

Abstract
Porcine epidemic diarrhea (PED) has been endemic causing sporadic outbreaks in Thailand since 2007. In 2014–2015, several herds had experienced severe PED outbreaks and the reason of the re-current outbreaks was unknown. Whether or not the introduction of exotic strains or continual evolution of existing PEDV, genetic analyses would provide a more understanding in its evolutionary pattern. In the study, 117 complete spike gene sequences of Thai PED virus (PEDV) collected from 2008 to 2015 were clustered along with 95 references of PEDV spike sequences, and analyzed with the US sequences dataset (n = 99).
The phylogenetic analysis demonstrated that Thai PEDV spike sequences were genetically diverse and had been influenced by multiple introduction of exotic strains. Although Thai PEDV have been evolved into 6 subgroups (TH1–6), Subgroup TH1 strains with the unique 9 nucleotides (CAA GGG AAT) insertion between 688th–689th position of spike (changing amino acid from N to TREY) insertion has become the dominant subgroup since 2014. Thai PEDV spike gene have higher evolutionary rate compare to that of the US sequences. One contributing factor would be the intra-recombination between subgroups. Thailand endemic strain should be assigned into new subclade of G2 (Thai pandemic variant).
Keywords: Porcine epidemic diarrhea virus, Epidemiology, Evolutionary rate, Thailand
Graphical abstract
Highlights
-
•
Thai isolates of PEDV developed their own clusters separated from other countries.
-
•
There were multiple introductions of PEDV causing outbreaks in Thailand.
-
•
Thai isolates have higher evolution than US isolates determined by the analysis in this study.
-
•
There were 2 clades of PEDV, TH1 and TH2, which mainly causes the outbreaks in Thailand.
-
•
There was a unique insertion of spike gene of Thai isolates between 2013-2015.
1. Introduction
Porcine epidemic diarrhea (PED) is a devastating enteric disease in pigs, characterized by vomiting and acute watery diarrhea leading to death due to severe dehydration. Pigs at all ages are susceptible to PED infection, but mortality is higher in younger pigs, especially in piglets at younger than a week of age. Since its first recognition in the late 1970s (Chasey and Cartwright, 1978, Pensaert and De Bouck, 1978), PED has continued to cause a severe economic impact in swine industry worldwide. At present, PED has been reported as re-emerging disease worldwide (Chen et al., 2014, Dennis et al., 2015, Lee et al., 2014, Lin et al., 2014, Masuda et al., 2015, Ojkic et al., 2015, Theuns et al., 2015, Vui et al., 2015).
PED virus (PEDV), a causative agent of PED, is an enveloped single-stranded positive-sense RNA virus belonging to the genus Alphacoronavirus, family Coronaviridae, order Nidovirales. At present, two PEDV variants including genogroups 1 and 2 are currently recognized (Lee, 2015, Sun et al., 2015). The PEDV genome is consisting of seven open reading frames (ORFs) organizing in order as following; ORF1a and ORF1b, spike (S), ORF3, envelope (E), membrane (M), and nucleoprotein (N) (Kocherhans et al. 2001). Two-thirds of the genome is occupied by ORF1a and ORF1b, which encode nonstructural proteins. The other 4 structural proteins including S, E, M, and N are located downstream of the ORF1a/1b gene. The S protein is a glycosylated protein involving with viral pathogenesis and be further divided into S1 and S2 domains. The S protein attaches to the host cellular receptors resulting to virus entry by membrane fusion (Bosch et al. 2003) and contains domain that stimulates the production of neutralizing antibodies (Chang et al., 2002, Cruz et al., 2006, Cruz et al., 2008, Duarte and Laude, 1994, Sun et al., 2008, Sun et al., 2007). The S gene is divergent and important for understanding the genetic relatedness of PEDV field strains, the epidemiological status of the virus and vaccine development (Li et al., 2012, Park et al., 2011, Sun et al., 2012). ORF3 encodes an accessory protein located between S and E. In addition, the ORF3 gene is the only accessory gene, and has been an important determinant of virulence in PEDV (Park et al. 2008). The vaccine-derived strains have unique deletion of 17 amino acids at position 82 to 99 (Park et al. 2008). The other E and M genes are associated with viral envelope formation and release. The N protein is the predominant antigen produced in coronavirus-infected cells, making it a major viral target (Kocherhans et al. 2001).
