expression in glioma cells that supports previous findings of the unstable nature of EGFRvIII expression throughout the course of disease. [68][69] While ACT IV was conducted, a separate Phase II clinical trial (ReACT) explored the clinical efficacy of rindopepimut with bevacizumab combination therapy, and analysis revealed significantly higher mOS in the rindopepimut with bevacizumab arm over the KLH control arm. [70] In addition to rindopepimut, immunization with a live-attenuated Listeria-based vaccine, ADU-623, is currently being tested in an ongoing Phase I study (NCT01967758). While EGFRvIII remains attractive as a tumor-specific target, the propensity of tumor escape and outgrowth in EGFRvIII therapies underscores the need to adjust the approach for this vaccine modality.
Isocitrate dehydrogenase (IDH) mutations are presented in approximately 80% of WHO grade 2 or 3 LGGs, with the majority of the mutations being R132H mutations in the IDH1 variant. In contrast to EGFRvIII, IDH mutations encompasses nearly 100% of tumor cells when present, making it a truly homogenously-expressed TSA. [67] Preclinical studies utilizing murine models expressing humanized MHC class I and MHC class II loaded with IDH1-R132H-derived 15-mer peptides elicited antigen-specific CD4+ T helper 1 cell-mediated immune responses. [22] This led to two ongoing Phase I clinical trials of IDH1-R132H-mutated peptide vaccines, NOA-16 (NCT02454643) and RESIST (NCT02193347). NOA-16 investigates the efficacy of the vaccine concurrently with topical imiquimod in patients with IDH1-R132H-mutated grade III and IV gliomas, and RESIST evaluates adjuvant treatment with GM-CSF and Montanide ISA-51 in patients with recurrent grade II gliomas. In the advent of this homogenously-expressed glioma-specific antigen, these trials may pave way to a new approach for treating patients with IDH-mutant gliomas.
Within the majority of diffuse midline gliomas (DMGs), a driver mutation in the histone H3 gene H3F3A confers a single amino acid mutation, K27M, in both histone 3 variant 1 (H3.1K27M) and variant 3 (H3.3K27M) [28][71] Studies conducted by our group led to the identification of the novel H3.3K27M p26–35 epitope (H3.3K27M26–35). Similarly to IDH-mutations, more than 70% of diffuse midline pontine gliomas (DIPGs) and the majority of DMGs harbor the H3.3K27M mutation, and given that the abysmal mOS of children with DIPG remains under one year, the identification of one of very few tumor-specific target underlines a rare and significant milestone in combating both diseases. Our in vitro studies demonstrated that patient-derived peripheral blood mononuclear cells (PBMCs) stimulated with H3.3K27M26–35 expressed increased levels of IFNγ compared to PBMCs stimulated with wild-type H3.3. [72] This paved way to a Phase I clinical trial (NCT02960230) evaluating the K3.3K27M-specific peptide vaccine with adjuvant Poly ICLC in pediatric K27M-mutated gliomas.
Lastly, a trend towards personalized vaccines have surfaced to answer the limitations in durability and efficacy of targeting TAAs. Personalized vaccines target neoantigens with the goal of potentiating an anti-tumor immune response without the risk of cross reactivity and systematic immune tolerance, and two prominent trials have demonstrated efficacy in this approach. Utilizing a computational pipeline for predicting HLA-binding of epitopes in conjunction with whole exome sequencing (WES), a Phase I study (NCT02287428) was conducted to investigate the feasibility and timing of personalized neoantigen cancer vaccine (NeoVax) with pembrolizumab in newly diagnosed O6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma. Single-cell TCR analysis provided evidence of neoantigen-specific T-cell infiltration within the tumor microenvironment and an increase in the number of tumor-infiltrating T-cells in patients who did not receive dexamethasone. [73] The Glioma Actively Personalized Vaccine Consortium (GAPVAC) have additionally conducted a multicenter Phase I clinical trial (NCT02149225) that incorporates data from HLA-ligandome analyses to evaluate the safety, tolerability, and immunogenicity of actively personalized vaccines (APVACs) in newly diagnosed glioblastoma patients. The approach adopted by Hilf et al. exploits the full repertoire of tumor antigens, and its inclusion of TAAs increased the number of actionable epitopes. [74] In both clinical studies, promising evidence of tumor-reactive T-cell infiltration with memory phenotypes and neoantigen-specific clonal expansion accentuates the therapeutic potential in the future of personalized cancer vaccines.
