

Abstract
Spontaneous DNA-PKcs deficiencies in animals result in a severe combined immunodeficiency (SCID) phenotype because DNA-PKcs is required to activate Artemis for V(D)J recombination coding end hairpin opening. The impact on signal joint formation in these spontaneous mutant mammals is variable. Genetically engineered DNA-PKcs null mice and cells from them show a > 1,000-fold reduction in coding joint formation and minimal reduction in signal joint formation during V(D)J recombination. Does chemical inhibition of DNA-PKcs mimic this phenotype? M3814 (also known as Nedisertib) is a potent DNA-PKcs inhibitor. We find here that M3814 causes a quantitative reduction in coding joint formation relative to signal joint formation. The sequences of signal and coding junctions were within normal limits, though rare coding joints showed novel features. The signal junctions generally did not show evidence of resection into the signal ends that is often seen in cells that have genetic defects in DNA-PKcs. Comparison of the chemical inhibition findings here with the known results for spontaneous and engineered DNA-PKcs mutant mammals is informative for considering pharmacologic small molecule inhibition of DNA-PKcs in various types of neoplasia.
Keywords: Immunoglobulin gene rearrangement, ArtemisV(D)J recombination, Site-Specific recombination
1. Introduction
There has been increasing interest in targeting DNA dependent protein kinase (DNA-PK) as a means to treat cancer because of its role in DNA repair (Harnor et al., 2017). DNA-PK is comprised of a Ku (consisting of Ku70 and Ku80) and the kinase catalytic domain (DNA-PKcs). Upon binding to a duplex DNA end, DNA-PKcs phosphorylates itself, which results in a conformational change, which in turn regulates the activity of Artemis (Ma and Lieber, 2002; Gu et al., 2010). DNA-PKcs forms a stable complex with Artemis in vivo even if DNA is not present (Ma et al., 2002). Artemis intrinsically has exonuclease activity but gains endonuclease activity when in complex with activated DNA-PKcs (Li et al., 2014). DNA-PKcs and Artemis are key enzymes in nonhomologous end joining (NHEJ) and V(D)J recombination (Lieber, 2010). Inhibiting DNA-PKcs to impair cells from repairing DNA damage is one possible strategy for inducing death in cancer cells, if there is an adequate therapeutic window.
DNA-PKcs inhibitors have been described over the past twenty years. Wortmannin is a naturally occurring, irreversible inhibitor of DNA-PKcs (IC50 = 120 nM) (Izzard et al., 1999). However, wortmannin more potently inhibits PI3K activity (IC50 = 4.2 nM) and thus, is not adequately selective (Powis et al., 1994). Synthetic inhibitors, such as LY294002, were developed against PI3K, but its analogs steadily improved in potency and selectivity for DNA-PKcs (Vlahos et al., 1994). NU7441 was among the most potent and most selective of these. NU7441 had an IC50 of 14 nM against DNA-PKcs and exhibited over 100-fold selectivity against related enzymes such as mTOR and PI3K (1.7 μM and 5 μM IC50, respectively) (Leahy et al., 2004; Zhao et al., 2006). As promising as they seem, many of these early DNA-PKcs inhibitors suffered from poor pharmacokinetic properties, such as metabolic lability (Davidson et al., 2013). Since then, small molecule inhibitors with improved properties have been developed against DNA-PKcs. CC-115, AZD7648 and M3814 are three DNA-PKcs inhibitors currently in or entering clinical trials. CC-115 inhibits mTOR and DNA-PKcs at 21 and 13 nM IC50, respectively, with 40-fold selectivity over other PIKK family members (Tsuji et al., 2017). Phase I/II trials are in progress (NCT02833883, NCT01353625, NCT02977780). AZD7648 inhibits IR-induced DNA-PKcs S2056 autophosphorylation with an IC50 of 92 nM in A549 cells and has shown synergistic effects with other drugs such as doxorubicin. A Phase I/II trial is currently being organized (NCT03907969). M3814 (also known as MSC2490484A or Nedisertib) has an IC50 of 46 nM against DNA-PKcs and sensitizes cells to radiation therapy (Harnor et al., 2017). Multiple Phase I trials for M3814 have been completed or are currently recruiting (NCT03724890, NCT03770689, NCT02516813, NCT02316197, NCT03983824).
It is interesting to compare chemical inhibition of DNA-PKcs with genetically deficient cells. Genetic mutants can arise spontaneously in nature or be engineered. Engineered DNA-PKcs null mice have diminished coding joint formation but normal signal joint formation (Taccioli et al., 1993; Gao et al., 1998a; Kurimasa et al., 1999). Spontaneous DNA-PKcs deficiencies in mice, horses, dogs, and humans have been previously described (Figs. 1 & 2). In scid mice, DNA-PKcs is truncated 83 amino acids from the C-terminus by a nonsense mutation that forms an ochre stop codon (Blunt et al., 1996). A drop is observed in both DNA-PKcs expression and kinase activity, and coding joint formation is severely diminished whereas signal joint formation is less affected (Blunt et al., 1995). Equine DNA-PKcs deficiency is caused by 5-bp deletion that creates a frameshift and a stop codon, truncating the enzyme 967 amino acids from the C-terminus, which spans the entire phosphatidylinositol 3-kinase domain (Wiler et al., 1995; Shin et al., 1997). In this equine scid, there is no detectable DNA-PKcs expression or kinase activity, and both signal and coding joint formation are diminished (Shin et al., 1997). Canine DNA-PKcs deficiency is caused by a nonsense mutation that truncates 517 amino acids at the C-terminal end of the protein (Meek et al., 2001; Bell et al., 2002). DNA-PKcs expression and kinase activity are both diminished in Jack Russell terriers (Meek et al., 2001). This DNA-PKcs deficiency leads to markedly reduced coding and signal joints, to an intermediate degree between murine and equine SCID.
Fig. 1. Spontaneous DNA-PKcs Mutants.

A summary of DNA-PKcs deficiencies that have been reported in various species. (WT = wild type; signal and coding joint data gathered from designated references (Blunt et al. (1996); Wiler et al. (1995); Shin et al., 1997; Meek et al., 2001; Woodbine et al. (2013); Lieber et al. (1988b)).
Fig. 2. Engineered DNA-PKcs Mutants.