In Thailand, PED was first emerged in 2007 and the variant responsible for the outbreaks was of genogroup 2 (Temeeyasen et al. 2014). At present, PED has developed into an endemic stage in which many herds experience recurrent outbreaks and both PEDV genogroups 1 and 2 are currently existing in the population (Cheun-Arom et al. 2015). In 2014–2015, several herds reported the severe diarrhea outbreaks. Whether or not the novel introduction or continual development of PEDV in Thai swine farms causing the outbreak is not known and the information on PEDV evolutionary dynamics in Thailand has been deficiency.
Since increasing of computer performance and development of graphic user interface of BEAST format support software, BEAST packages become fast, flexible and reliable tools for molecular sequences analysis based on Bayesian framework (Drummond and Rambaut 2007). Our study was aimed to investigate the genetic diversity of PEDV clinical samples and determine the evolutionary rate of PEDV in Thailand. The genetic analysis was based on the complete spike gene of Thai PEDV collected from 2008 to 2015 along with that of reference sequences from other countries provided in GenBank®. The combination of the sequences with time of isolation analyzing with BEAST can provide us the estimated time of divergences and the evolutionary rate of suspected group of population. The results would provide us the information on the evolutionary dynamics of the viruses in Thailand.
2. Materials and methods
2.1. Source of specimens and PEDV isolation
A total of 117 Thai PEDV spike gene sequences including 99 PEDV clinical samples that were genetically characterized during 2014–2015 together with 18 Thai sequences previously reported (Temeeyasen et al. 2014) (Table S1) were used in the genetic analyses. The 115 PEDV sequences were collected from 24 pig farms located in Ratchaburi, Nakhon Pathom, Saraburi, Lopburi, Nakhon Ratchasima, Buriram, Chonburi, Chanthaburi and ChaCherngsao from 2008 to 2015 that experienced PED outbreaks (55 outbreaks) whereas another 2 sequences are not the field strains, the AVCT12 (LC053455) and a vaccine strain sequence from the vaccine using in the farm that experienced the outbreak of EAS (KR610991). In each outbreak, intestinal samples were collected from 3- to 4-day-old piglets that displayed the clinical features associated with PED, including vomiting and watery diarrhea. Intestinal samples were minced into small pieces and suspended in PBS and clarified by centrifugation. The supernatant was filtered through 0.22 μm filters and stored at − 80 °C until use. The clarified supernatants were subjected to RT-PCR.
2.2. Cloning, plasmid purification and sequence determination
Sequencing was performed as previously reported (Temeeyasen et al. 2014). Briefly, total RNA was extracted from the supernatant using the Nucleospin®RNA Virus kit (Macherey-Nagel Inc., PA, USA) in accordance with the manufacturer's instructions. cDNA was synthesized from the extracted RNA using M-MuLV Reverse Transcriptase (New England BioLabs Inc., MA, USA).
PCR amplification was performed on the cDNA. To amplify the complete S genes, PCR amplification was performed using previously reported primers (Lee et al. 2010) and using Platinum® Tag DNA polymerase High Fidelity (Invitrogen, CA, USA). The PCR products were cloned into plasmid vectors for the subsequent transformation of Escherichia coli cells by using a commercial kit (pGEM-T® Easy Vector System I (Promega, WI, USA), and the controls were included at all stages of cloning and transformation. Bacterial transformant colonies were growth in Luria Broth (LB) agar for 18 h and randomly selected from each sample for plasmid purification using the Nucleospin® Plasmid kit (Macherey-Nagel Inc., PA, USA), and the 10 selected colonies were grown in LB broth for 24 h and subjected to plasmid isolation and sequencing. Sequencing reactions were performed by third party (1st BASE DNA Sequencing Services, Singapore).
