

Abstract
Elucidation of natural selection signatures and relationships with phenotype spectra is important to understand adaptive evolution of modern humans. Here, we conducted a genome-wide scan of selection signatures of the Japanese population by estimating locus-specific time to the most recent common ancestor using the ascertained sequentially Markovian coalescent (ASMC), from the biobank-based large-scale genome-wide association study data of 170,882 subjects. We identified 29 genetic loci with selection signatures satisfying the genome-wide significance. The signatures were most evident at the alcohol dehydrogenase (ADH) gene cluster locus at 4q23 (PASMC = 2.2 × 10−36), followed by relatively strong selection at the FAM96A (15q22), MYOF (10q23), 13q21, GRIA2 (4q32), and ASAP2 (2p25) loci (PASMC < 1.0 × 10−10). The additional analysis interrogating extended haplotypes (integrated haplotype score) showed robust concordance of the detected signatures, contributing to fine-mapping of the genes, and provided allelic directional insights into selection pressure (e.g., positive selection for ADH1B-Arg48His and HLA-DPB1*04:01). The phenome-wide selection enrichment analysis with the trait-associated variants identified a variety of the modern human phenotypes involved in the adaptation of Japanese. We observed population-specific evidence of enrichment with the alcohol-related phenotypes, anthropometric and biochemical clinical measurements, and immune-related diseases, differently from the findings in Europeans using the UK Biobank resource. Our study demonstrated population-specific features of the selection signatures in Japanese, highlighting a value of the natural selection study using the nation-wide biobank-scale genome and phenotype data.
Keywords: biobank analysis, natural selection signature, genome-wide selection search, phenome-wide approach
Introduction
Throughout long history of humans, demography and surrounding environment have left footprints in genomic sequences of the individuals in a population scale (Sabeti et al. 2006). Elucidation of such sequence-encoded information provides us a clue to understand global evolutionary signatures of modern human populations, which is often linked to population-specific genetic risk of human complex traits (Sabeti et al. 2006). Recent efforts to construct large-scale genome data have developed population-representative catalogs of genetic variants and their allele frequency spectra from diverse ancestries (Walter et al. 2015; Lek et al. 2016; Liu et al. 2018). Interpretation of such big data by applying sophisticated methods of statistical and population genetics has successfully revealed genomic loci under extensive natural selection signatures, as well as implication of the key phenotypes that might have been involved in evolution (e.g., skin pigmentation at SLC24A5 in Africans [Crawford et al. 2017], adaptation to agriculture at FADS1 in Europeans [Mathieson S and Mathieson I 2018], and hair morphology at EDAR in east Asians [Wu et al. 2016]).
The methods to explore selective sweeps embedded in the human genome sequences have been originally developed to examine diversity in derived allele frequency spectra among ancestries (Tajima’s D [Tajima 1989] and F-statistics [FST; Weir and Cockerham 1984]). Then, a variety of frameworks have been introduced 1) to enhance statistical power by interrogating extended haplotypes (integrated haplotype score [iHS; Voight et al. 2006; Johnson and Voight 2018] and cross-population extended haplotype homozygosity [Sabeti et al. 2007]), 2) to fine-map the variants responsible for the selective sweeps (composite of multiple signals [Grossman et al. 2010]), 3) to assign functional annotations to selection signatures (site frequency spectra [Moon and Akey 2016]), and 4) to expand the time phases corresponding to the selection from older ages to very recent ages (singleton density score [SDS; Field et al. 2016]). One unsettled point was, however, the efficient methodology to utilize the biobank-scale large genome data. Recently, the national biobank projects have constructed high-resolution genetic and phenotypic data of hundreds of thousands of participants, which could capture comprehensive population-specific spectra of the variants with phenome-wide association lists (Bycroft et al. 2018; Kanai et al. 2018; Hirata et al. 2019). However, the previous methodologies to examine natural selection signatures were mostly developed to handle genome data with relatively smaller sample sizes, and sometimes not applicable to such big data due to massive computational burdens (Szpiech and Hernandez 2014). Thus, a novel framework to powerfully conduct a genome-wide scan of the selection sweeps by fully utilizing biobank-scale large genome data has been warranted.
Recently, Palamara et al. developed a novel method named the ascertained sequentially Markovian coalescent (ASMC), which estimate the locus-specific coalescence times (time to most recent common ancestor [TMRCA]) for pairs of two homologous chromosomes in a genome-wide manner utilizing hidden Markov models (Palamara et al. 2018). Compared with the previous methods utilizing the whole-genome sequencing (WGS) data (Li and Durbin 2011; Terhorst et al. 2017), ASMC can estimate the coalescence times based only on the single nucleotide polymorphism (SNP) microarray data with the achievement of orders of magnitude faster computing, which is scalable for analyzing hundreds of thousands of individuals. Application of ASMC to the biobank-scale genome data such as UK Biobank has successfully detected 12 genetic loci with extensively high density of recent coalescence times as a signature of recent positive selection in Europeans (Palamara et al. 2018). ASMC could also provide insights into temporal aspects of the detected evolutionary events. Considering that worldwide populations have separately experienced demographic and natural selection history (Liu et al. 2017; Nakayama et al. 2017; Okada et al. 2018), its application to additional non-European populations should be warranted.
In this study, we report natural selection signatures in the Japanese population by applying ASMC to the large-scale genome-wide association study (GWAS) data (n > 170,000). In parallel, we calculated iHS, another measure to detect selective sweeps. Further, we quantitatively assessed overlap of the observed natural selection signatures with the risk variants of the modern human disease phenotypes to elucidate underlying impacts of evolution in Japanese.
Results
Japanese GWAS Data with >170,000 Unrelated Individuals
In this study, we utilized the large-scale GWAS data of the Japanese individuals enrolled in the BioBank Japan (BBJ) project, a nation-wide hospital-based cohort of the Japanese population (Hirata et al. 2017). As described previously (Kanai et al. 2018; Hirata et al. 2019), we obtained the GWAS data genotyped with the high-density SNP microarrays and applied stringent quality control (QC) filters, which yielded genotype data of 485,296 autosomal SNPs with minor allele frequency ≥0.01 for the unrelated 170,882 Japanese individuals. We then conducted phasing of the GWAS genotype data to obtain genome-wide haplotype data. As previously reported (Takeuchi et al. 2017; Okada et al. 2018), the principal component analysis (PCA) plot indicated that the Japanese population consisted of two subclusters (i.e., “Hondo” and “Ryukyu-Ainu” clusters), whereas we included all the individuals in this study (supplementary fig. 1, Supplementary Material online).
