

NALCN is the voltage-sensing, pore-forming subunit of a constitutively active, extracellular calcium-regulated complex.
Abstract
The sodium leak channel (NALCN) is essential for survival in mammals: NALCN mutations are life-threatening in humans and knockout is lethal in mice. However, the basic functional and pharmacological properties of NALCN have remained elusive. Here, we found that robust function of NALCN in heterologous systems requires co-expression of UNC79, UNC80, and FAM155A. The resulting NALCN channel complex is constitutively active and conducts monovalent cations but is blocked by physiological concentrations of extracellular divalent cations. Our data support the notion that NALCN is directly responsible for the increased excitability observed in a variety of neurons in reduced extracellular Ca2+. Despite the smaller number of voltage-sensing residues in NALCN, the constitutive activity is modulated by voltage, suggesting that voltage-sensing domains can give rise to a broader range of gating phenotypes than previously anticipated. Our work points toward formerly unknown contributions of NALCN to neuronal excitability and opens avenues for pharmacological targeting.
INTRODUCTION
Many neurons display a basal Na+ conductance at rest that is involved in the regulation of resting membrane potential (RMP), spontaneous firing, and pacemaking activity (1–3). The sodium leak channel (NALCN) contributes to this tonic current in certain types of neurons and plays a critical role in their excitability (4–9). There are now multiple lines of evidence in support of the physiological significance of NALCN. First, NALCN knockout is lethal within a day after birth in mice owing to disrupted respiratory rhythm (4). Second, changes in NALCN expression and/or function have been implicated in other physiological processes such as motor function, pain sensitivity, and circadian rhythm in animals (6, 10–14). Last, accumulating evidence shows that mutations in NALCN cause severe congenital neurodevelopmental disorders in humans (15, 16).
NALCN represents the sole member of a distinct branch of the four-domain ion channel family, which includes the extensively studied voltage-gated sodium and calcium channels (NaVs and CaVs) (17). These channels are made up of four homologous domains (DI to DIV), each with six transmembrane segments (S1 to S6). Functionally, the voltage-sensing domains (VSDs), formed by S1 to S4 of each domain, detect changes in membrane potential and induce the opening or closing of the ion-conducting pore domain, formed by S5 and S6. Unlike NaVs and CaVs, however, the fundamental functional and pharmacological properties of NALCN remain largely unexplored because of poor heterologous expression (18–21). In this study, we found that the robust functional expression of NALCN requires UNC79, UNC80, and FAM155A, three ubiquitous neuronal proteins that have been independently shown to interact with NALCN (5, 11, 12, 22). This enabled the first in-depth functional characterization of the NALCN channel complex and unequivocally defines NALCN as permeable to small monovalent cations, but potently blocked by divalent cations through a direct pore-blocking mechanism. Furthermore, we show that NALCN does not form a simple ohmic leak channel but displays voltage-dependent gating mediated primarily by positive charges in the S4 of DI and DII.
RESULTS
Co-expression of NALCN, UNC79, UNC80, and FAM155A results in robust currents
Whether NALCN alone can generate leak currents in heterologous expression systems has remained a matter of debate (4, 18–21, 23). Here, we found that the expression of NALCN alone did not result in detectable responses to a wide range of voltage steps in human embryonic kidney (HEK)–293T cells or Xenopus laevis oocytes (Fig. 1A and fig. S1). We thus set out to test whether functional expression of NALCN, like that of some of the CaVs (24), is dependent on the presence of additional auxiliary proteins. We found that functional expression of NALCN in both heterologous systems requires UNC79, UNC80, and FAM155A (Fig. 1A and fig. S1). Furthermore, rat NALCN (4), human FAM155B, and mouse FAM155A (12) orthologs can functionally substitute human NALCN and FAM155A, respectively (fig. S1B). We also tested the effect of co-expressing FAM155A and FAM155B with NALCN, UNC79, and UNC80 but did not find any apparent functional differences (fig. S1B). UNC79 and UNC80 are large, potentially disordered proteins (2635 and 3258 amino acids, respectively) that are highly conserved among animals (25, 26). They contain no recognizable functional domains, and their subcellular localizations remain unclear. By contrast, FAM155A (458 amino acids) contains at least one transmembrane segment and a conserved cysteine-rich domain (CRD) (12, 27). The Caenorhabditis elegans and mouse orthologs have previously been suggested to reside in the endoplasmic reticulum (ER) and facilitate NALCN trafficking. Here, however, we were able to co-immunoprecipitate UNC79, UNC80, and FAM155A with NALCN (fig. S2B). Therefore, we refer to the NALCN-UNC79-UNC80-FAM155A combination as the NALCN channel complex henceforth.
Fig. 1. Functional expression of NALCN requires UNC79, UNC80, and FAM155A.
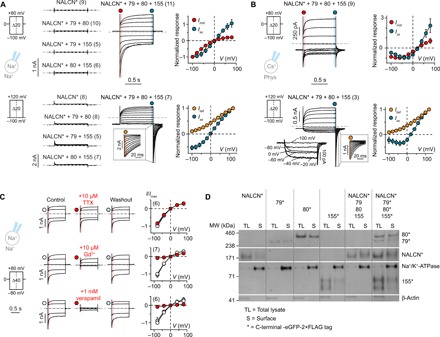
(A and B) Whole-cell patch-clamp recordings from HEK-293T cells expressing NALCN-eGFP-2×FLAG (NALCN*) alone or in different combinations with UNC79 (79), UNC80 (80), and FAM155A (155) under (A) symmetrical Na+ and (B) more physiological conditions using voltage-step protocols shown on the left. Normalized I-V plots highlighting the different current components of NALCN* + 79 + 80 + 155 are shown on the right. The instantaneous current (Iinst; red) was measured in the beginning of a voltage-step change, immediately after the transient current settled. The steady-state current (Iss; blue) was measured at the end of a voltage step. Insets show tail currents (Itail; orange) immediately after repolarization to −100 mV. (C) Current responses (left) and Iinst-V plots (right, normalized to the control current) in the absence and presence of TTX, Gd3+, or verapamil under symmetrical Na+ conditions. Data in (A) to (C) are shown as mean ± SD; gray dashed lines indicate 0 nA; numbers in parentheses indicate number of individual cells used for recordings. (D) Western blot of total lysate and surface fraction proteins extracted from HEK-293T cells expressing the indicated constructs (see also fig. S2A).
