

Abstract
Tissue-nonspecific alkaline phosphatase (TNAP) is necessary for skeletal mineralization by its ability to hydrolyze the mineralization inhibitor inorganic pyrophosphate (PPi), which is mainly generated from extracellular ATP by ectonucleotide pyrophosphatase phosphodiesterase 1 (NPP1). Since children with TNAP deficiency develop bone metaphyseal auto-inflammations in addition to rickets, we hypothesized that TNAP also exerts anti-inflammatory effects relying on the hydrolysis of pro-inflammatory adenosine nucleotides into the anti-inflammatory adenosine. We explored this hypothesis in bone metaphyses of 7-day-old Alpl+/− mice (encoding TNAP), in mineralizing hypertrophic chondrocytes and osteoblasts, and non-mineralizing mesenchymal stem cells (MSCs) and neutrophils, which express TNAP and are present, or can be recruited in the metaphysis. Bone metaphyses of 7-day-old Alpl+/− mice had significantly increased levels of Il-1β and Il-6 and decreased levels of the anti-inflammatory Il-10 cytokine as compared with Alpl+/+ mice. In bone metaphyses, murine hypertrophic chondrocytes and osteoblasts, Alpl mRNA levels were much higher than those of the adenosine nucleotidases Npp1, Cd39 and Cd73. In hypertrophic chondrocytes, inhibition of TNAP with 25 μM of MLS-0038949 decreased the hydrolysis of AMP and ATP. However, TNAP inhibition did not significantly modulate ATP- and adenosine-associated effects in these cells. We observed that part of TNAP proteins in hypertrophic chondrocytes was sent from the cell membrane to matrix vesicles, which may explain why TNAP participated in the hydrolysis of ATP but did not significantly modulate its autocrine pro-inflammatory effects. In MSCs, TNAP did not participate in ATP hydrolysis nor in secretion of inflammatory mediators. In contrast, in neutrophils, TNAP inhibition with MLS-0038949 significantly exacerbated ATP-associated activation and secretion of IL-1β, and extended cell survival. Collectively, these results demonstrate that TNAP is a nucleotidase in both hypertrophic chondrocytes and neutrophils, and that this nucleotidase function is associated with autocrine effects on inflammation only in neutrophils.
Keywords: tissue-nonspecific alkaline phosphatase, ATP, inflammation, nucleotidase, hypophosphatasia
1. Introduction
Ossification proceeds by two different processes [1]. Long bones are formed by endochondral ossification, during which growth plate cartilage is first mineralized and progressively replaced by new bone. Alternatively, flat bones grow by intramembranous ossification where there is no need for a cartilage template. In both processes, extracellular matrix mineralization is a relatively simple biological process that merely requires the coexpression of two broadly expressed proteins, a fibrillar collagen (type II collagen in growth plate cartilage and type I collagen in bone) and tissue-nonspecific alkaline phosphatase (TNAP) [2]. Mutations in the genes encoding type II collagen are associated with different forms of abnormal growth known as type II collagenopathies [3], and those that occur in type I collagen genes with osteogenesis imperfecta [4]. On the other hand, mutations in ALPL, the gene encoding human TNAP, lead to hypophosphatasia (HPP). Based on disease severity, HPP has been divided into 5 major subtypes: the perinatal form, the infantile form, the childhood form, the adult form and odontohypophosphatasia [5, 6]. The perinatal form is lethal at birth or soon after. In stillborns, bones can appear nearly devoid of minerals. In the infantile form, symptoms usually appear during the first six months, in particular with radiographic evidence of widespread demineralization and rachitic changes in the metaphyses, often leading to lethality. The childhood form is associated with dental problems, delayed walking and bone fractures. The adult forms manifest by osteomalacia and premature tooth loss [6]. The pro-mineralizing function of TNAP relies on its ability to hydrolyze extracellularly the mineralization inhibitor inorganic pyrophosphate (PPi) [2, 7]. Intracellularly formed PPi can be exported by the transporter progressive protein ankylosis (ANK), but the major fraction of extracellular PPi is likely generated extracellularly from adenosine triphosphate (ATP) by the transmembrane enzyme ectonucleotide pyrophosphatase phosphodiesterase −1 (NPP1) [8]. TNAP, attached by a GPI-anchor to the membrane of mineralizing cells, is transferred with its GPI anchor to the extracellular collagen matrix in the membrane of so-called matrix vesicles (MVs) to initiate collagen mineralization [9].
TNAP has at least one other substrate and function. TNAP dephosphorylates pyridoxal phosphate into pyridoxal to participate in vitamin B6-dependent reactions, in particular GABA synthesis [6]. This function explains in part why both HPP patients and Alpl−/− mice experience epileptic seizures [6, 10]. PPi and pyridoxal phosphate are probably not the only TNAP substrates in vivo since in vitro, TNAP has the ability to hydrolyze many different phosphorylated compounds [11]. Moreover, the expression of TNAP in many different locations such as liver, kidney, blood or brain suggests that TNAP exerts other functions, possibly by hydrolyzing other substrates. Thorough examination of the symptoms of the childhood form of HPP suggests a role for TNAP in inflammation. Indeed, cases of chronic recurrent multifocal osteomyelitis (CRMO), a sterile auto-inflammatory disease [12], have been reported independently by the groups of MP Whyte and H Girschick in unrelated HPP children who suffered chronic, multifocal, periarticular pain and soft tissue swelling [5, 13]. Magnetic resonance imaging (MRI) evidenced bone marrow edema in these children, and non-steroidal anti-inflammatory drugs diminished their pain [5, 13–16]. More recently, whole-body MRI of four children with the childhood form of HPP revealed bone metaphysis inflammation in all of them [14]. It was therefore proposed that chronic bone inflammation should be considered an additional feature of HPP [5].
CRMO inflammation in childhood HPP develops at the metaphysis [14]. This is where TNAP is the most active during growth, being expressed by mineralizing hypertrophic chondrocytes and mature osteoblasts. We hypothesized that metaphysis inflammation in HPP is due to the pathological persistence of high extracellular ATP, normally hydrolyzed by TNAP by these cells. ATP can indeed be secreted by virtually all cells, through transporters such as connexins and pannexins, in response to inflammatory molecules, mechanical stress, or danger signals [17]. ATP then binds to P2 purinergic receptors to trigger intracellular signaling pathways leading to pro-inflammatory responses. ATP for instance binds to P2X7 receptor to activate the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, which cleaves pro-interleukin-1β (pro-IL-1β) into mature IL-1β and triggers its secretion through the cleavage and activation of the pore-forming protein gasdermin D [18]. ATP also binds to P2Y receptors to delay the apoptosis of neutrophils [19] and activate their migration [20]. Two ubiquitous ectonucleotidases, CD39 and CD73, are involved in adenosine nucleotide dephosphorylation to control inflammation in a time- and location-dependent manner [17]. CD39 removes two Pi from ATP to generate adenosine monophosphate (AMP), which is further hydrolyzed into adenosine by CD73. Adenosine then binds to P1 receptors to activate anti-inflammatory responses. Several recent articles suggested that TNAP may be able to hydrolyze adenosine nucleotides in vivo. In the blood, TNAP is suspected to limit inflammation by its ability to dephosphorylate AMP into adenosine [21]. TNAP also appears to generate adenosine in the brain and the spinal cord [10, 22]. In our study, we explored the putative anti-inflammatory functions of TNAP using mouse and human mesenchymal and hematopoietic cells, wild-type Alpl+/+ mice, and TNAP-deficient Alpl+/− mice. To our knowledge, bone inflammation has never been explored in TNAP-deficient mice, but it was reported that Alpl−/− mice suffer from allodynia and hyperalgesia [23].
2. Materials and methods
2.1. Animals and sample collection
Alpl+/+ and Alpl+/− mice [24] were bred in the CERCO animal facilities in accordance with the Guide for the Care and Use of Laboratory Animals and the guidelines of the local institutional animal care and use committee. Study was approved by the Regional (Midi Pyrénées) Ethics Committee (MP/06/79/11/12). Femurs and tibias were extracted from 7-day-old male animals (8 Alpl+/+ and 3 Alpl+/−) in the morning. Immediately after mouse dissection, femurs and tibias were frozen in liquid nitrogen. Metaphysis-containing bone samples were grinded in a liquid nitrogen cooled mortar, and RNA was extracted as described below. Mice genotypes were specified a posteriori using tail samples as previously published [10].
2.2. Cell cultures
Primary osteoblasts and chondrocytes were isolated from newborn (4–6 days) SWISS mice by successive enzymatic digestions of calvaria, and articular cartilage from femoral head and knee, respectively [25]. Animal experimentations were conducted according to French and European laws and approved by our local ethic committee (approval numbers A 69266 0501 and BH2012–63). The animal procedures were performed conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Cells were routinely cultured at 37°C in a humidified atmosphere with 5% of CO2 in Dulbecco’s Modified Eagle Medium (DMEM) (4.5 g/L glucose) supplemented with 10% (v/v) fetal calf serum (FCS), penicillin (100 U/mL), streptomycin (100 μg/mL), 20 mmol/L Hepes, and 2 mmol/L L-glutamine. Culture media were changed every three days. To stimulate cell differentiation, ascorbic acid (50 μg/mL) was added in both cell culture at confluence; to induce mineralization, β-glycerophosphate (10 mM) was added at confluence in chondrocytes and 6 days after confluence in osteoblasts [25].
