

Abstract
The extracellular signal-regulated kinase (ERK) pathway is an essential component of developmental signaling in metazoans. Previous models of pathway activation suggested that dissociation of activated dually phosphorylated ERK (dpERK) from MAPK/ERK kinase (MEK), a kinase that phosphorylates ERK, and other cytoplasmic anchors, is sufficient for allowing ERK interactions with its substrates. Here, we provide evidence for an additional step controlling ERK’s access to substrates. Specifically, we demonstrate that interaction of ERK with its substrate Capicua (Cic) is controlled at the level of ERK phosphorylation, whereby Cic binds to dpERK much stronger than to unphosphorylated ERK, both in vitro and in vivo. Mathematical modeling suggests that the differential affinity of Cic for dpERK versus ERK is required for both down-regulation of Cic and stabilizing phosphorylated ERK. Preferential association of Cic with dpERK serves two functions: it prevents unproductive competition of Cic with unphosphorylated ERK and contributes to efficient signal propagation. We propose that high-affinity substrate binding increases the specificity and efficiency of signal transduction through the ERK pathway.
INTRODUCTION
The extracellular signal-regulated kinase (ERK) is the final component of the Raf-MAPK/ERK kinase-ERK (Raf-MEK-ERK) signaling module, which functions downstream of receptor tyrosine kinases (RTKs) and controls multiple cellular processes, including cell proliferation and differentiation (Lemmon and Schlessinger, 2010; Futran et al., 2013). Activated dually phosphorylated ERK (dpERK) relays pathway activation to the cell via phosphorylation of multiple substrates (Futran et al., 2013). ERK interactions with substrates are typically mediated by two docking domains, the D-site recruitment site (DRS, also known as the common docking, or CD domain), which binds the conserved D-site motif in the substrates, and the F-site recruitment site (FRS), which binds to the substrates containing the FXF motif (also known as the DEF motif) (Jacobs et al., 1999; Sharrocks et al., 2000; Tanoue et al., 2000; Futran et al., 2013).
To understand how ERK carries out its multiple cellular functions, it is critical to establish how pathway activation affects the ability of ERK to recognize its substrates. It is thought that unphosphorylated ERK is sequestered in the cytoplasm by MEK and other cytosolic scaffold and anchor proteins (Roskoski, 2012). Phosphorylation of ERK by MEK results in the release of dpERK from complexes with MEK and other cytoplasmic anchors, enabling subsequent association with substrates in the nucleus and the cytoplasm (Fukuda et al., 1997; Adachi et al., 1999). Whether ERK activation contributes to substrate selection is not well understood. It has been proposed that formation of dpERK results in a conformational change that allows binding of the FRS motif in ERK to the FXF motif in substrate proteins such as ELK1 (Lee et al., 2004). However, a subsequent study found that ERK phosphorylation results in reduced binding to ELK1 (Burkhard et al., 2011).
One of the key ERK targets in Drosophila and mammals is the high mobility group–box transcriptional repressor Capicua (Cic), which controls tissue patterning and organ growth (Jimenez et al., 2000; Astigarraga et al., 2007; Tseng et al., 2007; Ajuria et al., 2011; Grimm et al., 2012; Lim et al., 2013; Jin et al., 2015; Yang and Veraksa, 2017). In Drosophila, Cic phosphorylation and down-regulation is involved in most developmental contexts that are under ERK control (Jimenez et al., 2012). In humans, mutations in CIC have been implicated in the neurodegenerative disease spinocerebellar ataxia type 1 (Lam et al., 2006; Fryer et al., 2011) and in the majority of oligodendroglioma cases, as well as other cancers (Simon-Carrasco et al., 2017; Tanaka et al., 2017; Tan et al., 2018). In all contexts studied so far in Drosophila, Cic functions as a transcriptional repressor whose activity is inhibited in response to ERK activation (Jimenez et al., 2012). Recent evidence suggests that Cic phosphorylation by ERK leads to rapid relief of repression through interference with DNA binding or interactions with corepressors, followed by slower export from the nucleus and eventual proteolytic degradation (Astigarraga et al., 2007; Grimm et al., 2012; Lim et al., 2013, 2015). Because all of the proposed modes of Cic inactivation are dependent on the ERK-mediated phosphorylation of Cic, it is essential to determine how ERK associates with Cic. Previous studies identified a region in Drosophila Cic (the C2 motif) that mediates its binding to ERK (Astigarraga et al., 2007; Futran et al., 2015); however, these studies used the inactive (unphosphorylated) form of ERK.
Here, we report that Cic interacts with activated, dually phosphorylated ERK with a much higher affinity compared with unphosphorylated ERK. Our data suggest that preferential Cic-dpERK interaction prevents unproductive competition of Cic with unphosphorylated ERK, when both Cic and ERK are localized in the same cellular compartment. Furthermore, higher affinity of Cic for dpERK may be required for efficient signal propagation, as it contributes to pathway output (down-regulation of Cic) and increases the steady-state level of dpERK. Based on this, we propose that activation-dependent association with Cic is an integral part of ERK signaling dynamics that serves as an additional checkpoint to regulate the specificity and efficiency of signal propagation.