2.3. Sequence analyses
2.3.1. Phylogenetic analysis
The nucleotide and deduced amino acid sequences were aligned using the software, MAFFT (Multiple Alignment using Fast Fourier Transform) (Katoh et al. 2002). A phylogenetic analysis was performed based on the nucleotide sequences of complete spike genes of 117 Thai PEDV spike sequences collected from 2008 to 2015 along with 94 other PEDV spike sequences available in GenBank® (Table S1) from previous study (Lee 2015) using the MEGA6.0 program (Tamura et al. 2013). A phylogenetic tree was constructed using Bayesian Markov chain Monte Carlo (BMCMC) with best-fit substitution model, TN93 + G + I and base frequency was set as empirical, all other settings were left as default. The analysis was performed until ESS > 200, the genogroup described in other study (Lee 2015) was used to classify and compare with the sequences in this study.
2.3.2. Evolutionary analysis
The evolutionary analysis of 115 Thai PEDV spike sequences except of those two which not the field strains as described in previous part were performed analysis in comparison 99 US PEDV spike sequences (Table S2) using a BMCMC method implemented in the program BEAST v1.8.3 (Drummond and Rambaut, 2007, Drummond et al., 2012). The best fitted evolutionary model GTR + G + I was determined by maximum likelihood model selection function of MEGA 6.0 (Tamura et al. 2013) and was used in all BEAST analyses. The coalescent Bayesian skyline tree prior (Drummond et al., 2002, Drummond et al., 2005) and empirical base frequencies were applied under three different models for rate variation among branches: the strict molecular clock model (STR), the uncorrelated lognormal relaxed-clock model (LOG), and the uncorrelated exponential relaxed-clock model (EXP) (Drummond et al. 2006). For each analysis, 200 million states were applied at least with logged every 10,000 states, the states must be increased in order to gain ESS > 200. If there was another independent run, the log and tree data will be combined with the first 10% discarded as burn-in using LogCombinerv1.8.3, and, the resulting files were displayed using Tracer 1.6.0 (Rambaut and Drummond 2013). Maximum clade credibility trees were annotated using TreeAnnotator 1.8.3 and phylogenetic tree with timeline, estimated divergences, posterior probability and 95% HPD displays were generated using FigTree 1.4.2 (Rambaut and Drummond 2014).
2.3.3. Phylogeographic distribution
Phylogeographic distribution of the endemic subgroup was also estimated using BEAST 1.8.4 under TN93 + G + I substitute model. 600 million with logged 10,000 states was performed at least for subgroup TH1 and TH2. All other settings were performed as described in previous part. The results were converted and displayed in GoogleEarth (Lozano-Fuentes et al. 2008) using SPREAD 1.0.7 (Bielejec et al. 2011). The sequences which have 100% identity from the same location and time of the outbreak were excluded.
2.3.4. Recombinant analysis
Recombination events among the sequences (n = 117) were analyzed using automated RDP, GENECONV, BOOTSCAN, MaxChi, CHIMAERA, and SISCAN provided by RDP4 software (Martin et al. 2015). The evidence events with known potential parents and recombinants were displayed.