MSCs from 4 donors were used [a 34-year old female and 22-, 23- and 36-year-old males (Lonza, Walkersville, USA; certified positive for CD29, CD44, CD105 and CD166, and negative for CD14, CD34 and CD45)]. MSCs were seeded at a density of 5,000 cells per cm2 and routinely cultured in DMEM containing 10% (v/v) FCS, penicillin (100 U/mL), streptomycin (100 μg/mL), 20 mmol/L Hepes, and 2 mmol/L L-glutamine. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2 in air. Cells were subcultured at approximately 80–90% confluence with trypsin/EDTA. To induce osteoblast differentiation, medium was replaced at confluence by an osteogenic medium, consisting of DMEM with 10% FCS, containing 10 nM of 1,25(OH)2D3, 50 μg/mL of ascorbate and 10 mM of β-GP [26].
Neutrophils were obtained from peripheral blood from 4 healthy adult donors (one 45 year-old male and 3 females aged 36, 54 and 55). Blood cells were separated using a density gradient centrifugation (Pancoll human, P04–60500, PAN Biotech) as described by the manufacturer. Red blood cells and neutrophils were then separated in the presence of 3% dextran and red blood cells were finally lysed (BD Pharm lyse, BD Bioscience). Neutrophils were cultured in DMEM containing 2% of FCS and treated with 0.5 μg/mL of lipopolysaccharide (LPS) O111:B4 from Escherichia coli (from Sigma) for 3 hours and then with 2 mM of ATP for 45 min.
In all cells, TNAP inhibition was achieved with 25 μM MLS-0038949 (from Merck) [27], since we determined in preliminary experiments that this dose was efficient to fully inhibit the hydrolysis of para-nitrophenylphosphate (pNPP) by bone cells. ARL-67156 was added at 100 μM to inhibit CD39 and NPP1 [28] and AOPCP [adenosine-5’-O-[(phosphonomethyl) phosphonic acid)] at 100 μM to inhibit CD73 [29]. ARL-67156 and AOPCP were from Santa-Cruz.
2.3. RNA extraction, reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNAs were extracted from grinded bones using the TRI reagent (Sigma), and from cells using the NucleoSpin RNA II kit (Macherey-Nagel) following the manufacturers’ protocols. DNA was eliminated from bone RNA by DNAse from Roche. 1 μg of RNA was retro-transcribed into cDNA with Superscript II reverse transcriptase (Life Technologies) and quantitative PCR was performed using a CFX-96 system (Biorad). Primers and PCR conditions are given in Table 1. The obtained products were checked by sequencing. Relative quantification was performed using the 2−ΔCq method by the CFX Manager software (Biorad).
Table 1: Summary of primers used.
Shown are the mouse (m) and human (h) primer sequences (F: forward; R: reverse), annealing temperatures (Ta), base pair (bp) lengths of the corresponding PCR products, and GenBank accession numbers.
| Gene | GenBank | Ta (°C) | Sequence | Length (bp) |
|---|---|---|---|---|
| mGapdh | NM_008084.3 | 60 | F: 5’- GGCATTGCTCTCAATGACAA-3’ | 200 |
| R: 5’-TGTGAGGGAGATGCTCAGTG-3’ | ||||
| mAlpl | NM_007431.3 | 60 | F: 5’-CAAAGGCTTCTTCTTGCTGGT-3’ | 258 |
| R: 5’-AAGGGCTTCTTGTCCGTGTC-3’ | ||||
| mBglap | NM_001037939.2 | 60 | F: 5’-AAGCAGGAGGGCAATAAGGT-3’ | 364 |
| R: 5’-CGTTTGTAGGCGGTCTTCA-3’ | ||||
| mCol10a1 | NM_009925.4 | 60 | F: 5’-CAAACGGCCTCTACTCCTCTGA-3’ | 129 |
| R: 5’-CGATGGAATTGGGTGGAAAG-3’ | ||||
| mIl-10 | NM_010548.2 | 60 | F: 5’-CCCTTTGCTATGGTGTCCTTTC-3’ | 100 |
| R: 5’-GATCTCCCTGGTTTCTCTTCCC-3’ | ||||
| mIl-1β | NM_008361.4 | 62 | F: 5’-GGGCCTCAAAGGAAAGAATC-3’ | 152 |
| R: 5’-CCACTTTGCTCTTGACTTCTATC-3’ | ||||
| mIl-6 | NM_031168 | 60 | F: 5′-GTCACAGAAGGAGTGGCTA-3′ | 193 |
| R: 5’-AGAGAACAACATAAGTCAGATACC-3’ | ||||
| mIL1-ra | NM_031167 | 60 | F: 5′-GGGATACTAACCAGAAGACC-3’ | 157 |
| R: 5’-GACAGGCACAGCTTGCCCCC-3’ | ||||
| mTnf-α | NM-013693 | 60 | F: 5′-TGGGACAGTGACCTGGACTGT-3’ | 67 |
| R: 5’-TTCGGAAAGCCCATTTGAGT-3’ | ||||
| mNpp1 | NM_001308327.1 | 60 | F: 5′-CGGACGCTATGATTCCTTAGA-3′ | 93 |
| R: 5’-AGCACAATGAAGAAGTGAGTCG-3’ | ||||
| mCd39 | NM_001304721.1 | 60 | F: 5’-AGGTGAAGAGATTTTGCTCCAA-3’ | 101 |
| R: 5’-TTTGTTCTGGGTCAGTCCCAC-3’ | ||||
| mCd73 | NM_011851.4 | 60 | F: 5’-GGACATTTGACCTCGTCCAAT-3’ | 191 |
| R: 5’-GGGCACTCGACACTTGGTG-3’ | ||||
| hRPLP0 | NM_001002.3 | 60 | F: 5’-CGACCTGGAAGTCCAACTAC-3’ | 289 |
| R: 5’-AGCAACATGTCCCTGATCTC-3’ | ||||
| hGAPDH | NM_002046.5 | 60 | F: 5’-GTTCCAATATGATTCCACCC-3’ | 487 |
| R: 5’-AGGGATGATGTTCTGGAGAG-3’ | ||||
| hALPL | NM_000478.4 | 60 | F: 5’-CAAAGGCTTCTTCTTGCTGGT-3’ | 258 |
| R: 5’-AAGGGCTTCTTGTCTGTGTC-3’ | ||||
| hNPP1 | NM_006208.2 | 60 | F: 5’-CGATTTTGCCGATTGAGGATT-3’ | 146 |
| R: 5’-AAACTGGTGCTGGGAAAGAAGACA-3’ | ||||
| hCD39 | NM_001776.5 | 60 | F: 5’-CTGATTCCTGGGAGCACATC-3’ | 99 |
| R: 5’-CTGGGATCATGTTGGTCAGG-3’ | ||||
| hCD73 | NM_002526.3 | 60 | F: 5’-CTCCTCTCAATCATGCCGCT-3’ | 221 |
| R: 5’-CAAATGTGCCTCCAAAGGGC-3’ |
2.4. Measurement of ATP and AMP hydrolysis
Osteoblasts and chondrocytes were cultured for 16 days as described above. Then, cells were incubated in Tris-HCl buffer (pH = 7.4) containing MgCl2 (1 mM), CaCl2 (1 mM) and the substrate (10 μM ATP or 25 μM AMP) at room temperature. Aliquots were collected at different time points and Pi release was determined by Malachite Green assay as described elsewhere [30]. The results were expressed as μM of Pi by comparing to a standard curve of Pi, and initial reaction rates were determined from the curves representing Pi concentrations versus time.
2.5. Quantification of extracellular ATP and intracellular cAMP
Osteoblasts and chondrocytes were differentiated as indicated above. Culture media were removed and replaced with serum-free DMEM with or without TNAP inhibitor (25 μM MLS-0038949). Aliquots of media were collected every 5 minutes and extracellular ATP levels were measured using the Promega ATP assay kit (ENLITEN Luciferase/Luciferin reagent) and read in the luminometer Fluoroskan Ascent® 1506450 (ThermoLabsystems). After differentiation, chondrocytes were cultured for 24 h in DMEM containing 0.1% BSA instead of FCS. AMP and Ro 20–1724, a phosphodiesterase inhibitor, were then added to the medium. At different time points, intracellular cAMP was measured by ELISA (Enzo Life Sciences), according to the manufacturer’s instructions.
2.6. Measurement of TNAP activity
For the determination of TNAP activity using pNPP as substrate [30], cells were harvested in 0.2% (V:V) Nonidet P-40 and disrupted by sonication. TNAP specific activity was expressed as nmol of p-nitrophenolate formed/min/mg of protein.
2.7. MV purification
MVs were purified as described in details [31]. Hypertrophic chondrocytes were incubated with collagenase from Clostridium histolyticum (200 U/mL, type IA; Sigma) in a synthetic cartilage lymph (SCL) buffer at pH 7.4, at 37°C for 3 h. The digests were centrifuged at 800 g and 30,000 g during 30 min at 4°C to remove cell debris and microsomes, respectively. The supernatants were centrifuged at 250,000 g during 30 min to pellet the MVs, followed by re-suspension in SCL buffer (pH 7.4). In addition to these collagen-associated MVs, extracellular collagen-free vesicles and exosomes were isolated according to the protocol by Wuthier and collaborators [32]. After culture the medium was collected and centrifuged at 1,000 g during 30 min to remove apoptotic bodies. Collagen-free vesicles were harvested from the supernatant by centrifugation at 100,000 g for 30 min at 4°C.