ASMC Detected Abundant Natural Selection Signatures in Japanese
By applying the ASMC software, we estimated locus-specific TMRCA in a genome-wide manner for the Japanese GWAS data. As a parameter, we utilized the population-specific demographic model (Terhorst et al. 2017) estimated from the previously constructed deep WGS data of the Japanese population (n = 1,276; Okada et al. 2018). We computed a statistic DRCT, which reflects the density of recent coalescence within the past T generations for each window bin with 0.05 cM. We focused on DRC150, which is sensitive to detect signals of positive selection signatures approximately within the past 20,000 years (Palamara et al. 2018). By fitting the genome-wide DRC150 values into a gamma distribution, we obtained the P values representing the significance of the natural selection signatures (=PASMC) for the 71,320 bins (a quantile–quantile plot for supplementary fig. 2a, Supplementary Material online, and a Manhattan plot for fig. 1a).
Fig. 1.

Genome-wide natural selection signatures of the Japanese population. Manhattan plots of the genome-wide natural selection signatures obtained from the GWAS data of 170,882 Japanese individuals. The y-axis indicates the –log10(P) of a genome-wide selection signatures calculated by using (a) ASMC for each bin with 0.05cM (=PASMC) and (b) iHS for each SNP (=PiHS), respectively. The horizontal gray line represents the genome-wide significance threshold based on Bonferroni correction of the numbers of the evaluated bins or SNP.
As a result, we observed the 29 genetic loci which satisfied the genome-wide significance threshold considering multiple comparisons of the number of the tested bins (PASMC < 0.05/71,320 = 7.0 × 10−7; table 1), conspicuously expanding the genome-wide catalog of selective sweeps in Japanese. The most significant natural selection signature was observed at the alcohol dehydrogenase (ADH) gene cluster locus at 4q23 (PASMC = 2.2 × 10−36, 99.528–100.979 Mb at chr 4), which is known as one of the loci under strongest selection pressure in east Asian populations (Galinsky et al. 2016; Koganebuchi et al. 2017; Okada et al. 2018). Within the ADH cluster, the strongest selection pressure was observed at the bin of 100.137–100.244 Mb, which included ADH1A and ADH1B (fig. 2). The FAM96A locus at 15q22, the MYOF locus at 10q23 and 13q21, the GRIA2 locus at 4q32, and the ASAP2 locus at 2p25 demonstrated relatively strong selection signals (PASMC < 1.0 × 10−10; supplementary fig. 3, Supplementary Material online). The loci previously reported to be under selection pressure in the Japanese population (i.e., EDAR, the ADH cluster, the major histocompatibility complex [MHC] region, and ALDH2 [Fujimoto et al. 2008; Koganebuchi et al. 2017; Okada 2018; Okada et al. 2018]) also showed significant selection signatures in our results independently obtained from ASMC. Regarding the loci under selection reported in the Han Chinese population and other east Asian populations (Liu et al. 2013, 2018; Chiang et al. 2018), we found overlap of the signature at PSMB2, SLC44A5, and FADS2 (PASMC = 2.0 × 10−8, 1.3 × 10−8, and 0.0030), as well as the MHC region and ADH cluster. The DOCK9 locus at 13q32 under selection in Chinese was not replicated (PASMC = 0.27), whereas we observed selection signature in DOCK8 at 9p24 which belongs to the same gene family as DOCK9 in Japanese (PASMC = 6.4 × 10−10). The immune-related loci reported in the Chinese and other Asian populations (e.g., CR1 at 1q32, the IGH cluster at 14q32, and LILRA3 at 19q13) were not replicated (PASMC > 0.05 for the bins including these genes; Hirayasu et al. 2008; Chiang et al. 2018; Liu et al. 2018). The loci under selection specifically in Europeans (i.e., LCT and TLR; Field et al. 2016; Palamara et al. 2018) did not show enrichment of selection signals in our results (PASMC > 0.05 for the bins including these genes). These results suggested a population-specific feature of the natural selection signatures in the Japanese population.
Table 1.
Genetic Loci with Significant Natural Selection Signatures in the Japanese Population Detected by ASMC.
| Chr | Position (Mb) | Cytoband | P ASMC | Gene(s) |
|---|---|---|---|---|
| 1 | 35.635–36.412 | 1p34 | 2.0 × 10−8 | PSMB2 |
| 1 | 75.623–76.469 | 1p31 | 1.3 × 10−8 | SLC44A5 |
| 1 | 193.833–193.994 | 1q31 | 4.1 × 10−7 | CDC73 |
| 2 | 8.983–9.798 | 2p25 | 9.7 × 10−11 | ASAP2 |
| 2 | 17.139–17.791 | 2p24 | 2.0 × 10−8 | RAD51AP2 |
| 2 | 38.239–38.623 | 2p22 | 8.8 × 10−10 | CYP1B1 |
| 2 | 108.430–109.524 | 2q12 | 6.0 × 10−10 | EDAR |
| 2 | 125.760–126.395 | 2q14 | 3.5 × 10−7 | CNTNAP5 |
| 2 | 158.772–158.880 | 2q24 | 4.3 × 10−7 | UPP2 |
| 2 | 211.547–211.903 | 2q34 | 5.1 × 10−9 | CPS1 |
| 2 | 213.033–213.295 | 2q34 | 4.2 × 10−8 | ERBB4 |
| 3 | 134.389–134.622 | 3q22 | 6.3 × 10−8 | EPHB1 |
| 4 | 28.250–28.870 | 4p15 | 5.6 × 10−9 | MIR4275 |
| 4 | 99.528–100.979 | 4q23 | 2.2 × 10−36 | The ADH cluster |
| 4 | 122.149–122.362 | 4q27 | 2.5 × 10−7 | QRFPR |
| 4 | 158.116–159.173 | 4q32 | 4.1 × 10−11 | GRIA2 |
| 6 | 18.713–19.444 | 6p22 | 1.1 × 10−9 | MIR548A1 |
| 6 | 29.736–30.075 | 6p21 | 7.2 × 10−8 | The MHC region |
| 7 | 112.095–112.500 | 7q31 | 9.5 × 10−8 | LSMEM1 |
| 8 | 139.643–139.676 | 8q24 | 4.9 × 10−7 | COL22A1 |
| 9 | 0.204–0.399 | 9p24 | 6.4 × 10−10 | DOCK8 |
| 10 | 94.357–95.174 | 10q23 | 6.2 × 10−13 | MYOF |
| 11 | 24.896–25.467 | 11p14 | 3.6 × 10−8 | LUZP2 |
| 12 | 1.207–1.549 | 12p13 | 1.9 × 10−7 | ERC1 |
| 12 | 111.746–113.238 | 12q24 | 7.0 × 10−8 | ALDH2 |
| 13 | 63.401–64.618 | 13q21 | 1.1 × 10−11 | — |
| 13 | 74.493–74.862 | 13q22 | 2.2 × 10−8 | KLF12 |
| 15 | 63.733–65.208 | 15q22 | 5.9 × 10−13 | FAM96A |
| 20 | 30.578–31.091 | 20q11 | 2.5 × 10−7 | CCM2L |
Note.—Genetic loci with genome-wide significant natural selection signatures are shown (PASMC < 0.05/71,320 bins = 7.0 × 10−7). A gene nearest to the top bin of each region is indicated.