3. Results
3.1. Phylogenetic and genetic analyses
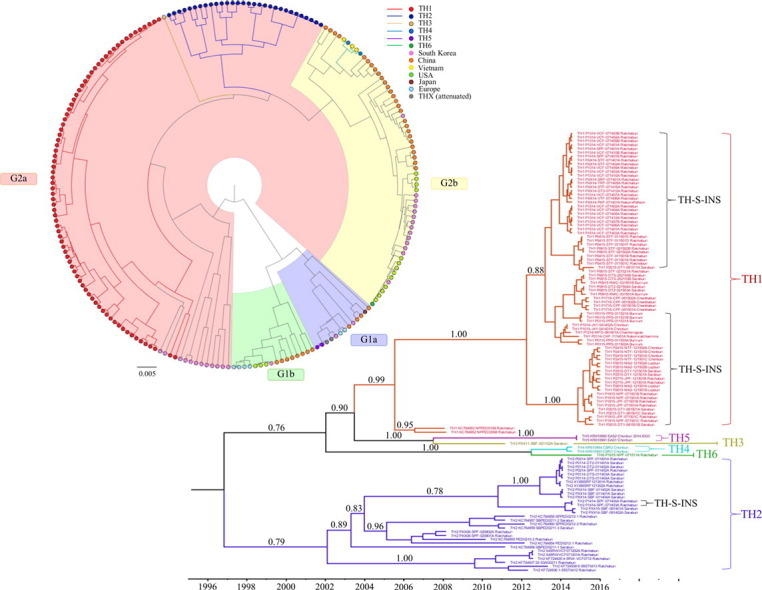
All PEDV samples were grouped based on spike gene sequence data using clades previously reported (Lee 2015) in which PEDV evolved into 2 separated clades including G1 and G2. Each clades was further divided into 2 subclades including subclades G1a, G1b, G2a and G2b. Subclade G1a included mainly the first period of PEDV found in Belgium, UK, (CV777, CH/S, Br1/87) and attenuated strains of vaccine from China and Korea. Subclade G1b included mostly the strains of China together with Korea, USA and Europe strains that were reported during 2014–2015. G2 was further evolved into G2a and G2b. G2a included strains from China, Korea and Thailand. G2b included mainly PEDV strains from US, and from countries which reported the detection of US-like PEDV.
Thai PEDV are classified into 6 subgroups including TH1–TH6 (Fig. 1 ). Based on spike gene identity and the clades previously described (Lee 2015), most of Thai sequences belong to G2a clade. Among these 6 subgroups, the genetic differences are > 3% between subgroups, we therefore classified 3 subgroups of Thai PEDV to G2a including TH1, TH2 and TH3. Subgroups TH1 (n = 79) and TH2 (n = 30) were the two dominant groups in Thailand. TH1 included the first Thai PEDV strain that was published in 2008 along with the latest samples we found in 2015. In the endemic group 1 (TH1), there were 26 samples from 19 outbreaks with the 9 nucleotides insertion of CAA GGG AAT (TH-S-INS) when aligned with the reference sequence (NC_003436; position 688th–689th) which caused the amino acid changed from N to T (229 of the reference) and insertion of REY at the site between 229th–230th. TH3 only contained one sample in 2011 which might be a new introduction of the virus or the recombinant one. However, the sequence seems to be a recombinant one because it shares 97% identity with some sequences in both G1 and G2 group when blasting with nucleotide BLAST.
Fig. 1.

The phylogenetic tree based on complete spike gene of Thai PEDV sequences along with reference sequences (Lee 2015). PEDV are evolved into two genogroups including G1 and G2. G1 and G2 are each further divided into two sub-groups including G1a, G1b, G2a and G2b. TH1–TH6 represent Thai PEDV subgroups of the samples collected during 2008 to 2015. Numbers above branches denote posterior probability values.
Subgroups TH4 and TH6 were in genogroup G2b. TH4 was applied for CBR1 and CBR2 (KR610993 and KR610994) sequences in July 2014, these sequences belong to G2b and very close to Vietnam sequences (99% identity) in 2013, so we can conclude them as a new introduction of the exotic strains. TH6 is the latest subgroup which contained the new introduction of the exotic strain (P1915-NPF-071511A) in 2015 that belong to G2b or US-like subgroup.