2.8. Western-blot and ELISAs
Neutrophils were cultured and treated as described above. Cell culture supernatants were collected, centrifuged at 2,000 g for 5 min and analyzed for IL-1β secretion by ELISA [Immunotools Gmbh (Friesoythe, Germany)]. Cell lysates (10 μg protein) were separated by SDS-PAGE, transferred onto Hybond-P membranes (GE Healthcare Life Sciences), and subjected to Western blot analysis. Briefly, for the analysis of IL-1β, the primary antibody incubation was carried out for 4 h at room temperature with a 1:1,000 dilution of the mouse monoclonal antibodies (Mab201, RD Systems). Second antibody incubation was carried out with a 1:5,000 dilution of anti-mouse immunoglobulin G antibody conjugated to HRP (Sigma). For Western blot analysis of MVs, exosomes, apoptotic bodies (ABs) and cell lysates, 20 μg of proteins were loaded and separated. Western blots were performed by incubation with primary antibodies at 1:1,000 dilution against TNAP (ab218574, Abcam), CD73 (ab137595, Abcam), or CD9 (ab92726, Abcam) overnight at 4°C, followed by incubating with a 1:5,000 dilution of peroxidase-conjugated secondary anti-rabbit antibody (Sigma). In all studies, immunostained bands were detected with the Enhanced Chemiluminescence method (ECL select, GE Healthcare).
2.9. Determination of neutrophil viability and death
Neutrophil viability after treatment with 25 μM MLS-0038949 for 24 h was determined using the MTT [3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] assay. MLS-0038949 cytotoxicity in the same conditions was estimated with the Cytotoxicity Detection kit from Roche based on the measurement of lactate dehydrogenase (LDH) activity released from damaged cells.
2.10. Statistical analysis
All cell experiments were repeated independently at least three times and up to 9 times (indicated in the legends). Results are expressed as mean ± the standard error of the mean (SEM). Statistical analysis was performed with Past 3.22. The Shapiro-Wilk test was used to determine data distribution and the F test to determine whether variances were equal or not. Depending on the results of these tests, paired or unpaired Student’s t-test, or Welch’s t-test or Wilcoxon test were used. The differences between groups were considered significant with * at p<0.05, ** at p<0.01, and *** at p<0.001.
3. Results
3.1. Metaphysis-containing bone fragments from Alpl+/− mice have increased levels of inflammatory cytokines
In order to explore the possible anti-inflammatory function of TNAP, we were first interested to determine whether metaphysis inflammation in childhood HPP [14] is associated with alterations in specific inflammatory cytokines. Childhood HPP is however a rare disease, which makes virtually impossible to obtain bone biopsies from children with HPP and the corresponding control biopsies. We therefore used Alpl+/− mice, since Alpl−/− mice die at birth or soon after. We measured an approximately 50% decrease in alkaline phosphatase activity (Fig. 1A) and in Alpl mRNA levels (Fig. 1B) in Alpl+/− as compared with Alpl+/+ mice. Moreover, we confirmed that Alpl+/− metaphyses have decreased levels of Bglap encoding osteocalcin and of Col10a1 encoding the α1 chain of type X collagen, as also recently reported in the same mice [33] and in cultured hypertrophic chondrocytes in which TNAP was inhibited [34]. These results are in agreement with reports indicating that Alpl−/− mice have disorganized growth plates, characterized by an arrest in chondrogenesis [35, 36]. We then measured the levels of inflammatory markers, first focusing on the pro-inflammatory Il-1β and Il-6 and the anti-inflammatory Il-10, which are thought to be dysregulated in CRMO [37]. We measured significantly increased levels of Il-1β and Il-6 in Alpl+/− bones as compared to Alpl+/+ bones. Levels of Il-1β and Il-6 were positively associated (r=0.7, p=0.015), which is coherent with IL-1β being a major inducer of IL-6. We also measured reduced levels of the anti-inflammatory cytokine Il-10. Finally, we didn’t observe differences in the levels of several other inflammatory cytokines and chemokines, including Il-1Ra encoding IL-1 receptor antagonist, Tnf-α (Fig. 1B) or Il-8 for instance (data not shown). Collectively, these data confirm that the metaphyses of Alpl+/− bones are characterized by disturbed chondrocyte maturation, and further indicate that they may also be characterized by dysregulated inflammatory cytokine expression.
Figure 1: Disturbed levels of hypertrophic chondrocyte markers and inflammatory cytokines in the metaphysis-containing bone fragments of Alpl+/− mice.
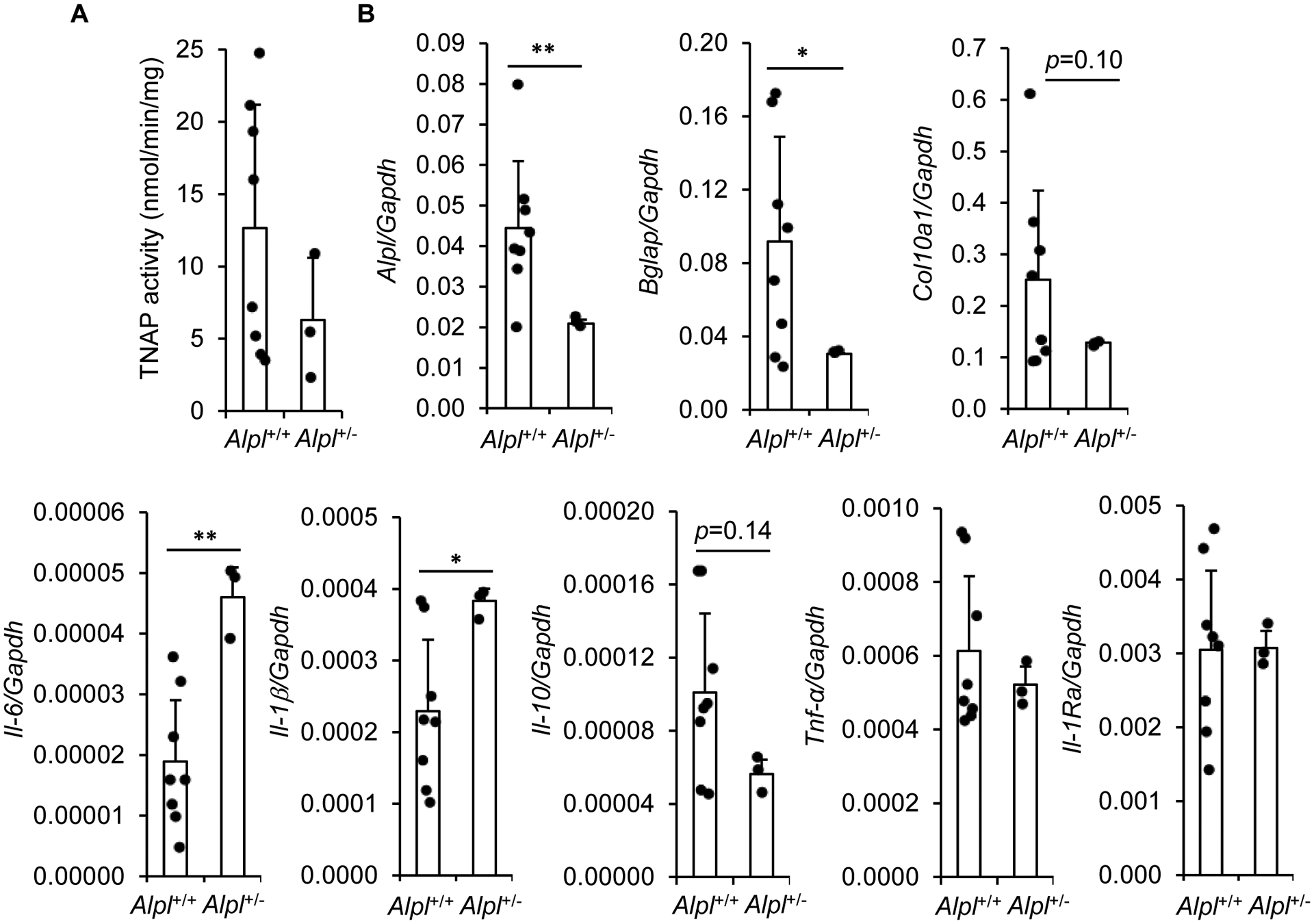
A: TNAP activity in the metaphysis-containing femurs and tibias of 7-day-old Alpl+/+ and Alpl+/− mice. B: RT-qPCR analysis of mRNA levels in the metaphysis-containing femurs and tibias of 7-day-old Alpl+/+ and Alpl+/− mice. * indicates a statistical difference with p<0.05; ** a difference with p<0.01.
