

Highlights
-
•
We used HTS to identify a lead compound that inhibited hRSV replication in vitro.
-
•
Analog compounds were characterized for structure activity relationships.
-
•
An hRSV lead compound-resistant mutant had a mutation in the hRSV G protein.
-
•
The lead compound activity in different cell types correlates with G protein function.
-
•
The lead compound did not reduce RSV pathogenicity in a cotton rat model.
Keywords: hRSV, Antiviral, SAR, G protein, Heparin
Abstract
Human respiratory syncytial virus (hRSV) is a highly contagious Paramyxovirus that infects most children by age two, generating an estimated 75,000–125,000 hospitalizations in the U.S. annually. hRSV is the most common cause of bronchiolitis and pneumonia among infants and children under 1 year of age, with significant mortality among high-risk groups. A regulatory agency-approved vaccine is not available, and existing prophylaxis and therapies are limited to use in high-risk pediatric patients; thus additional therapies are sorely needed. Here, we identify a series of benzimidazole analogs that inhibit hRSV infection in vitro with high potency, using a previously-reported high-throughput screening assay. The lead compound, SRI 29365 (1-[6-(2-furyl)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl]methyl-1H-benzimidazole), has an EC50 of 66 μM and a selectivity >50. We identified additional compounds with varying potencies by testing commercially-available chemical analogs. Time-of-addition experiments indicated that SRI 29365 effectively inhibits viral replication only if present during the early stages of viral infection. We isolated a virus with resistance to SRI 29365 and identified mutations in the transmembrane domain of the viral G protein genomic sequence that suggested that the compound inhibits G-protein mediated attachment of hRSV to cells. Additional experiments with multiple cell types indicated that SRI 29365 antiviral activity correlates with the binding of cell surface heparin by full-length G protein. Lastly, SRI 29365 did not reduce hRSV titers or morbidity/mortality in efficacy studies using a cotton rat model. Although SRI 29365 and analogs inhibit hRSV replication in vitro, this work suggests that the G-protein may not be a valid drug target in vivo.
1. Introduction
Human RSV (hRSV), which belongs to the family Paramyxoviridae, is the most important cause of infectious pulmonary disease in human infants, and is a major causative agent of respiratory tract infections among children worldwide. In the U.S. in 2003–2005, the estimated indirect and direct impact of hospitalizations due to hRSV infections was $650 million (Palmer et al., 2010). Infants, immune-compromised children, or those with underlying respiratory disorders have a significantly higher risk of developing severe hRSV infections that can be complicated by opportunistic viral pneumonia and respiratory distress. Elderly and immune-compromised individuals are also susceptible to complications from hRSV infections, thereby highlighting the importance of medical intervention in the form of prophylactic and antiviral therapies (Byrd and Prince, 1997).
Existing prophylaxis and antiviral therapies for acute hRSV infections in infants are limited to a humanized monoclonal antibody that is reserved for use in high risk pediatric patients and ribavirin (Olszewska and Openshaw, 2009). Multiple potential antivirals are under clinical investigation (Cianci et al., 2004, Pryde et al., 2013, Yu et al., 2004, Yu et al., 2007), including two currently in clinical trials. These are a nucleoside RNA polymerase inhibitor AL-8176 (Wang et al., 2015) and a fusion inhibitor GS-5806 (Mackman et al., 2015). However, none of these have been approved by government agencies as therapies or are available to the research community as tools to study the life cycle of the virus or host/virus interactions. Therefore, significant research and medical needs will be met by the discovery of additional probes and potential therapeutics for hRSV (Noah et al., 2010a, Noah et al., 2010b).
hRSV is a negative sense, single-stranded, non-segmented RNA virus with a genome of approximately 15 kb with ten viral genes that encode 11 viral proteins. These are NS1 and NS2, which inhibit type I interferon activity; N, which is the nucleocapsid protein; M, which encodes the matrix protein; and SH, G and F (fusion) proteins, which are incorporated into the viral coat. The G protein is a post-translationally glycosylated surface protein and also recognizes cell surface components (e.g., heparin) during viral attachment. The F protein mediates entry of the virus into the cell cytoplasm and also promotes the formation of syncytia. Typically, antibodies directed at the F protein are neutralizing because this protein is conserved in both subtypes of RSV, while the G protein varies in sequence considerably between hRSV A and B subtypes. The M2 gene encodes the elongation factor M2-1 and transcription factor M2-2. The L gene encodes the viral RNA polymerase, and the phosphoprotein P, which is a cofactor for L function (Olszewska and Openshaw, 2009).
In this study, we used an in vitro infection model of hRSV in HEp-2 cells to search the ChemBridge Small Molecule Library for lead therapeutic antiviral candidates. We identified and characterized a series of compounds that inhibit hRSV replication in vitro. We developed a mutant virus with resistance to the lead inhibitor compound, sequenced the mutant virus, and identified mutations in the N-terminus of the viral G protein that implicated the G protein as the target of compound inhibition. Further experiments with multiple cell lines (A549 and Vero E6) indicated that the lead compound was most effective in cell lines express a high percentage of full-length G protein and again suggested that the compound target was the viral G protein. However, in vivo studies with the lead compound did not reduce viral titers or extend the lives of cotton rats in a lethal hRSV challenge model. Our data suggest that, while targeting the function of G protein with small molecule inhibitors effectively inhibits hRSV replication in vitro, this strategy is cell type-dependent and may not be viable for therapeutic intervention in vivo.
2. Materials and methods
2.1. High-throughput screening
The inhibition of the cell death (cytopathic effect, CPE) caused by hRSV infection in HEp-2 cells was used as the primary assay to identify the antiviral effects of compounds screened (Noah et al., 2010a, Noah et al., 2010b).