Subgroup TH5 was in genogroup G1a. TH5 belongs to the latter group of samples in October 2014, EAS1 and EAS2 (KR610991 and 610992) which the first group of strain that belong to G1 clade that we found in Thailand. The Thai PEDV in this genogroup were genetically distinct from PEDV strains recently emerged in European countries. These strains were close to the vaccine strain so we did isolation the virus vaccine using in the farms experienced to the outbreak of the strain, and including of an AVCT12, both the attenuated strains are assigned into THX subgroup in this study.
Nucleotide and amino acid sequences of TH1–6 represent sequences and nearest identity to each subgroup are provided in Fig. S3, Fig. S4 in Supplementary materials.
3.2. Evolutionary analysis
The evolutionary rates were calculated for 4 datasets of PEDV groups including overall (all field strains of Thailand; n = 115), endemic (predominant TH1 and Th2 subgroups; n = 109), TH1 (n = 79), and TH2 (n = 30). In general, the higher evolutionary rate is present in Thai PEDV strains than in the US sequences represent. In comparison among Thai strains, subgroup TH1 (predominant endemic subgroup) had higher rate of substitution than TH2 subgroup (Fig. 2 ; Table S3).
Fig. 2.

Evolutionary rate comparison between overall (OA), endemic (EN), TH1 and TH2 of Thai dataset, and, US dataset. The results were show in substitution rate per site per year of strict (STR), uncorrelated relaxed logarithm (LOG) and exponential (EXP) prior clock.
The divergence times among Thai PEDV sequences were also estimated using BEAST exponential relaxed-clock method. All Thai PEDV sequences share a common ancestor that presented in year 1999, with 95% HPD interval of 1987–2004 (Fig. 3 , Table S3). The TH2 subgroup seems to evolve independently from all other samples. The emergence of TH2 common ancestor was estimated to be around year 2002 whereas the common ancestor of all other samples arose approximately in 2003. The TH1, TH3, and TH5 subgroups have a shared ancestor which presented around 2004. The TH1 subgroup emerged around 2005–2006 and the TH3/TH5 subgroups evolved later in 2007. The TH4 and TH6 subgroups descended from their common ancestor which emerged recently in 2012.
Fig. 3.

Phylogenetic tree with time and their estimated divergences of PEDV in Thailand based on spike gene. Red, blue, yellow, aqua, purple and green colors were represented for TH1, TH2, TH3, TH4, TH5 and TH6, respectively. TH-S-INS labeled parenthesis located the sequences with the unique insertion. Node labels denote the time of estimated divergences whereas branch labels denote the posterior probability values.
3.3. Phylogeographic distribution
Due to the ESS values, exponential molecular clock model was chosen to display the geographic distribution of PEDV in Thailand as shown in Fig. 4 . Based on the phylogeographical analysis, there was geographic separation between PEDV stains in Thailand. TH2 subgroup was confined mainly in the western regions. In contrast, TH1 subgroup was found across Thailand, especially the northeast region of Thailand, and has become the dominant subgroup since 2014.
Fig. 4.

Geographic distribution of TH1 (left panel), TH2 (center panel) and TH3–6 (right panel) estimated by the location and time of the outbreaks in Thailand during 2008–2015.
Since its emergence in 2007, PED has been endemic in the western region of Thailand including Ratchaburi and Nakhon Pathom provinces and sporadic outbreaks have been reported in the eastern region including Chonburi, Chanthaburi and ChaCherngsao provinces. However, farms in the eastern regions of Thailand experienced PED outbreaks in 2012–2013 and the sequencing results demonstrated the emergence of the TH1 group which could be responsible for the outbreak. The spread of the TH1 group to the northeast region could be due to the transportation of culled sows.
3.4. Recombinant analysis
A total of 22 recombination events based on spike, with known parents were detected from RDP4 sequence analysis as shown in Fig. S1 (left panel). Recombination rate plot (Fig. S2) located 5 recombination sites (cutoff with > 0.01 Rho/bp), position 1–117 (0.06131 Rho/bp), position 1048–1206 (0.05912 Rho/bp), position 2224–2266 (0.02536 Rho/bp), position 3070–3116 (0.01947 Rho/bp), and position 4102–4142 (0.06236). The recombination events and their patterns are show in Fig. S1 (right panel).