3.2. TNAP is a nucleotidase in hypertrophic chondrocytes
We then investigated whether TNAP is an ectonucleotidase in osteoblasts and hypertrophic chondrocytes. We first compared the levels of transcripts encoding TNAP with those of CD39, NPP1 and CD73 (Fig. 2A). We found that TNAP was the most highly expressed ectonucleotidase in metaphysis-containing bone fragments of 7-day-old wild-type mice (Fig. 2B). Moreover, in hypertrophic chondrocytes and osteoblasts differentiated for 16 days [25], the levels of Alpl were much higher than those of Npp1, Cd39 and Cd73 (Figures 2C and 2D respectively). These results suggested that TNAP might be involved in ATP and AMP hydrolysis in hypertrophic chondrocytes and osteoblasts. To address this question, we measured the hydrolysis of exogenously added AMP and ATP in hypertrophic chondrocytes or osteoblasts treated or not with the TNAP inhibitor MLS-0038949 [27], the CD73 inhibitor AOPCP [29], and the inhibitor of both NPP1 and CD39 ARL-67156 [28] (Fig. 2A). We first checked that 100 μM of AOPCP or 100 μM of ARL-67156 did not inhibit the hydrolysis of pNPP, whereas 25 μM of MLS-0038949 totally prevented this hydrolysis (data not shown). This result indicated that AOPCP and ARL-67156 have no inhibitory effect on TNAP. Inhibition of TNAP or CD39/NPP1 in chondrocytes slightly reduced the hydrolysis of exogenously added ATP, and this reduction was more pronounced when the three enzymes were inhibited together (Fig. 3A). To ascertain that TNAP participates in ATP hydrolysis in hypertrophic chondrocytes, we measured the extracellular levels of ATP that was exported from the cytoplasm to the extracellular compartment after a cellular stress. After a change to new medium without FCS, extracellular ATP levels peaked at 5 minutes and returned to basal levels after 20 minutes (Fig. 3C). TNAP inhibition with MLS-0038949 significantly increased extracellular ATP concentrations. Finally, TNAP inhibition with MLS-0038949 and CD73 inhibition with AOPCP both significantly reduced the hydrolysis of exogenously added AMP and again this reduction was strengthened by the concomitant use of both inhibitors (Fig. 3E). These data indicate that in hypertrophic chondrocyte cultures, TNAP participates in both ATP and AMP hydrolysis. In contrary, TNAP inhibition with MLS-0038949 in osteoblasts did not reduce the hydrolysis of exogenously added ATP (Fig. 3B), nor did it increase the levels of extracellular ATP in osteoblasts cultures (Fig. 3D). Moreover, TNAP inhibition neither reduced the hydrolysis of exogenously added AMP (Fig. 3F), suggesting that in osteoblasts, TNAP is not a strong ectonucleotidase.
Figure 2: expression of TNAP, and ectonucleotidases in metaphysis-containing bone fragments, hypertrophic chondrocytes and osteoblasts.
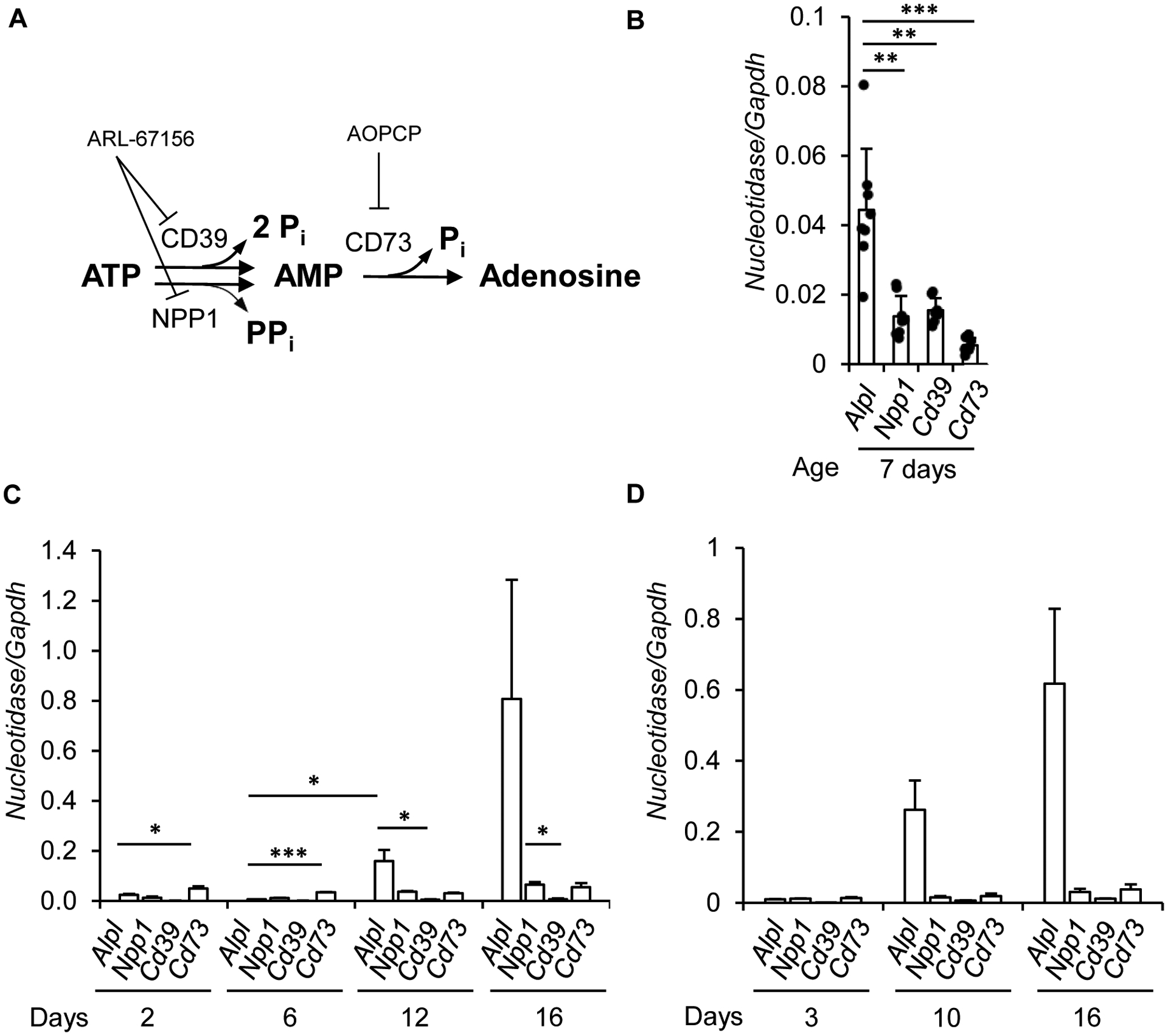
A: enzymes known to be involved in ATP hydrolysis into adenosine, and their inhibitor. B: RT-qPCR analysis of nucleotidase-encoding mRNA levels in the metaphysis-containing femurs and tibias of 7-day-old wildtype mice. Mouse primary chondrocytes (C, n=4 independent experiments) and primary osteoblasts (D, n=3) were cultured as detailed in the Materials and methods section and mRNA levels were quantified by RT-qPCR. * indicates a statistical difference with p<0.05; ** a difference with p<0.01; *** a difference with p<0.001.
Figure 3: ectonucleotidase activity of TNAP in hypertrophic chondrocytes and osteoblasts.
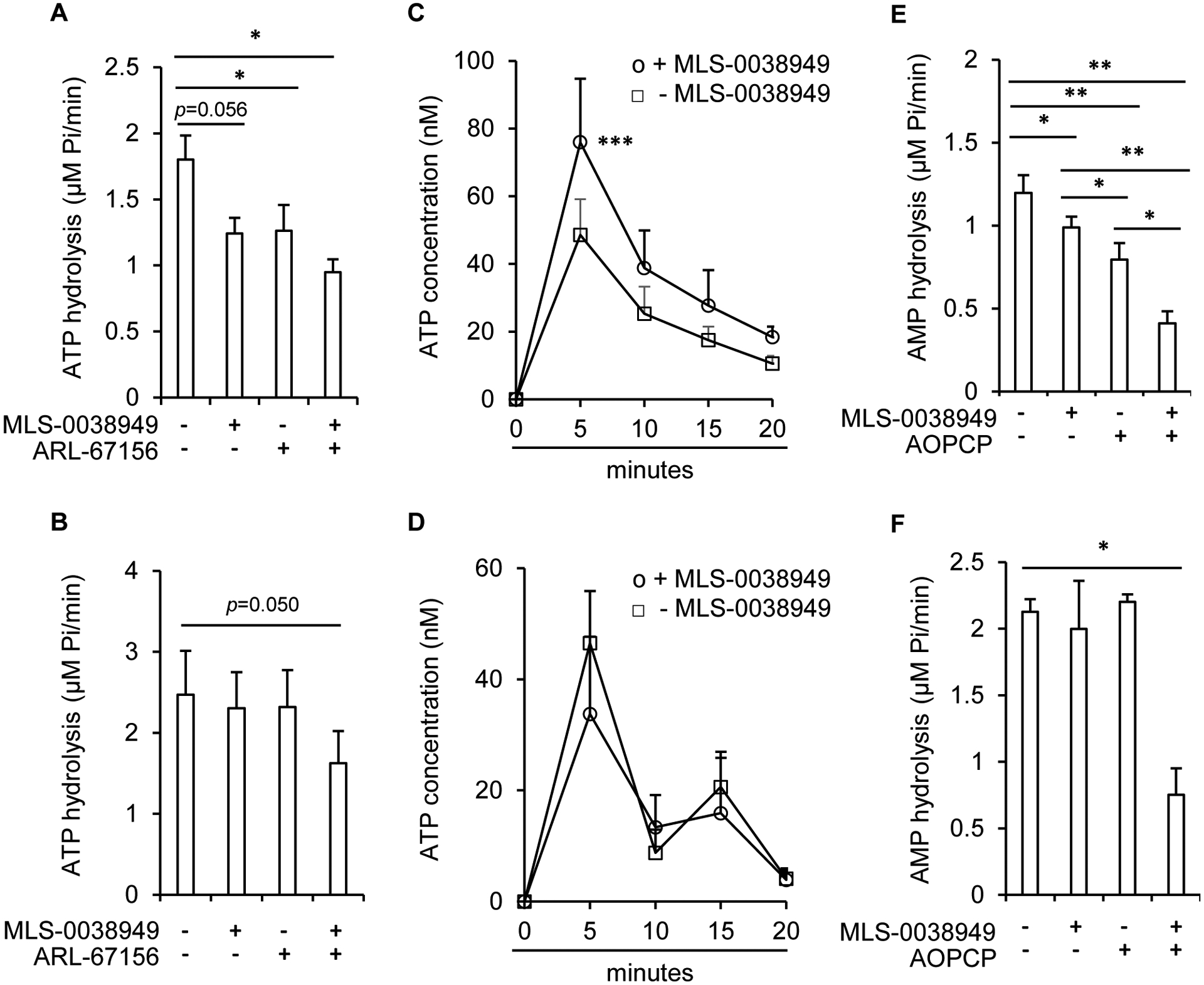
Mouse primary chondrocytes (A, C and E) and primary osteoblasts (B, D and F) were cultured as detailed in the Materials and methods section for 16 days. Hydrolysis of the substrate (10 μM ATP or 25 μM AMP) into Pi was measured by the Malachite green assay in the presence or absence of 25 μM of MLS-0038949 and 100 μM of ARL-67156 (A, n=5 and B, n=4), or in the presence or absence of 25 μM of MLS-0038949 and 100 μM of AOPCP (E, n=5 and F, n=4). Endogenously released ATP was quantified with the bioluminescent Luciferin-Luciferase reaction in presence (ο) or absence (□) of MLS-0038949 (C, n=5 and D, n=3). * indicates a statistical difference with p<0.05; ** a difference with p<0.01; *** a difference with p<0.001.