2.2. Cell and virus culture
All cells and hRSV strains were purchased from American Tissue Culture Type (Manassas, VA). HEp-2 cells (ATCC CCL-23), A549 cells (ATCC CCL-185), and Vero E6 cells (ATCC CRL-1586) were cultured in EMEM/10% FBS/1% Pen/Strep/L-glutamine (Invitrogen, Grand Island, NY). Cells were maintained as adherent cell lines at 37 °C in a humidified 5% CO2 atmosphere and were passaged as needed and harvested from flasks using Tryp-LE (Gibco). Human respiratory syncytial virus, strain Long (ATCC VR-26) was purchased and amplified for this study. Virus culture media was EMEM/2% FBS/1% Pen/Strep/l-glutamine. Virus stock from the supplier was diluted to 1:1000 and added to a T-75 flask containing virus culture media. The flask was incubated at 37 °C, 5% CO2, and 90% relative humidity After 72 h, the virus culture media was transferred to a 50 mL conical tube and cell debris pelleted at 1000 RPM, 10 min, at 4 °C. FBS was added to a final concentration of 10% (v/v) each for preservation and the supernatant was aliquoted (0.5 mL per tube), flash-frozen in dry ice/methanol, and stored at −80 °C. After amplification, virus stocks titers were quantified in HEp-2 cells by serial dilution to determine TCID50, and the titer of the virus was 1.0 × 107 TCID50/mL. The TCID50 of the virus was determined using ten half-log-fold serial dilutions, from 10−2 to 10−7, in a microtitration assay. HEp-2 cells were plated into a 96-well plate (10,000 cells/well) and infected with the serial dilutions of hRSV. The microtitration plates were incubated at 37 °C, 5% CO2, and 90% relative humidity for four days, after which the TCID50 of the cultured virus stock was determined.
2.3. Cytoprotection and cytotoxicity assays
The cell viability assay measured the extent of virus-induced cell death (cytopathic effect, CPE) in the presence of a concentration range of test compound (Noah et al., 2007). For hRSV, HEp-2 or A549 cells were used in the assay. Cells were plated into 96-well plates for the analysis of CPE. Test compounds were added immediately after infection and were present throughout the infection. For the standard antiviral assay, test wells and virus control wells received 100 TCID50s of hRSV (MOI = 0.01). Test compound cytotoxicity was assayed in parallel in wells that had the same compound concentrations, but without the addition of virus. Four days later, the cell viability in all wells was determined, and the concentration of test compound that reduced virus-induced CPE by 50% (EC50) was calculated. The cytotoxic concentration of test compound that reduced cell numbers by 50% (CC50) was determined, and the selectivity index of each compound (SI = CC50/EC50) was calculated. Unless otherwise stated, all experiments were performed in triplicate.
2.4. Titer reduction assays
Titer reduction assays measured the difference in viral titer between non-treated and test compound-treated cells. A compound that inhibits viral replication results in a reduction in progeny virus compared to virus cell controls. HEp-2, A549, or Vero E6 cells were infected with hRSV Long strain or mutant hRSV at an MOI of 0.01 in the presence of 5 μM SRI 29365. The viral titers were calculated using the TCID50 method (Noah et al., 2007). Statistical analysis of data was performed using a t-test (two-sample assuming equal variances) of the difference of TCID50 values between wt and resistant mutant viruses in the presence of SRI 29365. Samples were performed in quadruplicate.
2.5. Assay endpoint read
Following the four day incubation period, the assay plates were equilibrated to room temperature for 30 min. An equal volume (100 μL) of Cell Titer-Glo reagent (Promega Inc. Madison WI) was added to each microplate well and the plates were incubated for an additional 15 min at room temperature (Noah et al., 2007). Measurement of hRSV-induced CPE is based on quantitation of ATP, an indicator of metabolically active cells. The procedure involves adding the single reagent (CellTiter-Glo reagent) directly to previously cultured, subconfluent cells in media. This induces cell lysis and the production of a bioluminescent signal (half-life greater than 5 h, depending on the cell type) that is proportional to the amount of ATP present (which is a biomarker for viability). The test plates were then read for luminescence in a Perkin Elmer Envision multi-label plate reader (Perkin Elmer, Waltham, Massachusetts) with an integration time of 0.1 s.
2.6. Time-of-addition assays
Sri 29365 (5 μM) was added to plates containing A549 cells at timepoints of −1, 0, 1, 2, 4, 8 h post-viral infection of the cells. Cells were infected at a timepoint of zero hours with 1 MOI (10,000 TCID50s) of hRSV Long strain. Following a four-day incubation period, CellTiter-Glo reagent was added (as described above) and the percentage of viable cells (relative to uninfected controls) was calculated for each time point to determine the timepoint at which compound addition no longer sustained cell viability above 50%.
2.7. Resistant mutant virus generation and sequencing
hRSV Long strain was passaged in 96-well microplates in the presence of doubling concentrations (0.2–3.2 μM) of SRI 29365 for five passages, and then amplified for an additional five passages in the presence of 5 μM of SRI 29365. The cultures from each passage were pooled, diluted 100-fold, and used to inoculate the subsequent culture. As a control, hRSV Long strain was also passaged in the absence of SRI 29365 for the same number of passages. At the end of ten passages, the wt and putative mutant viruses were amplified one additional time in a T-25 flask, harvested, and aliquoted for storage and analysis. SRI 29365 (5 μM) was included in the mutant virus amplification. For sequencing, 1 mL of virus culture was used to isolated viral RNA (phenol:ether:ethanol precipitation). cRNA was produced (SuperScript® III One-Step RT-PCR System # 12574-018, Life Technologies). Sequencing of both cDNA strands was performed using the primers reported in Rebuffo-Scheer et al. (2011).
2.8. Compounds for SAR studies
Compound analogs were purchased from Chembridge (San Diego, CA) in 30–50 mg samples. These were dissolved in 100% DMSO to a concentration of 20–40 mM for stocks and used for antiviral testing.