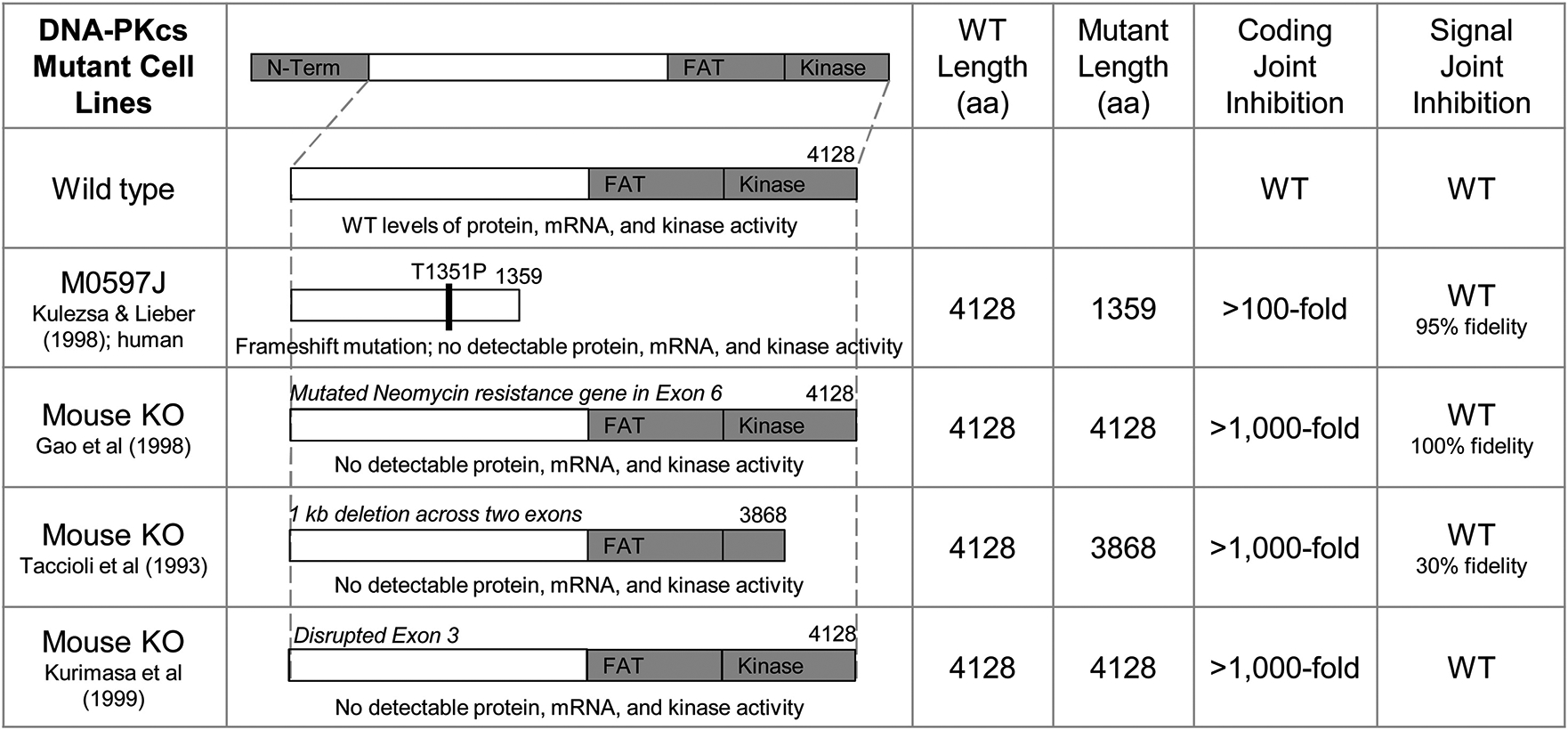
A summary of genetically engineered DNA-PKcs null cell lines. (WT = wild type; signal and coding joint data gathered from designated references (Taccioli et al. (1993); Kurimasa et al. (1999); Gao et al. (1998b); Kulesza and Lieber (1998)).
Two instances of spontaneous DNA-PKcs deficiency have been reported in humans. In the first patient, a missense mutation (L3062R) in the FAT domain caused a block in B-cell differentiation as is typically seen when Artemis or RAG are defective, indicating a primary defect in V(D)J recombination (van der Burg et al., 2009a; van der Burg et al.,2009b). DNA-PKcs expression and kinase activity were not affected in this case, which stands in contrast to observations in spontaneous and engineered DNA-PKcs deficiency in various animal species, where kinase activity and expression were lost. The presence of long coding joint P-nucleotides in this SCID patient suggests that faulty Artemis hairpin opening was the mechanism of disease, even though the defect itself was found in DNA-PKcs (van der Burg et al., 2009b). In the second patient, an A3574 V mutation presented with a more severe phenotype, with global neurological impairment in addition to classical SCID complications (Woodbine et al., 2013). This patient had low DNA-PKcs expression and kinase activity, resulting in impaired DNA repair and severely diminished coding joint formation. In all these mammalian examples, the detrimental mutations impacted the C-terminal end of DNA-PKcs. With such a large enzyme, it is not surprising that there are a wide variety of mechanisms by which DNA-PKcs function may be inhibited, and each has its own unique disease features.
In this study, we determine the extent to which the chemical inhibition of DNA-PKcs is similar or different relative to the spontaneous or engineered knockouts. We have used a gel-based assay to confirm DNA-PKcs inhibition by M3814 in a purified biochemical system. A human cellular V(D)J recombination assay was used to probe mechanistically whether the inhibition of DNA-PKcs would affect signal and/or coding joint formation. Our results indicate a decrease in coding joint formation relative to signal joint formation upon treatment with M3814, similar to several genetic knockouts of DNA-PKcs, but with interesting qualitative differences in the junctional sequences (Gao et al., 1998a; Kurimasa et al., 1999; Lieber et al., 1988a).
2. Materials and methods
2.1. Chemicals
M3814 (aka Nedisertib) was obtained from ChemieTek (Catalog # CT-M3814). The compound was solvated in DMSO to a stock concentration of 10 mM and divided into smaller aliquots (50–100 μL) to decrease the incidence of freeze-thaw cycles. Aliquots were stored at −20 °C and protected from light until use.