Fig. 2.

Regional plots of the natural selection signatures at the ADH cluster locus. Regional plots of the significant natural selection signatures observed at the ADH cluster locus at 4q23. Regional enrichment of (a) DRCT for recent coalescence events, (b) ASMC for each bin with 0.05 cM (=PASMC, upper), iHS for each SNP (=PiHS, middle), and SDS for each SNP (=PSDS, bottom), along with (c) the gene positions. The y-axes in (b) indicate the –log10(P) of genome-wide selection signatures. The horizontal gray lines represent the genome-wide significance thresholds.
To validate the results, we conducted a replication study using two independent Japanese cohorts; the Nagahama cohort (n = 1,591; see URLs) and the Japan Biological Informatics Consortium (JBIC; n = 1,209; Hirata, Hirota, et al. 2018; Hirata et al. 2019). Among the 29 loci with genome-wide significance in BBJ, 26 loci satisfied the nominal significance (PASMC < 0.05) in both of the replication cohorts (supplementary table 1, Supplementary Material online). When we confined the BBJ individuals to those included in the main Hondo cluster (n = 160,994), the genome-wide DRC150 estimates indicated high concordance with those from all the BBJ individuals (r = 0.9995; supplementary fig. 4, Supplementary Material online). These results empirically demonstrated the robustness of our genome-wide ASMC natural selection signature in Japanese.
iHS Analysis Revealed Selection Signatures at ADH and MHC in Japanese
As a comparative approach, we applied iHS to our Japanese GWAS data, one of the classical but robust methods designed to examine allelic discrepancy in extended haplotype lengths. iHS focuses on the selection signatures within the past 20,000–30,000 years (Voight et al. 2006; Johnson and Voight 2018), of which the time phase is similar to that of ASMC. Although parallel calculation of genome-wide haplotype lengths requires massive computing resources, introduction of multithreading computation enabled its application to a large-scale GWAS data representing the populations (Szpiech and Hernandez 2014). By standardizing the genome-wide iHS z-scores, we obtained the P values representing the significance of the selection signatures (=PiHS) for the 475,072 SNPs (a quantile–quantile plot for supplementary fig. 2b, Supplementary Material online, and a Manhattan plot for fig. 2b). The iHS analysis provided the two loci of the ADH cluster (PiHS = 7.3 × 10−8 at rs1442493; described in detail later) and the MHC region (PiHS = 9.0 × 10−16 at rs6930052), which satisfied the genome-wide significance threshold, considering multiple comparison of the number of the tested SNPs (PASMC < 0.05/475,072 = 1.1 × 10−7; table 2). The lead SNP allele within the MHC region rs6930052-T with iHS z-score = 8.04 was in tight linkage disequilibrium (LD) with the HLA-DPB1*04:01 allele (r2 = 0.88; Hirata et al. 2019), on which strong recent positive selection acted locally within the Japanese archipelago (Kawashima et al. 2012). In addition to concordance of the strongly selected genetic loci among these two independent algorithms, SNPs included in the loci detected by ASMC showed enrichment of iHS selection signals. In total, 4.58-fold increase of the mean iHS χ2 values was observed when compared with the genome-wide estimates. When we assessed each locus separately, 27 of the 29 loci demonstrated increased mean iHS χ2 values (>1.00; supplementary table 1, Supplementary Material online).
Table 2.
Genetic Loci with Significant Natural Selection Signatures in the Japanese Population Detected by iHS.
| rsID | Chr | Position | Cytoband | Ancestral/Derived Allele | Freq.a | iHS z-Scorea | P iHS | Region |
|---|---|---|---|---|---|---|---|---|
| rs1442493 | 4 | 100321365 | 4q23 | G/A | 0.799 | 5.38 | 7.3 × 10−8 | The ADH clusters |
| rs6930052 | 6 | 32990481 | 6p21 | T/A | 0.952 | −8.04 | 9.0 × 10−16 | The MHC region |
Note.—Genetic loci with genome-wide significant natural selection signatures are shown (PiHS < 0.05/475,072 SNP = 1.1 × 10−7).
Corresponding to the derived allele.
In the ADH cluster locus, we also observed nominally significant positive selection pressure at the well-known functional missense variant of ADH1B, which is associated with lower alcohol consumption, as well (rs1229984-A [Arg48His], PiHS = 7.1 × 10−4 with iHS z-score = 3.39, r2 = 0.34 with rs1442493 in Japanese; fig. 2b; Koganebuchi et al. 2017; Okada 2018). The previous Japanese SDS study demonstrated that this locus was under very recent selection pressure (PSDS = 7.1 × 10−4 at rs1229984; fig. 2b; obtained from Okada et al. [2018]). These three methods reflect differential time scales of natural selection (approximately the past around 3,000, 20,000, and 30,000 years for SDS, ASMC by DRC150, and iHS, respectively). When we observed time-series shifts of the DRC values among the 29 loci detected by ASMC (fig. 3), the ADH cluster region has been the locus under the strongest selection pressure in the past 300 generations (i.e., ∼10,000 years). We note that the SLC44A5 locus at 1p31 (Liu et al. 2013) was under the strongest selection at the generation before the selection on ADH was dominant. These results suggest that the ADH cluster region was under longitudinal selection pressure in the evolutional history of Japanese. To explore biological function of the region, we assessed tissue-specific expression profiles of the ADH1B gene obtained from the GTEx database (see URLs). High expression profiles of ADH1B were observed in liver, as well as adipose and breast, suggesting relation with alcohol metabolism (supplementary fig. 5, Supplementary Material online).
Fig. 3.