3.3. TNAP nucleotidase activity in hypertrophic chondrocytes is not associated with an autocrine anti-inflammatory function
We next investigated whether TNAP in hypertrophic chondrocytes is able to reduce the pro-inflammatory effects of ATP. Treatment of hypertrophic chondrocytes with 100 μM of ATP triggered a peak in Il-6 expression that returned to basal levels after about 2 hours (Fig. 4A). Pretreating cells with TNAP and/or CD39/NPP1 inhibitors slightly increased Il-6 expression in response to ATP, but their effects were not significant. We next hypothesized that TNAP may exert anti-inflammatory effects via the production of adenosine rather than the lowering of ATP concentration. Adenosine is known to mediate its effects by binding to G-protein associated receptors acting on intracellular cAMP production by adenylate cyclase [38]. We observed that treatment of hypertrophic chondrocytes with adenosine significantly increased the content of intracellular cAMP (data not shown). We therefore investigated whether TNAP inhibition might block AMP dephosphorylation into adenosine and alter the production of cAMP. Figure 4B shows that AMP dose-dependently increased cAMP production but that TNAP inhibition had modest and non-significant inhibitory effects. Collectively, these data indicated that despite being much more expressed than other nucleotidases, TNAP inhibition modestly decreased ATP and AMP hydrolysis and had no convincing anti-inflammatory effects in hypertrophic chondrocytes. We hypothesized that these weak effects may be related to the fact that part of TNAP proteins are not kept at the cell membrane in hypertrophic chondrocytes but sent in MVs to induce collagen mineralization [9]. We addressed this question by performing western-blot experiments to determine TNAP levels in cell lysates and in MVs. Figure 4C shows that part of TNAP proteins is transferred from the cell membrane to MVs. This transfer from hypertrophic chondrocytes to MVs is confirmed by the strong TNAP activity measured in MVs, and appears to be specific for MVs (Fig. 4D). Indeed, if the exosome fraction was enriched in the exosome marker CD9 [39], its TNAP levels and enzymatic activity were much weaker than in the MV fraction. Similarly, apoptotic bodies released by hypertrophic chondrocytes were not enriched in TNAP and had a weak TNAP activity. In contrast to TNAP, CD73 was not sent to MVs, but remained at the cell membrane. CD39, which was very weakly expressed in hypertrophic chondrocytes (Fig. 2C), could not be detected by western-blots in these preparations. Finally, it was tempting to speculate that TNAP might exert anti-inflammatory effects only when hypertrophic chondrocytes are stimulated by inflammation, but this is probably not the case since inflammatory stimulation of these cells with IL-1β strongly decreased TNAP expression, while increasing that of CD73 (Fig. 4E).
Figure 4: absence of anti-inflammatory effects of TNAP in hypertrophic chondrocytes.
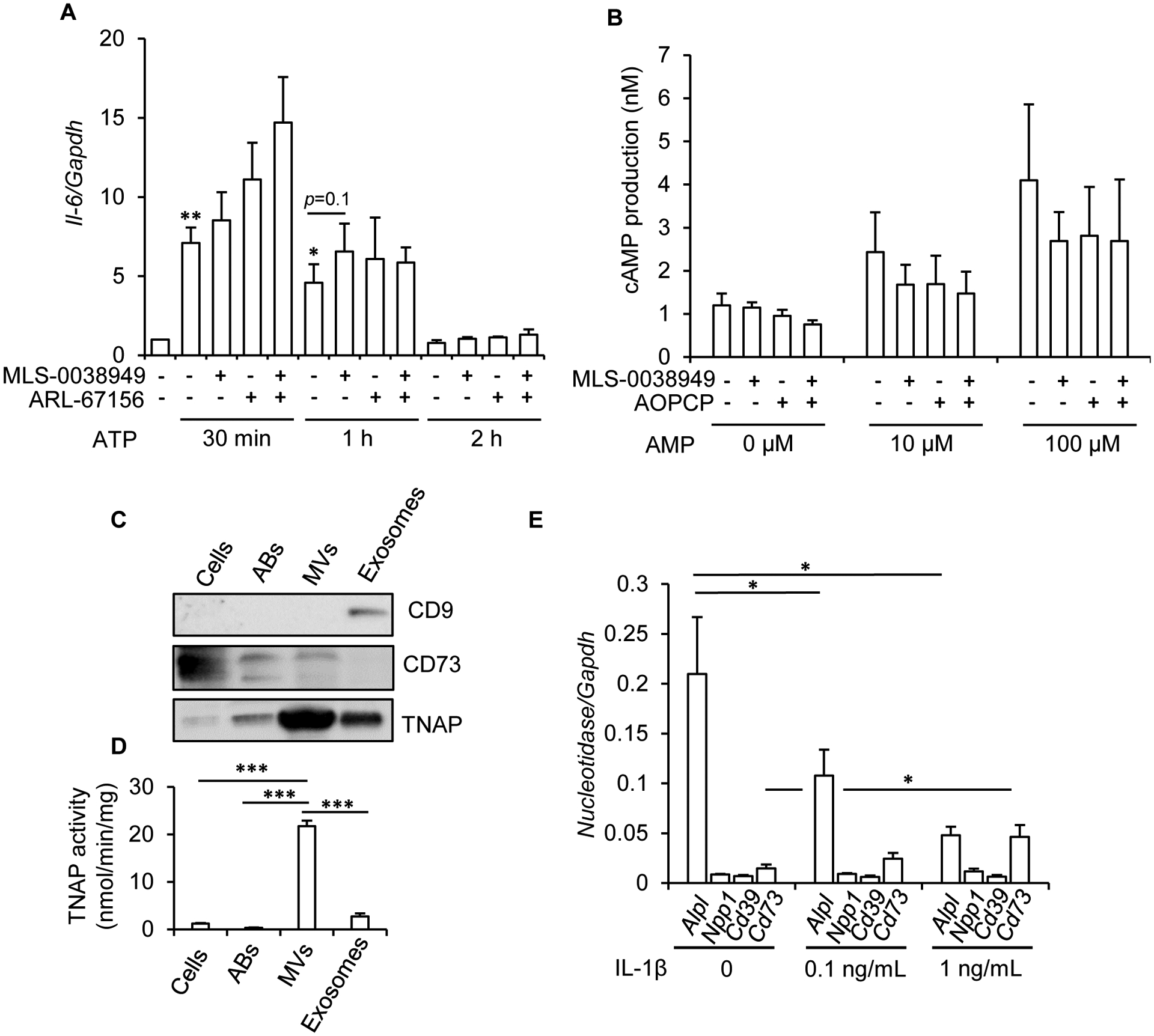
Mouse primary chondrocytes were differentiated into hypertrophic chondrocytes for 16 days as detailed in the Materials and methods section. A: effect of MLS-0038949 and/or ARL-67156 on ATP-stimulated Il-6 expression (n=8). To eliminate the inter-experimental variations in Il-6 levels, Il-6/Gapdh ratio in untreated cells was set to 1. B: effect of MLS-0038949 and/or AOPCP on AMP-stimulated intracellular cAMP production (n=4). C: western-blot analysis of TNAP, CD73, and CD9 in hypertrophic chondrocyte cells, and in apoptotic bodies (ABs), MVs and exosomes released by these cells (n=3, a representative result is shown; the same quantity of protein was used in all conditions). D: TNAP activity in hypertrophic chondrocyte cells, and in ABs, MVs and exosomes released by these cells (n=4). E: effect of a 24-h treatment with IL-1β on nucleotidase levels, as determined by RT-qPCR (n=4). * indicates a statistical difference with p<0.05; ** a difference with p<0.01, and *** a difference with p<0.001.