Heat Shock Protein Vaccines
Immunogenic properties of heat shock proteins (HSPs) have directed increasing attention towards the potential of HSP vaccines. HSPs, also known as chaperone proteins, function to stabilize proteins, resolve protein aggregates, and reassemble salvageable misfolded protein aggregates in response to cellular insults. [75] From an immunological standpoint, however, HSPs have the capacity to either stimulate antitumor immune responses as carriers for antigenic peptides or act as natural immunogens. When complexed with an associated peptide, these HSP complexes are capable of undergoing the endogenous MHC class I pathway to ultimately elicit a CD8+ cytotoxic response. [76] Heat shock protein 70 (HSP70), in particular, has been shown to be overexpressed and present in a majority of solid tumors. [77] Within glioblastomas, studies have demonstrated express elevated constitutive and inducible levels of HSP27, HSP72, HSP73, and HSP90 [78][79], and GBM released exosomes have also exhibited HSP27, HSP60, HSP70, and HSP90 presentation. [79]
Of the major HSP families, HSP70 and HSP90 have garnered the most attention as facilitators of antigen uptake, processing, and presentation onto MHC class I and MHC class II. Studies showing the increased transcription of HSP70 mRNA in correlation with glioma grade [80] and the involvement of HSP90 substrates (e.g. EGFRvIII, hTERT, P53, MAPK) in key tumor initiation and proliferation signaling pathways [81] have all led to the induction of multiple pre-clinical and clinical trials utilizing HSP vaccines. Exogenously delivered and purified HSP70 was shown to be capable of triggering the translocation of its intracellular analog in cancer cells to the cell surface [82], and a complementary pre-clinical study in a rat glioblastoma model demonstrated significant reduction of tumor progression and increased cytotoxic activity of NK cells and CD8+ T lymphocytes following prolonged intra-tumoral delivery of exogenous HSP70. [83] A different approach utilizing superparamagnetic iron oxide nanoparticles (SPIONs) coupled to HSP70 to deliver immunogenic peptides from tumor lysates to dendritic cells has demonstrated the induction of tumor-specific CD8+ T lymphocytes responses in a pre-clinical glioma model. [84] Clinical studies have demonstrated efficacy and relatively safety in patients who have undergone surgical resection and intra-tumoral delivery of recombinant HSP70. Of the 12 patients in the study, 1 patient showed a partial response while 1 patient demonstrated a complete response accompanied by an enhanced Th1-cell-mediated response. [82]
Autologous antigenic peptides chaperoned by HSP glycoprotein-96 comprises HSP-peptide-complex-96 (HSPPC-96), which is utilized in a large majority of HSP vaccine trials. 11 of 12 high grade glioma patients demonstrated a tumor-specific peripheral immune response to HSPPC-96 vaccination (either 25 ug HSPPC-96 every 2 weeks totaling 4 vaccinations or 25 ug HSPPC-96 weekly totaling 4 vaccinations) in a Phase I dose escalation trial. [85] A subsequent open label Phase II multicenter clinical trial in recurrent glioblastoma patients treated with 25 ug HSPPC-96 weekly for 4 weeks followed by biweekly treatment showed a median and 6 month overall survival of 42.6 weeks and 29.3%, respectively. [86] These results have sparked multiple ongoing clinical trials: A completed Phase II single arm study (NCT00905060) investigating the application of autologous HSPPC-96 in newly diagnosed adult GBM patients following tumor resection and adjuvant radiation therapy and temozolomide has data with pending publication. The Alliance for Clinical Trials in Oncology (ALLIANCE) has also sponsored a multi-institutional trial (NCT01814813) in cases of recurrent GBM that aims to explore the efficacy, tolerability, and safety of HSPPC-96 as an adjuvant therapy with bevacizumab. One additional Phase I trial (NCT02100822) is currently studying the safety and efficacy of autologous HSP glycoprotein-96 in patients with newly diagnosed supratentorial gliomas.
RNA Vaccines
Nucleic acid-based vaccines are conventionally based on the utilization of DNA or RNA encoding the antigen of interest. The application of DNA and RNA combines both the efficacy of in situ expression of antigens with the safety inactivated and subunit vaccines, and as a result, nucleic acid-based vaccines have emerged as a promising alternative to conventional vaccine approaches. The use of RNA vaccines holds several advantages over its DNA counterpart. Firstly, RNA modalities eliminate the issue of possible plasmid DNA vaccine integration into the host genome, and while DNA vaccinations require traversal of both the plasma and nuclear membrane for transcription, rapid cellular uptake of RNA vaccines into the cytoplasm is sufficient for expression. [87] Secondly, RNA vaccines serve as a safer approach: mRNA is naturally degraded by normal cellular processes and has no potential risk of infection or insertional mutagenesis due to its non-infectious and non-integrating properties. Design modifications in the delivery methods or down-modulation of the inherent mRNA immunogenicity can further increase the safety profile and highlights the flexibility in RNA vaccine engineering. [88][89] Finally, high yields of in vitro transcription reactions allow the rapid and inexpensive production of RNA vaccines. [90]
Among its various applications, recent advances and directions in RNA-based cancer vaccines have been made in stride following the first proof-of-concept studies highlighting the feasibility of the approach decades prior. [91][92] Dendritic cells play a central role in antigen presentation, and as a result, dendritic cells electroporated with mRNA have comprised the majority of RNA vaccine clinical trials that investigate its efficacy against glioblastomas. Two completed Phase I/II studies (NCT00890032) (NCT00846456) evaluated the feasibility and safety of brain tumor stem cell (BTSC)-specific mRNA-loaded dendritic cell vaccines with one study (NCT00846456) demonstrating the induction of an immune response in all seven patients without adverse autoimmune events or side effects. Studies that are currently recruiting include a Phase I/II trial (NCT02649582) that explores the overall and progression-free survival of new diagnosed glioblastoma patients treated with autologous Wilms’ tumor 1 (TW1) messenger mRNA-loaded dendritic cell vaccination and adjuvant temozolomide treatment following resection and a Phase II trial (NCT02465268) determining the efficacy of glioblastoma patients with investigational dendritic cell vaccine, pp65 DC. While there exist ongoing clinical trials investigating dendritic cells loaded with mRNAs encoding cytomegalovirus (CMV) proteins (NCT00626483) (NCT00639639), anti-viral therapy has not yet definitively improve outcomes for GBM patients. [93] Moreover, a recent consensus analysis using three recently developed pathogen pipelines (CaPSID, P-DiP, and SEPATH) could not detect CMV in 146 medulloblastoma, 89 pilocytic astrocytoma, 41 glioblastoma, and 18 oligodendroglioma samples. [94]
Advances made in the delivery formal and route of administration has furthered exemplified the outcome efficiency of RNA vaccines in inducing T-cell-mediated responses. As opposed to traditional tumor-associated-antigen encoding mRNAs, certain intratumoral mRNA vaccination approaches aim to activate tumor-resident T-cells in situ using immune stimulatory molecules. One particular study utilizing either naked mRNA or protamine-stabilized mRNA encoding a non-tumor related gene GLB1 demonstrated impair tumor growth upon its intratumoral introduction into a glioblastoma mouse model. [95] Furthermore, a recent 2020 study highlights the flexibility of RNA vaccine design by coupling its application with CAR T-cell therapy. This study adapted the introduction of intravenously administered liposomal antigen-encoding RNA (RNA-LPX) that led to the stimulation of adoptively transferred CAR-T cells, improved CAR T-cell engraftment, and regression of solid tumors at subtherapeutic CAR T-cell doses. [96]
Adoptive Cell Therapy
The functional advantage of adoptive cell therapy (ACT) lies in its ability to manipulate and activate lymphocytes in vitro to release them from the inhibitory factors that exist in the less favorable microenvironment. [97] ACT incorporates the ex vivo culture, cellular engineering, and infusion of lymphocytes to potentiate anti-tumor, anti-viral, or anti-inflammatory effects in patients. The primary forms of ACT can be broadly classified as tumor-infiltrating lymphocytes (TILs), T-cell receptor (TCR) T-cells, and CAR T-cells. Here, we discuss the breakthroughs and challenges that emerged in the ACT field, with a focus on the current state of the field in addressing the complex neuroanatomical and immunosuppressive nature of brain tumors.