RESULTS AND DISCUSSION
ERK activation is required and sufficient for high-affinity interaction with Cic
Previous studies of interactions between ERK and Cic used unphosphorylated forms of ERK in binding experiments (Astigarraga et al., 2007; Futran et al., 2015). For testing whether activation of ERK affects its interaction with Cic, V5-tagged Cic (Cic-V5) expressed in Drosophila S2 cells was immobilized on streptavidin beads and incubated with bacterially expressed, purified rat ERK2, which was converted into the active form by coexpression with active MEK (Figure 1A). ERK2 phosphorylation resulted in a much more robust interaction with Cic (Figure 1A). We observed a similar result with the fly ERK protein using extracts from S2 cells expressing either Drosophila ERK alone or ERK in combination with MEK and Raf, which is sufficient to induce dpERK formation (Tipping et al., 2010), and streptavidin binding peptide (SBP)-tagged Cic (Figure 1B). Activation of ERK by MEK by dual phosphorylation is therefore sufficient to convert it into a form that has a higher affinity for Cic compared with unphosphorylated ERK.
FIGURE 1:

ERK phosphorylation in the activation loop is required and sufficient to induce strong binding to Cic. (A) An in vitro binding assay in which bacterially expressed purified rat ERK2 and dpERK2 were incubated with beads bound with Cic-V5 purified from S2 cells, analyzed by Western blotting. Cic-V5 strongly prefers dpERK2 over unphosphorylated ERK2. (B) An in vitro binding assay in which protein lysates from S2 cells cotransfected with Drosophila ERK-Flag, Raf, and MEK (dpERK-Flag) or transfected only with ERK-Flag were incubated with beads bound with separately expressed Cic-SBP, analyzed by Western blotting. Cic-SBP strongly prefers dpERK over unphosphorylated ERK. (C) Coimmunoprecipitation between Cic and ERK mutants in Drosophila S2 cells, analyzed by Western blotting with the indicated antibodies. Blocking the formation of dually phosphorylated ERK (dpERK) results in a lower affinity for Cic. Asterisks in B and C indicate endogenous (untagged) dpERK present in S2 cells. (D) Summary of Cic interactions with ERK and dpERK. Cic preferentially associates with dpERK.
To determine whether full activation of ERK is required for its increased affinity for Cic, we studied ERK/Cic binding using the T198A and Y200F ERK mutants that disrupt the TEY phosphorylation motif targeted by MEK (Canagarajah et al., 1997). As shown in Figure 1C, mutation of either residue impaired the binding between Cic and Drosophila ERK-Flag when both were coexpressed in S2 cells, relative to the wild-type enzyme, suggesting that full ERK activation and formation of dpERK is required for its highest affinity for Cic. We note that in this experiment coexpression of ERK with Cic resulted in the stabilization of the activated form of ERK, likely due to the effect of shielding of dpERK from the action of phosphatases in the Cic-dpERK complex (Kim et al., 2011). Collectively, these studies reveal that ERK phosphorylation is required and sufficient to induce a high-affinity interaction with Cic (Figure 1D).
Preferential binding of Cic to dpERK in vivo
To determine whether Cic preferentially associates with dpERK in vivo, we studied the binding between Cic-Venus (Grimm et al., 2012), which is expressed from genomic regulatory sequences at the endogenous level, and the endogenous ERK protein in 0- to 4-h Drosophila embryos. At this stage, dpERK is formed specifically at the embryonic termini downstream of RTK Torso activation (Gabay et al., 1997). Immunoprecipitation of Cic-Venus resulted in copurification of endogenous ERK, which also gave a strong dpERK signal (Figure 2A). To test whether dpERK is limiting in this assay, we generated additional dpERK via maternal expression of a dominant-active form of MEK, MEKE203K (Goyal et al., 2017a). In these embryos, dpERK was produced throughout the embryo, including the middle region, where dpERK is normally absent (Figure 2, B and C), while the total ERK level was unchanged (Figure 2A and Supplemental Figure S1). Remarkably, under these conditions, a higher amount of total ERK was associated with Cic-Venus, consistent with a higher overall level of dpERK in these embryos (Figure 2A). We note that the level of Cic-Venus was lower in MEKE203K-overexpressing embryos (Figure 2A, GFP signal), likely because of excessive degradation due to ERK hyperactivity (Goyal et al., 2017b). However, we still observed an increase in the amount of total ERK in our coimmunoprecipitation experiment, despite it being pulled down by this lower amount of Cic-Venus. In summary, Cic has a higher affinity for dpERK in vivo, and the amount of ERK bound to Cic is limited by the level of dpERK produced downstream of RTK activation.
FIGURE 2:
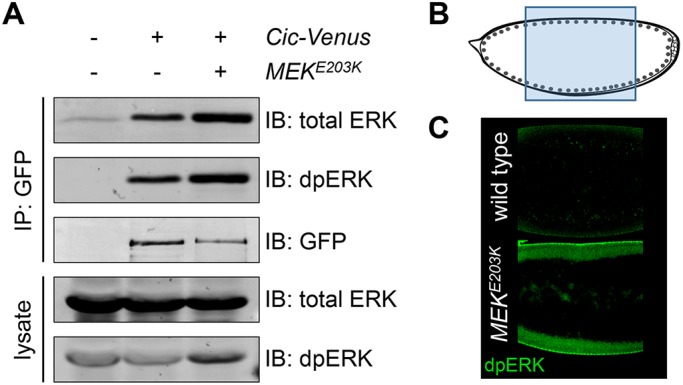
Cic preferentially associates with dpERK in vivo. (A) Extracts from embryos expressing Cic-Venus alone or Cic-Venus with activated MEK (MEKE203K) were immunoprecipitated with anti–green fluorescent protein (GFP) beads and analyzed by Western blotting. Up-regulation of ERK signaling is sufficient to increase ERK association with Cic in vivo. (B, C) Overexpression of activated MEK (MEKE203K) results in an increase of dpERK formation in the middle of the embryo. (B) Schematic diagram of the embryo, indicating the location of the images shown in C.