In patch-clamp experiments, we found that HEK-293T cells expressing all components of the NALCN channel complex showed low seal resistances (Rm ~ 100 megohms) after breaking into whole-cell mode (fig. S1D). Despite the low seal resistances, a feature typically associated with nonspecific ohmic leak current in electrophysiological studies (20), we observed clear voltage-dependent changes in current in response to both depolarizing and hyperpolarizing voltage steps, irrespective of the holding potential (HP) (Fig. 1, A and B, and fig. S1A). The gating behavior of the NALCN complex observed is unusual in a few aspects. First, we did not observe full channel closure within the tested voltage range, indicating that the complex is constitutively active (Fig. 1, A and B). Second, the complex is differentially modulated by voltage, with depolarization eliciting noninactivating current and hyperpolarization eliciting large inward currents that deactivate rapidly but incompletely. Third, the inward component during hyperpolarizing steps is much more pronounced under symmetrical Na+ conditions than under more physiological conditions (Fig. 1, A and B), suggesting that ionic species can affect channel function. Fourth, a NaV-like, bell-shaped current-voltage (I-V) relationship between −100 and 0 mV, with a maximal inward current around −40 mV, was detected for the NALCN complex under a more physiological recording condition (Fig. 1B). Overall, the gating phenotype is distinct from that of other canonical voltage-gated ion channels (VGICs), which typically populate one or more closed (or inactivated) states upon hyper- or depolarization. We also tested for potential involvement of the Src family of kinases (SFKs) in the function of NALCN channel complex, as SFKs have been suggested to underlie NALCN activation (22). However, we found that neither the SFK activator peptide nor the SFK inhibitors (PP1 and SU 6656) showed any apparent functional effects during patch-clamping experiments (fig. S1, E and F), indicating that SFKs are not required for the NALCN-mediated currents observed in our study.
Next, we investigated whether the limited pharmacological profile established for NALCN thus far is preserved when the channel is co-expressed with UNC79, UNC80, and FAM155, i.e., insensitivity to tetrodotoxin (TTX) and inhibition by low micromolar concentrations of Gd3+ and high micromolar concentrations of the CaV inhibitor verapamil. We found that TTX application had no effect on current amplitude, while both Gd3+ and verapamil showed inhibitory effects (Fig. 1C). We also assessed the subcellular localization of NALCN, UNC79, UNC80, and FAM155A and found that all four proteins showed membrane localization when expressed alone or together in HEK-293T cells (Fig. 1D and fig. S2A). To determine functionally critical regions of these proteins, we expressed full-length NALCN with a series of truncated constructs for UNC79, UNC80, and FAM155A in X. laevis oocytes and measured the resulting currents. We found that robust function required the presence of virtually full-length UNC79 and UNC80 proteins, although short truncations were tolerated at the C and N terminus, respectively (fig. S3, A and B). In the case of FAM155A, the presence of the first putative transmembrane domain and the CRD was absolutely required for function, while deletion of a second putative transmembrane domain was less detrimental (fig. S3C). To determine whether the lack of function was due to impaired cell surface expression, we also assessed the subcellular localization of the truncated proteins. Somewhat unexpectedly, we detected clear membrane localization for all truncated constructs (fig. S4). These results raise the possibility that UNC79, UNC80, and FAM155A are integral or peripheral membrane proteins that are at least, in part, exposed to the extracellular side of the cell membrane. However, given the current severe lack of knowledge on the topology of these proteins and the possibility that the -eGFP (enhanced green fluorescent protein)–2×FLAG tag may affect the subcellular localization of fusion proteins, further studies are necessary to clarify these results in the future. Together, our data suggest that although NALCN can traffic to the membrane by itself, co-expression with UNC79, UNC80, and FAM155A is a prerequisite for the formation of a functional NALCN channel complex.
The NALCN channel complex is selective for monovalent cations
To define the ion selectivity profile of the NALCN channel complex, we first determined the current-carrying ions under bi-ionic conditions. We found that current directionality and reversal potentials (Erevs) were sensitive to substitution of either extracellular or intracellular Na+ with the large cation N-methyl-d-glucamine (NMDG+), but not to replacement of extracellular Cl− with the large anion methanesulfonate (MS−) (Fig. 2A), consistent with permeability for cations and not anions. Next, we assessed the permeability of different cations [Li+, K+, Cs+, TEA+ (tetraethylammonium), Ca2+, Mg2+, and Ba2+] by having equimolar concentrations of test cations on the extracellular side [150 mM for monovalent cations (X+); 110 mM for divalent cations (X2+)] and the impermeable NMDG+ (150 mM) on the intracellular side. We observed voltage-dependent currents in the presence of extracellular Li+, K+, or Cs+, but not with TEA+ or X2+ (Ca2+, Mg2+, Ba2+; Fig. 2B). The maximal current amplitudes of Li+, K+, and Cs+ elicited at −80 mV, however, were smaller compared to Na+ (57 ± 8%, 89 ± 11%, and 76 ± 11% of Na+ current, respectively). To measure the Erevs of the permeable cations, we ran a ramp protocol from −80 to +80 mV. The permeability ratios (PNa/PX) calculated on the basis of the Erevs revealed a permeability sequence of Na+ ≈ Li+ > K+ > Cs+ (Fig. 2C). This permeability sequence was altered to Na+ ≈ Li+ ≈ K+ > Cs+ when the putative SF motif of NALCN (EEKE) was mutated to that of NaVs (DEKA; Fig. 2D).
Fig. 2. The NALCN channel complex is selective for small monovalent cations.
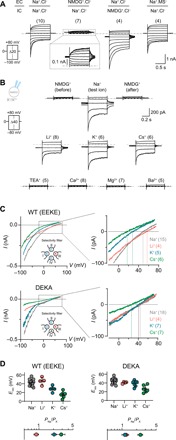
(A) Representative current traces of HEK-293T cells expressing NALCN* + 79 + 80 + 155 under various bi-ionic conditions in response to the voltage protocol shown. EC, extracellular; IC, intracellular. (B) Representative current traces obtained with intracellular NMDG+ and different extracellular mono- or divalent cations using voltage-step protocols shown on the left. (C) The top panel shows currents obtained with a voltage ramp protocol (−80 to 80 mV) with different test ions for both wild-type (WT) [EEKE selectivity filter (SF), top] and the DEKA SF mutant (bottom) using intracellular NMDG+ solution; insets show only −20- to +80-mV range. (D) Reversal potentials of different test ions and their permeability relative to Na+; data are shown as mean ± SD; gray dashed lines indicate 0 nA; numbers in parentheses indicate number of individual cells used for recordings.
The NALCN pore is blocked by extracellular divalent cations
To investigate whether X2+ are merely impermeable or directly blocking the channel, we exposed cells to extracellular solutions containing 120 mM Na+ in the absence and presence of 1 mM Ca2+, Mg2+, or Ba2+. We found that all three X2+ inhibited Na+ currents to varying degrees, with Ca2+ being the most potent, followed by Mg2+ and Ba2+ (83 ± 10%, 59 ± 11%, and 37 ± 11% inhibition of Iinst respectively; Fig. 3A). We also determined the sensitivity of Ca2+ inhibition by measuring current responses in the presence of a broad range of extracellular [Ca2+] (1 nM to 10 mM) under symmetrical Na+ conditions. This resulted in a half maximal inhibitory concentration (IC50) of 319 μM [95% confidence interval (CI), 192 to 692 μM] for Iinst and 132 μM (95% CI, 70 to 350 μM) for Iss (Fig. 3B). The inhibition by X2+ appeared to be limited to the extracellular side, as the presence of 1 mM free Ca2+ in the intracellular recording solution did not have any apparent effects on the current phenotype (fig. S5A).