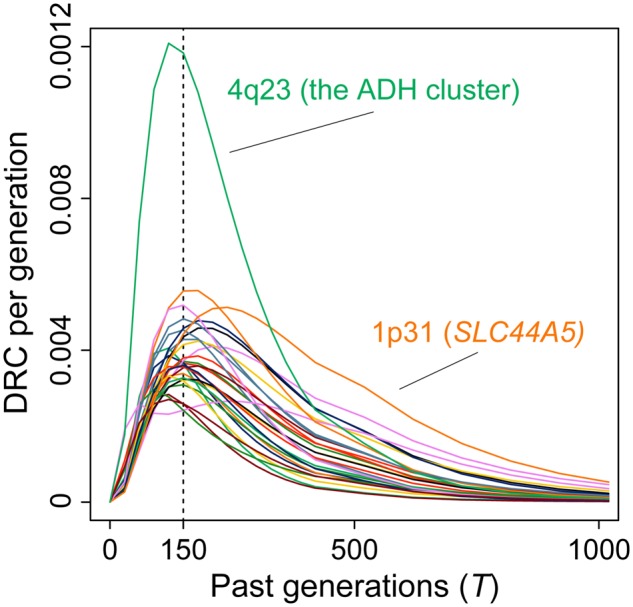
Time-series shifts of the DRC values among the loci with natural selection signature. Time-series shifts of the DRC values (per generation) among the 29 loci with significant natural selection signature in the Japanese population detected by ASMC. The x-axis indicates the past generations, and the y-axis indicates the DRC values.
Phenome-Wide Enrichment of the Trait-Associated Variants with Selection Signatures
Genetic variants associated with the human phenotypes have been targets of natural selection pressure through evolutional history of each population. Although it would be difficult to directly observe the past phenotypic events related to adaptation, one can indirectly estimate the phenotypes that drove selection by examining enrichment of the GWAS-identified trait-associated variants with selection signatures (Field et al. 2016; Okada et al. 2018; Palamara et al. 2018). Thus, we conducted a phenome-wide enrichment analysis of the ASMC and iHS selection signatures on the lead variants that were associated with human complex traits in Japanese satisfying the genome-wide significance threshold (P < 5.0 × 10−8; Kanai et al. 2016). We curated the 2,190 Japanese trait-associated variants of the 105 phenotypes which consist of 35 diseases and 70 quantitative measurements classified into 14 categories (anthropometric [n = 2], behavior [n = 4], blood pressure [n = 4], echocardiographic [n = 4], electrolyte [n = 5], hematological [n = 13], kidney function [n = 4], liver function [n = 6], metabolic [n = 6], other biochemical [n = 7], pharmacogenetics [n = 2], protein [n = 5], reproductive aging [n = 4], and skin pigmentation [n = 4]).
Abundant selection signatures detected by ASMC enabled the phenome-wide analysis to identify in total 37 traits, of which the associated variants showed significant enrichments with selection, vastly expanding the findings from the previous efforts (P < 0.05/105 = 4.8 × 10−4; fig. 4a and supplementary table 2, Supplementary Material online; Okada et al. 2018). The strongest phenotypic enrichments in selection were observed for drinking-related behavior (P = 2.9 × 10−41 for alcohol drinking dose and P = 2.0 × 10−34 for alcohol drinking history) and for esophageal cancer supposed as a consequence of drinking alcohol (P = 2.9 × 10−41; Abnet et al. 2018). These results were mostly driven by strong selection signatures at the functional missense SNPs involved in alcohol metabolism (Arg47His at ADH1B and Glu504Lys at ALDH2), which are specifically observed in east Asian populations due to population-specific positive selection pressure (Okada 2018). These two loci explain 7.4% and 1.6% of the phenotype variances of alcohol drinking dose and esophageal cancer in east Asians, respectively, and selection enrichment on these traits became nonsignificant when these two loci were removed. Further, enrichments in reproductive aging (P = 2.4 × 10−21 for menopause), anthropometric traits (P = 3.4 × 10−21 for body mass index and P = 5.6 × 10−8 for height), kidney function (P = 3.8 × 10−13 for serum creatinine, P = 7.9 × 10−12 for estimated glomerular filtration rate, P = 9.8 × 10−6 for uric acid, and P = 2.5 × 10−4 for blood urea nitrogen), and other hematological and biochemical measurements such as lipids, electrolyte, and protein, were observed. As novel findings beyond the previous studies in Japanese (Okada et al. 2018), we newly identified selection enrichments in immune-related diseases (P = 6.3 × 10−6 for adult asthma, P = 7.9 × 10−6 for psoriasis, P = 2.0 × 10−5 for Stevens–Johnson syndrome for cold medicine, P = 2.3 × 10−5 for ulcerative colitis, P = 3.7 × 10−5 for rheumatoid arthritis, P = 2.4 × 10−4 for Behcet’s disease, and P = 2.8 × 10−4 for systemic lupus erythematosus). In contrast, no enrichment was observed in skin pigmentation (P > 0.13), for which selection pressure was often observed in Europeans and Africans (Crawford et al. 2017; Palamara et al. 2018). The DRC150 statistic computed using ASMC does not provide allelic direction of the selection pressure but is expected to capture the signature of positive selection rather than negative selection. Our phenome-wide selection enrichment scan, however, cannot be interpreted as providing a directional effect of selection on phenotypic values.
Fig. 4.

Overlap between ASMC natural selection signatures and genetic risk of modern human phenotypes. Enrichment of ASMC natural selection signatures of in the GWAS-identified trait-associated risk variants in (a) the Japanese population and (b) the European population (UK Biobank). For each trait, inflation of the selection ASMC DRC150 is indicated along with the x-axis, and –log10(P) of enrichment is plotted along with the y-axis. The horizontal gray lines represent significance thresholds based of Bonferroni correction on the numbers of the evaluated traits.
We then conducted the comparative phenome-wide enrichment analysis in the European population using the UK Biobank resource. We integrated the previously reported genome-wide ASMC natural selection signature (n = 113,851; Palamara et al. 2018) and the phenome-wide GWAS lead SNPs of the UK Biobank GWAS (Canela-Xandri et al. 2018). We observed significant enrichment in 222 of the 639 traits (P < 0.05/639 = 7.8 × 10−5; fig. 4b and supplementary table 3, Supplementary Material online). The strongest phenotypic enrichment was observed in dietary habits (P = 7.9 × 10−53 for bread intake and P = 2.3 × 10−33 for cereal intake). Enrichment in anthropometric traits (P = 1.8 × 10−44 and 3.0 × 10−43 for left and right hand grip strength, respectively, P = 8.4 × 10−42 for walking pace, P = 1.7 × 10−31 for trunk fat, P = 3.9 × 10−30 for height at age 10, and P = 1.1 × 10−29 for height), immune-related diseases (P = 1.4 × 10−44 for connective tissue disorder, P = 3.0 × 10−41 for spondylopathies, and P = 1.2 × 10−37 for psoriasis), and hematological traits (P = 9.4 × 10−31 for red blood cell and P = 1.2 × 10−30 for monocyte) was also observed. Interestingly, traits belonging to the same category of the dietary habits (i.e., drinking for Japanese and bread for Europeans) demonstrated the strongest enrichment in both populations, suggesting the prominent roles of dietary habits in natural selection pressure in a population-specific way.