Preferential association of Cic with dpERK prevents competition with ERK and contributes to efficient signal propagation
Previous quantitative models of dpERK interactions with substrates assumed that dpERK is largely nuclear (Kim et al., 2011). However, several studies showed that both Cic and dpERK can localize in the cytoplasm in developing tissues (Roch et al., 2002; Astigarraga et al., 2007; Coppey et al., 2008; Grimm et al., 2012). We studied ERK and Cic localization in Drosophila cultured S2 cells under the basal conditions of limited ERK phosphorylation or in cotransfection with Raf and MEK, which generates large amounts of dpERK (Tipping et al., 2010). Transfection of Cic alone resulted in a predominantly cytoplasmic localization, with some nuclear signal (Figure 3, A–A″). Cotransfection of ERK with Raf and MEK led to the formation of dpERK, which was also localized mostly in the cytoplasm (Figure 3, B–B‴). Upon cotransfection of Cic with ERK, Raf, and MEK, the Cic and dpERK signals were still primarily cytoplasmic, with detectable but low nuclear staining (Figure 3, C–C‴). These data showed that Cic, ERK, and dpERK can coexist in the cytoplasm in S2 cells, which agrees with previously reported localization of Cic and dpERK in both the nucleus and the cytoplasm in vivo (Roch et al., 2002; Astigarraga et al., 2007; Coppey et al., 2008; Grimm et al., 2012). Subcellular localization of the two major ERK phosphatases in Drosophila is consistent with the importance of cytoplasmic regulation of dpERK: both Mkp3 and PTP-ER are cytoplasmic proteins (Karim and Rubin, 1999; Molnar and de Celis, 2013).
FIGURE 3:

Localization studies of Cic, ERK, and dpERK in S2 cells, and ERK phosphorylation analysis. In A–C‴, transfected expression constructs are shown on the left, and staining signals are shown on the individual panels. In A and C, dashed lines indicate nuclear boundaries. Scale bars, 5 µm. (A–A″) Cic-V5 was predominantly cytoplasmic, with some nuclear distribution. (B–B‴) When cotransfected with MEK and Raf, both ERK-Flag and dpERK signals were mostly cytoplasmic but also showed nuclear localization. (C–C‴) When Cic-V5 and ERK-Flag were cotransfected with Raf and MEK, the Cic-V5 and dpERK signals remained predominantly cytoplasmic, with some nuclear distribution. (D) Extracts from S2 cells transfected with vector control, ERK alone, or ERK together with MEK and RAF were analyzed by Western blotting. A representative blot of three independent experiments is shown. (E) Quantification of results in D. No significant down-regulation of unphospho-ERK signal was observed with cotransfection of Raf and MEK, despite a detectable up-regulation of dpERK. n = 3; n.s., not significant (p > 0.05).
We hypothesized that preferential association of Cic with dpERK may be important for preventing competition between ERK and dpERK for binding to Cic, when both ERK forms and Cic are localized in the same cellular compartment (cytoplasm). Such competition would occur if ERK was still present in significant amount after pathway activation. To determine whether the production of dpERK in the cells that are cotransfected with ERK, Raf, and MEK is limited, we analyzed extracts from S2 cells expressing ERK alone or ERK together with MEK and RAF by Western blotting with an antibody specific for unphosphorylated ERK (unphospho-ERK). We did not observe a significant down-regulation of the unphospho-ERK signal upon cotransfection with Raf and MEK, despite detecting robust formation of dpERK (Figure 3, D and E), suggesting that dpERK is present in limited amounts in these cells, and unphosphorylated ERK is still the predominant form. We also compared the normalized ratios of unphospho-ERK to total ERK by immunofluorescence of transfected S2 cells. These measurements showed that, upon cotransfection with Raf and MEK, the amount of unphosphorylated ERK was reduced from 1 to 0.75 (a 25% decrease), again indicating only limited dpERK formation in cells (Supplemental Figure S2).
To explore the consequences of colocalization of Cic, ERK, and dpERK, we formulated a mathematical model in which Cic and both the unphosphorylated and phosphorylated forms of ERK are localized in the same compartment (Figure 4A; see Supplemental Materials and Methods for model details). The model describes the conversion of an inactive form of ERK (E) to its active dually phosphorylated form, dpERK (E*), in the presence of active enzyme (MEK) that phosphorylates ERK and phosphatases that dephosphorylate ERK. Active enzyme E* binds to and promotes the degradation of its substrate (S), which is continuously synthesized and also undergoes intrinsic degradation when free or in any complex. Inactive enzyme E also binds S, but does not cause its degradation. In this model, an outcome of successful signal propagation is a reduction in the level of S (Figure 4A).