2.2. DNA-PKcs kinase assay
A p53 peptide was used as a phosphorylation substrate for DNA-PKcs. Each 10 μl reaction contains 20 nM DNA-PKcs, 250 μM p53 peptide, 1 μM dsDNA (to activate DNA-PKcs), and 80 nM [γ−32P]ATP in 25 mM Tris−HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, and 5 % sucrose. Reactions were incubated at 37 °C for 10 min. The reaction was stopped by the addition of an appropriate volume of 6X SDS Loading Buffer (0.35 M Tris, 30 % glycerol, 10 % SDS, 603 mM DTT, and 0.012 % bromophenol blue) and incubation at 95 °C for 5 min. Reactions were resolved with a 15 % SDS-PAGE at 90 V for 75 min.
2.3. Artemis nuclease assay and oligonucleotides
Full-length Artemis (Gu et al., 2010; Li et al., 2014) and DNA-PKcs (Ma et al., 2002; Chan et al., 1996) were purified as previously described. A 1:1 ratio of Artemis and DNA-PKcs was incubated together on ice for 10 min prior to addition of M3814 and other reaction components. This pre-incubation step ensures that the Artemis is active upon encountering its DNA substrate. The Artemis substrate, called ZE16, is fluorescently labeled at the 5′ end, and the first four nucleotides are modified with phosphorothioate bonds (*) to prevent nuclease de-gradation (5′-FAM-T*T*T*T*TT TTG CCA GCT GAC GCG CGT CAG CTG GC-3′). This substrate anneals to create an 8-nt 5′ overhang and a 12-bp double-stranded hairpin portion. Each 10 μL reaction contains 500 nM fluorescently labeled substrate, 100 μM ATP, and 10 nM Artemis:DNA-PKcs complex in 25 mM Tris−HCl, pH 8.0, 10 mM KCl, 10 mM MgCl2, and 1 mM DTT. Reactions were incubated at 37 °C for 30 min. The reaction was stopped by the addition of an equal volume of 1:1 40 % glycerol: formamide and incubation at 95 °C for 5 min. Reaction products were resolved with an 18 % denaturing PAGE at 70 W for 90 min.
The radiolabeled version of the Artemis nuclease assay uses a similar substrate (ZE16) as described above but without the fluorescent group at the 5′ end (5′-T*T*T*T*TT TTG CCA GCT GAC GCG CGT CAG CTG GC-3′). In this case, each 10 μL reaction contains 40 nM radiolabeled substrate, 200 μM ATP, and 10 nM Artemis: DNA-PKcs complex in 25 mM Tris−HCl, pH 8.0, 10 mM KCl, 10 mM MgCl2, and 1 mM DTT. Reactions were incubated at 37 °C for 50 min. The reaction was stopped by the addition of an equal volume of loading dye (98 % for-mamide, 20 mM EDTA, 0.025 % bromophenol blue, and 0.025 % xylene cyanol) and incubation at 95 °C for 5 min. Reactions were resolved with a 12 % denaturing PAGE at 300 V for 120 min. A 32 nt marker oligonucleotide called GW35 (5′-FAM TT TTT TTA CTG AGT CCT ACA GAA GGA TCG TAG-3′) and an 8 nt marker oligonucleotide called FAM-8 T (5′-FAM-T*T*T*T*T*T*TT-3′) were used to aid gel mobility position. The ZE16 nicked hairpin product contains all of the nucleotides of ZE16 except that Artemis has nicked 2 nt 3′ of the hairpin tip, and the nucleotides GTCAGCTGGC are no longer present on that product.
For assays of wortmannin and NU7441 inhibition, the radiolabeled substrate (SL23) sequence was 5′-T*T*TT*TTTGCCAGCTGACGC/GCGT CAGCTGGC-3′, where the / marks the hairpin tip, and the nicked hairpin product is missing the following 3′ nucleotides 5′ GTCAGCTGGC.
2.4. Cellular V(D)J recombination assay
The cellular V(D)J recombination assay measures replication and recombination on extrachromosomal DNA transfected into V(D)J positive cells (Gauss and Lieber, 1992a; Gauss and Lieber, 1993). Pre-B-ALL cells (697) were used in a cellular V(D)J recombination assay as previously described (Gauss and Lieber, 1992a; Gauss and Lieber, 1993). The plasmids designed for this assay confer ampicillin (Amp) resistance upon transformation in bacteria. Once these plasmids undergo recombination, they form either a signal joint (SJ) or coding joint (CJ) and gain an additional resistance to chloramphenicol (Cam). The 697 cells were transfected with these plasmids via DEAE-dextran-electroporation (Gauss and Lieber, 1992b) and then harvested for analysis after 48 h. This assay allows for the determination of % replication (%r) and % recombination (%R). Replication is determined by taking the ratio of DpnI-sensitive (DpnI Amp, DA) plasmids versus total plasmid (mock Amp, mA) population (DA/A). Recombination is determined by taking the ratio of the Amp (A) colonies versus AmpCam (AC) colonies and dividing by % replication. Typically, in untreated cells, the CJ/SJ ratio is about 0.3 (Gauss and Lieber, 1993).
Briefly, the plasmids used here generate either a signal joint (SJ) or coding joint (CJ) upon recombination (Gauss and Lieber, 1993). Mid-log phase 697 cells were transfected with one of these plasmid substrates using DEAE-dextran electroporation and incubated at 37 °C with 5 % CO2 for 48 h. In this time, a fraction of the substrates experience rearrangement via V(D)J recombination. Plasmids were recovered by alkaline lysis and transformed into DH10B cells, which were then plated on LB agar plates with ampicillin (Amp, 100 μg/mL) or ampicillin and chloramphenicol (AmpCam, 100 μg/mL and 25 μg/mL, respectively). Plasmid substrates were initially amplified in E. coli and thus are DpnI-sensitive. Plasmids that replicated in the mammalian cells post-transfection during the 48-hr incubation are not dam methylated and are DpnI-resistant. Mock- (mA) and DpnI-digested (DA) plasmids were transformed into DH10B and plated on LB Amp to determine % replication (%r, ratio of colonies on DA versus colonies on mA). Plasmid substrates confer Amp resistance while the recombinant plasmid is resistant to both Amp and Cam. Plasmids were transformed into DH10B and plated on LB Amp (A) and AmpCam (AC) to determine % recombination (%R, ratio of colonies on AC versus colonies on A). Additionally, AmpCam colonies were sequenced to determine junction fidelity. Single colonies were grown in LB overnight, and plasmids were extracted via standard isolation methods. Both signal and coding joint plasmids were sequenced (Genewiz) using the following primer: 5′-GTT GAC AAT TAA TCA TCG AAC TAG TTA ACT-3′.