Tumor-infiltrating lymphocytes
The difficulty of expanding a sufficient TILs and the exhaustive nature of lymphocytes at the tumor bed limits the efficacy of TILs within brain tumors. TILs require highly immunogenic and accessible tumors for effective expansion, and besides melanoma, no other cancers have allowed sufficient expansion of TILs from their respective tumor samples. [98] Furthermore, while CD8+ TILs have demonstrated exceptional trafficking to tumor targets in extracranial sites, murine studies have demonstrated its inability to replicate its in vitro efficacy against tumors in the brain. [99] These results have been recapitulated in patients treated with infusion of autologous TILs. While disease stabilization and no serious toxicity were demonstrated in both studies, TIL treatment of patients with recurrent malignant gliomas [100] and leptomeningeal disease (LMD) [101] provided inconclusive evidence of durable and prolonged survival. [100] Administration of autologous TILs in a patient with LMD from metastatic melanoma stabilized progression but was insufficient in regressing metastatic deposits in the brain, liver, lung, and peritoneal and retroperitoneal lymph nodes. [101] In an attempt to reactivate exhausted T lymphocytes at the tumor bed, two studies by Plautz et al. involved the infusion of T-cells harvested from patients vaccinated with irradiated autologous tumor cells and expansion ex vivo with anti-CD3 antibody and Staphylococcal enterotoxin A. While both studies demonstrated objective clinical [102][103], neither studies proved prolongation of survival in patients with glioma. These results demonstrate the immunosuppressive challenges of TIL therapy in brain tumors and highlight the need in improving the expansion of brain tumor-derived TILs and the maintenance of autologous TIL activation within the brain tumor microenvironment.
T-cell receptor-transduced T-cells
To redirect T-cell specificity to molecularly defined target antigens, genetically modified antigen-specific TCRs were introduced into T-cells to generate the first TCR-transduced T-cells. More specifically, the cDNAs of the α- and β-chain can be cloned from class I HLA-restricted TCRs of tumor-reactive cytotoxic T-cells and transferred into T-cells to redirect their specificity. Upon recognition of the target peptide bound to MHCs on the surface of antigen-presenting cells or tumor cells, these TCR T-cells can then mount a target-specific cytotoxic response. The first successful demonstration of ACT through genetically engineered lymphocytes utilized autologous T-cells transduced with human TCR recognizing MART-1 to treat patients with metastatic melanoma. [104]
However, off-tumor, on-target toxicity in a following study involving the adoptive transfer of high-avidity TCRs recognizing either MART-1 or gp100 melanoma-melanocyte antigens in 36 patients with metastatic melanoma hinted towards the first significant set of limitations for this therapy. [105] This challenge was further emphasized through two TCR-transduced T-cell therapies targeting the cancer-testis antigen MAGE-A3 separately induced neurological toxicity [106] and cross reactivity within cardiac tissue. [107] Furthermore, in a different study, all three patients with metastatic colorectal cancer treated with high-affinity TCR against the carcinoembryonic antigen experienced life-threatening colitis and colonic hemorrhage. [108] Together, these findings point towards the importance of identifying antigens that are uniquely expressed by tumor cells and not in healthy tissue.
This prevalence of cross reactivity with tumor-associated antigen-targeting TCR-transduced T-cell trials underscores the need to identify neoantigen targets for safe application of therapy – one of which is the H3.3K27M26–35 epitope identified by our group.[72] Isolation of cDNA for the α- and β-chains generated a high affinity TCR capable of recognizing the H3.3 K27M mutation in the context of HLA-A*02:01. T-cells transduced with this TCR demonstrated efficient targeting and cytotoxic behavior against HLA-A*02:01+ H3.3 K27M+ glioma cells in an antigen- and HLA-specific manner in preclinical in vivo models. While no TCR T-cell based therapies for gliomas have been initiated in clinical trials, alanine-scanning assays conducted by our group suggested the absence of any known human proteins that share the key amino acid residues required for TCR recognition of the H3.3K27M26–35 epitope. The potential mitigation of cross reactivity provides crucial evidence that supports the basis for developing T cell-based therapies targeting this neoepitope and highlights the monumental opportunity for ultimately pioneering TCR-transduced T cell therapies for gliomas.
Chimeric antigen receptor T-cells
CAR engineering combines transgene expression of a single-chain variable fragment (scFv) derived from the variable domains of antibodies with the signaling domains of the TCR and additional costimulatory domains to redirect the cytotoxic potential of T-cells against tumor cells that would otherwise be ignored. T-cells transduced with CARs are able to recognize tumor surface antigens directly without the need for MHC expression and costimulation, effectively overcoming the previous limitations of TCR T-cells. In addition to the recognition of surface antigens, the independence of CAR recognition from MHC restriction grants CAR T-cells with a fundamental antitumor advantage of recognizing tumors that have lost MHC-associated antigen presentation as a mechanism of immunoevasion. Conversely, this limits CAR T-cells to antigens that are intrinsically present on the extracellular surface.