3.4. TNAP is an anti-inflammatory nucleotidase in hematopoietic neutrophils, but not in mesenchymal cells
Since in hypertrophic chondrocytes, TNAP is involved in mineralization but not in inflammation, we hypothesized that TNAP might participate in inflammation in two non-mineralizing cells in the bone metaphysis: mesenchymal stem cells (MSCs) and/or neutrophils. Indeed, MSCs express TNAP before their differentiation into osteoblasts or chondrocytes [40], and it was recently reported that they may use TNAP to dephosphorylate ATP [33]. We checked and confirmed that MSCs indeed have a significant but weak TNAP activity before their differentiation into osteoblasts (Fig. 5A). TNAP activity in MSCs from 4 adult donors was 23.24 ± 8.82 nmol/min/mg, as compared to 2456.32 ± 226.9 nmol/min/mg in mouse hypertrophic chondrocytes, and 2208.83 ± 157.3 in mouse osteoblasts. Interestingly, TNAP activity in undifferentiated MSCs was increased upon treatment with IL-1β, while it was decreased in MSCs that had been previously differentiated into osteoblasts (Fig. 5A), confirming the inhibitory effect of IL-1β on TNAP in osteoblasts (Fig. 5B). Whereas TNAP was active in undifferentiated MSCs, its expression was nonetheless very weak as compared to that of NPP1 and CD73 (Fig. 5C), making unlikely that it significantly contributed to ATP and AMP hydrolysis in these cells. We indeed observed that TNAP inhibition with MLS-0038949 weakly reduced ATP hydrolysis, and only when MSCs had been pretreated for 4 days with IL-1β (Fig. 5D). However, inhibition of NPP1 with ARL-67156 was much more efficient to reduce ATP hydrolysis, indicating that TNAP contribution was quite modest as compared to that of NPP1. Finally, we failed to detect any anti-inflammatory function of TNAP in MSCs.
Figure 5: absence of anti-inflammatory effects of TNAP in MSCs.
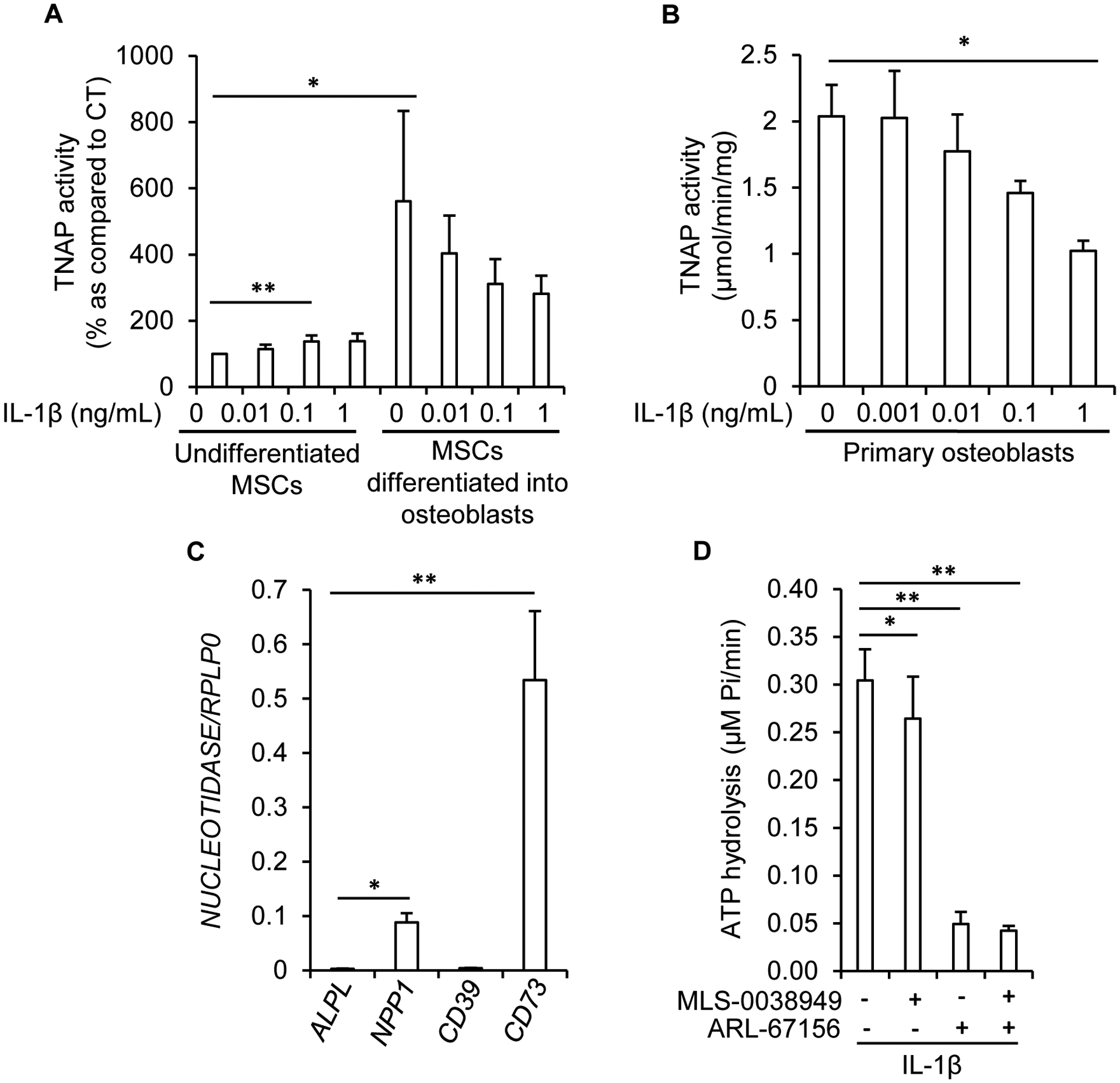
A: effect of a 7-day treatment with IL-1β on TNAP activity in MSCs left undifferentiated or differentiated into osteoblasts (n=6). To eliminate the inter-experimental variations, TNAP activity was set to 100% in untreated cells. B: effect of a 7-day treatment with IL-1β on TNAP activity in murine primary osteoblasts (n=3). C: quantification of nucleotidase mRNA levels in undifferentiated MSCs by RT-qPCR (n=8 independent experiments with MSCs from 4 different donors). D: in undifferentiated MSCs pre-treated for 4 days with 1 ng/mL of IL-1β, the hydrolysis of 10 μM of ATP into Pi was measured by the Malachite green assay in the presence or absence of 25 μM of MLS-0038949 and 100 μM of ARL-67156 (n=8 with MSCs from 4 different donors). * indicates a statistical difference with p<0.05; ** a difference with p<0.01.
In summary, hypertrophic chondrocytes, and maybe also MSCs, may use TNAP to hydrolyze ATP into adenosine, but the changes in ATP and adenosine levels do not seem to impact these mesenchymal cells in an autocrine action. We hypothesized that changes in ATP levels in the metaphysis of TNAP-deficient bones induce inflammation through the involvement of neutrophils. Indeed, neutrophils are often the first cells to be recruited in inflamed or damaged tissues, and ATP release in tissues is a strong signal that initiates neutrophil recruitment [41]. Moreover, neutrophils are the only hematopoietic cells that express TNAP [42]. We first confirmed that neutrophils from healthy donors had a much higher TNAP activity than peripheral blood mononuclear cells (PBMCs) from the same donors (Fig. 6A), and observed that ALPL expression was increased by a pro-inflammatory stimulation with LPS (Fig. 6B). Interestingly, TNAP was, together with CD39, the most highly expressed nucleotidase in neutrophils, which expressed no detectable NPP1 and very weak CD73 levels (Fig. 6C) [43]. We then explored whether TNAP participates in the control of IL-1β secretion, which was reported to be at least in part ATP- and NLRP3-dependent in neutrophils [44]. Figure 6D indicates that IL-1β secretion induced by a 3-hrs treatment with LPS was significantly increased by a short 15-min co-treatment with ATP, confirming that NLRP3 was likely involved. Interestingly, TNAP inhibition with MLS-0038949 significantly increased LPS and LPS/ATP stimulated IL-1β secretion (Fig. 6D). This stimulation of the mature 17 kDa IL-1β secretion by MLS-0038949 was accompanied by a reduction in the intracellular levels of the 31 kDa precursor form (Fig. 6D), strongly suggesting that TNAP inhibition increased IL-1β secretion by exacerbating ATP-associated NLRP3 activation [44]. TNAP-deficient neutrophils not only secreted more IL-1β, they also lived longer since TNAP inhibition by MLS-0038949 significantly increased cell viability and reduced cell death, as shown with MTT and LDH activity respectively (Fig. 6E). This seems in agreement with previous work reporting that ATP delays neutrophil apoptosis [19].
Figure 6: anti-inflammatory ectonucleotidase function of TNAP in neutrophils.