Recombination analysis results of PXX11-SBF-021102A obtained from RDP and CHIMERA are different. The RDP result showed that this sequence is the minor parent, and together with X45RWVCF071202A, they were recombined into SBPED0211-1 whereas the CHIMERA show that it is the recombinant sequence of X1356SRF121302A and SBPED0211-1. In the other hand, SBPED0211-1 has a highest potential evidence of recombination since it can be detected by RDP, GENECONV and BOOTSCAN, and SiScan. Recombination event 16 (Fig. S1) were occurred in the same farm experienced with several outbreaks.
4. Discussion
In this study, the genetic diversity and evolutionary dynamics of PEDV samples collected between 2008 and 2015 in Thailand were investigated. Based on the phylogenetic analysis of complete spike gene, several important findings involving the genetic evolution of PEDV in Thailand were unveiled. The findings included the multiple introductions of genetically distinct PEDV variants influencing the development of Thai PEDV strains. Thai endemic PEDV strains forming clades separated from other countries and undergoing high mutation rate. One mechanism involving in the high mutation rate was the recombination.
The multiple introduction included the presence of PEDV in G1a and G2b subclades. The introduction of G1a variant in Thailand was not surprised. A previous study demonstrated that those PEDV stains in G1a group were vaccine-like stains (Cheun-Arom et al. 2015). The emergence of PEDV variants in this group was due to the heavy use of modified live PEDV vaccines, both modified live (MLV) and killed virus (KV) vaccines during 2013–2014. At present, the use of MLV and KV in Thailand drastically decreased compared to 2013–2014. This was due to the efficacy of intramuscular vaccines in which provided a limited degree of success against PED. In addition, MLV was reported to cause PEDV outbreaks with mild clinical disease (Cheun-Arom et al. 2015). The emergence of a US-like variant as shown in G2b group (TH6), was indeed intriguing and this is the first report showing the emergence of US-like PEDV in Thailand. TH6 is the latest subgroup in which contained the new introduction of the exotic PEDV strain (P1915-NPF-071511A) in 2015 that belong to G2b or US-like subgroup. This sequence share 100% similarity with NIG-1/JPN/2014, GNM-2/JPN/2014, MIE-1/JPN/2014, PC21A, KUIDL-PED-2014-002, KUIDL-PED-2014-007, USA/Ohio75/2013, USA/Ohio69/2013, USA/Ohio60/2013, and USA/Indiana/17846/2013, suggesting the first US-like strain in Thailand. However, the abilities of this strain to develop into the endemic strain in the Thai swine industry was still questionable and further investigation is required to monitor the development of these exotic strains.
Whether or not multiple introduction influences the strain development of Thai PEDV in the study was not known. However, although multiple introduction was evident, those strains were yet to form their own clusters. In contrast, Thai PEDV developed its own cluster separated from other countries. PEDV variants that has been dominant in Thai swine farms are strains in G2a group and can be further divided into 2 subgroups including TH1 and TH2. We found that Thailand strains are unique and underwent mutation and recombination with some other strains which resulted in pandemic outbreak between 2014 and 2015. We identified the sequence insertion which is unique only in Thailand strains (TH-S-INS) and was mainly detected in TH1 subgroup particularly the samples in the outbreaks between 2014 and 2015. This insertion was also detected in 2 samples of TH2 subgroup which was sampled in 2014 and no sample that belong to subgroup TH2 was detected in 2015. These suggested that the strains in TH1 might underwent positive selection pressure somehow and some of them experienced an insertion mutation event as described in the result. The first sample which containing this insertion of TH2 was P1414-SPF-071403A, and, was collected in July 2014 whereas another 27 samples from 8 outbreaks which collected at the same month are TH1. P1314-VCF-071401A is the represent sequence of the samples that containing this insertion of TH1 at 99% identity.