Similarly to TCR-transduced T-cells, subsequent studies with CD19-specific, BCMA-specific, and CD22-specific CAR T-cells indicated cross-reactions in the form of cytokine release syndrome (CRS). [109][110]. While B-cell aplasia can be manageable, T-cell mediated toxicity on vital organs have been a major setback in targeting solid tumors. For instance, a patient treated with ERBB2/HER2 CAR T-cell therapy experienced pulmonary failure and CRS caused by off-target killing of ERBB2-positive cells expressed on the lung epithelium [111], and subsequent CAR T-cell trials targeting carbonic anhydrase IX antigen and CEACAM5 similarly resulted in hepatotoxicity and respiratory toxicity. [112][113] As such, the identification of antigens that arise from mutations present in tumor but absent in healthy tissues is an important milestone in solid tumor specificity.
CARs generated for brain tumors have primarily focused on receptors and growth factors presented on the glioma surface. IL-13Rα2 (NCT02208362), HER2 (NCT02442297), EphA2 (NCT02575261), and epidermal growth factor receptor variant III (EGFRvIII) (NCT01454596 and NCT02209376) have garnered predominant attention for generating CARs, but EGFRvIII remains as the only glioma-specific target amongst the other tumor-associated antigens. This variant of EGFR is a truncated receptor expressed in 20% of newly diagnosed glioblastoma patients solely on tumor cells and not healthy tissue. [114][115] Preclinical in vivo studies demonstrated target-specific cytotoxicity by EGFRvIII CAR T-cells [116][117], and in a Phase I clinical trial, all 10 recurrent glioblastoma patients treated with a single intravenous infusion of autologous EGFRvIII CAR T-cells experienced no off-tumor toxicities or cytokine release syndrome. [101] This finding proved the glioma-specific nature of EGFRvIII and its feasibility as a safe therapeutic target, but the trial itself failed to demonstrate objective clinical responses. Significant expansion and successful infiltration of CAR T-cells into the tumor site was observed in all patients, but only one patient of ten presented stable residual disease for over 18 months while no other objective radiographic response was documented. The recurrence of EGFRvIII-negative tumor cells underscores the challenge of antigen heterogeneity within tumors and the need to further identify novel neoantigens for combination CAR T-cell therapy. Despite antigen escape and the lack of CAR-T cell persistence being notable issues, the penetrance of CAR-T cells into the brain garners substantial potential in the approach, and new approaches aimed at countering heterogeneity, the immunosuppressive milieu, and imprecision have already commenced and provided encouraging results. [119–130] (Table 2)
Table 2: Advancements and ongoing approaches investigated in CAR T-cell therapy:
Breakthroughs in cell engineering technologies continue to engender novel CAR T-cell approaches that overcome the immunosuppressive and heterogenic nature of the glioblastoma tumor landscape.
| Further Novel Approaches in CAR T-cell Therapy | ||
|---|---|---|
| Approach | Examples | |
| Multiple Antigen-Targeting CARs | ||
| Split-signal CARs |
|
|
| Tandem CARs |
|
|
| Dual-receptor circuit CARs |
|
|
| Overcoming Tumor Microenvironment Immunosuppression by Modifying Activation Phenotypes | Co-expression of costimulatory molecules | |
| Inhibition of TGFβ |
|
|
| Immune checkpoint inhibitor blockade |
|
|
| Transduction of viral- antigen T-cell with CAR |
|
|
| Circumventing Tumor Escape by Facilitating Targeting of Antigen-loss Tumor Cell Variants | Co-expression of CD40 L |
|
| CAR BiTE cells |
|
|
CLOSING REMARKS
The neuroanatomical barriers, immunosuppressive state of the glioma microenvironment, and the perpetual risk of cross reactivity continues to present challenges for immunotherapy in the brain. The multi-layered complexity of the BBB alongside the array of immunosuppression modalities employed by the glioma microenvironment hinders T-cell infiltration and activation, and lethal on-tumor, off-target toxicity of targeting shared antigens underscores the need for identifying tumor-specific antigens for T-cell based strategies. However, leaps and bounds in the past decade have been made in modulating the tumor microenvironment and designing T-cell based therapies that overcome these challenges. A greater understanding of the tumor archetype has allowed us to identify key components in secreted factor-, cell intrinsic-, and myeloid cell-mediated immunosuppression that can be targeted and blocked. In-depth exploration of the glioma-associated genomic variations engendered the discovery of novel targetable neoantigens for T-cell based therapies. Altogether, the properties of the brain tumor microenvironment propose a daunting set of challenges unique to its own, but through novel pre-clinical strategies, experts have proven time and time again that immunotherapy can overcome these hurdles with just the right push.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- 1.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. (2017) Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 377:1345–1356. 10.1056/NEJMoa1709684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motzer RJ, Tannir NM, McDermott DF, et al. (2018) Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 378:1277–1290. 10.1056/NEJMoa1712126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, et al. (2014) Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med 371:1507–1517. 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Laetsch TW, Buechner J, et al. (2018) Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378:439–448. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medawar PB IMUINITY TO HOMOLOGOUS GRAFTED SKIN. III. THE FATE OF SKIN HOMOGRAFTS TRANSPLNTED TO THE BRAIN, TO SUBECUTANEOUS TISSUE, AN] TO THE ANTERIOR CHAMBER OF THE EYE. 14 [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson P-B, Perry VH, Gordon† S (1992) The acute inflammatory response to lipopolysaccharide in cns parenchyma differs from that in other body tissues. Neuroscience 48:169–186. 10.1016/0306-4522(92)90347-5 [DOI] [PubMed] [Google Scholar]