2.9. In Vivo studies
In vivo studies were performed to evaluate the effect of dosage on the antiviral efficacy of SRI 29365 when administered at T = −1, +6 and +24 h by intraperitoneal injection in the respiratory syncytial virus (RSV)-cotton rat (CR) model. The endpoints of this study were demonstration of reduced virus titers in the lung lavage (3 mL) and nasal wash (2 mL) fluids of the treated infected cotton rats (ca. 100 g in weight) compared to vehicle-treated infected cotton rats. Virus quantification was done by plaque reduction assay. Serum samples were obtained at time of infection (T = 0) and at time of sacrifice in all 24 cotton rats. Body weight was recorded at end of experiment (day +4), and animal body weight and sex distribution was selected so as to be as similar as possible across all groups at the start. RSV/A/Tracy (passage three grown in HEp-2 cells), 1.35 × 105 PFU intranasally (100 μL) to cotton rats lightly anesthetized with isoflurane. Stock virus concentration was 1.35 × 106 PFU/mL. Compound SRI 29365 (140 mg, Chembridge, San Diego, CA) was supplied as dry powder and was diluted in 7 mL of PBS (20 mg/mL). The preparation was vortexed and sonicated in a water bath until the solution appeared clear. After 30 min of sonication the solution had the appearance of an emulsion. The emulsion was used for the IP injections. All aliquots were stored at 4 °C. Dose volume was 2 mL/kg (0.2 mL/0.1 kg cotton rat). Plaque assays were performed using 24-well tissue cultures plates containing nearly confluent monolayers (20–40 × 104 cells/well) of HEp-2 cells prepared in 10% FBS 24 h prior to start of assay. At the start of each assay, dilutions (usually serial log10) were made of the test samples. A 0.2 mL sample of each was added to wells in duplicate and allowed to adsorb for 90 min with occasional gentle agitation. After the inoculum was removed, the monolayers were overlayed with 0.75% methylcellulose in 2% FBS-MEM containing antibiotics, vitamins and other nutrients. Tissue culture and positive virus controls were included in each assay. The plates were placed in a 36 °C, 5% CO2 incubator. On day 7, plates were stained with 0.01% crystal violet/10% formalin solution (1.5 mL/well) and allowed to sit for 24–48 h at room temperature. Wells were rinsed with water, and the visible plaques were counted. All of the plaques in wells containing between 20 and 100 plaques were enumerated, averaged and the virus titers calculated as total log10 PFU for nasal wash fluid or log10 PFU/g of tissue for lungs. The lower limit of detection by this method is 0.70 log10 total PFU or approximately 1.4 log10 PFU/g of lung, respectively.
3. Results
Previously published reports have detailed the development and performance of the hRSV high-throughput screen and the identification of hit compounds derived from the screening of the NIH Molecular Libraries chemical collection (Noah et al., 2010a, Noah et al., 2010b). The same screening assay and format was used here to screen 100,000 compounds from the ChemBridge Small Molecule Library (Chembridge, 2015). We selected compounds with EC50 values <20 μM and selective indices (SI) > 5 as hits. The primary and confirmatory screens resulted in 30 confirmed hits, with EC50s ranging from 0.66 to 20 μM and SIs ranging from 6 to 55. Compounds for further characterization were selected based on potency, their previous characterization, reactivity, and tractability for chemical optimization. Compounds characterized in previous hRSV screening campaigns (Matharu et al., 2014, Moore et al., 2012, Noah et al., 2010a, Noah et al., 2010b) were not further characterized here.
The most potent, novel confirmed compound from this HTS screen was (1-[6-(2-furyl)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl]methyl-1H-benzimidazole), the structure of which is shown in Table 1 . This compound has not been previously reported as effective against hRSV and was designated as SRI 29365. Based on confirmed and potent in vitro antiviral activity, the lack of previously-reported antiviral activity, ant the potential for chemical modification, SRI 29365 was chosen for further characterization to determine EC50, CC50, SI, structure–activity relationships, broad antiviral activity, mechanism of action, and in vivo potency.
Table 1.
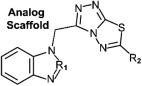
Structure activity relationships of SRI 29365. The lead compound and 16 active analog structures are shown, along with EC50 and selectivity values.
| SRI 29365 |
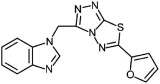 |
 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 = 0.66 μM | ||||||||||||||
| SI = 83 | ||||||||||||||
| Com# | Variable | EC50 μM | SI | Com# | Variable | EC50 μM | SI | Com# | Variable | EC50 μM | SI | |||
| 1 | R1 | CH | 0.23 | 391 | 7 | R1 | CH | 3.4 | 13 | 13 | R1 | N | 9.7 | 1.8 |
| R2 | R2 | R2 | ||||||||||||
| 2 | R1 | CH | 0.66 | 83 | 8 | R1 | N | 4.1 | >24 | 14 | R1 | CH | 11 | 2.4 |
| R2 | R2 | R2 | ||||||||||||
| 3 | R1 | CH | 0.88 | 34 | 9 | R1 | N | 7.5 | 13 | 15 | R1 | CH | 22 | >4.5 |
| R2 | R2 | R2 | ||||||||||||
| 4 | R1 | CH | 1.0 | 21 | 10 | R1 | CH | 8.1 | 3.3 | 16 | R1 | N | 27 | 1.9 |
| R2 | R2 | R2 | ||||||||||||
| 5 | R1 | CH | 1.6 | 41 | 11 | R1 | N | 8.5 | 12 | 17 | R1 | N | 31 | 3.2 |
| R2 | R2 | R2 | ||||||||||||
| 6 | R1 | N | 1.8 | 29 | 12 | R1 | CH | 8.6 | >11 | |||||
| R2 | R2 | |||||||||||||
3.1. SRI 29365 reduces hRSV-induced CPE in HEp-2 and A549 cells
To determine the EC50 of SRI 29365, a concentration range of the compound (100–0.05 μM) was added to HEp-2 and A549 cells, and then the cells were immediately infected with 100 TCID50s of hRSV. Compound cytotoxicity experiments (-virus) were performed in parallel to identify the compound CC50. Fig. 1 shows the concentration-dependent effects of SRI 29365 on both virus-induced CPE and cell viability in HEp-2 and A549 cells. The EC50 values were 0.66 and 1.2 μM in HEp-2 and A549 cells, respectively, with SI values of 83 and >83, respectively. The EC50 and SI values for SRI 29365 in HEp-2 cells are also indicated in Table 1.