Fig. 3. Divalent cation block of the NALCN channel complex is attenuated by mutations of side chains in the putative SF region.
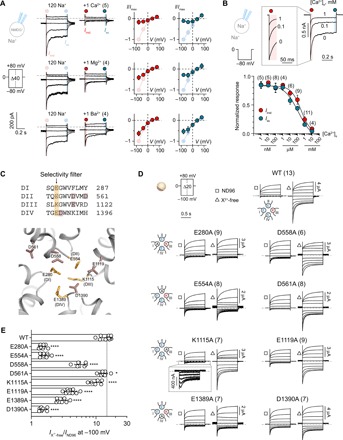
(A) Current responses of HEK-293T cells expressing NALCN* + 79 + 80 + 155 in the absence and presence of 1 mM Ca2+, Mg2+, or Ba2+. Normalized I-V plots illustrate the inhibitory effects of each divalent cation on Iinst and Iss. (B) Sample traces of a HEK-293T cell expressing NALCN* + 79 + 80 + 155 exposed to 0, 0.1, and 1 mM Ca2+ under symmetrical Na+ conditions when stepping from 0 to −80 mV. Inset shows Iinst on an expanded time scale. IC50 graph shows the potency of Ca2+ inhibition on both Iinst and Iss of NALCN-mediated current. (C) Top: Alignment of the selective filter (SF) region from the four homologous domains in NALCN. The putative SF motif EEKE is highlighted in orange and other negatively charged residues selected for charge neutralization are highlighted in dark pink. Bottom: Homology model of the putative SF region of NALCN based on the structure of CaV1.1 (Protein Data Bank ID: 5GJV). (D) Representative currents from X. laevis oocytes expressing WT NALCN or alanine mutants in response to step protocols from +80 to −100 mV (HP = 0 mV) in the presence (ND96; 1.8 mM Ca2+ and 1 mM Mg2+) and absence of divalent cations (X2+-free). (E) Fold increase in inward current elicited at −100 mV for WT NALCN and SF alanine mutants in response to removal of divalent cations. Data are shown as mean ± SD; *P < 0.05; ****P < 0.0001; one-way analysis of variance (ANOVA), Dunnett’s test (against WT); gray dashed lines indicate 0 nA; numbers in parentheses indicate number of individual cells used for recordings. See fig. S3.
We hypothesized that the net negative charge around the putative EEKE SF of NALCN (Fig. 3C) influences the sensitivity of X2+ block, analogous to what has been demonstrated for NaVs and CaVs (28, 29). To test this notion directly, we substituted eight charged side chains in the putative SF region of NALCN with alanine and compared the effects of removing X2+ between WT and mutant channel complexes expressed in X. laevis oocytes. While the WT channel complex showed a large increase (15-fold) in inward currents when X2+ (1.8 mM Ca2+ and 1 mM Mg2+ in ND96) were removed (Fig. 3, D and E), the alanine mutations affected the complex’s sensitivity to X2+ removal to varying degrees. E280A, E554A, and D1390A showed drastically reduced current increase (1.5-fold); D558A, E1119A, and E1389A showed moderately reduced current increase (three- to sixfold), while D561A and K1115A showed WT-like (>10-fold) sensitivity (Fig. 3, D and E, and fig. S5B). These experiments confirm the direct inhibitory effect of X2+ on NALCN and suggest that X2+ block the channel by interacting with negatively charged side chains around the putative EEKE SF. The C-terminal tail of NALCN has previously been implicated in an indirect Ca2+-sensing mechanism of NALCN that involves the calcium-sensing receptor (5). However, we found that C-terminally truncated NALCN variants (Δ1638 to 1738 and Δ1570 to 1738) are as sensitive to X2+ as the WT channel (fig. S5C), indicating that the direct effects of X2+ on NALCN are not dependent on its C terminus.
Delineating the molecular basis of voltage sensitivity in NALCN
The reduced number of positively charged side chains in the S4 segments of the VSDs of NALCN (Fig. 4A) has previously been suggested to be the cause of its lack of voltage sensitivity (4). Here, however, we observed clear voltage dependence in the NALCN channel complex (Fig. 1). A similar observation has recently been reported when NALCN and FAM155A were co-expressed in the neuronal cell line NG108-15, which endogenously expresses UNC79 and UNC80 (30). To determine the basis of this voltage sensitivity, we mutated positively charged side chains in S4 to glutamine. We found that neutralizing most, if not all, S4 positive charges in VSDI (R146Q, R152Q, and R155Q) or VSDII (R481Q, R484Q, and K487Q) resulted in channels that did not respond to voltage pulses at an HP of 0 mV, but were still sensitive to depolarizing pulses from an HP of −100 mV (Fig. 4B and fig. S6, A to C). In contrast, robust WT-like currents were still elicited in neutralized VSDIII (R989Q, R992Q, and R995Q) and VSDIV (R1310Q) mutants (Fig. 4B), indicating that the S4 segments in domains III and IV are not essential for the voltage sensitivity of NALCN. Next, we examined the function of single and double charge-neutralizing S4 mutations in VSDI and VSDII. Most single- and double-VSDI S4 mutants exhibited WT-like voltage sensitivity (Fig. 4C), activation time constants (when stepping from 0 to +80 mV in ND96; Fig. 4E), and deactivation time constants (when stepping from 0 to −100 mV in X2+-free buffer; Fig. 4F). While the R143Q + R146Q mutant exhibited WT-like voltage sensitivity (Fig. 4C), it had significantly larger activation time constants than WT (Fig. 4E). The R146Q + R152Q, similar to the triple R146Q + R152Q + R155Q mutant, did not respond to voltage pulses at an HP of 0 mV (Fig. 4C) but was still sensitive to depolarizing pulses from an HP of −100 mV (fig. S6, A and B). These results indicate that the presence of two arginine residues in the middle of the VSDI S4 is critical for voltage sensitivity. In VSDII S4, the R481Q mutation, but not the R484Q and K487Q single mutations, exhibited altered current kinetics compared to WT (Fig. 4, D and E). R481Q showed significantly larger activation time constants when stepping from 0 to +80 mV in ND96 (Fig. 4E). Moreover, of the 20 VSD mutations tested, R481Q is the only mutation that significantly slowed the deactivation time constants when stepping from 0 to −100 mV under the X2+-free condition (Fig. 4F). Neutralizing an additional VSDII charge on the background of R481Q rendered the channel irresponsive to voltage pulses at an HP of 0 mV (Fig. 4, D and E, and fig. S6, A and B). Together, these results suggest that charged S4 residues in both VSDI and VSDII of NALCN are involved in voltage sensing. Supporting evidence for the intrinsic voltage dependence of VSDI originates from the finding that isolated NALCN VSDI generates voltage-dependent currents (fig. S6D), reminiscent of what is observed with the isolated VSD of the Shaker potassium channel (31).