- 7.Stevenson PG, Hawke S, Sloan DJ, Bangham CRM (1997) The Immunogenicity of Intracerebral Virus Infection Depends on Anatomical Site. J VIROL 71:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russo MV, McGavern DB (2015) Immune Surveillance of the CNS following Infection and Injury. Trends in Immunology 36:637–650. 10.1016/j.it.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mrdjen D, Pavlovic A, Hartmann FJ, et al. (2018) High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48:380–395.e6. 10.1016/j.immuni.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 10.Thion MS, Low D, Silvin A, et al. (2018) Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 172:500–516.e16. 10.1016/j.cell.2017.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuntova P, Downey KM, Hegde B, et al. (2019) Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front Immunol 9:3062 10.3389/fimmu.2018.03062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engelhardt B (2012) Capture, crawl, cross: the T cell code to breach the blood–brain barriers. 33:11. [DOI] [PubMed] [Google Scholar]
- 13.Baron JL (1993) Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. Journal of Experimental Medicine 177:57–68. 10.1084/jem.177.1.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galea I, Bernardes-Silva M, Forse PA, et al. (2007) An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med 204:2023–2030. 10.1084/jem.20070064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran EH, Hoekstra K, van Rooijen N, et al. (1998) Immune Invasion of the Central Nervous System Parenchyma and Experimental Allergic Encephalomyelitis, But Not Leukocyte Extravasation from Blood, Are Prevented in Macrophage-Depleted Mice. J Immunol 161:3767. [PubMed] [Google Scholar]
- 16.Dohgu S, Takata F, Yamauchi A, et al. (2005) Brain pericytes contribute to the induction and upregulation of blood–brain barrier functions through transforming growth factor-β production. Brain Research 1038:208–215. 10.1016/j.brainres.2005.01.027 [DOI] [PubMed] [Google Scholar]
- 17.Wyss-Coray T, Borrow P, Brooker MJ, Mucke L (1997) Astroglial overproduction of TGF-b 1 enhances inflammatory central nervous system disease in transgenic mice. 6 [DOI] [PubMed] [Google Scholar]
- 18.Roszman T, Elliott L, Brooks W (1991) Modulation of T-cell function by gliomas. Immunology Today 12:370–374. 10.1016/0167-5699(91)90068-5 [DOI] [PubMed] [Google Scholar]
- 19.Bloch O, Crane CA, Kaur R, et al. (2013) Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clinical Cancer Research 19:3165–3175. 10.1158/1078-0432.CCR-12-3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chae M, Peterson TE, Balgeman A, et al. (2015) Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro-Oncology 17:978–991. 10.1093/neuonc/nou343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown CE, Alizadeh D, Starr R, et al. (2016) Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375:2561–2569. 10.1056/NEJMoa1610497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schumacher T, Bunse L, Pusch S, et al. (2014) A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512:324–327. 10.1038/nature13387 [DOI] [PubMed] [Google Scholar]
- 23.Okada H, Butterfield LH, Hamilton RL, et al. (2015) Induction of Robust Type-I CD8+ T-cell Responses in WHO Grade 2 Low-Grade Glioma Patients Receiving Peptide-Based Vaccines in Combination with Poly-ICLC. Clinical Cancer Research 21:286–294. 10.1158/1078-0432.CCR-14-1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollack IF, Jakacki RI, Butterfield LH, et al. (2016) Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro Oncol 18:1157–1168. 10.1093/neuonc/now026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Platten M, Reardon D (2018) Concepts for Immunotherapies in Gliomas. Semin Neurol 38:062–072. 10.1055/s-0037-1620274 [DOI] [PubMed] [Google Scholar]
- 26.Jackson CM, Kochel CM, Nirschl CJ, et al. (2016) Systemic Tolerance Mediated by Melanoma Brain Tumors Is Reversible by Radiotherapy and Vaccination. Clinical Cancer Research 22:1161–1172. 10.1158/1078-0432.CCR-15-1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heimberger AB, Sun W, Hussain SF, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: Case study. 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang SS, Bandopadhayay P, Jenkins MR (2019) Towards Immunotherapy for Pediatric Brain Tumors. Trends in Immunology 40:748–761. 10.1016/j.it.2019.05.009 [DOI] [PubMed] [Google Scholar]
- 29.Quail DF, Bowman RL, Akkari L, et al. (2016) The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352:aad3018–aad3018. 10.1126/science.aad3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takenaka MC, Gabriely G, Rothhammer V, et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 22:729–740. 10.1038/s41593-019-0370-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opitz CA, Litzenburger UM, Sahm F, et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203. 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- 33.Wainwright DA, Balyasnikova IV, Chang AL, et al. (2012) IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clinical Cancer Research 18:6110–6121. 10.1158/1078-0432.CCR-12-2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beatty GL, O’Dwyer PJ, Clark J, et al. First-in-Human Phase 1 Study of the Oral Inhibitor of Indoleamine 2,3-dioxygenase-1 Epacadostat (INCB024360) in Patients With Advanced Solid Malignancies. 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichimura K (2012) Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol 29:131–139. 10.1007/s10014-012-0090-4 [DOI] [PubMed] [Google Scholar]
- 36.Kohanbash G, Carrera DA, Shrivastav S, et al. (2017) Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 127:1425–1437. 10.1172/JCI90644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berghoff AS, Kiesel B, Widhalm G, et al. (2017) Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 19:1460–1468. 10.1093/neuonc/nox054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei J, Barr J, Kong L-Y, et al. Glioblastoma Cancer-Initiating Cells Inhibit T-Cell Proliferation and Effector Responses by the Signal Transducers and Activators of Transcription 3 Pathway. Molecular Cancer Therapeutics 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hussain SF, Kong L-Y, Jordan J, et al. (2007) A Novel Small Molecule Inhibitor of Signal Transducers and Activators of Transcription 3 Reverses Immune Tolerance in Malignant Glioma Patients. Cancer Res 8 [DOI] [PubMed] [Google Scholar]