Fig. 1.
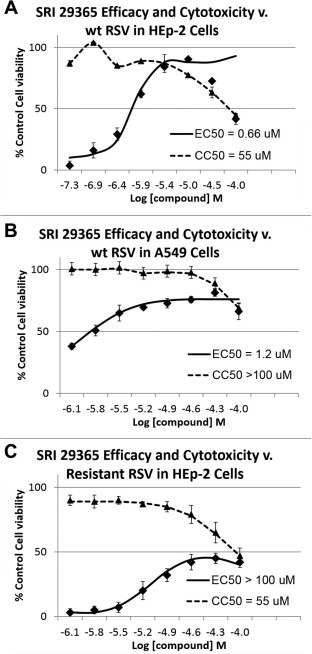
EC50 and CC50 plots for SRI 29365 in HEp-2 and A549 cells. EC50 and CC50 values were calculated for SRI 29365 against wt hRSV in either (A) HEp-2 cells or (B) A549 cells. In panel C, the concentration-dependent antiviral and cytotoxic responses were calculated for SRI 29365 against wt hRSV in HEp-2 cells. An eight point concentration dose response cytoprotection assay was performed (compound concentration range of 100–0.04 μg/mL) in the presence of hRSV-infected cells and the percent of cell viability (compared to uninfected, undrugged cell controls) was plotted v. test compound concentration (solid line). Each concentration point was performed in triplicate. In parallel, the same concentration range of test compound was added to uninfected cells to determine compound-induced cytotoxicity (dotted line). EC50/CC50 values were converted to Log M (molar) concentrations (x-axis). Dose–response curves were plotted, and EC50 values were calculated using IDBS’ XLfit function 205 [y = A + ((B − A)/(1 + ((C/x)^D)))] with minimum (A) and maximum (B) parameters set at 0 and 100 respectively.
The antiviral efficacy of SRI 29365 against other RNA viruses was explored using additional in vitro CPE assay models. The compound was tested against human metapneumovirus and Nipah virus (paromyxoviruses), Venezuelan equine encephalitis virus (alphavirus), Dengue 2 virus (flavivirus), human influenza virus (orthomyxovirus), and SARS CoV (coronavirus). No significant reduction in CPE due to antiviral activity was observed for any of these viruses.
3.2. Chemical analogs of SRI 29365 also reduce virus-induced CPE
To define structure–activity relationships for SRI 29365, 71 commercially-available compounds with structural similarities to SRI 29365 were purchased and tested in the CPE assay for efficacy against hRSV in HEp-2 cells. Of these compounds, 16 reduced virus-induced CPE, with EC50 values ranging from 0.23 to 31 μM, enabling structure–activity relationships to be determined. Table 1 shows SRI 29365 and the structural variation of the effective chemical analogs. Table S1 shows the structures of all 71 compounds tested, commercial source, EC50, CC50, and SI data. Of the tested compounds, only one compound (Table 1. Compound 1; 1-[6-(2-thienyl)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl]methyl-1H-benzimidazole) had greater potency (lower EC50 and higher SI) than SRI 29365. However, SRI 29365 was chosen as the lead compound based on potency and supplier availability, and all further characterization experiments were performed with this compound.
3.3. Structure activity relationship analysis
Of the 17 compounds shown in Table 1 that effectively reduced RSV-induced CPE in HEp 2 cells, two had high activity (SI > 50), five were moderately active (SI > 20) and ten had low activity (SI < 20). Structural variance occurred at positions R1 and R2 only, but significant differences in compound potency were observed with changes in these positions (Table 1). A structure–activity relationship analysis indicates that a carbon atom at the R1 position is more favorable than an imine at the same position (Compound 2 is more potent than Compound 8). Compound 1 had the greatest potency (lowest EC50 and highest SI), indicating that a thiofuran at the R2 position is more potent than when a furan (Compound 2, SRI-29365) is substituted. An R2 aromatic ring substitution results in even less potent compounds (Compounds 5 and 6). Further modification of the aromatic ring with halogens (chlorine or fluorine) decreases potency further. The benzimidazole scaffold and its analogs were stable in assay conditions, during storage in DMSO below −70 °C, and during general manipulation. The SRI 29365 structure does not contain moieties that are known generally to be reactive.
3.4. Time-of-addition experiments suggest that SRI 29365 inhibits an early step in virus infection
Time-of-addition experiments were performed to determine the stage of the hRSV replication cycle that was inhibited by SRI 29365. Using an MOI of 1 (∼10,000 TCID50s), HEp-2 cells were infected with hRSV. SRI 29365 (5 μM) was added at designated time points and CPE was measured 48 h post-infection. Fig. 2 shows that SRI 29365 prevented CPE only when added at or before 2 h post-infection. Addition of SRI 29365 after 2 h post-infection did not prevent greater than 50% of virus-induced CPE, indicating that compound inhibits an early step within the first 2 h of the virus replication cycle.
Fig. 2.
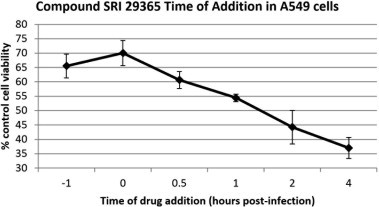
Time-of-addition assays for SRI 29365. SRI 29365 (5 μM) was added to 96-well microplates containing a confluent monolayer of HEp-2 cells at timepoints of −1, 0, 1, 2, 4, 8 h, post viral infection of the cells with 1 MOI of hRSV Long strain. After a four-day incubation, cell viability was determined (compared to uninfected, undrugged cell controls) (n = 3). The graph shows the change in cell viability as the compound addition timepoint changes, and demonstrates that SRI 29365 did not reduce CPE more than 50% if added at 2 h post-infection.