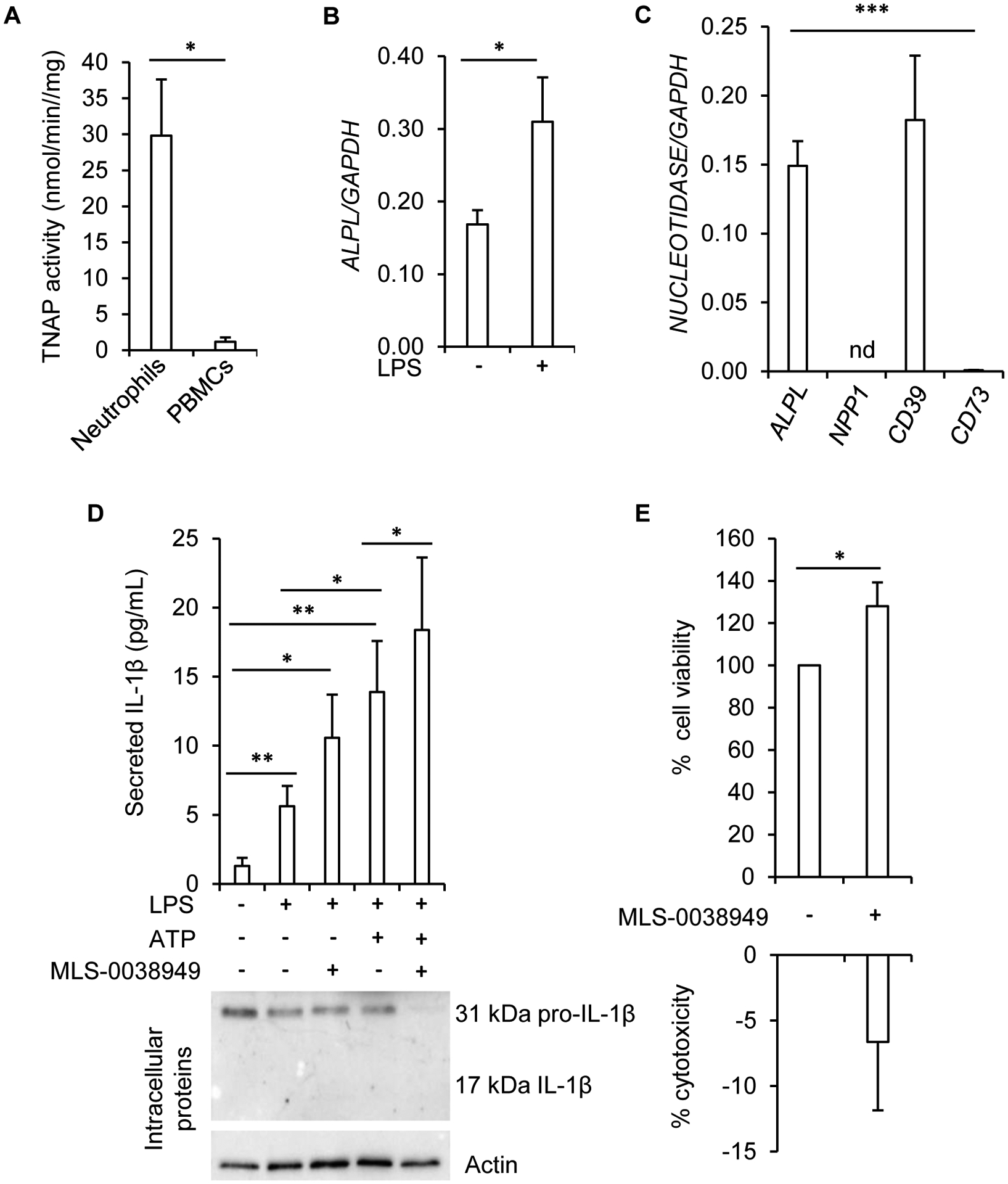
A: TNAP activity measured in freshly isolated human neutrophils (n=5 for neutrophils and n=3 for PBMCs). B: effect of 0.5 μg/mL of LPS on ALPL mRNA levels as determined by RT-qPCR (n=6 independent experiments from 4 different donors). C: RT-qPCR analysis of nucleotidase transcripts in freshly isolated human neutrophils (n=5; nd: not detected). D: quantification by ELISA of extracellular levels of IL-1β in neutrophils primed with 0.5 μg/mL of LPS for 3 h and further stimulated with 2 mM of ATP during 45 min (n=9 independent experiments from 4 different donors). A representative western-blot experiment of intracellular IL-1β is shown. E: quantification of cell viability and cell death with MTT and LDH assays respectively in cells treated with 25 μM of MLS-0038949 for 24 h (n=7 independent experiments from 4 different donors). * indicates a statistical difference with p<0.05; ** a difference with p<0.01; *** a difference with p<0.001.
4. Discussion
Alpl−/− mice have disturbed hypertrophic chondrocyte maturation and growth plate cartilage development [35–36]. Results of our study indicate that Alpl+/− mice not only have reduced levels of RNA encoding type X collagen and osteocalcin in their bone metaphysis, but also have increased levels of Il-1β and Il-6. Interestingly, it was recently reported that experimental pancreatitis induced more Il-1β and Il-6 in the pancreas of Alpl+/− mice as compared to that of Alpl+/+ mice [45]. This suggests that TNAP deficiency up-regulates these cytokines in several tissues. Since there was a significant positive association between Il-1β and Il-6 levels in the metaphyses, and since IL-1β is usually upstream of IL-6 in the inflammatory cascade, it is likely that TNAP deficiency led to IL-1β activation, which then induced IL-6. It is almost certain that TNAP deficiency in hypertrophic chondrocytes, or in other mesenchymal cells, does not induce autocrine effects that result in increased IL-1β secretion. Indeed, although Il-1β mRNA levels were increased in hypertrophic chondrocytes and MSCs in response to pro-inflammatory stimulation with TNF-α, we always failed to detect any intracellular cleavage of the precursor form of IL-1β, and any secretion of active IL-1β by ELISA or Western-blot (data not shown). It is likely that NLRP3 is not functional in mesenchymal cells and that IL-1β is activated and secreted by other mechanisms. Indeed, NLRP3-activating mutations in myeloid cells but not in chondrocytes or mesenchymal precursors trigger chronic inflammation and growth place dysplasia, that mimic the cryopyrin-associated periodic syndrome (CASP, due to NLRP3 activating mutations) [46].
Our hypothesis is that neutrophils, rather than cells from the mesenchymal lineage, are activated by supraphysiological ATP levels. As already mentioned above, ATP release in damaged tissues is a strong signal that initiates neutrophil recruitment [41]. Moreover, neutrophils express TNAP, which is more expressed in these cells in response to inflammatory stimulation. When TNAP was chemically inhibited in neutrophils, intracellular levels of the IL-1β precursor form were decreased, and extracellular levels of the cleaved mature IL-1β form were increased. These results are in agreement with the recent report that neutrophils from Alpl+/− mice secrete more IL-6 than those from wildtype mice [45]. Therefore, we think reasonable to hypothesize that the increased expression of Il-1β and Il-6 in the bone metaphysis of Alpl+/− mice occurs in neutrophils or is induced by neutrophils.
Neutrophils are likely responsible for increased cytokine expression in the metaphysis of Alpl+/− mice, and we think that the signal that induces their recruitment and activation in the metaphysis is the abnormally elevated levels of extracellular ATP resulting from TNAP deficiency in hypertrophic chondrocytes. Indeed, TNAP inhibition in hypertrophic chondrocytes resulted in significantly higher extracellular ATP levels. Since the hypertrophic zone is where the growth plate becomes vascularized, these increased ATP levels are likely to induce the recruitment of neutrophils [41].
Finally, in the particular context of childhood hypophosphatasia, the fact that IL-1β inhibits TNAP expression in mineralizing hypertrophic chondrocytes and osteoblasts suggests that a vicious cycle takes place where TNAP deficiency in mineralizing cells induces neutrophil recruitment, which further exacerbates TNAP deficiency in mineralizing cells and accelerates rickets. More generally, considering the importance of neutrophils in blood and tissue inflammation [47], such a function of TNAP as an anti-inflammatory nucleotidase deserves much consideration.
Acknowledgements:
this work was performed thanks to financial support from the “Hypophosphatasie Europe” Foundation, and from the “Société Française de Rhumatologie”. We are particularly grateful to Steve Ursprung, president of the “Hypophosphatasie Europe” Foundation, for his constant support and help.