- 40.Facoetti A, Nano R, Zelini P, et al. (2005) Human Leukocyte Antigen and Antigen Processing Machinery Component Defects in Astrocytic Tumors. Clin Cancer Res 9 [DOI] [PubMed] [Google Scholar]
- 41.Yeung JT, Hamilton RL, Ohnishi K, et al. LOH in the HLA Class I Region at 6p21 Is Associated with Shorter Survival in Newly Diagnosed Adult Glioblastoma. Clinical Cancer Research 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The quantity of naturally processed peptides stably bound by HLA-A*0201 is significantly reduced in the absence of tapasin. Tissue Antigens 6 [DOI] [PubMed] [Google Scholar]
- 43.Thuring C HLA class I is most tightly linked to levels of tapasin compared with other antigen-processing proteins in glioblastoma. BRITISH JOURNAL OF CANCER 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chongsathidkiet P, Jackson C, Koyama S, et al. (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24:1459–1468. 10.1038/s41591-018-0135-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quail DF, Joyce JA (2017) The Microenvironmental Landscape of Brain Tumors. Cancer Cell 31:326–341. 10.1016/j.ccell.2017.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, Feng X, Herting CJ, et al. (2017) Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res 77:2266–2278. 10.1158/0008-5472.CAN-16-2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18:225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- 48.Massara M, Persico P, Bonavita O, et al. (2017) Neutrophils in Gliomas. Front Immunol 8:1349 10.3389/fimmu.2017.01349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nitta T, Sato K, Allegretta M, et al. (1992) Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Research 571:19–25. 10.1016/0006-8993(92)90505-4 [DOI] [PubMed] [Google Scholar]
- 50.Wiencke JK, Koestler DC, Salas LA, et al. (2017) Immunomethylomic approach to explore the blood neutrophil lymphocyte ratio (NLR) in glioma survival. Clin Epigenet 9:10 10.1186/s13148-017-0316-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang J, Piao Y, Holmes L, et al. (2014) Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clinical Cancer Research 20:187–198. 10.1158/1078-0432.CCR-13-1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Achyut BR, Shankar A, Iskander ASM, et al. (2015) Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Letters 369:416–426. 10.1016/j.canlet.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broz ML, Binnewies M, Boldajipour B, et al. (2014) Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26:638–652. 10.1016/j.ccell.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eagles M, Nassiri F, Badhiwala J, et al. (2018) Dendritic cell vaccines for high-grade gliomas. TCRM Volume 14:1299–1313. 10.2147/TCRM.S135865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alban TJ, Alvarado AG, Sorensen MD, et al. (2018) Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 3:e122264 10.1172/jci.insight.122264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kohanbash G, McKaveney K, Sakaki M, et al. (2013) GM-CSF Promotes the Immunosuppressive Activity of Glioma-Infiltrating Myeloid Cells through Interleukin-4 Receptor-. Cancer Research 73:6413–6423. 10.1158/0008-5472.CAN-12-4124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujita M, Kohanbash G, Fellows-Mayle W, et al. (2011) COX-2 Blockade Suppresses Gliomagenesis by Inhibiting Myeloid-Derived Suppressor Cells. Cancer Research 71:2664–2674. 10.1158/0008-5472.CAN-10-3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohkuri T, Ghosh A, Kosaka A, et al. (2014) STING Contributes to Antiglioma Immunity via Triggering Type I IFN Signals in the Tumor Microenvironment. Cancer Immunology Research 2:1199–1208. 10.1158/2326-6066.CIR-14-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arbour KC, Mezquita L, Long N, et al. (2018) Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients With Non–Small-Cell Lung Cancer. JCO 36:2872–2878. 10.1200/JCO.2018.79.0006 [DOI] [PubMed] [Google Scholar]
- 60.Ajlan A, Thomas P, Albakr A, et al. (2017) Optimizing bevacizumab dosing in glioblastoma: less is more. J Neurooncol 135:99–105. 10.1007/s11060-017-2553-2 [DOI] [PubMed] [Google Scholar]
- 61.Reardon DA, Gokhale PC, Klein SR, et al. (2016) Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunology Research 4:124–135. 10.1158/2326-6066.CIR-15-0151 [DOI] [PubMed] [Google Scholar]
- 62.Coley WB (1910) The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med 3:1–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hodges TR, Ott M, Xiu J, et al. (2017) Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-Oncology 19:1047–1057. 10.1093/neuonc/nox026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weller M, Kaulich K, Hentschel B, et al. (2014) Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy: EGFRvIII mutation and prognosis of glioblastoma. Int J Cancer 134:2437–2447. 10.1002/ijc.28576 [DOI] [PubMed] [Google Scholar]
- 65.Heimberger AB, Crotty LE, Friedman AH, et al. (2002) Dendritic Cells Pulsed with a Tumor-specific Peptide Induce Long-lasting Immunity and Are Effective against Murine Intracerebral Melanoma. 50:9. [DOI] [PubMed] [Google Scholar]
- 66.Schuster J, Lai RK, Recht LD, et al. (2015) A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology 17:854–861. 10.1093/neuonc/nou348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weller M, Butowski N, Tran DD, et al. (2017) Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. The Lancet Oncology 18:1373–1385. 10.1016/S1470-2045(17)30517-X [DOI] [PubMed] [Google Scholar]
- 68.van den Bent MJ, Gao Y, Kerkhof M, et al. (2015) Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology 17:935–941. 10.1093/neuonc/nov013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Felsberg J, Hentschel B, Kaulich K, et al. (2017) Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR -Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin Cancer Res 23:6846–6855. 10.1158/1078-0432.CCR-17-0890 [DOI] [PubMed] [Google Scholar]