FIGURE 4:

Mathematical model of signaling from dpERK (E*) to Cic (S). Simulations were carried out under the conditions of colocalization of Cic, ERK, and dpERK in the same compartment. See text and Supplemental Materials and Methods for model details. (A) Diagram of main reactions. (B) Steady-state concentrations of the total amounts (free and in complexes) of E* and S for various values of β3 and β4. Smaller β3 indicates stronger substrate binding to E, while smaller β4 indicates stronger substrate binding to E*. In all panels, β4 = 10−2. Strong preferential association of E* with S (large value of β3/β4) is required for efficient degradation of S (blue curve) and generation of E* (red curve). (C) Steady-state concentrations of E* and S for different values of S0, the steady-state amount of S in the absence of input to the pathway. Here, β3 = 10−1. Low concentrations of S (Cic) result in inefficient formation of E* (dpERK). (D) Transients of E* and S for selected values of β3, corresponding to the vertical lines in B (β3 = 10−3, 10−2, and 10−1). The time courses were initialized from the steady state when there is no input, then at τ = 0, an input was introduced until τ = 5 (gray patch), at which time the input was removed, and the system relaxed back toward the initial steady state. (E, F) Model predictions of the steady-state dependence of e*tot and stot on β3 and β4, for β2 = 10−2 and with all other parameters the same as in B and C. Substantial activation of e*tot and degradation of stot requires that both β4 ≤ β3 and β4 ≤ β2.
We investigated the effects of the relative strengths of binding of substrate (S) to the inactive (E) and active (E*) forms of enzyme, which are controlled by the binding parameters β3 and β4, respectively. The β parameters in this model are Michaelis–Menten constants rescaled by the total ERK concentration and indicate the concentrations of unmodified (β3) or modified (β4) enzyme at which unbound substrate concentration falls due to binding or degradation. Therefore, smaller values of β3 and β4 indicate stronger interactions of Cic with the corresponding form of ERK; that is, smaller β3 indicates stronger binding of S to E, whereas smaller β4 indicates stronger binding of S to E*. At steady state, varying the ratio of β3/β4 revealed that strong preferential association of E* with S (large values of β3/β4) is required for efficient substrate down-regulation (Figure 4B, blue curve). In principle, inactive E could interact with S and compete with active E*. However, under this condition, the pathway cannot function efficiently, as shown by an inability of S to be degraded under small values of β3/β4 (Figure 4B, blue curve). Our simulations also showed that preferential association of S with E* is required even for the generation of significant amounts of active enzyme, E* (Figure 4B, red curve). As an extension of this requirement, only a small amount of activated enzyme is generated when concentration of S is low (Figure 4C). Dynamic simulations have confirmed the findings of steady-state analysis: efficient degradation of S and generation of E* was achievable at β3/β4 values approaching 10 (Figure 4D). A more complete exploration of the parameter space further confirmed these observations (Figure 4, E and F). Thus, preferential association of Cic with dpERK is required for efficient signal propagation in the presence of significant levels of unphosphorylated ERK, both in terms of the effects of active ERK on substrate degradation and for establishing high levels of ERK activation.
Cic-ERK interactions are critical for setting dpERK level and signaling in vivo
Given the importance of Cic-ERK interactions for ERK pathway activity, we investigated the effects of disrupting Cic-ERK binding at the level of ERK in vivo. Previous biochemical studies identified specific residues near the DRS domain in mammalian ERK that contribute to its interactions with the C2 domain of Drosophila Cic (Futran et al., 2015). One such residue is H123, whose mutation to alanine in mammalian ERK2 reduced the binding affinity to the C2 domain (Futran et al., 2015). This histidine residue is conserved and corresponds to H138 in Drosophila ERK. To test for the functional importance of this residue in vivo, we generated SBP-tagged variants of ERK that carried either the single H138A mutation, the activating mutation in the DRS domain, Sevenmaker (ERKSem, D334N), which impairs ERK binding to phosphatases (Bott et al., 1994; Brunner et al., 1994; Tanoue et al., 2000), or a combination of H138A and D334N within the same polypeptide. These ERK-SBP variants were expressed in vivo using the wing pouch MS1096-GAL4 driver (Capdevila and Guerrero, 1994). The development of wing veins is promoted by the activation of the epidermal growth factor receptor and dpERK, which relieves Cic-mediated repression of vein-specific genes and allows vein formation (Roch et al., 2002; Ajuria et al., 2011). Overexpression of wild-type ERK-SBP did not alter the normal venation pattern (Figure 5A), while overexpression of ERKSem-SBP promoted formation of ectopic veins (Figure 5B). This phenotype is likely caused by ectopic inhibition of Cic function by overactive ERK, outside the normal areas of vein formation (Roch et al., 2002; Ajuria et al., 2011). Interestingly, while overexpression of ERKH138A-SBP alone did not alter the normal venation pattern (Figure 5C), this mutation in combination with Sevenmaker within the same polypeptide (ERKSem/H138A-SBP) strongly suppressed the ability of the double-mutant protein to induce ectopic veins (Figure 5D). Therefore, the H138A mutation is completely dominant over the activating mutation ERKSem, when tested in cis within the same protein.
FIGURE 5:

The H138A mutation in cis suppresses the ability of ERKSem to induce ectopic veins. (A–D) Wings from adult female flies expressing indicated constructs under the control of the MS1096-GAL4 driver: (A) UAS-ERK-SBP, (B) UAS-ERKSem-SBP, (C) UAS-ERKH138A-SBP, and (D) UAS-ERKSem/H138A-SBP. Arrowheads in B mark ectopic veins. (E) Western blot showing ERK-SBP and dpERK levels in embryo extracts expressing indicated ERK isoforms under the control of the da-GAL4 driver.