3. Results
3.1. Chemical inhibition of DNA-PKcs inhibits kinase activity
M3814 is stated to be a potent DNA-PKcs inhibitor with an IC50 of 46 nM (Harnor et al., 2017), but there is currently no published primary data illustrating this. We were interested in establishing this point using kinase assays. A kinase assay using p53 peptide as the phosphorylation substrate was used to determine whether M3814 inhibits DNA-PKcs kinase activity. A range of concentrations up to 1.6 μM M3814 was tested. [γ−32P] ATP was used to detect phosphorylation. Inhibition of kinase activity was determined by comparing the amount of phosphorylated-p53 product in each sample relative to the DMSO control (Fig. 3). Here, we show a gradual, dose-dependent inhibition of kinase activity that correlates with the increase in M3814 concentration used in each sample. The IC50 for this kinase assay is roughly 50 nM, which is very close to the reported IC50 of 46 nM (Harnor et al., 2017). Thus, chemical inhibition of DNA-PKcs inhibits its kinase activity.
Fig. 3. M3814 Inhibits DNA-PKcs Kinase Activity.
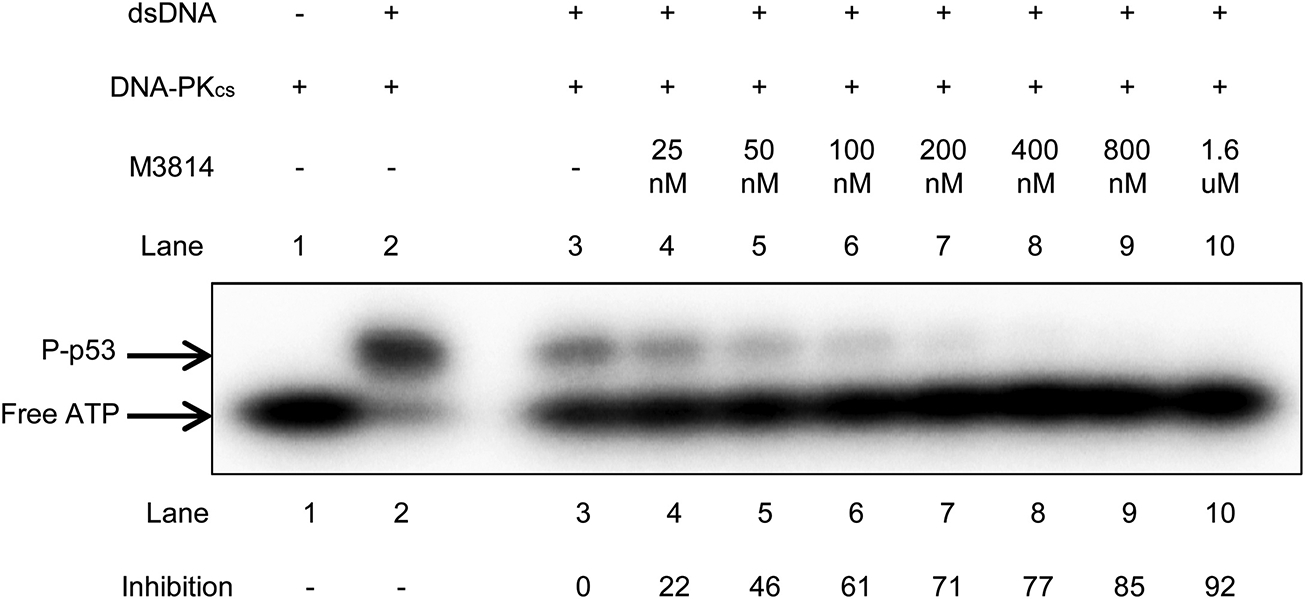
A p53 peptide was used to measure DNA-PKcs kinase activity in the presence of M3814. DNA-PKcs (20 nM) was incubated with p53 peptide (250 μM), 1 μM dsDNA, and 80 nM [γ−32P] ATP at37 °C for 10 min. in 25 mM Tris−HCl (pH 7.5), 10 mM MgCl2,10 mM DTT, and 5 % sucrose. Inhibition was measured by comparing the amount of P-p53 in each sample relative to the DMSO control (Lane 3).
3.2. Chemical inhibition of DNA-PKcs inhibits biochemical Artemis activity
We used a purified Artemis nuclease assay to confirm DNA-PKcs inhibition by M3814. Artemis cleaves single-strand to double-strand junctions or boundaries. The substrate for this assay has an 8-nt 5′ overhang and a 12-bp duplex region that has a hairpin at one end. Artemis cleaves the 5′ overhang off, thereby making that DNA end blunt. Activated Artemis also nicks the hairpin end of the substrate 2 nt 3′ of the hairpin tip.
Artemis is complexed with DNA-PKcs under physiological conditions (Ma et al., 2002), and thus DNA-PKcs inhibition should cause Artemis inhibition as well (Ma et al., 2002; Goodarzi et al., 2006). Full-length Artemis is active in biochemically purified systems containing Mg2+ when DNA-PKcs and ATP are present. Other DNA-PKcs inhibitors have been tested in this assay (Supp. Fig. 1), and each compound was able to inhibit Artemis activity. Here, we show that the inhibition of DNA-PKcs by M3814 is able to inhibit the ability of Artemis to cleave its substrate (Fig. 4). This inhibition is detected with as low as 50 nM M3814. Thus, chemical inhibition of DNA-PKcs with ≥50 nM M3814 inhibits Artemis endonuclease activity.
Fig. 4. M3814 Inhibits Artemis:DNA-PKcs Complex.