- 70.Reardon DA, Schuster J, Tran DD, et al. (2015) ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. JCO 33:2009–2009. 10.1200/jco.2015.33.15_suppl.2009 [DOI] [Google Scholar]
- 71.Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, et al. (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. 10.1007/s00401-012-0998-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chheda ZS, Kohanbash G, Okada K, et al. (2018) Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med 215:141–157. 10.1084/jem.20171046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keskin DB, Anandappa AJ, Sun J, et al. (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565:234–239. 10.1038/s41586-018-0792-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hilf N, Kuttruff-Coqui S, Frenzel K, et al. (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565:240–245. 10.1038/s41586-018-0810-y [DOI] [PubMed] [Google Scholar]
- 75.Becker J, Craig EA (1994) Heat-shock proteins as molecular chaperones. Eur J Biochem 219:11–23. 10.1111/j.1432-1033.1994.tb19910.x [DOI] [PubMed] [Google Scholar]
- 76.Blachere NE, Li Z, Chandawarkar RY, et al. (1997) Heat Shock Protein–Peptide Complexes, Reconstituted In Vitro, Elicit Peptide-specific Cytotoxic T Lymphocyte Response and Tumor Immunity. The Journal of Experimental Medicine 186:1315–1322. 10.1084/jem.186.8.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shevtsov M, Multhoff G (2016) Heat Shock Protein–Peptide and HSP-Based Immunotherapies for the Treatment of Cancer. Front Immunol 7:. 10.3389/fimmu.2016.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hermisson M, Strik H, Rieger J, et al. (2000) Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology 54:1357 10.1212/WNL.54.6.1357 [DOI] [PubMed] [Google Scholar]
- 79.Graner MW, Cumming RI, Bigner DD (2007) The Heat Shock Response and Chaperones/Heat Shock Proteins in Brain Tumors: Surface Expression, Release, and Possible Immune Consequences. Journal of Neuroscience 27:11214–11227. 10.1523/JNEUROSCI.3588-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beaman GM, Dennison SR, Chatfield LK, Phoenix DA (2014) Reliability of HSP70 (HSPA) expression as a prognostic marker in glioma. Mol Cell Biochem 393:301–307. 10.1007/s11010-014-2074-7 [DOI] [PubMed] [Google Scholar]
- 81.Graner MW, Bigner DD (2005) Chaperone proteins and brain tumors: Potential targets and possibletherapeutics. Neuro-Oncology 7:260–278. 10.1215/S1152851704001188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shevtsov M, Kim A, Samochernych A, et al. (2014) Pilot study of intratumoral injection of recombinant heat shock protein 70 in the treatment of malignant brain tumors in children. OTT 1071 10.2147/OTT.S62764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shevtsov MA, Komarova EY, Meshalkina DA, et al. (2014) Exogenously delivered heat shock protein 70 displaces its endogenous analogue and sensitizes cancer cells to lymphocytes-mediated cytotoxicity. Oncotarget 5:. 10.18632/oncotarget.1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shevtsov MA, Nikolaev BP, Yakovleva LY, et al. (2015) 70-kDa heat shock protein coated magnetic nanocarriers as a nanovaccine for induction of anti-tumor immune response in experimental glioma. Journal of Controlled Release 220:329–340. 10.1016/j.jconrel.2015.10.051 [DOI] [PubMed] [Google Scholar]
- 85.Crane CA, Han SJ, Ahn B, et al. (2013) Individual Patient-Specific Immunity against High-Grade Glioma after Vaccination with Autologous Tumor Derived Peptides Bound to the 96 KD Chaperone Protein. Clinical Cancer Research 19:205–214. 10.1158/1078-0432.CCR-11-3358 [DOI] [PubMed] [Google Scholar]
- 86.Bloch O, Crane CA, Fuks Y, et al. (2014) Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology 16:274–279. 10.1093/neuonc/not203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ulmer JB, Mason PW, Geall A, Mandl CW (2012) RNA-based vaccines. Vaccine 30:4414–4418. 10.1016/j.vaccine.2012.04.060 [DOI] [PubMed] [Google Scholar]
- 88.Karikó K, Muramatsu H, Welsh FA, et al. (2008) Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Molecular Therapy 16:1833–1840. 10.1038/mt.2008.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thess A, Grund S, Mui BL, et al. (2015) Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Molecular Therapy 23:1456–1464. 10.1038/mt.2015.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pardi N, Hogan MJ, Porter FW, Weissman D (2018) mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov 17:261–279. 10.1038/nrd.2017.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Conry RM, LoBuglio AF, Wright M, et al. Characterization of a Messenger RNA Polynucleotide Vaccine Vector’. 5 [PubMed] [Google Scholar]
- 92.Boczkowski D, Nair SK, Snyder D, Gilboa E (1996) Dendritic Cells Pulsed with RNA are Potent Antigen-presenting Cells In Vitro and In Vivo [DOI] [PMC free article] [PubMed]
- 93.Rahman M, Dastmalchi F, Karachi A, Mitchell D (2019) The role of CMV in glioblastoma and implications for immunotherapeutic strategies. OncoImmunology 8:e1514921 10.1080/2162402X.2018.1514921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zapatka M, Borozan I, Brewer DS, et al. (2018) The landscape of viral associations in human cancers. Cancer Biology [Google Scholar]
- 95.Scheel B, Aulwurm S, Probst J, et al. (2006) Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol 36:2807–2816. 10.1002/eji.200635910 [DOI] [PubMed] [Google Scholar]
- 96.Reinhard K, Rengstl B, Oehm P, et al. (2020) An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 367:446–453. 10.1126/science.aay5967 [DOI] [PubMed] [Google Scholar]
- 97.Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68. 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balch CM (1990) Patterns of Human Tumor-Infiltrating Lymphocytes in 120 Human Cancers. Arch Surg 125:200 10.1001/archsurg.1990.01410140078012 [DOI] [PubMed] [Google Scholar]
- 99.Saris SC, Spiess P, Lieberman DM, et al. (1992) Treatment of murine primary brain tumors with systemic interleukin-2 and tumor-infiltrating lymphocytes. Journal of Neurosurgery 76:513–519. 10.3171/jns.1992.76.3.0513 [DOI] [PubMed] [Google Scholar]