The inability of ERKSem/H138A-SBP to induce ectopic veins might be interpreted solely through loss of binding to Cic, which is expected for the H138A mutation (Futran et al., 2015). However, it is possible that a reduction of binding to Cic would also lead to a reduction in steady-state level of dpERK due to the action of phosphatases (Kim et al., 2011), which would contribute to the inability of the double-mutant protein to induce ectopic veins. To distinguish between these possibilities, we compared steady-state phosphorylation levels of various SBP-tagged ERK mutants. Protein extracts from dissected larval wing disks did not provide sufficient dpERK for analysis on Western blots. We therefore analyzed dpERK levels of the ERK-SBP variants in Drosophila embryos using the da-GAL4 driver, which is expressed ubiquitously. Because all ERK-SBP variants were injected as a matching set into the same genomic location and provided equivalent levels of total ERK (Figure 5E), expression in the embryo can be used as a valid method of comparison of dpERK levels. The level of phosphorylation (dpERK signal) of the ERKSem-SBP protein was much higher than that of the wild-type ERK-SBP or ERKH138A-SBP (Figure 5E). Interestingly, the level of phosphorylation of the double-mutant ERKSem/H138A-SBP protein was much lower than that of ERKSem-SBP (Figure 5E). This result reveals two important properties of the mutant ERK proteins. First, it shows that the ERKSem protein is still susceptible to inactivation by phosphatases, because a reduction in Cic binding due to the H138A mutation resulted in a reduction in steady-state phosphorylation of ERKSem/H138A-SBP. Second, it suggests that the inability of the ERKSem/H138A-SBP protein to induce the ectopic vein phenotype is due to a combination of both reduced binding to Cic and a strong reduction in phosphorylation. These results underscore the importance of Cic-ERK interactions in vivo and highlight their importance for maintaining high levels of dpERK, which is required for pathway activity (seen here as formation of ectopic wing veins).
Conclusion
Previous studies have shown that substrates of ERK may have preferences for binding to either the active or the inactive form of ERK (Lee et al., 2004; Burkhard et al., 2011) and have mapped the dynamic ERK interactome (von Kriegsheim et al., 2009). Here, we established that phosphorylated ERK (dpERK) interacts with its substrate Cic more strongly than unphosphorylated ERK. Our data suggest that the preferred binding of Cic to dpERK is functionally important at two levels: first, it prevents a possible competition between ERK and dpERK for binding to Cic, when these proteins are localized in the same cellular compartment; second, it may allow for efficient signal propagation when only a small proportion of ERK is converted to dpERK. In support of the latter, we have shown that only a small fraction of ERK gets phosphorylated upon activation with Raf and MEK in cultured S2 cells (Figure 3 and Supplemental Figure S2), consistent with our previous results in vivo (Johnson et al., 2017).
In Drosophila, ERK-mediated Cic phosphorylation and down-regulation is involved in most developmental contexts that are under ERK control. Drosophila Cic interacts with dpERK through the C2 docking site, which is not well conserved in other species (Astigarraga et al., 2007; Futran et al., 2015). C2 domain-mediated down-regulation is therefore a unique property of fly Cic; however, the exact mechanism of Cic down-regulation upon dpERK binding is not fully understood. It likely involves phosphorylation of multiple sites in Cic upon binding of dpERK through the C2 domain (S.P., unpublished data). One of the potential mechanisms of Cic down-regulation is proteolytic degradation (Roch et al., 2002; Astigarraga et al., 2007; Suisse et al., 2017). A stronger binding of Cic to dpERK may thus represent a mechanism to ensure efficient phosphorylation and subsequent degradation of Cic, possibly still in the dpERK-bound state. This would ensure that dpERK is available for phosphorylating other substrates like Bicoid and Hunchback during embronic patterning, after Cic protein levels are reduced (Ronchi et al., 1993; Löhr et al., 2009; Kim et al., 2010). We have shown by mathematical modeling that preferential association of Cic with dpERK also contributes to the maintenance of a steady-state level of dpERK, which in turn is required for pathway output (i.e., Cic degradation), which is critical for the patterning of the embryonic termini (Astigarraga et al., 2007). Together, our results suggest that activation-induced high-affinity binding of dpERK to Cic is an important part of ERK signaling dynamics that can increase both the specificity and efficiency of signaling.