According to the evolutionary rate analysis, we found that the overall group of Thailand sequences (TH1–6; n = 115) evolved with relatively greater substitution rate compared to the US sequences when estimating under lognormal and exponential relaxed clock models. The US sequences showed higher substitution rate under the strict clock model; however, this rate was less than that of Thailand endemic group (TH1–2; 109). TH1 subgroup showed distinctly higher evolutionary rate than other analyzed groups. Its substitution rate was over than two fold compared to the endemic or TH2 group and was over than 5 fold compared to the US group as determined by uncorrelated relaxed-clock models. This event may suggests that there is any positive pressure on this subgroup.
Besides influenced by the multiple introduction, the relatively greater evolutionary rate of Thai PEDV was partially due to recombination as supported by the results of the recombination analysis. In comparison between TH1 and the TH2 groups, TH1 is more in population parted in the events whereas TH2 is more in number of recombination evidences. These results may explain that TH1 recombinants were chosen to be the subgroup that suitable to cause an outbreaks whereas TH2 is keep undergoing recombination but not usually cause an outbreaks. TH2 might be the predominant group which could adapt to survive and persist in the host without triggering symptom development, therefore some were not included in the study. On the other hand, TH2 might not be preferred for proliferation in the host. However, we did not perform active surveillance to take the samples from subclinical herds so we could not make a final conclusion. For TH3, we did further analysis with the dataset that using in phylogenetic analysis, and, there is possible that PXX11-SBF-021102A is the recombinant of NPPED2008 (KC764952) and Chinju99 (AV167585) of G2 and G1 genogroup as major and minor parents, respectively. This information supported why TH3 subgroup is locate between genogroup 1 and 2 (Fig. 1), and, very close and share the same estimated divergence to TH5 subgroup (Fig. 3). The results of geographic distribution show that the western part of Thailand (mainly in Ratchaburi) is the endemic area of most strains, however, TH2 seem to be limited in these area whereas TH1 is spreading from west-central-east of Thailand. Another new introductions (TH3–6) were denoted by the pinpoint since they could not performed analysis. These results together with previous described suggested that TH1 is becoming the predominant subgroup which endemic in Thailand since 2014.
5. Conclusion
Thai PEDV was genetically diverse and the genetic development has been influenced by the multiple introduction of exotic strains. Such introductions were not developed into epidemic stage. In contrast, Thai PEDV strains developed their own subgroups separated from other countries and underwent relatively higher evolutionary than the US sequences. There was an evidence of intra-recombination between sub-groups that can be one factor to accelerate the evolutionary rate of Thai PEDV. Samples in the sub-group with high evolutionary rate become dominant causing not only geographic separation, but also influence the development of other subgroup as the unique insertion could be found in both TH1 and TH2 whereas the identity between subgroup is > 97%. We did provided the information of P1314-VCF-071401A, P1414-SPF-071403A, PXX11-SBF-021102A, P1915-NPF-071511A and PXX08-SPF-020801A on GenBank® as following accession number: KX981897, KX981898, KX981899, KX981900 and KY000559, respectively. PXX08-SPF-020801A was the first strain in TH2 genogroup found in 2008.
Author contributions
C.J.S. and D.N. conceived and designed the experiments; C.J.S., G.T., P.K., T.T. and J.P. performed the experiments; C.J.S. and D.N. analyzed the data; C.J.S., T.T. and A.T. contributed reagents/materials/analysis tools; C.J.S. and D.N. composed the manuscript.
Conflicts of interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Acknowledgments
This research was financially supported by the National Research Council of Thailand and Agricultural Research Development Agency (Public organization; PRP5805021650). Partial funding was provided by Special Task Force for Activating Research (STAR), Swine Viral Evolution and Vaccine Research (SVEVR), Chulalongkorn University (GSTAR 59-013-31-007). The first author is supported by the Royal Golden Jubilee PhD program (PHD/0204/2557) Scholarship of the Thailand Research Fund (TRF) and the Ratchadaphiseksomphot Endowment Fund 2013 of Chulalongkorn University (CU-56-527-HR).