- 100.Quattrocchi KB, Miller CH, Cush S, et al. Pilot Study of Local Autologous Tumor Infiltrating Lymphocytes for the Treatment of Recurrent Malignant Gliomas. 17 [DOI] [PubMed] [Google Scholar]
- 101.Glitza IC, Haymaker C, Bernatchez C, et al. (2015) Intrathecal Administration of Tumor-Infiltrating Lymphocytes Is Well Tolerated in a Patient with Leptomeningeal Disease from Metastatic Melanoma: A Case Report. Cancer Immunology Research 3:1201–1206. 10.1158/2326-6066.CIR-15-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Plautz GE, Barnett GH, Miller DW, et al. (1998) Systemic T cell adoptive immunotherapy of malignant gliomas. Journal of Neurosurgery 89:42–51. 10.3171/jns.1998.89.1.0042 [DOI] [PubMed] [Google Scholar]
- 103.Plautz GE, Miller DW, Barnett GH, et al. T Cell Adoptive Immunotherapy of Newly Diagnosed Gliomas. 11 [PubMed] [Google Scholar]
- 104.Morgan RA, Dudley ME, Wunderlich JR, et al. (2006) Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. 314:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnson LA, Morgan RA, Dudley ME, et al. (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–546. 10.1182/blood-2009-03-211714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morgan RA, Chinnasamy N, Abate-Daga D, et al. (2013) Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy: Journal of Immunotherapy 36:133–151. 10.1097/CJI.0b013e3182829903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cameron BJ, Gerry AB, Dukes J, et al. (2013) Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Science Translational Medicine 5:197ra103–197ra103. 10.1126/scitranslmed.3006034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parkhurst MR, Yang JC, Langan RC, et al. (2011) T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Molecular Therapy 19:620–626. 10.1038/mt.2010.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fry TJ, Stetler-Stevenson M, Shah NN, et al. (2015) Clinical Activity and Persistence of Anti-CD22 Chimeric Antigen Receptor in Children and Young Adults with Relapsed/Refractory Acute Lymphoblastic Leukemia (ALL). Blood 126:1324–1324. 10.1182/blood.V126.23.1324.132426153519 [DOI] [Google Scholar]
- 110.Ali SA, Shi V, Maric I, et al. (2016) T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 128:1688–1700. 10.1182/blood-2016-04-711903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morgan RA, Yang JC, Kitano M, et al. (2010) Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Molecular Therapy 18:843–851. 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lamers CH, Sleijfer S, van Steenbergen S, et al. (2013) Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Molecular Therapy 21:904–912. 10.1038/mt.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thistlethwaite FC, Gilham DE, Guest RD, et al. (2017) The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 66:1425–1436. 10.1007/s00262-017-2034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal Antibodies against EGFRvIII Are Tumor Specific and React with Breast and Lung Carcinomas and Malignant (¿liornas. 10 [PubMed] [Google Scholar]
- 115.Gupta P, Han S-Y, Holgado-Madruga M, et al. (2010) Development of an EGFRvIII specific recombinant antibody. BMC Biotechnol 10:72 10.1186/1472-6750-10-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ohno M, Ohkuri T, Kosaka A, et al. (2013) Expression of miR-17–92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer 1:21 10.1186/2051-1426-1-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Johnson LA, Scholler J, Ohkuri T, et al. (2015) Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 7:275ra22–275ra22. 10.1126/scitranslmed.aaa4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Rourke DM, Nasrallah MP, Desai A, et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9:eaaa0984 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hegde M, Corder A, Chow KK, et al. (2013) Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma. Molecular Therapy 21:2087–2101. 10.1038/mt.2013.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bielamowicz K, Fousek K, Byrd TT, et al. (2018) Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 20:506–518. 10.1093/neuonc/nox182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kloss CC, Condomines M, Cartellieri M, et al. (2013) Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31:71–75. 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grada Z, Hegde M, Byrd T, et al. (2013) TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Molecular Therapy - Nucleic Acids 2:e105 10.1038/mtna.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roybal KT, Rupp LJ, Morsut L, et al. (2016) Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164:770–779. 10.1016/j.cell.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang L, Kerkar SP, Yu Z, et al. (2011) Improving Adoptive T Cell Therapy by Targeting and Controlling IL-12 Expression to the Tumor Environment. Molecular Therapy 19:751–759. 10.1038/mt.2010.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Krenciute G, Prinzing BL, Yi Z, et al. (2017) Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 5:571–581. 10.1158/2326-6066.CIR-16-0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Foster AE, Heslop HE (2008) Antitumor Activity of EBV-specific T Lymphocytes Transduced With a Dominant Negative TGF-b Receptor. J Immunother 31:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sengupta S, Katz SC, Sengupta S, Sampath P (2018) Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Letters 433:131–139. 10.1016/j.canlet.2018.06.035 [DOI] [PubMed] [Google Scholar]
- 128.Cherkassky L, Morello A, Villena-Vargas J, et al. (2016) Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. Journal of Clinical Investigation 126:3130–3144. 10.1172/JCI83092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kuhn NF, Purdon TJ, van Leeuwen DG, et al. (2019) CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 35:473–488.e6. 10.1016/j.ccell.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lorrey SJ, Sanchez-Perez L, Fecci PE (2019) Rescuing imperfect antigens for immuno-oncology. Nat Biotechnol 37:1002–1003. 10.1038/s41587-019-0248-2 [DOI] [PubMed] [Google Scholar]