Fig. 4. NALCN voltage sensitivity primarily arises from S4 charges in domains I and II.
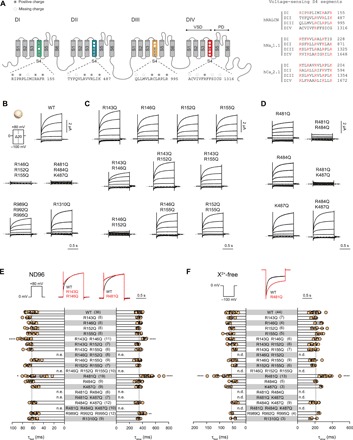
(A) Left: Schematic illustration of the structural topology of NALCN, which consists of four domains (DI to DIV) connected via intracellular linkers. Each domain contains six transmembrane segments (S1 to S6), with S1 to S4 forming the VSDs and S5 to S6 forming the pore domains. The S4 segments typically carry several positively charged residues important in voltage sensing. The conserved and missing (compared to canonical VSDs) positively charged residues in each S4 segment of NALCN are highlighted. Right: Alignment of the S4 segments of the four homologous domains in hNALCN, hNaV1.1, and hCaV2.1. Positively charged side chains are highlighted in red. (B) Current traces and Iss-V plots from X. laevis oocytes expressing WT and charge-neutralized mutants in S4 of DI to DIV using the indicated protocol. (C and D) Representative traces of single and double charge-neutralized mutants in S4 of VSDI (C) and VSDII (D). (E and F) Slow and fast time constants of depolarization-elicited currents (0 to +80 mV) in ND96 (E) and hyperpolarization-elicited currents (0 to −100 mV) in X2+-free buffer (F) for WT and VSD mutants. Superimposed traces of WT (black) and selected VSD mutants (red) are shown above the bar graphs. Data are shown as mean ± SD; *P < 0.05; **P < 0.01; ****P < 0.0001; one-way ANOVA, Dunnett’s test (against WT); n.e., no effect; n.d., not determined; gray dashed lines indicate 0 nA; numbers in parentheses indicate number of individual cells used for recordings.
DISCUSSION
UNC79, UNC80, and FAM155A are crucial for NALCN function
Two decades have passed since NALCN was first cloned, but functional expression of the channel in heterologous systems is still hampered by issues such as low levels of current (14, 18, 32) or absence of NALCN-specific currents (19–21, 23). Given the ample evidence supporting overlapping spatiotemporal gene expression profiles for NALCN, UNC79, UNC80, and FAM155A in Drosophila and mouse (8, 33); their functional interdependence in Drosophila (6, 34), C. elegans (11, 12, 35, 36), and mouse (5, 22); and the physical interactions between these proteins in vitro and in vivo (5, 12, 22), we set out to test the effect of co-expressing these proteins in different heterologous systems. We found that the robust, reproducible functional expression of NALCN is critically dependent on the presence of UNC79, UNC80, and FAM155A/B (Fig. 1A and fig. S1).
UNC79 and UNC80 are widely expressed in the brain, where they form a complex with NALCN and regulate its localization and function (5, 11, 34). While UNC79 is not absolutely required for NALCN activity in mouse hippocampal neurons [NALCN-like currents were observed in the absence of UNC79 (5)], both proteins are needed for NALCN function in Drosophila (34), the neuronal cell line NG108-15 (30), HEK-293T cells, and X. laevis oocytes (Fig. 1 and fig. S1), highlighting potential interspecies differences in the regulatory roles of these proteins. FAM155A is pivotal for the axonal localization of NALCN in C. elegans (12). On the basis of the proposed ER residency of the C. elegans and mouse homologs (12), it was suggested that FAM155A acts as a chaperone to facilitate NALCN folding and delivery to the cell membrane. By contrast, we were able to detect plasma membrane localization of the human FAM155A, even when expressed alone (Fig. 1D). Whether this discrepancy in subcellular localization is due to species differences requires further investigation. Overall, our data indicate that NALCN function and not membrane localization per se is critically dependent on the presence of UNC79, UNC80, and FAM155. Furthermore, the co-immunoprecipitation of UNC79, UNC80, and FAM155A with NALCN (fig. S2B) reinforces the notion that NALCN predominantly functions in complex with other proteins (26).
NALCN is the voltage-sensing, pore-forming subunit of the complex
NALCN was considered to be voltage insensitive owing to the reduced number of voltage-sensing residues in the S4 segments compared to typical VGICs. Here, we found that NALCN, when co-expressed with UNC79, UNC80, and FAM155, exhibits an unusual modulation by membrane voltage (Fig. 1). This highlights how voltage sensor domains can, depending on the context, contribute to gating in previously unanticipated ways and thus contributes to our understanding of how this widely encountered motif can modulate ion channel function (37). We show that the voltage dependence originates from positively charged residues in the S4 segments of DI and DII of NALCN, as charge-neutralizing substitutions affected voltage sensitivity (Fig. 4). NALCN had been suggested to be a nonselective cation channel that poorly discriminates between X+ and X2+. This lack of ion selectivity is often attributed to the EEKE SF motif of NALCN, which is a hybrid between the Na+-selective DEKA and the Ca2+-selective EEEE. Here, we found that NALCN is moderately selective among the permeable X+, but does not conduct X2+. The X+ selectivity profile is altered when the EEKE motif was mutated to DEKA (Fig. 2C). Furthermore, some charge-neutralizing mutations around the putative channel pore also affected current rectification and phenotype (Fig. 3D and fig. S5B). For instance, the K1115A mutant displays a unique “hooked” phenotype (Fig. 3D), consistent with the notion that this side chain lines the ion permeation pathway. Together, our results strongly support the idea for NALCN to be the voltage-sensing, pore-forming subunit of the complex.