We also assessed overlap between the phenotype-associated variants and iHS selection signatures in Japanese and identified six traits with significant enrichment (P < 4.8 × 10−4; supplementary fig. 6 and table 4, Supplementary Material online). The most significant enrichment was observed for aspartate aminotransferase (P = 3.5 × 10−9). Enrichment for drinking-related behavior (P = 8.1 × 10−4 for alcohol drinking dose and P = 0.0026 for alcohol drinking history), esophageal cancer (P = 8.1 × 10−4), and immune-related diseases (P = 1.4 × 10−6 for ulcerative colitis and P = 4.5 × 10−6 for Takayasu’s arteritis) was also observed. We considered that, as ASMC detected more loci with significant natural selection, enrichment was observed in a larger number of the complex human traits in ASMC than iHS. Further, the differences in the methodology to screen selection signature and target timescale could explain discrepancy of the phenotypic spectra between these two methods.
Alcohol and Nutrition Metabolisms-Related Pathways Implicated in Selection
To have further insights into biological backgrounds in evolution of Japanese, we conducted functional interpretation of the genes within the loci under significant selection signatures (PASMC < 7.0 × 10−7; n = 135). We thus performed a molecular pathway analysis. We observed implication of the pathways related to alcohol and nutrition metabolisms, such as ethanol oxidation (Padjusted = 8.6 × 10−13 by Reactome), ADH activity (Padjusted = 1.2 × 10−10 by Gene Ontology molecular function), fatty acid degradation (Padjusted = 1.3 × 10−9 by Kyoto Encyclopedia of Genes and Genomes and Padjusted = 7.7 × 10−10 by WikiPathways), and noradrenaline and adrenaline degradation (Padjusted = 5.7 × 10−6 by HumanCyc; supplementary table 5, Supplementary Material online). These results again highlight that 1) phenome-wide spectrum driven by natural selection pressure could be differently characterized in each population and 2) the Japanese population have experienced adaptation processes represented by alcohol and nutrition metabolisms, which were different from those observed in European or African ancestry.
Discussion
In this study, we conducted a genome-wide scan of natural selection signatures in the Japanese population using the large-scale GWAS data with >170,000 subjects, which is one of the largest efforts to date in non-European ancestry. Estimation of locus-specific TMRCA from the SNP microarray data using ASMC demonstrated enough statistical power to detect 29 loci with significant selection signatures, which conspicuously expanded the previous findings. Additional analysis using iHS showed robustness of the analytic results, contributing to the fine-mapping of the genes responsible for selection drive, and provided allelic directional insights (e.g., positive selection for ADH1B-Arg48His and HLA-DPB1*04:01). Our study clearly highlighted a value of natural selection study using the nation-wide biobank-scale genome data supported by application of multiple analytical methods.
The phenome-wide enrichment analysis with the selection signatures demonstrated that a variety of categories of the modern human phenotypes have been involved in the adaptation of the Japanese population. Comparative analysis with the European population demonstrated that the identified phenotype spectra were population specific, which was highlighted by the striking enrichment for alcohol and nutrition-related phenotypes in Japanese. Our scan also identified involvement of anthropometric and biochemical clinical measurements, as well as immune-related diseases.
Adaptation of the Japanese (or neighboring east Asian) population with the alcohol-related phenotypes has been mostly observed for the two genetic loci of ADH1B and ALDH2 (Koganebuchi et al. 2017; Okada 2018; Okada et al. 2018). These two loci showed relatively similar magnitude of selection pressure in the past 3,000 years assessed with rare variant distributions obtained by WGS data of Japanese (Okada et al. 2018). In this study, we revealed that the ADH1B region has been under longitudinal selection pressure throughout the evolutional history of Japanese, and in the older ages (e.g., the past 20,000–30,000 years), selection pressure was much evident at ADH1B rather than ALDH2. Natural selection pressure on ADH1B was also observed in worldwide populations, which was not the case for ALDH2 (Galinsky et al. 2016; Johnson and Voight 2018). These results suggest the history of selection pressure, initially preceded by ADH1B and recently followed by ALDH2, which should provide novel insights into long-standing discussions on the physiological and environmental origin of these genes to drive evolutions (Oota et al. 2004). On the other hand, their phenotypic origin on selection pressure in Japanese remains elusive. In addition to alcohol-related phenotypes, associations of these loci with human behaviors have been recently reported (e.g., risk-taking behaviors and assortative mating at ADH1B rs1229984; Howe et al. 2019; Linner et al. 2019). Causal effects of genetically determined alcohol drinking behaviors on disease risk and mortality are also known (Kiiskinen et al. 2020; Millwood et al. 2019). Recent large-scale studies in Japanese reported contribution of the ADH1B and ALDH2 functional variants on dietary consumptions of wider ranges of traditional foods and beverages, as well as all-cause mortality rates (Matoba et al. 2020; Sakaue et al. 2020). Further follow-up studies utilizing the cohorts with deep phenotypes are warranted.
As potential limitations of our study, the P values for ASMC (and iHS) were estimated approximately according to the expected null distribution. This concern has been mitigated in our work by adopting a conservative Bonferroni significance threshold, and by our use of an empirical distribution of test statistics to fit null model parameters, which is conservative due to overdispersion that is likely caused by the underlying presence of undetected loci undergoing recent positive selection. Further theoretical approaches to more robustly estimate empirical distribution of statistics, such as mixture of gamma distributions corresponding to the both null and alternative hypotheses, should be warranted. Since the BBJ cohort consisted of the disease-affected individuals, we would note that its potential effect on selection screening might be different from that in healthy cohort. Accuracy of haplotype phasing, and its heterogeneity among genome-wide regions, could also affect the selection scan results. Next, there exist concerns on overlap between selection and trait-associated genetics in the light of potential artifacts due to population stratification, especially in the field of polygenic analysis (Novembre and Barton 2018). For example, polygenic selection signals observed for the height GWAS meta-analysis are now considered to be partly driven by residual population structure (Berg et al. 2019; Sohail et al. 2019). As our phenome-wide selection enrichment analysis has only considered conclusively trait-associated (i.e., genome-wide significant) lead SNPs, rather than all genome-wide variants considering polygenicity, we believe the risk for such a confounder to be minimal in our results.