MATERIALS AND METHODS
Expression constructs, cell culture, and immunoprecipitations
Construction of C-terminally tagged full-length pMT-Cic-V5 and pMK33-Cic-SBP was described in Yang et al. (2016). Construction of C-terminally Flag-tagged Drosophila ERK(Rolled)-SBP was described in Yang and Veraksa (2017). Construction of amino terminally tagged HA-Raf, and carboxy terminally tagged MEK-V5 and ERK-Flag was described in Tipping et al. (2010). Site-directed mutagenesis was performed using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs) on pMT-ERK-Flag following the manufacturer’s protocol to generate ERKSem, ERKH138A, and ERKSem/H138A, which were then subcloned into pUAST-attB-SBP. Drosophila S2 cells were used for all cell-based assays. Cells were cultured at 25°C in standard Schneider’s S2 medium with 10% fetal bovine serum (Life Technologies) and 5% Pen/Strep (Invitrogen). For stable expression in S2 cells, a pMK33-Cic-SBP construct was transfected by using Effectene transfection reagent (Qiagen), and stable cell lines were selected in the presence of 300 μg/ml hygromycin (Sigma). Transient DNA transfections were performed using Effectene transfection reagent (Qiagen). At 24 h after transfection with indicated plasmids, cells were induced with 0.35 mM CuSO4 and incubated overnight to allow expression of the protein. Cells were harvested and then lysed with default lysis buffer (50 mM Tris, pH 7.5, 125 mM NaCl, 5% glycerol, 0.2% IGEPAL CA-630, 1.5 mM MgCl2, 1 mM dithiothreitol, 25 mM NaF, 1 mM Na3VO4, 1 mM EDTA) containing 2x Complete protease inhibitor (Roche). Cleared cell lysates were incubated with anti-V5 beads (Sigma) or Streptavidin beads (Pierce) at 4°C for 2 h. Beads were washed three times with default lysis buffer, and the protein complexes were eluted with SDS sample buffer. Immunoprecipitations using Cic-Venus–expressing embryos followed a similar protocol using GFP-Trap beads (Chromotek). All experiments were carried out at least twice, and representative results are shown.
Immunoblotting and immunostaining
Protein complexes were resolved on 8% SDS protein gels and transferred onto Millipore Immobilon-FL PVDF Transfer Membranes with 0.45-μm pores. Primary antibodies used for Western blots were as follows: mouse anti-dpERK 1:1000 (Sigma), rabbit anti–total ERK 1:1000 (Cell Signaling Technology), mouse anti-V5 1:1000 (Sigma), rabbit anti-Flag 1:1000 (Sigma), and mouse anti-SBP 1:1000 (Santa Cruz Biotechnology). Secondary antibodies used were as follows: IRDye 800CW donkey anti-rabbit immunoglobulin G (IgG) 1:20,000 (LI-COR) and IRDye 680CW donkey anti-mouse IgG, 1:20,000 (LI-COR). Primary antibodies used for S2 cell staining were as follows: mouse anti-dpERK 1:500 (Sigma), rabbit anti-V5 1:500 (Sigma), rabbit anti-Flag 1:500 (Sigma), and mouse anti–nonphosphorylated ERK 1:500 (Sigma). Secondary antibodies used were as follows: donkey anti-rabbit Alexa Fluor 488 1:500 (Invitrogen) and goat anti-mouse Alexa Fluor 555 1:500 (Invitrogen). Stained cells were mounted with Prolong Gold anti-fade mounting reagent with 4′,6-diamidino-2-phenylindole (Life Technologies), and images were acquired with Zeiss LSM 880 confocal microscope. Embryo staining was performed as in Lim et al. (2015). Rabbit antibody to total ERK (1:100 dilution) was used as primary antibody, Alexa Fluor 488 anti-rabbit conjugate (1:500 dilution; Invitrogen) or Alexa Fluor 568 anti-rabbit conjugate 1:500 (Invitrogen) was used as secondary antibody. All experiments were carried out at least twice, and representative results are shown. All statistical analyses were performed using Student’s t test.
Purification of unphosphorylated and phosphorylated ERK from bacteria
For the expression of ERK2, the plasmid encoding tagged rat ERK2 in pQE80 was transformed into Escherichia coli BL21(DE3)-competent cells. Overnight cultures were subcultured into 1 l LB (Luria–Bertani) medium supplemented with 100 μg/ml ampicillin to a starting OD600 of 0.02, and cultures were grown at 37°C with agitation at 250 rpm until they reached an OD600 of 1.0. Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and cultures were grown at 22°C for 6 h with agitation at 250 rpm. Bacterial cell pellets were harvested by centrifugation and stored at −20°C. For the expression of phosphorylated ERK2 (dpERK2), the plasmids encoding tagged rat ERK2 in pQE80 and tagged constitutively active (CA)-MEK1 (MEK1-G7B; Mansour et al., 1996) in pBAD33 were transformed into E. coli BL21(DE3)-competent cells. Overnight cultures were subcultured into 1 l LB medium supplemented with 100 μg/ml ampicillin and 25 μg/ml chloramphenicol to a starting OD600 of 0.02, and cultures were grown at 37°C with agitation at 250 rpm. At an OD600 of 0.8, 0.1% l-arabinose was added, and the temperature was shifted to 22°C. At OD600 1.0, ERK2 protein expression was induced with 1 mM IPTG, and cultures were grown at 22°C for 6 h with agitation at 250 rpm. Bacterial cell pellets were harvested by centrifugation and stored at −20°C. For all protein purifications, cell pellets were resuspended in 40 ml of 10 mM imidazole, 300 mM NaCl, and 50 mM NaH2PO4 (pH 8) and lysed by treatment with lysozyme and sonication on ice. Cell debris was removed by centrifugation, and the supernatant was sterile filtered. All proteins were purified from clarified lysate using Ni-NTA agarose resin (Qiagen) following the manufacturer’s recommendations. ERK2 was buffer exchanged into 50 mM HEPES, 100 mM NaCl, 20 mM MgCl2, and 10% glycerol (pH 7.4) using PD-10 desalting columns (Bio-Rad). Aliquots of 5–50 μl were snap frozen in liquid nitrogen and stored at −80°C.