The NALCN channel complex reconstitutes the extracellular [Ca2+]-sensitive NSCC
A nonselective cation current (NSCC) that is activated by the lowering of extracellular [Ca2+] ([Ca2+]e) has been detected in different types of neurons, such as the chick dorsal root ganglia (38), mouse hippocampal (5, 39), neocortical nerve terminals (40), and dopaminergic neurons (9). The NSCC depolarizes the RMP, allowing the continuous firing of action potentials. The mechanism(s) underlying this phenomenon are not well understood, but the NALCN channel complex may play a role, as NALCN- and UNC79-knockout neurons are insensitive to drops in [Ca2+]e (5, 9). This NALCN-mediated effect has been suggested to occur via an indirect mechanism that is initiated and transduced by the G protein–coupled Ca2+-sensing receptor (5). Here, however, we show that the NALCN channel complex–mediated current, without co-expression of GPCRs, is directly blocked by [Ca2+]e, as alanine mutations of several negatively charged residues in the putative pore of NALCN markedly reduced the sensitivity to [Ca2+]e (Fig. 3, D and E). Furthermore, the NALCN channel complex displays notably similar biophysical properties compared to the neuronal NSCC: (i) marked increase in inward Na+ current (>10-fold; Figs. 1 and 3) following the reduction in [Ca2+]e, with a similar sensitivity to [Ca2+]e (5, 39, 40); (ii) selective permeability to small X+, with a small preference for Na+ over K+, followed by Cs+ (Fig. 2) (39); (iii) insensitivity to TTX (1–3); and (iv) inhibition by polyvalent cations in the rank order of potency: Gd3+ > Ca2+ > Mg2+ and Ba2+ (Figs. 1C and 3A) (38, 39).
Physiological implications
The contribution of NALCN to the RMP of neurons and hence their excitability is undisputed. To perform the task as a regulator of the RMP, it is necessary for NALCN to respond dynamically to the potential difference across the plasma membrane. The unique gating behavior of the NALCN channel complex, along with the modulation by extracellular X2+ through a direct pore-blocking mechanism, accentuates its suitability to perform this task. The constitutive activity of the complex implies that a tonic inward Na+ conductance will be present even at rest, which may contribute to the depolarization of the RMP. While this Na+ conductance is likely to be small in the presence of normal [Ca2+]e and [Mg2+]e, it is expected to increase considerably following reductions in [Ca2+]e, such as during repetitive chemical or electrical stimulation, or under pathophysiological conditions (17), thus lowering the threshold for action potential generation. As such, the NALCN complex is predicted to act as an X2+ sensor that directly fine-tunes neuronal excitability in response to fluctuations of [X2+]e. Furthermore, the differential modulation of NALCN function by voltage means that the NALCN complex undergoes partial deactivation in response to hyperpolarization, which would allow Na+ influx to counter the negative shift in membrane potential. By contrast, during prolonged depolarization, K+ ions would leave the cell, contributing to the repolarization of the membrane potential. Overall, we postulate that the NALCN complex functions as a thermostat (or rheostat) that maintains the RMP at a desired value to determine the intrinsic excitability of a neuron.
MATERIALS AND METHODS
Molecular biology
Human NALCN*, UNC-79*, UNC-80*, FAM155A*, FAM155A•, and FAM155B*, and mouse FAM155A* complementary DNAs (cDNAs; * indicates the C-terminal -eGFP-2×FLAG tag; • indicates the C-terminal -MBP-2×FLAG tag) cloned between Hind III and Xho I sites in a modified pCDNA3.1(+) vector containing 3′-Xenopus globin untranslated region and a polyadenylation signal were generated using custom gene synthesis with codon optimization for Homo sapiens (GeneArt, Thermo Fisher Scientific). The tags were removed using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) to generate WT, untagged constructs. The rat NALCN construct was a gift from D. Ren at the University of Pennsylvania and was cloned into the same modified pCDNA3.1(+) vector between Kpn I and Xho I sites. NALCN pore mutants (Figs. 2 and 3) and nonsense mutations bearing UNC80 variants (figs. S3 and S4) were generated with site-directed mutagenesis using custom-designed primers (Eurofins Genomics) and PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies). Truncated NALCN, UNC-79*, UNC-80*, and FAM155A* constructs (figs. S3 to S5) were generated using the Q5 Site-Directed Mutagenesis Kit. The sequences of purified plasmid DNAs from transformed E. coli were verified by Sanger DNA sequencing (Eurofins Genomics). For expression in X. laevis oocytes, plasmid DNAs were linearized with Xba I restriction enzyme, from which capped RNAs were synthesized using the T7 mMessage mMachine Kit (Ambion). For expression in HEK-293T cells, plasmid DNAs purified using the NucleoBond Xtra Midi Plus kit (Macherey-Nagel) were used.
Two-electrode voltage-clamp electrophysiology
Ovarian lobes were surgically removed from female X. laevis frogs anesthetized in 0.3% tricaine (procedure approved by the Danish Veterinary and Food Administration; license number, 2014-15-0201-0031), divided into smaller clumps, and defolliculated by shaking at 200 rpm, 37°C in OR2 [82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, and 5 mM Hepes (pH 7.4) with NaOH] containing type I collagenase (1 mg/ml; Worthington Biochemical Corporation). Healthy-looking stage V-VI oocytes were isolated and injected with 60 to 70 ng of RNA in a volume of 41 to 46 nl using a Nanoliter 2010 injector (World Precision Instruments). The NALCN, UNC-79, UNC-80, and FAM155A RNAs were mixed in a ratio of 3:1:1:1. The same ratio was used when NALCN was replaced with rNALCN and when FAM155A was replaced with FAM155B or mFAM155A (fig. S1B). When one or more constructs are excluded (fig. S1A), an equivolume of nuclease-free water was added to keep the concentration of each RNA constant across different combinations. Injected cells were incubated in OR3 [50% (v/v) Leibovitz’s L-15 medium (Gibco), 1 mM glutamine, gentamicin (250 μg/ml), and 15 mM Hepes (pH 7.6) with NaOH] at 18°C, 140 rpm. Four to 5 days after RNA injection, two-electrode voltage-clamp measurements were performed on oocytes continuously perfused in ND96 recording solution [96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM Hepes (pH 7.4) with NaOH] or divalent cation-free ND96 [96 mM NaCl, 2 mM KCl, and 5 mM Hepes (pH 7.4) with NaOH] at room temperature using a Warner OC-725C Oocyte Clamp amplifier (Warner Instrument Corp, USA). Data were acquired using the pCLAMP 10 software (Molecular Devices) and a Digidata 1550 digitizer (Molecular devices), sampled at 10 kHz. Electrical powerline interference was filtered with a Hum Bug 50/60 Hz Noise Eliminator (Quest Scientific). Recording microelectrodes with resistances around 0.2 to 1.0 megohms were pulled from borosilicate glass capillaries (Harvard Apparatus) using a P-1000 Flaming/Brown Micropipette Puller System (Sutter Instrument) and were filled with 3 M KCl.