3.5. hRSV with resistance to SRI 29365 has mutations in the viral G protein gene
Sequential passaging of the wild-type virus under the selective pressure of increasing concentrations of SRI 29365 resulted in the development of a virus that was resistant to the antiviral effects of SRI 29365. This resistant virus was plaque purified and amplified in HEp-2 cells in the presence of 5 μM SRI 29365 for characterization. However, titration experiments with the resistant virus did not establish an EC50 for SRI-29365 (Fig. 1C). This is likely because the resistant mutant virus is >100-fold resistant to the wt virus EC50 concentration of SRI 29365. Because the selectivity of the SRI 29365 is <100 (83), the compound-induced CC50 is below the EC50 value for the mutant virus. However, because of the high resistance of the mutant hRSV, this virus was sub-cultured and amplified in the presence of 5 μM of SRI 29365, and a stock of the virus was prepared and stored. The viral RNA in 1 mL of the amplified mutant virus culture was isolated and cDNA was reverse-transcribed using hRSV-specific primers. The complete mutant virus genome was sequenced twice and a mutation was identified in the viral G protein gene that suggested that the antiviral mechanism of SRI 29365 was associated with hRSV G protein function. Fig. 3 A and B depict the regions of the viral G protein genetic sequence that contains the mutation and location of the mutation in the primary protein domain structure of the viral G protein. Even after multiple passages under compound selection, a mixed population of wt and mutant was observed in the G protein genetic sequence, but the virus was still >100-fold more resistant to SRI 29365 than the wt hRSV. The mutation (A63V) occurs in the N-terminal region of the G protein within the transmembrane domain. Fig. 3D shows the extended amino acid sequence of the transmembrane domain surrounding the point mutation. The region containing the mutation is conserved in both RSVA strain Long and RSVA strain Tracy (used for in vivo studies described below), but not in RSVB. SRI 29365 was not tested against RSVB.
Fig. 3.
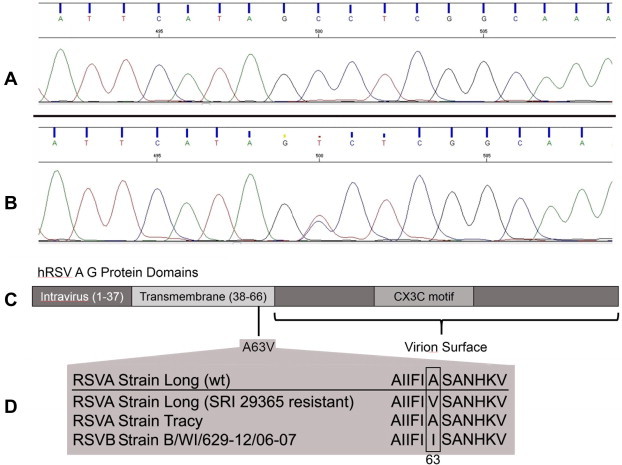
An A63V mutation in the hRSV G protein grants resistance to SRI 29365. Panel A shows the hRSV G protein genetic sequence trace of the wt hRSV and Panel B shows the genetic trace of the SRI 29365-resistant mutant hRSV, which shows a C → T mutation at position 500 in the sequence trace. Panel C shows the hRSV G protein domain structure and the location of the translated A63V amino acid mutation at the interface of the transmembrane domain and the virion surface domain. Panel D shows the amino acid sequence conservation of the region of the G protein transmembrane domain flanking residue A63 in RSVA Long (wild-type), RSVA Long (SRI-29365-resistant mutant), RSVA Tracy, and RSVB WI/629-12/06-07.
3.6. SRI 29365 antiviral efficacy correlates with truncated G protein expression and heparin-dependent infection in different cell types
The viral G protein mediates virus attachment by binding heparin molecules that are expressed on the cell surface (Krusat and Streckert, 1997), and requires full-length G protein to do so efficiently. It has been previously shown that the virus-associated expression of full-length G protein in cell culture is a factor of cell type (Kwilas et al., 2009). Viral replication in HEp-2 cells results in expression of full-length G protein (90 kDa), whereas replication in A549 cells or Vero E6 cells results in the expression of a truncated G protein (45–55 kDa) that is missing the C-terminus. Replication of hRSV in HEp-2 cells is dependent on the binding of G protein to cell surface heparin glycosaminoglycans. However, replication in A549 cells or Vero cells produces hRSV that infects cells by a heparin-independent mechanism, most likely because the truncated G protein does not contain a fully functional heparin binding domain (Kwilas et al., 2009). We used this cell-dependent difference in expressed G protein length to measure the antiviral efficacy of SRI 29365 in the three different cell types to determine if the compound was more effective against hRSV with full-length G protein. All three cell types were infected with the wild-type or the SRI 29365-resistant hRSV mutant in the presence of 5 μM SRI 29365, and the reduction in virus (TCID50 reduction assay) was determined. Fig. 4 shows the compound-mediated reduction in viral titers for both wt and mutant virus in HEp-2, A549, and Vero E6 cells, relative to infected (- compound) controls. Against wt hRSV, the compound reduced virus TCID50 values by 300-fold (P < 0.003), but only 10-fold (P < 0.0005) in A549 cells, and less than 10-fold (P < 0.00001) in Vero E6 cells. The antiviral activity of SRI 29365 against wt hRSV is cell-type specific and strongly correlates with the expression of full-length G protein, implicating the G protein as the compound target. Against the mutant hRSV, the compound reduced virus TCID50 values by less than 10-fold in all cell types. Furthermore, in the presence of SRI 29365, a 2.0 log reduction in wt hRSV titer v. 0.5 log reduction in compound-resistant hRSV titer was observed in HEp-2 cells. However, SRI-29365 reduced titers of both viruses less than 0.5 log in Vero E6 cells, indicating that the compound has little effect on either wt or mutant virus titers when a truncated G protein is expressed.