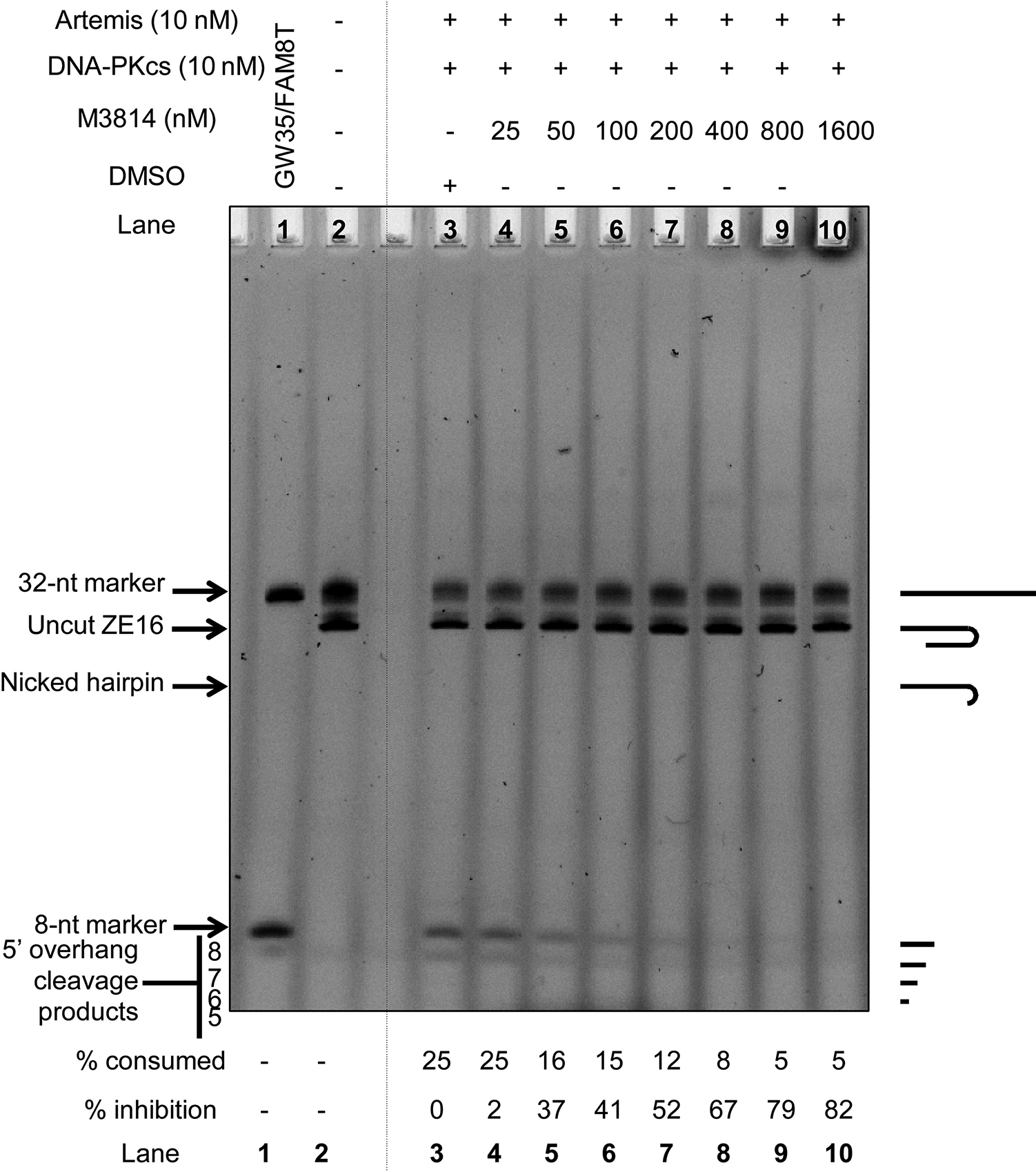
A fluorescently-labeled substrate (500 nM) was used to determine whether M3814 can inhibit Artemis (10 nM) by inhibiting DNA-PKcs (10 nM) (4). Reactions were performed in 25 mM Tris−HCl, pH 8.0, 10 mM KCl, 10 mM MgCl2, and 1 mM DTT incubated for 30 min at 37 °C. DNA-PKcs was treated with M3814, and inhibition was measured by comparing substrate-to-product conversion relative to the DMSO control (Lane 3). A visual description of substrates and products is provided on the right-hand side of the figure, and the sequences are in the Methods.
3.3. Chemical inhibition of DNA-PKcs reduces coding joint formation in a dose-dependent manner
The human pre-B 697 cells were treated with 0.5 μM, 1 μM, or 1.5 μM M3814 during transfection with V(D)J recombination substrates, and the drug was maintained during the 48 h incubation (Table 1). In order for substantial amounts of V(D)J recombination to be observed in this assay, the SV40 replicons on these substrates must drive their extrachromosomal replication. If DNA replication is reduced, as evidenced by reduced substrate replication in this assay, then this is one measure of a general metabolic toxicity of a drug or chemical compound.
Table 1. Inhibits V(D)J Recombination in 697 Cells.
Artemis: DNA-PKcs is required to open double-stranded DNA hairpins generated during V(D)J recombination. Inhibiting DNA-PKcs would prevent this action and result in a decrease in CJ formation and perhaps SJ formation in V(D)J recombination. This assay measures % replication and % recombination in 697 cells upon treatment with 0.5 μM, 1 μM, and 1.5 μM M3814. Values marked with an asterisk (*) are significant compared to the DMSO control (p < 0.05).
| Signal Joint | Coding Joint | ||||
|---|---|---|---|---|---|
| M3814 | % replication | % recombination | % replication | % recombination | |
| 0.5 μM | − | 4.0 | 7.0 | 2.8 | 1.02 |
| + | 5.6 | 3.2 | 6.7 | 0.18* | |
| 1 μM | − | 6.2 | 7.9 | 10.3 | 1.5 |
| + | 7.4 | 1.1* | 10.1 | < 0.004* | |
| 1.5 μM | − | 16.7 | 4.0 | 26.1 | 0.6 |
| + | 7.7 | 0.34* | 8.1* | 0.003* | |
No effect on substrate replication was observed with 0.5 μM M3814 treatment (Table 1). SJ and CJ recombination were also not affected.
Treating with 1 μM M3814 did not affect substrate replication but did cause a significant inhibition of V(D)J recombination (Table 1). SJ formation was reduced 8-fold while CJ formation was reduced more severely by over 300-fold.
Treating with 1.5 μM M3814 caused a 12-fold reduction in SJ, and a 200-fold reduction in CJ (Table 1). Thus, our data indicate that M3814 is capable of inhibiting coding joint formation in V(D)J recombination by acting on DNA-PKcs. The inhibition of signal joint formation is clear, but the SJ inhibition is not as severe as for coding joint formation.