HEK-293T cell culture and transfection
HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin-streptomycin (10,000 U/ml; Thermo Fisher Scientific) at 37°C in a 5% CO2 humidified growth incubator. Cells between passages 6 and 20 were used for experiments and were tested for mycoplasma (Eurofins Genomics) five times throughout this study. For patch-clamp experiments, cells reaching 40 to 60% confluency in 35-mm cell culture dishes were transiently transfected with constructs of interest using LipoD293 ver. II (tebu-bio) or PEI (Polyethylenimine) 25K (Polysciences) 20 to 24 hours before recording. Depending on the transfection reagent used, a total of 2.5 μg (for LipoD293) or 5 μg (for PEI) of cDNAs was used. The NALCN*, UNC-79, UNC-80, and FAM155A cDNAs were mixed in the mass ratio of 2:1:1:1. When one or more constructs are excluded from the combination, an equal amount of empty vector was added to keep the total cDNA amount constant. For mock-transfected cells, empty vector and eGFP were mixed in a 24:1 ratio. For Western blot experiments (Fig. 1D and figs. S2A and S4), cells reaching 20 to 40% confluency in 35-mm cell culture dishes were transiently transfected with constructs of interest using LipoD293 2 days before the cells were harvested. A total of 0.8 μg of cDNAs was used, with the NALCN*, UNC-79, UNC-80, and FAM155A cDNAs mixed in the mass ratio of 1:1:1:1. For co-immunoprecipitation experiments (fig. S2B), ~2 to 2.5 million HEK-293T cells were plated on 100-mm cell culture dishes ~30 hours before transfection. A total of 12 to 20 μg of DNA was used, with the NALCN* or 2×FLAG-eGFP-eGFP-2×FLAG, UNC79-2×FLAG, UNC80-2×FLAG, and FAM155A• cDNAs mixed in the mass ratio of 1:1:1:1.
Patch-clamp electrophysiology
On the day of the experiment, transfected HEK-293T cells were seeded on poly-l-lysine–coated glass coverslips at least 3 hours before recording. Cells were voltage-clamped at room temperature in the whole-cell configuration using an Axopatch 200B amplifier (Molecular Devices). Data were acquired using the pCLAMP 10 software (Molecular Devices) and an Axon Digidata 1550A digitizer (Molecular Devices) at 10 kHz. Patch pipettes were pulled from Kwik-Fil 1.5/1.12 [outer diameter (OD)/inner diameter (ID), in millimeters] borosilicate glass capillaries (World Precision Instruments) and fire-polished to resistances around 3.0 to 8.5 megohms. A custom-built glass perfusion tool with four adjacent barrels (OD/ID 0.45/1.60, in millimeters; CM Scientific) controlled by an MXPZT-300R solution switcher (Siskiyou) was used to rapidly exchange extracellular solutions.
For symmetrical Na+ conditions (Fig. 1A), extracellular solution contained 150 mM NaCl, 10 mM Hepes, and 30 mM d-(+)-glucose (pH 7.4) with NaOH, ~325 mosmol/liter, and intracellular solution contained 136 mM NaCl, 10 mM NaF, 5 mM EGTA, 10 mM Hepes, and 2 mM Na2ATP (adenosine 5′-triphosphate) (pH 7.2) with NaOH, ~309 mosmol/liter. For a more physiological condition (Fig. 1B), extracellular solution contained 150 mM NaCl, 5 mM KCl, 0.5 mM CaCl2, 1.2 mM MgCl2, 10 mM Hepes, and 13 mM d-(+)-glucose (pH 7.4) with NaOH, ~320 mosmol/liter, and intracellular solution contained 140 mM CsCl, 10 mM CsF, 5 mM EGTA, 10 mM Hepes, and 2 mM Na2ATP (pH 7.2) with CsOH, ~304 mosmol/liter. For ion-selectivity experiments (Fig. 2) (i) involving monovalent cations, the extracellular solution that contained 150 mM XCl, 10 mM Hepes, and d-(+)-glucose was added accordingly to achieve osmolarity ~325 mosmol/liter, and the pH was adjusted to 7.4 with XOH, where X indicates the cation of interest; (ii) involving divalent cations, the extracellular solution that contained 110 mM XCl2, 10 mM Hepes, and d-(+)-glucose was added accordingly to achieve osmolarity ~325 mosmol/liter, and the pH was adjusted to 7.4 with X(OH)2, where X indicates the cation of interest; (iii) the intracellular solution contained 150 mM NMDG, 5 mM EGTA, 10 mM Hepes, 2 mM Na2ATP, and 30 mM d-(+)-glucose (pH 7.2) with HCl, ~310 mosmol/liter. For the determination of Ca2+ IC50 (Fig. 3B), extracellular Na+ solutions containing 1 nM to 1 mM free Ca2+ were made by mixing Ca2+-free stock [150 mM NaCl, 10 mM Hepes, and 5 mM EGTA (pH 7.4) adjusted with NaOH] and 1 mM free Ca2+ stock [150 mM NaCl, 10 mM Hepes, 5 mM EGTA, and 6 mM Ca(OH)2 (pH 7.4 adjusted) with NaOH] at the ratio calculated according to the WEBMAXC STANDARD calculator (https://web.stanford.edu/~cpatton/webmaxcS.htm). Solution containing 10 mM Ca2+ was prepared by adding CaCl2 from a 1 M stock solution. d-(+)-Glucose was added accordingly to all extracellular solutions to achieve osmolarity ~325 mosmol/liter. The intracellular solution contained 136 mM NaCl, 10 mM NaF, 5 mM EGTA, 10 mM Hepes, and 2 mM Na2ATP (pH 7.2) with NaOH, ~309 mosmol/liter.
Experiments involving SFK-targeting peptide or compounds (fig. S1, E to F) were performed under symmetrical Na+ conditions. The Src family activator peptide (Santa Cruz Biotechnology; catalog #: sc-3052) was dissolved in nuclease-free water to make a 1 mM stock solution and was diluted to a final concentration of 1 μM in the intracellular solution: 136 mM NaCl, 10 mM NaF, 5 mM EGTA, 10 mM Hepes, and 2 mM Na2ATP (pH 7.2) with NaOH, ~309 mosmol/liter. The SFK inhibitors PP1 (catalog #: P0040) and SU 6656 (catalog #: S9692) were dissolved in sterile-filtered dimethyl sulfoxide (DMSO; Sigma Aldrich, catalog #: D2650) to make 50 and 20 mM stock solutions, respectively. PP1 and SU 6656 were then diluted to 5 μM in the extracellular solution: 150 mM NaCl, 10 mM Hepes, and 30 mM d-(+)-glucose (pH 7.4) with NaOH, ~325 mosmol/liter. The final concentrations of DMSO in the extracellular solutions were around 0.1 to 0.025% v/v.
To prevent nonspecific leaks from affecting the accuracy of our results, we routinely checked for loose seals by exposing cells to NMDG-only extracellular solution before and/or after experiments (Fig. 2B). Cells that showed steady-state inward current >10 pA at −80 mV in the absence of permeable ions were discarded. Voltage ramp protocol used to determine Erevs (Fig. 2C) is as follows: whole-cell mode was first achieved in symmetrical NMDG+ solutions, and cells were exposed to NaCl solution for 200 ms before a 1-s voltage step (0 to −80 mV) was applied to obtain a steady-state current; at the end of the voltage step, a 200-ms ramp (−80 to +80 mV) was run before returning to 0 mV; cells were allowed to rest in NDMG solution for ~10 s before the same protocol was run in a solution containing a different ion of interest.