List of nonstandard abbreviations:
- ABs
apoptotic bodies
- ATP
adenosine triphosphate
- BGLAP
bone Gla protein (osteocalcin)
- CASP
cryopyrin-associated periodic syndrome
- CNO
chronic non-bacterial osteomyelitis
- CRMO
chronic recurrent multifocal osteomyelitis
- DIRA
deficiency of interleukin-1 receptor antagonist
- DMEM
Dulbecco’s Modified Eagle Medium
- GABA
Gamma-aminobutyric acid
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HPP
hypophosphatasia
- IL
interleukin
- MRI
magnetic resonance imaging
- MSC
mesenchymal stem cell
- MV
matrix vesicle
- NLRP3
NOD-like receptor family, pyrin domain containing 3
- NPP1
ectonucleotide pyrophosphatase phosphodiesterase 1
- PBMC
peripheral blood mononuclear cell
- pNPP
paranitrophenylphosphate
- PPi
inorganic pyrophosphate
- RPLP0
ribosomal protein lateral stalk subunit P0
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- TNAP
tissue-nonspecific alkaline phosphatase
Footnotes
Declaration of interests: none
5 References
- [1].Aghajanian P, Mohan S, The art of building bone: emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification, Bone Res 6 (2018) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Murshed M, Harmey D, Millán JL, McKee MD, Karsenty G, Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone, Genes Dev 19(9) (2005) 1093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Deng H, Huang X, Yuan L, Molecular genetics of the COL2A1-related disorders, Mutat Res Rev Mutat Res 768 (2016) 1–13. [DOI] [PubMed] [Google Scholar]
- [4].Bishop N, Bone Material Properties in Osteogenesis Imperfecta, J Bone Miner Res 31(4) (2016) 699–708. [DOI] [PubMed] [Google Scholar]
- [5].Girschick HJ, Mornet E, Beer M, Warmuth-Metz M, Schneider P, Chronic multifocal non-bacterial osteomyelitis in hypophosphatasia mimicking malignancy, BMC Pediatr 7 (2007) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Whyte MP, Physiological role of alkaline phosphatase explored in hypophosphatasia, Ann N Y Acad Sci 1192 (2010) 190–200. [DOI] [PubMed] [Google Scholar]
- [7].Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL, Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization, Proc Natl Acad Sci U S A 99(14) (2002) 9445–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Orriss IR, Arnett TR, Russell RG, Pyrophosphate: a key inhibitor of mineralisation, Curr Opin Pharmacol 28 (2016) 57–68. [DOI] [PubMed] [Google Scholar]
- [9].Bottini M, Mebarek S, Anderson KL, Strzelecka-Kiliszek A, Bozycki L, Simão AMS, Bolean M, Ciancaglini P, Pikula JB, Pikula S, Magne D, Volkmann N, Hanein D, Millán JL, Buchet R, Matrix vesicles from chondrocytes and osteoblasts: Their biogenesis, properties, functions and biomimetic models, Biochim Biophys Acta 1862 (2018) 532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cruz T, Gleizes M, Balayssac S, Mornet E, Marsal G, Millán JL, Malet-Martino M, Nowak LG, Gilard V, Fonta C, Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis, J Neurochem 140(6) (2017) 919–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Buchet R, Millán JL, Magne D, Multisystemic functions of alkaline phosphatases, Methods Mol Biol 1053 (2013) 27–51. [DOI] [PubMed] [Google Scholar]
- [12].Ferguson PJ, El-Shanti HI, Autoinflammatory bone disorders, Curr Opin Rheumatol 19(5) (2007) 492–8. [DOI] [PubMed] [Google Scholar]
- [13].Whyte MP, Wenkert D, McAlister WH, Mughal MZ, Freemont AJ, Whitehouse R, Baildam EM, Coburn SP, Ryan LM, Mumm S, Chronic recurrent multifocal osteomyelitis mimicked in childhood hypophosphatasia, J Bone Miner Res 24(8) (2009) 1493–505. [DOI] [PubMed] [Google Scholar]
- [14].Beck C, Morbach H, Wirth C, Beer M, Girschick HJ, Whole-body MRI in the childhood form of hypophosphatasia, Rheumatol Int 31(10) (2011) 1315–20. [DOI] [PubMed] [Google Scholar]
- [15].Girschick HJ, Seyberth HW, Huppertz HI, Treatment of childhood hypophosphatasia with nonsteroidal antiinflammatory drugs, Bone 25(5) (1999) 603–7. [DOI] [PubMed] [Google Scholar]
- [16].Girschick HJ, Schneider P, Haubitz I, Hiort O, Collmann H, Beer M, Shin YS, Seyberth HW, Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia, Orphanet J Rare Dis 1 (2006) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Antonioli L, Pacher P, Vizi ES, Haskó G, CD39 and CD73 in immunity and inflammation, Trends Mol Med 19(6) (2013) 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gombault A, Baron L, Couillin I, ATP release and purinergic signaling in NLRP3 inflammasome activation, Front Immunol 3 (2012) 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vaughan KR, Stokes L, Prince LR, Marriott HM, Meis S, Kassack MU, Bingle CD, Sabroe I, Surprenant A, Whyte MK, Inhibition of neutrophil apoptosis by ATP is mediated by the P2Y11 receptor, J Immunol 179(12) (2007) 8544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG, ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors, Science 314(5806) (2006) 1792–5. [DOI] [PubMed] [Google Scholar]
- [21].Pettengill M, Robson S, Tresenriter M, Millán JL, Usheva A, Bingham T, Belderbos M, Bergelson I, Burl S, Kampmann B, Gelinas L, Kollmann T, Bont L, Levy O, Soluble ecto-5’-nucleotidase (5’-NT), alkaline phosphatase, and adenosine deaminase (ADA1) activities in neonatal blood favor elevated extracellular adenosine, J Biol Chem 288(38) (2013) 27315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Street SE, Kramer NJ, Walsh PL, Taylor-Blake B, Yadav MC, King IF, Vihko P, Wightman RM, Millán JL, Zylka MJ, Tissue-nonspecific alkaline phosphatase acts redundantly with PAP and NT5E to generate adenosine in the dorsal spinal cord, J Neurosci 33(27) (2013) 11314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Millán JL, Whyte MP, Alkaline Phosphatase and Hypophosphatasia, Calcif Tissue Int 98(4) (2016) 398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Narisawa S, Fröhlander N, Millán JL, Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia, Dev Dyn 208(3) (1997) 432–46. [DOI] [PubMed] [Google Scholar]
- [25].Bougault C, El Jamal A, Briolay A, Mebarek S, Boutet MA, Garraud T, Le Goff B, Blanchard F, Magne D, Brizuela L, Involvement of sphingosine kinase/sphingosine 1-phosphate metabolic pathway in spondyloarthritis, Bone 103 (2017) 150–158. [DOI] [PubMed] [Google Scholar]
- [26].Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian JC, Hardouin P, Magne D, TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells, Life Sci 84(15–16) (2009) 499–504. [DOI] [PubMed] [Google Scholar]
- [27].Kiffer-Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, Cosford ND, Dahl R, Farquharson C, Hoylaerts MF, Macrae VE, Millán JL, Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification, J Bone Miner Res 28(1) (2013) 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lévesque SA, Lavoie EG, Lecka J, Bigonnesse F, Sévigny J, Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases, Br J Pharmacol 152(1) (2007) 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bhattarai S, Freundlieb M, Pippel J, Meyer A, Abdelrahman A, Fiene A, Lee SY, Zimmermann H, Yegutkin GG, Sträter N, El-Tayeb A, Müller CE, α,β-Methylene-ADP (AOPCP) Derivatives and Analogues: Development of Potent and Selective ecto-5’-Nucleotidase (CD73) Inhibitors, J Med Chem 58(15) (2015) 6248–63. [DOI] [PubMed] [Google Scholar]
- [30].Harder KW, Owen P, Wong LK, Aebersold R, Clark-Lewis I, Jirik FR, Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase beta (HPTP beta) using synthetic phosphopeptides, Biochem J 298 (Pt 2) (1994) 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Buchet R, Pikula S, Magne D, Mebarek S, Isolation and characteristics of matrix vesicles, Methods Mol Biol 1053 (2013) 115–24. [DOI] [PubMed] [Google Scholar]
- [32].Wuthier RE, Chin JE, Hale JE, Register TC, Hale LV, Ishikawa Y, Isolation and characterization of calcium-accumulating matrix vesicles from chondrocytes of chicken epiphyseal growth plate cartilage in primary culture, J Biol Chem 260(29) (1985) 15972–9. [PubMed] [Google Scholar]
- [33].Liu W, Zhang L, Xuan K, Hu C, Liu S, Liao L, Li B, Jin F, Shi S, Jin Y, Alpl prevents bone ageing sensitivity by specifically regulating senescence and differentiation in mesenchymal stem cells, Bone Res 6 (2018) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fakhry M, Roszkowska M, Briolay A, Bougault C, Guignandon A, Diaz-Hernandez JI, Diaz-Hernandez M, Pikula S, Buchet R, Hamade E, Badran B, Bessueille L, Magne D, TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability, Biochim Biophys Acta 1863(3) (2017) 643–653. [DOI] [PubMed] [Google Scholar]
- [35].Liu J, Nam HK, Campbell C, Gasque KC, Millán JL, Hatch NE, Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia, Bone 67 (2014) 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millán JL, MacGregor GR, Whyte MP, Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia, J Bone Miner Res 14(12) (1999) 2015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hofmann SR, Kapplusch F, Girschick HJ, Morbach H, Pablik J, Ferguson PJ, Hedrich CM, Chronic Recurrent Multifocal Osteomyelitis (CRMO): Presentation, Pathogenesis, and Treatment, Curr Osteoporos Rep 15 (2017) 542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K, Pharmacology of Adenosine Receptors: The State of the Art, Physiol Rev 98(3) (2018) 1591–1625. [DOI] [PubMed] [Google Scholar]
- [39].Lee Y, El Andaloussi S, Wood MJ, Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy, Hum Mol Genet 21(R1) (2012) R125–34. [DOI] [PubMed] [Google Scholar]
- [40].Estève D, Galitzky J, Bouloumié A, Fonta C, Buchet R, Magne D, Multiple Functions of MSCA-1/TNAP in Adult Mesenchymal Progenitor/Stromal Cells, Stem Cells Int 2016 (2016) 1815982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P, Intravascular danger signals guide neutrophils to sites of sterile inflammation, Science 330(6002) (2010) 362–6. [DOI] [PubMed] [Google Scholar]
- [42].Rambaldi A, Terao M, Bettoni S, Tini ML, Bassan R, Barbui T, Garattini E, Expression of leukocyte alkaline phosphatase gene in normal and leukemic cells: regulation of the transcript by granulocyte colony-stimulating factor, Blood 76(12) (1990) 2565–71. [PubMed] [Google Scholar]
- [43].Corriden R, Chen Y, Inoue Y, Beldi G, Robson SC, Insel PA, Junger WG, Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine, J Biol Chem 283(42) (2008) 28480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E, Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP, Nat Commun 7 (2016) 10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gámez-Belmonte R, Hernández-Chirlaque C, Sánchez de Medina F, Martínez-Augustin O, Experimental acute pancreatitis is enhanced in mice with tissue nonspecific alkaline phoshatase haplodeficiency due to modulation of neutrophils and acinar cells, Biochim Biophys Acta Mol Basis Dis 1864(11) (2018) 3769–3779. [DOI] [PubMed] [Google Scholar]
- [46].Wang C, Xu CX, Alippe Y, Qu C, Xiao J, Schipani E, Civitelli R, Abu-Amer Y, Mbalaviele G, Chronic inflammation triggered by the NLRP3 inflammasome in myeloid cells promotes growth plate dysplasia by mesenchymal cells, Sci Rep 7(1) (2017) 4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER, Neutrophil kinetics in health and disease, Trends Immunol 31(8) (2010) 318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]