In conclusion, our study to utilize large-scale biobank-driven genome data of the Japanese population identified abundant selection signatures and their involvements in modern phenotypes. The findings highlighted population-specific and time phase-dependent features of the selection signatures, which would warrant further studies incorporating additional ancestries followed by trans-ethnic comparisons.
Materials and Methods
Characteristics of the Subjects
We enrolled a total of 170,882 individuals of Japanese ancestry for genome-wide natural selection screening. Of these, 169,994 individuals were obtained from BBJ, and 888 were obtained from Epstein-Barr virus transformed B-lymphoblast cell lines of Japanese individuals established by the Japan Pharma SNP Consortium. The BBJ subjects were affected with any of the 45 diseases (Hirata et al. 2017), and the Pharma SNP Consortium subjects were healthy controls. In the replication study, we enrolled 1,591 Japanese healthy participants from the Nagahama cohorts (see URLs) and 1,209 Japanese participants enrolled from JBIC (Hirata, Hirota, et al. 2018; Hirata et al. 2019). Subjects who were identified as non-Japanese origin either by self-reporting or by PCA were excluded, as described elsewhere (Kanai et al. 2018; Hirata et al. 2019). All the participants provided written informed consent as approved by the ethical committee of the institutes. This study was approved by the ethical committee of Osaka University Graduate School of Medicine.
Characteristics of the GWAS Data
We obtained BBJ GWAS genotype data of the unrelated Japanese subjects (n = 170,882). Details of the data processing were described elsewhere (Kanai et al. 2018; Hirata et al. 2019). Briefly, the subjects were genotyped using Illumina HumanOmniExpressExome BeadChip or in combination of Illumina HumanOmniExpress and HumanExome BeadChips. The population structure of the BBJ participants depicted by PCA is indicated in supplementary figure 1, Supplementary Material online. The genotype data were followed by the stringent QC filters, which yielded dense coverage of the genome-wide autosomal chromosome SNP with minor allele frequency ≥0.01 (n = 485,296). The individuals in the Nagahama cohort and JBIC were genotyped using Illumina Human610-Quad and HumanCoreExome-12v1, respectively and processed in the same manner as the BBJ cohort (431,648 and 257,803 autosomal SNP after QC, respectively).
Genome-Wide Selection Signature Scan Using ASMC
We estimated locus-specific TMRCA in a genome-wide manner by applying ASMC to the GWAS genotype data, as described elsewhere (Palamara et al. 2018). We estimated the population-specific demographic model by applying SMC++ (version 1.8.0) to the previously constructed deep WGS data of the Japanese population (n = 1,276; Okada et al. 2018). The GWAS genotype data were phased separately for each short or long autosomal chromosome arm as haplotype data using Eagle (version 2.3), and then split into 50 batches (3,417 or 3,418 samples per batch). For each phased batch, we applied ASMC with options --majorMinorPosteriorSums, --mode array and generation time intervals ranging from 30 to 2,000 and demographic model made by SMC++. Then, we merged all batches using MergePosteriorSums.jar which was included in ASMC. The output was normalized so that the posterior sums to 1 for each site and average values were calculated for each bin of 0.05 cm. Finally, DRC150 was calculated. Since genome-wide DRCT statistics are known to follow a gamma distribution, we fitted the single null gamma distribution of the DRCT statistics in our data set based on maximum likelihood estimate by conservatively excluding those in the previously known variants with significant selection signatures in the Japanese or Asian populations (±5 Mb of the MHC region or ALDH2, or ±500 kb of the other detected loci; Hirayasu et al. 2008; Liu et al. 2017, 2018; Chiang et al. 2018; Okada et al. 2018). We obtained one-tailed P values of the genome-wide bins according to the DRC150 values and the fitted gamma distribution (=PASMC). We set the significance threshold by considering the Boferroni correction based on the number of the tested bins (α = 0.05).
Genome-Wide Selection Signature Scan Using iHS
Using the phased BBJ GWAS haplotype data, we calculated genome-wide natural selection signatures based on iHS (Voight et al. 2006; Johnson and Voight 2018) separately for each short or long chromosome arm, using the multithread computing option of selscan (--threads, version 1.1.0b; Szpiech and Hernandez 2014). The genome-wide iHS z-scores were standardized through normalization within each derived allele frequency bin (bin widths = 0.01). The variants in the previously known loci with selection signatures in Japanese or Asians were also excluded in the process of normalization fitting (Hirayasu et al. 2008; Liu et al. 2017, 2018; Chiang et al. 2018; Okada et al. 2018). We estimated two-tailed P values of the SNP according to the normalized z-scores (=PiHS). We set the significance threshold by considering the Boferroni correction based on the number of the assessed SNP (α = 0.05). Summary statistics of the SDS selection signals in Japanese were obtained from the previous study (Okada et al. 2018).
GTEx Transcriptomic Data Analysis
Tissue-specific expression profiles of the ADH1B gene was obtained from the GTEx Portal database (see URLs).
Phenome-Wide Selection Signature Analysis Using the Trait-Associated SNP
We assessed enrichment of the natural selection signatures in the Japanese population within the variants associated with human complex traits in a phenome-wide manner. We collected a list of the independent sets of the variants identified by the GWAS conducted for the Japanese population that satisfied the genome-wide significance threshold of P < 5.0 × 10−8 (Kanai et al. 2016). In addition to curation of the public and internal databases that archive the trait-associated variants (e.g., the GWAS Catalog database; see URLs), we conducted manual curation of the literature to obtain the variant list (Okada et al. 2014; Matsuo et al. 2016; Akiyama et al. 2017; Hirata, Hirota, et al. 2018; Hirata, Koga, et al. 2018; Kanai et al. 2018; Sakaue et al. 2018; Suzuki et al. 2019; Masuda et al. 2020; Matoba et al. 2020). The references in which the variant lists were originally obtained were listed as supplementary table 6, Supplementary Material online. In the phenome-wide screening, we did not include the traits for which only a single risk variant has been reported.
Regarding enrichment analysis of the selection signatures by ASMC, we obtained the sum of the DRC150 values of the bins where the trait-associated variants were included for each of the traits. We then estimated enrichment P values based on reproducing property of a gamma distribution (i.e., the sum of the k DRC150 values also follows a gamma distribution with a shape parameter of k). The enrichment analysis in the UK Biobank resource was conducted by integrating the previously reported genome-wide ASMC DRC150 values (Palamara et al. 2018) and the phenome-wide GWAS lead SNPs of the UK Biobank GWAS downloaded from the GeneAtlas database (Canela-Xandri et al. 2018; see URLs).