Cell surface biotinylation and Western blots
Cell surface proteins were purified using the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific) based on the manufacturer’s instructions with a few modifications: (i) washing was performed with ice-cold phosphate-buffered saline (PBS)–CM (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1 mM CaCl2, and 1 mM MgCl2); (ii) biotin was dissolved in ice-cold PBS-CM to a final concentration of 1.25 mg/ml; and (iii) the quenching buffer consisted of PBS-CM, supplemented with 200 mM glycine. Cells were lysed in lysis buffer [100 mM tris-HCl, 150 mM NaCl (pH 7.4), 0.1% SDS, and 1% Triton X-100] supplemented with 1:100 Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) under gentle agitation on ice for 30 min. Denatured samples from both the total lysates and surface fractions were separated on NuPAGE 3 to 8% tris-acetate protein gels (Thermo Fisher Scientific) at 200 V for 40 min. The HiMark Pre-Stained Protein Standard (30 to 460 kDa; Thermo Fisher Scientific) was used as protein molecular weight reference. Samples were transferred onto an Invitrolon polyvinylidene difluoride membrane activated with 10% methanol using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific). The membranes were then incubated in blocking buffer [1 × tris-buffered saline (TBS) supplemented with fish gelatin (4.5 g/liter), casein (1 g/liter), and 0.02% sodium azide] for 1 hour at room temperature (RT) before they were incubated with the primary antibodies [rabbit anti-FLAG (701629; Invitrogen), mouse anti–β-actin (sc-47778; Santa Cruz Biotechnology), and mouse anti-Na+/K+-ATPase (05-369; EMD Merck Millipore)] at 4°C overnight. Next, the membranes were washed 5 × 2 min with TBST [20 mM tris-HCl, 150 mM NaCl, and 0.1% Tween 20 (pH 7.5)] and incubated with the secondary antibodies [IRDye 800CW goat–anti-rabbit (925-32211; LI-COR Biosciences) and IRDye 680RD goat–anti-mouse (926–68070; LI-COR Biosciences)] for 1 hour at RT in the dark. Last, the membranes were washed 5 × 2 min in TBST before being imaged using a PXi gel imaging station (Syngene).
Co-immunoprecipitation and Western blots
HEK-293T cells were harvested ~24 hours after transfection and washed twice in ice-cold PBS. Cells were lysed in lysis buffer [100 mM tris-HCl, 150 mM NaCl (pH 7.4), 0.1% SDS, and 1% Triton X-100] supplemented with 1:100 Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) with end-over-end rotation at 4°C for 1 hour. Lysates were cleared by centrifugation at 10,000g for 10 min, after which the supernatant was transferred onto equilibrated GFP-Trap Magnetic Agarose Beads (ChromoTek) with end-over-end rotation at 4°C for 2 hours. The magnetic beads were washed four times with lysis buffer before incubation in 2× SDS sample buffer (supplemented with 100 mM dithiothreitol) at 60°C for 20 min to elute bound proteins. Samples from cleared total lysates, supernatant (after incubation with magnetic beads), wash, and elution steps were detected with the rabbit anti-FLAG (701629; Invitrogen) antibody using the Western blot technique (described in detail above).
Data analysis
Raw current traces were generally filtered at 500 to 800 Hz (8-pole Bessel low-pass filter) before data analysis. Current traces were subjected to data reduction (substitute average by a factor of 5) for illustration. Data were presented as mean ± SD. Statistical comparisons were performed using GraphPad Prism (version 8.1, GraphPad Software), and the specific tests used were mentioned in the text where relevant. For ion-selectivity experiments performed with Na+ intracellular solution (Fig. 2A), liquid junction potential (LJP) was measured with reference to the Na+ intracellular solution and corrected after recording. The measured LJP values for extracellular NMDG+ and MS− solutions were −5.3 and 4.2 mV, respectively. For ion-selectivity experiments performed with NMDG+ intracellular solution (Fig. 2), LJP was measured with reference to the NMDG intracellular solution and corrected after recording. The measured LJP values for extracellular NaCl, LiCl, KCl, and CsCl solutions were 4.5, 2.6, 8.4, and 7.4 mV, respectively. Relative ion permeabilities (PNa/PX) were calculated using the Goldman-Hodgkin-Katz equation PNa/PX = exp.(F(Erev(Na) − Erev(X))/RT), where F is Faraday’s constant, R is the gas constant, T = 274 K, and Erevs were measured from the −80- to +80-mV ramp (corrected for LJP). The homology model of human NALCN was generated using the Phyre2 Protein Fold Recognition Server; the model based on the Cav1.1 channel (Protein Data Bank ID: 5GJV) was selected for the presentation shown in Fig. 3C.