Fig. 4.
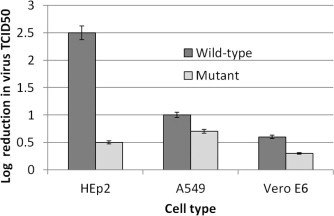
SRI 29365 has differential efficacy in HEp-2, A549, and Vero E6 cells. Confluent monolayers of HEp-2, A549, or Vero E6 cells with either the RSVA Long strain wild-type or SRI-29356-resistant mutant in the presence of 5 μM SRI 29365, and the reduction in virus titer (TCID50 reduction) was determined relative to infected (-compound) control cells (n = 4). The compound reduced wt hRSV virus TCID50 values by 300-fold, but only 10-fold in A549 cells, and less than 10-fold in Vero E6 cells. SRI 29365 reduced mutant hRSV virus TCID50 values by less than 10-fold in all cell types.
3.7. SRI 29365 does not reduce virus-induced pathology or titers in vivo
A cotton rat model for hRSV (strain A/Tracy) was used to test the in vivo potency of SRI 29365. In vitro potency against RSVA Tracy was not determined. Administration of SRI 29365 to infected animals resulted in no significant difference was observed in lung lobe weight, body weight, or RSV titers in nasal wash or lung lavage fluids. The in vitro pharmacokinetics of SRI 29365 have been partially investigated. Aqueous (media) solubility was determined (<400 uM) and aqueous stability half-life at 37 °C was measured (>60 h in cell culture media), with a cLogP = 2.35. Because the compound was not completely soluble at the concentration used for the high dose, this may account for the lack of in vivo activity. However, the lack of in vivo activity at the lower doses suggests that SRI 29365 is not effective at inhibiting viral replication in vivo.
4. Discussion
Although hRSV was identified decades ago, there is currently no approved vaccine. The first vaccine testing study caused disease enhancement with severe bronchiolitis and pneumonia, requiring hospitalization of 80% of vaccinated children after hRSV challenge, and two infant deaths (Delgado et al., 2009, Dudas and Karron, 1998, Karron et al., 1997). As a result, development of a vaccine has proceeded slowly and with great caution. Approved therapies for viral infections are ribavirin (Pelaez et al., 2009), and the prophylactic humanized monoclonal antibody Synagis (Palivizumab), which is administered only to high risk pediatric patients and infected immunocompromised patients (Ridky, 2014). Newer potential therapeutics are under investigation and reviewed elsewhere (Mackman et al., 2015, Olszewska and Openshaw, 2009, Wang et al., 2015), but the fact that none of these have been approved by regulatory agencies (due to economics and clinical testing complexities) underscores the need for a renewed search for novel, effective, and safe antivirals that are effective against hRSV.
Here, we used an in vitro HTS cytoprotection assay to identify a lead compound (SRI 29365) that effectively protected HEp-2 and A549 cells from hRSV-induced CPE. We established compound EC50 and selectivity and used chemical analogs of SRI 29365 to determine structure activity relationships. Using in vitro assays, SRI 29365 was not effective against other RNA viruses. Time-of-addition experiments indicated that SRI 29365 reduced hRSV-induced CPE only if added within 2 h of virus infection, suggesting that it inhibited an early virus replication step. A hRSV mutant that was resistant to inhibition by the compound was isolated and sequenced, and a mutation was identified in the sequence of the viral G protein, implicating the G protein as the putative target of inhibition. In vitro characterization of both the wt and mutant viruses in cell types that express either full-length or truncated G protein suggested that SRI 29365 inhibits viral G protein function. Lastly, an in vivo study was performed to determine the effectiveness of the compound at reducing hRSV titers in a challenged cotton rats, but SRI 29365 was largely insoluble in the chosen dose formulation and was not effective at reducing hRSV titers or pathology in the animal model.
The specific cell surface receptors used for infection by hRSVs are not known, but cell surface glycosaminoglycans, and in particular heparan sulfates, have been shown to be important for infection (Schlender et al., 2003). The hRSV G protein, is a type II transmembrane glycoprotein and is the major hRSV attachment protein (Kauvar et al., 2010). The role of the nominal attachment protein G is confined to facilitating infection in a non-species-specific manner, most probably by binding to cell surface glycosaminoglycans (Kauvar et al., 2010). It contains a single hydrophobic region which serves as a signal peptide and also as a membrane anchor. The RSV G protein is also an important contributor to the disease process. A conserved CX3C chemokine like motif in G likely contributes to the pathogenesis of disease (Harcourt et al., 2006). The G protein is evolutionarily conserved in all strains of RSV and has a central conserved cysteine noose region that contains a CX3C chemokine motif at amino acid positions 182–186 (Harcourt et al., 2006). Through this motif G protein binds to CX3CR1 present on various immune cells and affects immune responses to hRSV, as has been shown in a mouse model of RSV infection. However, very little is known of the role of the RSV CX3C–CX3CR1 interactions in human disease. Reports suggest that the hRSV G protein CX3C motif impairs innate and adaptive human immune responses and may be important to vaccine and antiviral drug development. (Chirkova et al., 2013). Lastly vaccination with this CX3C motif induces a protective immune response in mice (Jorquera et al., 2013).