Regarding enrichment analysis of the selection signatures by iHS, we obtained the sum of the squared values of the normalized iHS z-scores of the variants (or the proxy variants in LD when available; r2 > 0.5 in the Japanese WGS data; Okada et al. 2018), which was compared with the χ2 distribution with the degree of freedom equal to the number of the variants. Statistical analyses were done by using R statistical software (version 3.4.3) and python (version 3.6.6) with the scipy library (version 1.1.0).
Biological Annotation of the Genes within the Loci with Selection Signatures
For each locus with the genome-wide significant selection signatures detected by ASMC, we defined the boundary of the locus by collapsing the neighboring bins with significant signatures. We conducted the biological pathway analysis using Enrichr (Kuleshov et al. 2016). Among the 22 pathways and Gene Ontology classifications implemented in Enrichr, those indicating the most significant associations that satisfied the significance threshold considering multiple testing were highlighted (Bonferroni correction with α = 0.05). As suggested previously (Sakaue et al. 2018), we did not include the genes in the MHC region for the pathway analysis, considering complex LD structure of the variants and pivotal functional roles of the human leukocyte antigen (HLA) genes (Okada et al. 2015). Due to the small number of the loci outside the MHC region with significant signatures, we did not conduct the pathway analysis based on the iHS result.
URLs
The URLs for data presented herein are as follows:
ASMC, http://www.palamaralab.org/software/ASMC; last accessed October 31, 2019.
The Nagahama cohort GWAS data, https://humandbs.biosciencedbc.jp/hum0012-v1; last accessed October 31, 2019.
GTEx Portal, https://gtexportal.org/home/; last accessed October 31, 2019.
GWAS Catalog, https://www.ebi.ac.uk/gwas/; last accessed October 31, 2019.
GeneAtlas, http://geneatlas.roslin.ed.ac.uk; last accessed October 31, 2019.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This research was supported by the Tailor-Made Medical Treatment program (the BioBank Japan Project) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), the Japan Agency for Medical Research and Development (AMED), Bioinformatics Initiative of Osaka University Graduate School of Medicine, and Osaka University Center of Medical Data Science, Advanced Clinical Epidemiology Investigator’s Research Project. Y.Y. was supported by the Osaka University Medical Doctor Scientist Training Program. Y.O. was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (15H05911 and 19H01021) and AMED (JP19gm6010001, JP19ek0410041, JP19ek0109413, and JP19km0405211).
References
- Abnet CC, Arnold M, Wei WQ.. 2018. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology 154(2):360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, et al. 2017. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet. 49(10):1458–1467. [DOI] [PubMed] [Google Scholar]
- Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, Field Y, Boyle EA, Zhang X, Racimo F, Pritchard JK, et al. 2019. Reduced signal for polygenic adaptation of height in UK Biobank. Elife 8:e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela-Xandri O, Rawlik K, Tenesa A.. 2018. An atlas of genetic associations in UK Biobank. Nat Genet. 50(11):1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CWK, Mangul S, Robles C, Sankararaman S.. 2018. A comprehensive map of genetic variation in the world’s largest ethnic group—Han Chinese. Mol Biol Evol. 35(11):2736–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford NG, Kelly DE, Hansen MEB, Beltrame MH, Fan S, Bowman SL, Jewett E, Ranciaro A, Thompson S, Lo Y, et al. 2017. Loci associated with skin pigmentation identified in African populations. Science 358(6365):eaan8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field Y, Boyle EA, Telis N, Gao Z, Gaulton KJ, Golan D, Yengo L, Rocheleau G, Froguel P, McCarthy MI, et al. 2016. Detection of human adaptation during the past 2000 years. Science 354(6313):760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Kimura R, Ohashi J, Omi K, Yuliwulandari R, Batubara L, Mustofa MS, Samakkarn U, Settheetham-Ishida W, Ishida T, et al. 2008. A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum Mol Genet. 17(6):835–843. [DOI] [PubMed] [Google Scholar]
- Galinsky KJ, Bhatia G, Loh P-R, Georgiev S, Mukherjee S, Patterson NJ, Price AL.. 2016. Fast principal-component analysis reveals convergent evolution of ADH1B in Europe and East Asia. Am J Hum Genet. 98(3):456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Shylakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, et al. 2010. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327(5967):883–886. [DOI] [PubMed] [Google Scholar]
- Hirata J, Hirota T, Ozeki T, Kanai M, Sudo T, Tanaka T, Hizawa N, Nakagawa H, Sato S, Mushiroda T, et al. 2018. Variants at HLA-A, HLA-C, and HLA-DQB1 confer risk of psoriasis vulgaris in Japanese. J Invest Dermatol. 138(3):542–548. [DOI] [PubMed] [Google Scholar]
- Hirata J, Hosomichi K, Sakaue S, Kanai M, Nakaoka H, Ishigaki K, Suzuki K, Akiyama M, Kishikawa T, Ogawa K, et al. 2019. Genetic and phenotypic landscape of the major histocompatibility complex region in the Japanese population. Nat Genet. 51(3):470–480. [DOI] [PubMed] [Google Scholar]
- Hirata M, Kamatani Y, Nagai A, Kiyohara Y, Ninomiya T, Tamakoshi A, Yamagata Z, Kubo M, Muto K, Mushiroda T, et al. 2017. Cross-sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J Epidemiol. 27(3S):S9–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata T, Koga K, Johnson TA, Morino R, Nakazono K, Kamitsuji S, Akita M, Kawajiri M, Kami A, Hoshi Y, et al. 2018. Japanese GWAS identifies variants for bust-size, dysmenorrhea, and menstrual fever that are eQTLs for relevant protein-coding or long non-coding RNAs. Sci Rep. 8(1):8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayasu K, Ohashi J, Tanaka H, Kashiwase K, Ogawa A, Takanashi M, Satake M, Jia GJ, Chimge N-O, Sideltseva EW, et al. 2008. Evidence for natural selection on leukocyte immunoglobulin-like receptors for HLA class I in Northeast Asians. Am J Hum Genet. 82(5):1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe LJ, et al. 2019. Genetic evidence for assortative mating on alcohol consumption in the UK Biobank. Nat Commun 10(1):5039. [DOI] [PMC free article] [PubMed]
- Johnson KE, Voight BF.. 2018. Patterns of shared signatures of recent positive selection across human populations. Nat Ecol Evol. 2(4):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, et al. 2018. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 50(3):390–400. [DOI] [PubMed] [Google Scholar]
- Kanai M, Tanaka T, Okada Y.. 2016. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J Hum Genet. 61(10):861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima M, Ohashi J, Nishida N, Tokunaga K.. 2012. Evolutionary analysis of classical HLA class I and II genes suggests that recent positive selection acted on DPB1*04:01 in Japanese population. PLoS One 7(10):e46806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiiskinen T, et al. 2020. Genomic prediction of alcohol-related morbidity and mortality. Transl Psychiatry. 10:23. [DOI] [PMC free article] [PubMed]