Supplementary Material
Acknowledgments
We thank J. Colding, C. Chirvas, and C. Bulancea for technical assistance and L. Anson, H. Kurata, J. Cui, and members of the Pless laboratory for helpful comments on the manuscript. Funding: We acknowledge the Carlsberg Foundation (CF16-0504; S.A.P.), the Independent Research Fund Denmark (7025-00097A; S.A.P.), and the Lundbeck Foundation (R252-2017-1671; S.A.P.) for financial support. Author contributions: H.C.C., M.W., and S.A.P. designed the research. H.C.C., M.W., C.W., and L.P.R. performed the experiments. H.C.C., M.W., C.W., and L.P.R. analyzed the data. S.A.P. supervised the project. H.C.C. and S.A.P. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz3154/DC1
REFERENCES AND NOTES
- 1.Raman I. M., Gustafson A. E., Padgett D., Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J. Neurosci. 20, 9004–9016 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson A. C., Yao G. L., Bean B. P., Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J. Neurosci. 24, 7985–7998 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khaliq Z. M., Bean B. P., Pacemaking in dopaminergic ventral tegmental area neurons: Depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 30, 7401–7413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu B., Su Y., Das S., Liu J., Xia J., Ren D., The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129, 371–383 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Lu B., Zhang Q., Wang H., Wang Y., Nakayama M., Ren D., Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron 68, 488–499 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flourakis M., Kula-Eversole E., Hutchison A. L., Han T. H., Aranda K., Moose D. L., White K. P., Dinner A. R., Lear B. C., Ren D., Diekman C. O., Raman I. M., Allada R., A conserved bicycle model for circadian clock control of membrane excitability. Cell 162, 836–848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y., Abe C., Holloway B. B., Shu S., Kumar N. N., Weaver J. L., Sen J., Perez-Reyes E., Stornetta R. L., Guyenet P. G., Bayliss D. A., NALCN is a “leak” sodium channel that regulates excitability of brainstem chemosensory neurons and breathing. J. Neurosci. 36, 8174–8187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lutas A., Lahmann C., Soumillon M., Yellen G., The leak channel NALCN controls tonic firing and glycolytic sensitivity of substantia nigra pars reticulata neurons. eLife 5, e15271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philippart F., Khaliq Z. M., Gi/o protein-coupled receptors in dopamine neurons inhibit the sodium leak channel NALCN. eLife 7, e40984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lear B. C., Lin J.-M., Keath J. R., McGill J. J., Raman I. M., Allada R., The ion channel narrow abdomen is critical for neural output of the Drosophila circadian pacemaker. Neuron 48, 965–976 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Yeh E., Ng S., Zhang M., Bouhours M., Wang Y., Wang M., Hung W., Aoyagi K., Melnik-Martinez K., Li M., Liu F., Schafer W. R., Zhen M., A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C. elegans. PLOS Biol. 6, e55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie L., Gao S., Alcaire S. M., Aoyagi K., Wang Y., Griffin J. K., Stagljar I., Nagamatsu S., Zhen M., NLF-1 delivers a sodium leak channel to regulate neuronal excitability and modulate rhythmic locomotion. Neuron 77, 1069–1082 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Gao S., Xie L., Kawano T., Po M. D., Pirri J. K., Guan S., Alkema M. J., Zhen M., The NCA sodium leak channel is required for persistent motor circuit activity that sustains locomotion. Nat. Commun. 6, 6323 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Eigenbrod O., Debus K. Y., Reznick J., Bennett N. C., Sánchez-Carranza O., Omerbašić D., Hart D. W., Barker A. J., Zhong W., Lutermann H., Katandukila J. V., Mgode G., Park T. J., Lewin G. R., Rapid molecular evolution of pain insensitivity in multiple African rodents. Science 364, 852–859 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Köroğlu Ç., Seven M., Tolun A., Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J. Med. Genet. 50, 515–520 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Al-Sayed M. D., Al-Zaidan H., Albakheet A., Hakami H., Kenana R., Al-Yafee Y., Al-Dosary M., Qari A., Al-Sheddi T., Al-Muheiza M., Al-Qubbaj W., Lakmache Y., Al-Hindi H., Ghaziuddin M., Colak D., Kaya N., Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am. J. Hum. Genet. 93, 721–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren D., Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 72, 899–911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swayne L. A., Mezghrani A., Varrault A., Chemin J., Bertrand G., Dalle S., Bourinet E., Lory P., Miller R. J., Nargeot J., Monteil A., The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic β-cell line. EMBO Rep. 10, 873–880 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senatore A., Monteil A., van Minnen J., Smit A. B., Spafford J. D., NALCN ion channels have alternative selectivity filters resembling calcium channels or sodium channels. PLOS ONE 8, e55088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boone A. N., Senatore A., Chemin J., Monteil A., Spafford J. D., Gd3+ and calcium sensitive, sodium leak currents are features of weak membrane-glass seals in patch clamp recordings. PLOS ONE 9, e98808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan J. M., Peterson C. A., Fry W. M., Lack of current observed in HEK293 cells expressing NALCN channels. Biochimie Open 6, 24–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu B., Su Y., Das S., Wang H., Wang Y., Liu J., Ren D., Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature 457, 741–744 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J. H., Cribbs L. L., Perez-Reyes E., Cloning of a novel four repeat protein related to voltage-gated sodium and calcium channels. FEBS Lett. 445, 231–236 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Dolphin A. C., Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369–5390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senatore A., Spafford J. D., A uniquely adaptable pore is consistent with NALCN being an ion sensor. Channels (Austin) 7, 60–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cochet-Bissuel M., Lory P., Monteil A., The sodium leak channel, NALCN, in health and disease. Front. Cell. Neurosci. 8, 132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei J., Grishin N. V., Cysteine-rich domains related to frizzled receptors and Hedgehog-interacting proteins. Protein Sci. 21, 1172–1184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J., Elllnor P. T., Sather W. A., Zhang J.-F., Tsien R. W., Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature 366, 158–161 (1993). [DOI] [PubMed] [Google Scholar]
- 29.Schlief T., Schönherr R., Imoto K., Heinemann S. H., Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur. Biophys. J. 25, 75–91 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Bouasse M., Impheng H., Servant Z., Lory P., Monteil A., Functional expression of CLIFAHDD and IHPRF pathogenic variants of the NALCN channel in neuronal cells reveals both gain- and loss-of-function properties. Sci. Rep. 9, 11791 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J., Blunck R., The isolated voltage sensing domain of the Shaker potassium channel forms a voltage-gated cation channel. eLife 5, e18130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funato H., Miyoshi C., Fujiyama T., Kanda T., Sato M., Wang Z., Ma J., Nakane S., Tomita J., Ikkyu A., Kakizaki M., Hotta-Hirashima N., Kanno S., Komiya H., Asano F., Honda T., Kim S. J., Harano K., Muramoto H., Yonezawa T., Mizuno S., Miyazaki S., Connor L., Kumar V., Miura I., Suzuki T., Watanabe A., Abe M., Sugiyama F., Takahashi S., Sakimura K., Hayashi Y., Liu Q., Kume K., Wakana S., Takahashi J. S., Yanagisawa M., Forward-genetics analysis of sleep in randomly mutagenized mice. Nature 539, 378–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghezzi A., Liebeskind B. J., Thompson A., Atkinson N. S., Zakon H. H., Ancient association between cation leak channels and Mid1 proteins is conserved in fungi and animals. Front. Mol. Neurosci. 7, 15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lear B. C., Darrah E. J., Aldrich B. T., Gebre S., Scott R. L., Nash H. A., Allada R., UNC79 and UNC80, putative auxiliary subunits of the NARROW ABDOMEN ion channel, are indispensable for robust circadian locomotor rhythms in Drosophila. PLOS ONE 8, e78147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jospin M., Watanabe S., Joshi D., Young S., Hamming K., Thacker C., Snutch T. P., Jorgensen E. M., Schuske K., UNC-80 and the NCA ion channels contribute to endocytosis defects in synaptojanin mutants. Curr. Biol. 17, 1595–1600 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Humphrey J. A., Hamming K. S., Thacker C. M., Scott R. L., Sedensky M. M., Snutch T. P., Morgan P. G., Nash H. A., A putative cation channel and its novel regulator: Cross-species conservation of effects on general anesthesia. Curr. Biol. 17, 624–629 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Bezanilla F., How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 9, 323–332 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Hablitz J. J., Heinemann U., Lux H. D., Step reductions in extracellular Ca2+ activate a transient inward current in chick dorsal root ganglion cells. Biophys. J. 50, 753–757 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiong Z.-G., Lu W.-Y., MacDonald J. F., Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 7012–7017 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith S. M., Bergsman J. B., Harata N. C., Scheller R. H., Tsien R. W., Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron 41, 243–256 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz3154/DC1