We identified a compound that inhibits viral replication by the putative mechanism of interfering with the function the viral G protein. Since G protein binds to cell surface heparin molecules during virus attachment, one possible mechanism of SRI 29365 action is inhibition of G protein-mediated heparin binding. We demonstrated that the antiviral efficacy of the test compound SRI 29365 against wt hRSV is significantly reduced in a cell type (i.e., Vero E6) that expresses a truncated G protein that is not dependent on heparin binding for infection. We also showed that, in the presence of SRI 29365, the phenotypes of the wt and mutant hRSVs are different in HEp-2 cells, but similar in Vero E6 cells, suggesting that the truncation of the G protein (and possibly the loss of the heparin-binding domain) is linked to compound mechanism of action. Although hRSV primarily infects cells through heparin binding domains on the G protein, mutant or knockout viruses lacking the G gene do still infect cells and replicate in vitro and in vivo presumably by attachment through redundant heparin binding domains on the F protein (Karron et al., 1997). This observation was reciprocated by in vivo studies of SRI 29365, as SRI-29365 did not significantly reduce hRSV titers or pathology in a cotton rat infection model. Overall, this compound serves as an excellent in vitro inhibitor of hRSV replication, but it was not effective in vivo against RSVA strain Tracy. Since the A63 residue is conserved in both the RSVA Long and Tracy strains, it is expected that this compound would be effective against RSVA Tracy. The lack of in vivo confirmation of in vitro activity may be a result of compound formulation, or may be indicative that the viral G protein function is not a valid drug target for small molecule therapy. This might be due to the above stated observation that hRSV G protein knockout viruses still infect with sufficient efficacy to support robust virus replication. Lastly, residue A63 is not conserved in both A and B strains. In RSVB, residue 63 is an isoleucine, and it is unclear whether it would be susceptible to inhibition by SRI-29365 from the sequence alone.
Animal care and use statement
All research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the National Research Council of the National Academies Guide for the Care and Use of Laboratory Animals. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Acknowledgments
The project was initiated with the Southern Research Institute Strategic Initiatives Program and NIH contract # HHSN2722011000131 to JWN and NIH contract HHSN272201000004I to BEG.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2015.06.016.
Appendix A. Supplementary data
References
- Byrd L.G., Prince G.A. Animal models of respiratory syncytial virus infection. Clin. Infect. Dis. 1997;25(6):1363–1368. doi: 10.1086/516152. [DOI] [PubMed] [Google Scholar]
- Chembridge, 2015. Chembridge corporation: pre-selected diversity libraries for HTS and chemical biology. <http://www.chembridge.com/screening_libraries/diversity_libraries/> Accessed March 9, 2015.
- Chirkova T., Boyoglu-Barnum S., Gaston K.A., Malik F.M., Trau S.P., Oomens A.G., Anderson L.J. Respiratory syncytial virus G protein CX3C motif impairs human airway epithelial and immune cell responses. J. Virol. 2013;87(24):13466–13479. doi: 10.1128/JVI.01741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci C., Genovesi E.V., Lamb L., Medina I., Yang Z., Zadjura L., Yang H., D’Arienzo C., Sin N., Yu K.L., Combrink K., Li Z., Colonno R., Meanwell N., Clark J., Krystal M. Oral efficacy of a respiratory syncytial virus inhibitor in rodent models of infection. Antimicrob. Agents Chemother. 2004;48(7):2448–2454. doi: 10.1128/AAC.48.7.2448-2454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M.F., Coviello S., Monsalvo A.C., Melendi G.A., Hernandez J.Z., Batalle J.P., Diaz L., Trento A., Chang H.Y., Mitzner W., Ravetch J., Melero J.A., Irusta P.M., Polack F.P. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 2009;15(1):34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas R.A., Karron R.A. Respiratory syncytial virus vaccines. Clin. Microbiol. Rev. 1998;11(3):430–439. doi: 10.1128/cmr.11.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt J., Alvarez R., Jones L.P., Henderson C., Anderson L.J., Tripp R.A. Respiratory syncytial virus G protein and G protein CX3C motif adversely affect CX3CR1+ T cell responses. J. Immunol. 2006;176(3):1600–1608. doi: 10.4049/jimmunol.176.3.1600. [DOI] [PubMed] [Google Scholar]
- Jorquera P.A., Choi Y., Oakley K.E., Powell T.J., Boyd J.G., Palath N., Haynes L.M., Anderson L.J., Tripp R.A. Nanoparticle vaccines encompassing the respiratory syncytial virus (RSV) G protein CX3C chemokine motif induce robust immunity protecting from challenge and disease. PLoS ONE. 2013;8(9):e74905. doi: 10.1371/journal.pone.0074905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karron R.A., Buonagurio D.A., Georgiu A.F., Whitehead S.S., Adamus J.E., Clements-Mann M.L., Harris D.O., Randolph V.B., Udem S.A., Murphy B.R., Sidhu M.S. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. 1997;94(25):13961–13966. doi: 10.1073/pnas.94.25.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauvar L.M., Harcourt J.L., Haynes L.M., Tripp R.A. Therapeutic targeting of respiratory syncytial virus G-protein. Immunotherapy. 2010;2(5):655–661. doi: 10.2217/imt.10.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusat T., Streckert H.J. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 1997;142(6):1247–1254. doi: 10.1007/s007050050156. [DOI] [PubMed] [Google Scholar]