- Koganebuchi K, Haneji K, Toma T, Joh K, Soejima H, Fujimoto K, Ishida H, Ogawa M, Hanihara T, Harada S, et al. 2017. The allele frequency of ALDH2*Glu504Lys and ADH1B*Arg47His for the Ryukyu islanders and their history of expansion among East Asians. Am J Hum Biol. 29(2):e22933. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44(W1):W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2011. Inference of human population history from individual whole-genome sequences. Nature 475(7357):493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linner RK, et al. 2019. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 51:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Huang S, Chen F, Zhao L, Yuan Y, Francis SS, Fang L, Li Z, Lin L, Liu R, et al. 2018. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history. Cell 175(2):347–359. [DOI] [PubMed] [Google Scholar]
- Liu X, Lu D, Saw W-Y, Shaw PJ, Wangkumhang P, Ngamphiw C, Fucharoen S, Lert-itthiporn W, Chin-inmanu K, Chau TNB, et al. 2017. Characterising private and shared signatures of positive selection in 37 Asian populations. Eur J Hum Genet. 25(4):499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ong RT-H, Pillai EN, Elzein AM, Small KS, Clark TG, Kwiatkowski DP, Teo Y-Y.. 2013. Detecting and characterizing genomic signatures of positive selection in global populations. Am J Hum Genet. 92(6):866–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Low S-K, Akiyama M, Hirata M, Ueda Y, Matsuda K, Kimura T, Murakami Y, Kubo M, Kamatani Y, et al. 2020. Cross-trait genetic analysis of five gynecologic diseases in Japanese. Eur J Hum Genet. 28(1):95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson S, Mathieson I.. 2018. FADS1 and the timing of human adaptation to agriculture. Mol Biol Evol. 35(12):2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba N, et al. Forthcoming 2020. GWAS of 165,084 Japanese individuals identified nine loci associated with dietary habits. Nat Hum Behav. doi:10.1038/s41562-019-0805-1. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Yamamoto K, Nakaoka H, Nakayama A, Sakiyama M, Chiba T, Takahashi A, Nakamura T, Nakashima H, Takada Y, et al. 2016. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum Dis. 75(4):652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millwood IY, Walters RG, Mei XW, Guo Y, Yang L, Bian Z, Bennett DA, Chen Y, Dong C, Hu R, et al. 2019. Conventional and genetic evidence on alcohol and vascular disease aetiology: a prospective study of 500 000 men and women in China. Lancet 393(10183):1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S, Akey JM.. 2016. A flexible method for estimating the fraction of fitness influencing mutations from large sequencing data sets. Genome Res. 26(6):834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Ohashi J, Watanabe K, Munkhtulga L, Iwamoto S.. 2017. Evidence for very recent positive selection in Mongolians. Mol Biol Evol. 34(8):1936–1946. [DOI] [PubMed] [Google Scholar]
- Novembre J, Barton NH.. 2018. Tread lightly interpreting polygenic tests of selection. Genetics 208(4):1351–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. 2018. eLD: entropy-based linkage disequilibrium index between multiallelic sites. Hum Genome Var. 5(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Momozawa Y, Ashikawa K, Kanai M, Matsuda K, Kamatani Y, Takahashi A, Kubo M.. 2015. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat Genet. 47(7):798–802. [DOI] [PubMed] [Google Scholar]
- Okada Y, Momozawa Y, Sakaue S, Kanai M, Ishigaki K, Akiyama M, Kishikawa T, Arai Y, Sasaki T, Kosaki K, et al. 2018. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat Commun. 9(1):1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. 2014. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506(7488):376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oota H, Pakstis AJ, Bonne-Tamir B, Goldman D, Grigorenko E, Kajuna SLB, Karoma NJ, Kungulilo S, Lu R-B, Odunsi K, et al. 2004. The evolution and population genetics of the ALDH2 locus: random genetic drift, selection, and low levels of recombination. Ann Human Genet. 68(2):93–109. [DOI] [PubMed] [Google Scholar]
- Palamara PF, Terhorst J, Song YS, Price AL.. 2018. High-throughput inference of pairwise coalescence times identifies signals of selection and enriched disease heritability. Nat Genet. 50(9):1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES.. 2006. Positive natural selection in the human lineage. Science 312(5780):1614–1620. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature 449(7164):913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue S, Hirata J, Maeda Y, Kawakami E, Nii T, Kishikawa T, Ishigaki K, Terao C, Suzuki K, Akiyama M, et al. 2018. Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res. 46(22):11898–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue S, et al. Forthcoming 2020. Functional variants in ADH1B and ALDH2 are non-additively associated with all-cause mortality in Japanese population. Eur J Hum Genet. doi:10.1038/s41431-019-0518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, Chiang CW, Hirschhorn J, Daly MJ, Patterson N, et al. 2019. Signals of polygenic adaptation on height have been overestimated due to uncorrected population structure in genome-wide association studies. ELife 8:e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Akiyama M, Ishigaki K, Kanai M, Hosoe J, Shojima N, Hozawa A, Kadota A, Kuriki K, Naito M, et al. 2019. Identification of 28 novel susceptibility loci for type 2 diabetes in the Japanese population. Nat Genet. 51(3):379–386. [DOI] [PubMed] [Google Scholar]
- Szpiech ZA, Hernandez RD.. 2014. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 31(10):2824–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi F, Katsuya T, Kimura R, Nabika T, Isomura M, Ohkubo T, Tabara Y, Yamamoto K, Yokota M, Liu X, et al. 2017. The fine-scale genetic structure and evolution of the Japanese population. PLoS One 12(11):e0185487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst J, Kamm JA, Song YS.. 2017. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet. 49(2):303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK.. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4(3):e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, Lawson D, et al. 2015. The UK10K project identifies rare variants in health and disease. Nature 526(7571):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC.. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- Wu S, Tan J, Yang Y, Peng Q, Zhang M, Li J, Lu D, Liu Y, Lou H, Feng Q, et al. 2016. Genome-wide scans reveal variants at EDAR predominantly affecting hair straightness in Han Chinese and Uyghur populations. Hum Genet. 135(11):1279–1286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.