In vitro binding assays
S2 cells were transfected with pMT-Cic-V5 or empty vector. Transfected cells were preincubated with 2 μM PD0325901, an MEK inhibitor (Biotang) with dimethyl sulfoxide as vehicle, for 3 h before induction. Cells were induced with 0.35 mM CuSO4 and incubated overnight. Extracts prepared as described earlier (except additional IGEPAL was added to default lysis buffer for a final concentration of 0.4%), and were incubated with anti-V5 beads for 2 h at 4°C. After three washes, the Cic-V5-bound beads were incubated with 500 ng of purified ERK2 or dpERK2 in default lysis buffer for 2 h at 4°C. Bovine serum albumin was added in binding solution to the final concentration of 0.05%, to reduce nonspecific binding to the beads.
Drosophila melanogaster stocks
Fly stocks and crosses were maintained on standard yeast–cornmeal–agar medium at 25°C or 18°C. MS1096-GAL4 was from the Bloomington Drosophila Stock Center. UAS-MEKE203K, Histone-GFP, and P{matα4-GAL-VP16}67, used as a maternal driver, were described in Goyal et al. (2017a). Wild-type Drosophila ERK (rolled) as well as ERK mutants, C-terminally tagged with SBP, were subcloned into pUAST-attB, and transgenic lines were generated by inserting the constructs into the attP40 genomic site by using ϕC31-based integration system (Venken et al., 2006; Bischof et al., 2007). All constructs were sent for injection together and therefore provide a matching set for comparisons of expression levels. Cic-Venus uses genomic regulatory sequences for expression and was described in Grimm et al. (2012).
Wing phenotypes
Transgenic male flies were crossed with MS1096-GAL4 virgins, and the wings of the resulting female progeny were imaged with Olympus BX60 compound microscope using bright-field illumination and a 4× objective.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center and the Vienna Drosophila Resource Centre for their services. We are grateful to Gerardo Jimenez for fruitful discussions and comments on the article. This work was supported by National Institutes of Health grant HD085870 (to A.V. and S.Y.S.). L.Y. and S.P. were supported by a UMass Boston Sanofi Genzyme Doctoral Fellowship.
Abbreviations used:
- Cic
Capicua
- dpERK
dually phosphorylated ERK
- DRS
D-site recruitment site
- ERK
extracellular signal-regulated kinase
- FRS
F-site recruitment site
- IgG
immunoglobulin G
- IPTG
isopropyl β- d-1-thiogalactopyranoside
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- RTK
receptor tyrosine kinase
- SBP
streptavidin binding peptide
- Sem
Sevenmaker.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-07-0355) on January 8, 2020.
REFERENCES
- Adachi M, Fukuda M, Nishida E. (1999). Two co-existing mechanisms for nuclear import of MAP kinase: passive diffusion of a monomer and active transport of a dimer. EMBO J , 5347–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajuria L, Nieva C, Winkler C, Kuo D, Samper N, José Andreu M, Helman A, González-Crespo S, Paroush Z, Courey AJ, Jiménez G. (2011). Capicua DNA binding sites are general response elements for RTK signaling in Drosophila. Development , 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astigarraga S, Grossman R, Diaz-Delfin J, Caelles C, Paroush Z, Jimenez G. (2007). A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J , 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. (2007). An optimized transgenesis system for Drosophila using germ-line-specific phi C31 integrases. Proc Natl Acad Sci USA , 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott CM, Thorneycroft SG, Marshall CJ. (1994). The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett , 201–205. [DOI] [PubMed] [Google Scholar]
- Brunner D, Oellers N, Szabad J, Biggs WH, 3rd, Zipursky SL, Hafen E. (1994). A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell , 875–888. [DOI] [PubMed] [Google Scholar]
- Burkhard KA, Chen F, Shapiro P. (2011). Quantitative analysis of ERK2 interactions with substrate proteins: roles for kinase docking domains and activity in determining binding affinity. J Biol Chem , 2477–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. (1997). Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell , 859–869. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Guerrero I. (1994). Targeted expression of the signaling molecule decapentaplegic induces pattern duplications and growth alterations in Drosophila wings. EMBO J , 4459–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppey M, Boettiger AN, Berezhkovskii AM, Shvartsman SY. (2008). Nuclear trapping shapes the terminal gradient in the Drosophila embryo. Curr Biol , 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, Zoghbi HY. (2011). Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science , 690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Gotoh Y, Nishida E. (1997). Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J , 1901–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futran AS, Kyin S, Shvartsman SY, Link AJ. (2015). Mapping the binding interface of ERK and transcriptional repressor Capicua using photocrosslinking. Proc Natl Acad Sci USA , 8590–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futran AS, Link AJ, Seger R, Shvartsman SY. (2013). ERK as a model for systems biology of enzyme kinetics in cells. Curr Biol , R972–R979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay L, Seger R, Shilo BZ. (1997). MAP kinase in situ activation atlas during Drosophila embryogenesis. Development , 3535–3541. [DOI] [PubMed] [Google Scholar]
- Goyal Y, Jindal GA, Pelliccia JL, Yamaya K, Yeung E, Futran AS, Burdine RD, Schupbach T, Shvartsman SY. (2017a). Divergent effects of intrinsically active MEK variants on developmental Ras signaling. Nat Genet , 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal Y, Levario TJ, Mattingly HH, Holmes S, Shvartsman SY, Lu H. (2017b). Parallel imaging of Drosophila embryos for quantitative analysis of genetic perturbations of the Ras pathway. Dis Model Mech , 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm O, Sanchez Zini V, Kim Y, Casanova J, Shvartsman SY, Wieschaus E. (2012). Torso RTK controls Capicua degradation by changing its subcellular localization. Development , 3962–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. (1999). Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev , 163–175. [PMC free article] [PubMed] [Google Scholar]