This V(D)J cellular assay was also conducted on Nalm6 wildtype and Artemis−/− cells in the complete absence of any drug (Supp. Table 1). Here, the genetic ablation of Artemis did not significantly affect replication in any of the conditions tested. Signal joint formation was no different between wildtype and Artemis−/− cells. In wildtype cells, coding joint formation was about 8-fold lower than signal joint formation, consistent with the modest differences we have seen previously (Gauss and Lieber, 1993). However, a significant difference was observed in Artemis−/− cells, showing a marked > 300-fold less coding joint formation compared to signal joint formation (p < 0.00001). Thus, chemical inhibition of DNA-PKcs and genetic Artemis mutation both significantly reduce coding joint formation while only causing a mild reduction in signal joint formation.
3.4. Chemical inhibition of DNA-PKcs does not affect local resection at the signal or coding joint
In WT cells, signal ends in V(D)J recombination typicallyrarely show resection, and even in these cases, the resection is only 1 or 2 nts. But in DNA-PKcs deficient mice, Artemis is able to resect into signal ends more often, resulting in signal joints that manifest such local resection from either of the two signal ends (Lieber et al., 1988a; Touvrey et al., 2008).
Interestingly, here, upon DNA-PKcs chemical inhibition, signal joints were precise and did not show signal end resection (Table 2). Coding joints from the treated cells also showed the normal range of coding end resection seen in WT cells, which is < 15 nts from each coding end. One coding joint appeared to include the heptamer from the RSS on the anti-parallel strand (Table 2), for which we suggest possible models (Suppl. Figs. 2 & 3).
Table 2. Does Not Affect Junction Fidelity.
Recombinant plasmids containing signal or coding joints were sequenced to determine the fidelity of junction formation. Tables 2a and 2b show sequences collected from the 0.5 μM and 1.5 μM V(D)J recombination data sets, respectively.
| a. 0.5 micromolar M3814 V(D)J Recombination | |||
|---|---|---|---|
| Signal Junctions | |||
| Sample | CACTGTG | Junctional Additions | CACAGTG |
| 1 | CACTGTG | CACAGTG | |
| 2 | CACTGTG | CACAGTG | |
| 3 | CACTGTG | CACAGTG | |
| 4 | CACTGTG | CACAGTG | |
| 5 | CACTGTG | TCCCG | CACAGTG |
| 6 | CACTGTG | GG | CACAGTG |
| 7 | CACTGTG | TGGCTT | CACAGTG |
| Coding Junctions | |||
| Sample | TGCAGGTCGAC | Junctional Additions | GGATCCCCGGG |
| 1 | TGCAGGTC— | -GATCCCCGGG | |
| 2 | TGCAGGTC— | C | -GATCCCCGGG |
| 3 | TGCAGG—— | CC | -GATCCCCGGG |
| 4 | TGCAGGTCG– | CC | GGATCCCCGGG |
| 5 | TGCAGGTCG– | CC | GGATCCCCGGG |
| b. 1.5 micromolar M3814 V(D)J Recombination | |||
| Signal Junctions | |||
| Sample | CACTGTG | Junctional Additions | CACAGTG |
| 1 | CACTGTG | CACAGTG | |
| 2 | CACTGTG | GG | CACAGTG |
| 3 | CACTGTG | CC | CACAGTG |
| 4 | CACTGTG | GCC | CACAGTG |
| 5 | CACTGTG | CACAGTG | |
| 6 | CACTGTG | CACAGTG | |
| 7 | CACTGTG | G | CACAGTG |
| Coding Junctions | |||
| Sample | TGCAGGTCGAC | Junctional Additions | GGATCCCCGGG |
| 1 | TGCAGGTC— | -GATCCCCGGG | |
| 2 | TGCAGGTCGAC | ——CCCCGGG | |
| 3 | TGCAGGTCGAC | ——CCCCGGG | |
| 4 | TGCAGGTCGA- | —TCCCCGGG | |
| 5 | TGCAGGTCGAC | ——CCCCGGG | |
| 6 | TGCAGGTCGAC | CACTGTG | GGATCCCCGGG |
| 7 | TGCAGGTCGA- | —TCCCCGAG | |
| 8 | TGCAGGTCGAC | ——CCCCGTG | |
| 9 | TGCAGGTCGAC | GGATCCCCGGG | |
| 10 | TGCAGGTCGA- | —TCCCCGGG | |
| 11 | TGCAGGTCGAC | GGATCCCCGGG | |
| 12 | TGCAGGTC— | –ATCCCCGGG | |
| 13 | TGCAGGTC— | -GATCCCCGGG | |
| 14 | TGCAGGT—— | -GATCCCCGGG | |
| 15 | TGCAG——— | C | -GATCCCCGGG |
| 16 | TGCAGGTCGAC | ——CCCCGGG | |
| 17 | TGCAG———— | CCCT | GGATCCCCGGG |
| 18 | TGCAGGTCGAC | ——CCCCGGG | |
Thus, our data from this extrachromosomal assay indicate that the chemical inhibition of DNA-PKcs does mimic mammalian DNA-PKcs mutants in that coding joint formation is reduced more severely than signal joint formation. Though DNA-PKcs deficiency is often associated with greater than normal signal end resection, that is not the case here based on the fact that nearly all the signal junctions are precise.
4. Discussion
4.1. Summary of M3814 effects on signal and coding joint formation
Here, we study the chemical inhibition of DNA-PKcs with M3814. DNA-PKcs null cells have been shown to be radiosensitive (Gao et al.,1998b), as are cells treated with NU7441 (Goodarzi et al., 2006). However, studies of the effect of DNA-PKcs inhibitors on Artemis activity have been limited (Goodarzi et al., 2006), and we are aware of no studies that have used a DNA-PKcs inhibitor that compare biochemical and concomitant cellular V(D)J recombination assays.