- Kwilas S., Liesman R.M., Zhang L., Walsh E., Pickles R.J., Peeples M.E. Respiratory syncytial virus grown in Vero cells contains a truncated attachment protein that alters its infectivity and dependence on glycosaminoglycans. J. Virol. 2009;83(20):10710–10718. doi: 10.1128/JVI.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman R.L., Sangi M., Sperandio D., Parrish J.P., Eisenberg E., Perron M., Hui H., Zhang L., Siegel D., Yang H., Saunders O., Boojamra C., Lee G., Samuel D., Babaoglu K., Carey A., Gilbert B.E., Piedra P.A., Strickley R., Iwata Q., Hayes J., Stray K., Kinkade A., Theodore D., Jordan R., Desai M., Cihlar T. Discovery of an oral respiratory syncytial virus (RSV) fusion inhibitor (GS-5806) and clinical proof of concept in a human RSV challenge study. J. Med. Chem. 2015;58(4):1630–1643. doi: 10.1021/jm5017768. [DOI] [PubMed] [Google Scholar]
- Matharu D.S., Flaherty D.P., Simpson D.S., Schroeder C.E., Chung D., Yan D., Noah J.W., Jonsson C.B., White E.L., Aube J., Plemper R.K., Severson W.E., Golden J.E. Optimization of potent and selective quinazolinediones: inhibitors of respiratory syncytial virus that block RNA-dependent RNA-polymerase complex activity. J. Med. Chem. 2014;57(24):10314–10328. doi: 10.1021/jm500902x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B.P., Chung D.H., Matharu D.S., Golden J.E., Maddox C., Rasmussen L., Noah J.W., Sosa M.I., Ananthan S., Tower N.A., White E.L., Jia F., Prisinzano T.E., Aube J., Jonsson C.B., Severson W.E. (S)-N-(2,5-Dimethylphenyl)-1-(quinoline-8-ylsulfonyl)pyrrolidine-2-carboxamide as a small molecule inhibitor probe for the study of respiratory syncytial virus infection. J. Med. Chem. 2012;55(20):8582–8587. doi: 10.1021/jm300612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah, J.W., Severson, W., Chung, D.H., Moore, B., Jia, F., Xu, X., Maddox, C., Rasmussen, L., Sosa, M.I., Tower, N.A., Ananthan, S., White, E.L., Jonsson, C., Matharu, D.S., Golden, J.E., Prisinzano, T.E., Aube, J., 2010. A cell based HTS approach for the discovery of new inhibitors of RSV. Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD). [PubMed]
- Noah J.W., Severson W., Noah D.L., Rasmussen L., White E.L., Jonsson C.B. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007;73(1):50–59. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Noah, J.W., Severson, W.E., Chung, D.H., Moore, B., Jia, F., Xu, X., Maddox, C., Rasmussen, L., Sosa, M.I., Tower, N.A., Ananthan, S., Evans, C.W., White, E.L., Jonsson, C., Matharu, D.S., Flaherty, D.P., Simpson, D.S., Golden, J.E., Aube, J., 2010. Identification of a series of quinazolinediones as potent, selective, post-entry inhibitors of human respiratory syncytial virus (hRSV) via a cell-based high throughput screen and chemical optimization. Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD). [PubMed]
- Olszewska W., Openshaw P. Emerging drugs for respiratory syncytial virus infection. Expert Opin. Emerg. Drugs. 2009;14(2):207–217. doi: 10.1517/14728210902946399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer L., Hall C.B., Katkin J.P., Shi N., Masaquel A.S., McLaurin K.K., Mahadevia P.J. Healthcare costs within a year of respiratory syncytial virus among medicaid infants. Pediatric Pulmonol. 2010;45(8):772–781. doi: 10.1002/ppul.21244. [DOI] [PubMed] [Google Scholar]
- Pelaez A., Lyon G.M., Force S.D., Ramirez A.M., Neujahr D.C., Foster M., Naik P.M., Gal A.A., Mitchell P.O., Lawrence E.C. Efficacy of oral ribavirin in lung transplant patients with respiratory syncytial virus lower respiratory tract infection. J. Heart Lung Transplant. 2009;28(1):67–71. doi: 10.1016/j.healun.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde D.C., Tran T.D., Gardner I., Bright H., Stupple P., Galan S., Alsop L., Watson L., Middleton D.S., Dayal S., Platts M., Murray E.J., Parkinson T., Webster R. Non-benzimidazole containing inhibitors of respiratory syncytial virus. Bioorg. Med. Chem. Lett. 2013;23(3):827–833. doi: 10.1016/j.bmcl.2012.11.062. [DOI] [PubMed] [Google Scholar]
- Rebuffo-Scheer C., Bose M., He J., Khaja S., Ulatowski M., Beck E.T., Fan J., Kumar S., Nelson M.I., Henrickson K.J. Whole genome sequencing and evolutionary analysis of human respiratory syncytial virus A and B from Milwaukee, WI 1998–2010. PLoS ONE. 2011;6(10):e25468. doi: 10.1371/journal.pone.0025468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridky J. Medication highlights: new recommendations for palivizumab (Synagis) Neonatal Netw. 2014;33(6):358–359. [PubMed] [Google Scholar]
- Schlender J., Zimmer G., Herrler G., Conzelmann K.K. Respiratory syncytial virus (RSV) fusion protein subunit F2, not attachment protein G, determines the specificity of RSV infection. J. Virol. 2003;77(8):4609–4616. doi: 10.1128/JVI.77.8.4609-4616.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Deval J., Hong J., Dyatkina N., Prhavc M., Taylor J., Fung A., Jin Z., Stevens S.K., Serebryany V., Liu J., Zhang Q., Tam Y., Chanda S.M., Smith D.B., Symons J.A., Blatt L.M., Beigelman L. Discovery of 4′-chloromethyl-2′-deoxy-3′,5′-di-O-isobutyryl-2′-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J. Med. Chem. 2015;58(4):1862–1878. doi: 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- Yu K.L., Sin N., Civiello R.L., Wang X.A., Combrink K.D., Gulgeze H.B., Venables B.L., Wright J.J., Dalterio R.A., Zadjura L., Marino A., Dando S., D’Arienzo C., Kadow K.F., Cianci C.W., Li Z., Clarke J., Genovesi E.V., Medina I., Lamb L., Colonno R.J., Yang Z., Krystal M., Meanwell N.A. Respiratory syncytial virus fusion inhibitors. Part 4: optimization for oral bioavailability. Bioorg. Med. Chem. Lett. 2007;17(4):895–901. doi: 10.1016/j.bmcl.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Yu K.L., Zhang Y., Civiello R.L., Trehan A.K., Pearce B.C., Yin Z., Combrink K.D., Gulgeze H.B., Wang X.A., Kadow K.F., Cianci C.W., Krystal M., Meanwell N.A. Respiratory syncytial virus inhibitors. Part 2: benzimidazol-2-one derivatives. Bioorg. Med. Chem. Lett. 2004;14(5):1133–1137. doi: 10.1016/j.bmcl.2003.12.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.