- Jimenez G, Guichet A, Ephrussi A, Casanova J. (2000). Relief of gene repression by Torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev , 224–231. [PMC free article] [PubMed] [Google Scholar]
- Jimenez G, Shvartsman SY, Paroush Z. (2012). The Capicua repressor—a general sensor of RTK signaling in development and disease. J Cell Sci , 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Ha N, Fores M, Xiang J, Glasser C, Maldera J, Jimenez G, Edgar BA. (2015). EGFR/Ras signaling controls Drosophila intestinal stem cell proliferation via Capicua-regulated genes. PLoS Genet , e1005634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HE, Goyal Y, Pannucci NL, Schupbach T, Shvartsman SY, Toettcher JE. (2017). The spatiotemporal limits of developmental Erk signaling. Dev Cell , 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. (1999). PTP-ER, a novel tyrosine phosphatase, functions downstream of Ras1 to downregulate MAP kinase during Drosophila eye development. Mol Cell , 741–750. [DOI] [PubMed] [Google Scholar]
- Kim Y, Coppey M, Grossman R, Ajuria L, Jimenez G, Paroush Z, Shvartsman SY. (2010). MAPK substrate competition integrates patterning signals in the Drosophila embryo. Curr Biol , 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Paroush Z, Nairz K, Hafen E, Jiménez G, Shvartsman SY. (2011). Substrate-dependent control of MAPK phosphorylation in vivo. Mol Syst Biol , 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. (2006). ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell , 1335–1347. [DOI] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. (2004). Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell , 43–55. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. (2010). Cell signaling by receptor tyrosine kinases. Cell , 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Dsilva CJ, Levario TJ, Lu H, Schupbach T, Kevrekidis IG, Shvartsman SY. (2015). Dynamics of inductive ERK signaling in the Drosophila embryo. Curr Biol , 1784–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Samper N, Lu H, Rushlow C, Jimenez G, Shvartsman SY. (2013). Kinetics of gene derepression by ERK signaling. Proc Natl Acad Sci USA , 10330–10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhr U, Chung H-R, Beller M, Jäckle H. (2009). Antagonistic action of Bicoid and the repressor Capicua determines the spatial limits of Drosophila head gene expression domains. Proc Natl Acad Sci USA , 21695–21700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Candia JM, Matsuura JE, Manning MC, Ahn NG. (1996). Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry , 15529–15536. [DOI] [PubMed] [Google Scholar]
- Molnar C, de Celis JF. (2013). Tay bridge is a negative regulator of EGFR signalling and interacts with Erk and Mkp3 in the Drosophila melanogaster wing. PLoS Genet , e1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch F, Jiménez G, Casanova J. (2002). EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development , 993–1002. [DOI] [PubMed] [Google Scholar]
- Ronchi E, Treisman J, Dostatni N, Struhl G, Desplan C. (1993). Down-regulation of the Drosophila morphogen bicoid by the torso receptor-mediated signal transduction cascade. Cell , 347–355. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr (2012). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res , 105–143. [DOI] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A. (2000). Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci , 448–453. [DOI] [PubMed] [Google Scholar]
- Simon-Carrasco L, Grana O, Salmon M, Jacob HKC, Gutierrez A, Jimenez G, Drosten M, Barbacid M. (2017). Inactivation of Capicua in adult mice causes T-cell lymphoblastic lymphoma. Genes Dev , 1456–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suisse A, He D, Legent K, Treisman JE. (2017). COP9 signalosome subunits protect Capicua from MAPK-dependent and -independent mechanisms of degradation. Development , 2673–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Brunetti L, Rousseaux MWC, Lu HC, Wan YW, Revelli JP, Liu Z, Goodell MA, Zoghbi HY. (2018). Loss of Capicua alters early T cell development and predisposes mice to T cell lymphoblastic leukemia/lymphoma. Proc Natl Acad Sci USA , E1511–E1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Yoshimoto T, Nakamura T. (2017). A double-edged sword: the world according to Capicua in cancer. Cancer Sci , 2319–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Adachi M, Moriguchi T, Nishida E. (2000). A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol , 110–116. [DOI] [PubMed] [Google Scholar]
- Tipping M, Kim Y, Kyriakakis P, Tong M, Shvartsman SY, Veraksa A. (2010). β-arrestin Kurtz inhibits MAPK and Toll signaling in Drosophila development. EMBO J , 3222–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng AS, Tapon N, Kanda H, Cigizoglu S, Edelmann L, Pellock B, White K, Hariharan IK. (2007). Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr Biol , 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. (2006). P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science , 1747–1751. [DOI] [PubMed] [Google Scholar]
- von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, et al (2009). Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol , 1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Paul S, Trieu KG, Dent LG, Froldi F, Fores M, Webster K, Siegfried KR, Kondo S, Harvey K, et al (2016). Minibrain and wings apart control organ growth and tissue patterning through down-regulation of Capicua. Proc Natl Acad Sci USA , 10583–10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Veraksa A. (2017). Single-step affinity purification of ERK signaling complexes using the streptavidin-binding peptide (SBP) tag. Methods Mol Biol , 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.