We demonstrated that M3814 inhibited DNA-PKcs kinase activity (Fig. 3). In our purified biochemical nuclease assay, M3814-inhibited DNA-PKcs reduced Artemis conversion of substrate to product (Fig. 4). Thus, M3814 was able to inhibit the function of DNA-PKcs in regulating Artemis endonuclease activity in a purified system. In cellular assays of V(D)J recombination, consistent with the murine genetic KO and with some spontaneous scid mammalian mutants, M3814 markedly inhibited coding joint formation, even at the low end of clinically-utilized blood concentrations (1 μM). We find that signal joint formation was inhibited at slightly higher drug concentrations, but not as dramatically as coding joint formation (Table 1). DNA-PKcs-deficient human M059J cells were able to form signal joints at comparable levels to control cells, but they showed markedly reduced coding joint formation (Kulesza and Lieber, 1998). Chemically inhibiting DNA-PKcs with M3814 shows features that are intermediate between the various spontaneous and engineered animal models of DNA-PKcs deficiency. Like human and murine scid mutants, chemical inhibition has little or only small effects (8- to 12-fold) on signal joint formation efficiency. But coding joint efficiency is reduced substantially more (> 200-fold). This predominant reduction in coding joint formation is consistent with our data from genetic mutants of Artemis (Supp. Table 1). Additionally, signal end sequences are precisely preserved (Table 2). This is in contrast to observations in many DNA-PKcs deficient mammalian cells, where Artemis resects into the signal end (Touvrey et al., 2008). The quantitative effects of M3814 on signal and coding joint formation, while more similar to the case in human and murine DNA-PKcs mutant models, is different from that seen in the canine and equine spontaneous mutants, in which severe reductions in both coding and signal joint formation were seen (Wiler et al., 1995; Shin et al., 1997).
4.2. Biochemical and molecular biology complexities related to inhibition
The complexities of enzyme inhibition are substantial for this system. First, DNA-PKcs deficiency presents differently across various mammalian species. The degree of inhibition, mechanism of inhibition, and features of inhibition vary due to the location of spontaneous mutations within the DNA-PKcs gene and possibly on the species. Second, chemical inhibition of DNA-PKcs with a small molecule does not necessarily mimic observations found in genetic knockouts or spontaneous DNA-PKcs deficient species. Third, there may be multiple modalities for the small molecule inhibition of DNA-PKcs, and these may also result in variations in phenotype. It is important to be aware of these different possibilities especially when targeting a specific mechanism of action for therapeutic purposes.
Various forms of DNA-PKcs deficiency have been characterized in different species. In DNA-PKcs deficient mice, signal joint formation is unaffected while coding joint formation is diminished. In contrast, equine and canine DNA-PKcs deficiencies markedly reduce both signal and coding joint formation (Wiler et al., 1995; Shin et al., 1997). The DNA-PKcs deficiencies in these three species vary in protein expression levels and kinase activity as well as the causative mutation and degree of protein truncation present in each one. This observation is also observed for other NHEJ proteins. For example, the Ku80 C-terminus is not required for recruiting DNA-PKcs to DNA ends or activating DNA-PKcs, but a C-terminal deletion still causes sensitivity to ionizing radiation and diminished end joining ability (Weterings et al., 2009). In this case, decreased autophosphorylation at T2647 was observed whereas S2056, the classical DNA-PKcs autophosphorylation marker, was unaffected (Weterings et al., 2009). Since DNA-PKcs is such a large protein, there are many phosphorylation sites and a large surface area at which small molecule inhibitors may interact. It is not surprising that there are multiple modes of inhibition that could affect its function. Likewise, it follows that each of these modes may result in different phenotypes. For example, the missense mutation in one known human patient does not affect kinase expression, activity, or ability to recruit Artemis to DNA ends but it still causes a SCID phenotype. In contrast, the C-terminal truncation in mice, horses, and dogs causes a loss of expression and kinase activity. Furthermore, a different human DNA-PKcs mutation resulted in severe neurological impairment in addition to SCID, and unlike the first human DNA-PKcs deficient patient, there were only residual levels of protein expression and kinase activity. In considering DNA-PKcs inhibition as a potential means to treat cancer, it is important to keep such complexities in mind to guide drug development towards an optimal mechanism of action.
There is much more that needs to be understood about the structure and function of DNA-PKcs. One of the recorded human DNA-PKcs mutations presents just as classical SCID would, even though expression and kinase activity, and Artemis recruitment to DNA ends were unaffected (van der Burg et al., 2009a; van der Burg et al., 2009b). This implies that it is not the absence or impairment of DNA-PKcs itself but rather its indirect effect on Artemis function that causes the SCID phenotype. This also suggests that the ability of DNA-PKcs to form a complex with and recruit Artemis to DNA damaged ends is independent from its ability to regulate the activity of Artemis. This latter function is the critical one to inhibit in terms of preventing DNA repair in cancer cells. For example, M3814 is a DNA-PKcs inhibitor that likely affects kinase activity but may not affect the portion of the enzyme that interacts with Artemis. At this point, the detailed mechanism of action for M3814 has yet to be published by Merck/EMD Serono. In our purified biochemical system, where the Artemis: DNA-PKcs complex forms prior to adding M3814, inhibition of Artemis activity was detected upon addition of M3814 (Fig. 4). But in the vastly more complex environment of the cell, M3814 may not sufficiently inhibit DNA-PKcs to the point where cancer cells are selectively eliminated due to the accumulation of DNA damage.
It is also possible that while DNA-PKcs activity is inhibited relative to its full potential, the residual level of kinase activity post-M3814 treatment is sufficient for Artemis to function. This provides further justification for inhibiting the Artemis nuclease directly, from a therapeutic standpoint, instead of modulating the DNA-PKcs in hopes of attenuating Artemis activity. In support of this view, use of the M3814 chemical on human pre-B cells (Nalm6) that have an Artemis KO causes more cell death than either DNA-PKcs inhibition or lack of Artemis alone (Supp. Fig. 4).
4.3. Complexities of V(D)J recombination related to inhibition
We observed no resection into the signal ends in samples treated with the DNA-PKcs inhibitor. When DNA-PKcs is genetically deficient, resection into the signal joint is often observed, and it is substantial evidence that Artemis is responsible for this resection (Touvrey et al., 2008). One possible explanation of this is that chemical inhibition only affects a subset of the Artemis:DNA-PKcs complexes in the cell, whereas genetic mutants involve a change in all of the complexes in the cell. The signal joints that we observe may arise primarily from the subset of Artemis: DNA-PKcs complexes that were not inhibited.
We did, however, observe a preferential reduction in coding joint efficiency at the lower concentration of M3814 (0.5 μM), and a reduction in both signal and coding joint efficiency at the two higher concentrations (1 and 1.5 μM). From the earlier work demonstrating signal end resection in DNA-PKcs mutants and work demonstrating Artemis is responsible for the signal end resection, this had already suggested that both Artemis and DNA-PKcs are in proximity to the signal ends during V(D)J recombination (Touvrey et al., 2008). Now that we can observe a quantitative reduction in coding joint formation at low DNA-PKcs inhibitor concentrations and a reduction in both signal and coding joint formation at higher inhibitor concentrations, this adds to the view that Artemis and DNA-PKcs are in proximity to all four DNA ends during V(D)J recombination.
Supplementary Material
Acknowledgements
We thank Dr. Noritaka Adachi for the Nalm 6 Artemis wild type and Artemis null cells.
Funding
This work was supported by the National Institutes of Health (GM118009, CA196671, CA100504).
Abbreviations:
- SJ
Signal joint
- CJ
coding joint
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- IC50
inhibitory concentration for 50% inhibition
Footnotes
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.molimm.2020.01.018.
References
- Bell TG, et al. , 2002. Autosomal recessive severe combined immunodeficiency of JackRussell terriers. J. Vet. Diagn. Invest 14 (3), 194–204. [DOI] [PubMed] [Google Scholar]
- Blunt T, et al. , 1996. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc. Natl. Acad. Sci. U.S.A 93, 10285–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunt T, et al. , 1995. Defective DNA-Dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80, 813–823. [DOI] [PubMed] [Google Scholar]
- Chan DW, et al. , 1996. Purification and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem. Cell Biol 74, 67–73. [DOI] [PubMed] [Google Scholar]
- Davidson D, et al. , 2013. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front. Pharm 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, et al. , 1998a. A targeted DNA-PKcs-null mutation reveals DNA-PK independent functions for Ku in V(D)J recombination. Immunity 9, 367–376. [DOI] [PubMed] [Google Scholar]
- Gao Y, et al. , 1998b. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95, 891–902. [DOI] [PubMed] [Google Scholar]
- Gauss GH, Lieber MR, 1993. Unequal signal and coding joint formation in human V(D)J recombination. Mol. Cell. Biol 13, 3900–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss GH, Lieber MR, 1992a. The basis for the mechanistic bias for deletional over inversional V(D)J recombination. Genes Dev. 6, 1553–1561. [DOI] [PubMed] [Google Scholar]
- Gauss G, Lieber MR, 1992b. DEAE-dextran enhances electroporation of mammalian cells. Nucl. Acids Res 20, 6739–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, et al. , 2006. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 25 (16), 3880–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, et al. , 2010. DNA-PKcs regulates a single-stranded DNA endonuclease activity ofArtemis. DNA Repair (Amst) 9 (4), 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnor SJ, Brennan A, Cano C, 2017. Targeting DNA-Dependent protein kinase forCancer therapy. ChemMedChem 12 (12), 895–900. [DOI] [PubMed] [Google Scholar]
- Izzard RA, Jackson SP, Smith GC, 1999. Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res. 59 (11), 2581–2586. [PubMed] [Google Scholar]
- Kulesza P, Lieber MR, 1998. DNA-PK is essential only for coding joint formation in V(D)J recombination. Nucl. Acids Res 26, 3944–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimasa A, et al. , 1999. Catalytic subunit of DNA-PK: impact on lymphocyte development and tumorigenesis. Proc. Natl. Acad. Sci 96, 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy JJ, et al. , 2004. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett 14 (24), 6083–6087. [DOI] [PubMed] [Google Scholar]
- Li S, et al. , 2014. Evidence that the DNA endonuclease ARTEMIS also has intrinsic 5’-exonuclease activity. J. Biol. Chem 289 (11), 7825–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, et al. , 1988a. Lymphoid V(D)J recombination: nucleotide insertion at signal joints as well as coding joints. Proc. Natl. Acad. Sci 85, 8588–8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, et al. , 1988b. The defect in murine severe combined immune deficiency: joining of signal sequences but not coding segments in V(D)J recombination. Cell 55, 7–16. [DOI] [PubMed] [Google Scholar]
- Ma Y, Lieber MR, 2002. Binding of inositol hexakisphosphate (IP6) to Ku but not toDNA-PKcs. J. Biol. Chem 277, 10756–10759. [DOI] [PubMed] [Google Scholar]
- Ma Y, et al. , 2002. Hairpin opening and overhang processing by an Artemis:DNA-PKcs complex in V(D)J recombination and in nonhomologous end joining. Cell 108, 781–794. [DOI] [PubMed] [Google Scholar]
- Meek K, et al. , 2001. SCID in Jack Russell terriers: a new animal model of DNA-PKcs deficiency. J. Immunol 167 (4), 2142–2150. [DOI] [PubMed] [Google Scholar]
- Powis G, et al. , 1994. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 54 (9), 2419–2423. [PubMed] [Google Scholar]
- Shin EK, Perryman LE, Meek K, 1997. Evaluation of a test for identification ofArabian horses heterozygous for the severe combined immunodeficiency trait. J. Am. Vet. Med. Assoc 211 (10), 1268–1270. [PubMed] [Google Scholar]
- Taccioli GE, et al. , 1993. Impairment of V(D)J recombination in double-strand break repair mutants. Science 260, 207–210. [DOI] [PubMed] [Google Scholar]
- Touvrey C, et al. , 2008. Distinct effects of DNA-PKcs and Artemis inactivation on signal joint formation in vivo. Mol. Immunol 45 (12), 3383–3391. [DOI] [PubMed] [Google Scholar]
- Tsuji T, et al. , 2017. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 8 (43), 74688–74702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, et al. , 2009a. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest 119(1), 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, van Dongen JJ, van Gent DC, 2009b. DNA-PKcs deficiency in human: long predicted, finally found. Curr. Opin. Allergy Clin. Immunol 9 (6), 503–509. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, et al. , 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem 269 (7), 5241–5248. [PubMed] [Google Scholar]
- Weterings E, et al. , 2009. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol. Cell. Biol 29 (5), 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiler R, et al. , 1995. Equine severe combined immunodeficiency: a defect in V(D)J recombination and DNA-dependent protein kinase activity. Proc Natl Acad Sci U S A 92 (25), 11485–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, et al. , 2013. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Invest 123 (7), 2969–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. , 2006. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 66 (10), 5354–5362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.