Footnotes
Supplementary data associated with this article can be found in the online version, at doi: 10.1016/j.meegid.2017.02.014. These data include the Google maps of the most important areas described in this article.
Appendix A. Supplementary data
Fig. S1.

Recombination events with name of isolation parted in major and minor parents, and, their recombinants (left panel). And their recombination in this study provided with the ladder between 1st–4188th position (right panel).
Fig. S2.

Recombination rate plot provided with Rho/bp values, the number inside red box present the position in alignment that related with the plot.
Nucleotide sequences of TH1–6 represent sequences and their most closely related strain.
Amino acids sequences of TH1–6 represent sequences and their most closely related strain.
The phylogenetic tree conducted neighbor-joining using Tamura-Nei parameter, pairwise deletion, and, 1000 bootstraps. Grey colored label the strains that closest to each TH1–6 represent sequences.
Sequences using in this study for phylogenetic analysis provided with country, strain name, accession number and their genogroup in this study compare with other study.
Sequences of US samples using to compare their evolutionary analysis with Thai sequences in this study.
Evolutionary rate of each dataset and clock model performed in this study.
Google maps
The following KML file contains the Google maps of the most important areas described in this article.
KML file containing the Google maps of the most important areas described in this article.
References
- Bielejec F., Rambaut A., Suchard M.A., Lemey P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2912. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.H., Bae J.L., Kang T.J., Kim J., Chung G.H., Lim C.W., Laude H., Yang M.S., Jang Y.S. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol. Cells. 2002;14:295–299. [PubMed] [Google Scholar]
- Chasey D., Cartwright S. Virus-like particles associated with porcine epidemic diarrhoea. Res. Vet. Sci. 1978;25:255. doi: 10.1016/S0034-5288(18)32994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Li G., Stasko J., Thomas J.T., Stensland W.R., Pillatzki A.E., Gauger P.C., Schwartz K.J., Madson D., Yoon K.J., Stevenson G.W., Burrough E.R., Harmon K.M., Main R.G., Zhang J. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J. Clin. Microbiol. 2014;52:234–243. doi: 10.1128/JCM.02820-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheun-Arom T., Temeeyasen G., Srijangwad A., Tripipat T., Sangmalee S., Vui D.T., Chuanasa T., Tantituvanont A., Nilubol D. Complete genome sequences of two genetically distinct variants of porcine epidemic diarrhea virus in the eastern region of Thailand. Genome Announc. 2015;3 doi: 10.1128/genomeA.00634-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz D.J., Kim C.J., Shin H.J. Phage-displayed peptides having antigenic similarities with porcine epidemic diarrhea virus (PEDV) neutralizing epitopes. Virology. 2006;354:28–34. doi: 10.1016/j.virol.2006.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz D.J., Kim C.J., Shin H.J. The GPRLQPY motif located at the carboxy-terminal of the spike protein induces antibodies that neutralize porcine epidemic diarrhea virus. Virus Res. 2008;132:192–196. doi: 10.1016/j.virusres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis H., Maria J., Anja P., Mathias R., Julia S., Valerij A., Sandra B., Anne P., Horst S., Martin B., Dirk H. Comparison of porcine epidemic diarrhea viruses from Germany and the United States, 2014. Emerg. Infect. Dis. 2015;21:493. doi: 10.3201/eid2103.141165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:1–8. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Nicholls G., Rodrigo A., Solomon W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161 doi: 10.1093/genetics/161.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Ho S., Phillips M., Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006:4. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012 doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte M., Laude H. Sequence of the spike protein of the porcine epidemic diarrhoea virus. J. Gen. Virol. 1994;75(Pt 5):1195–1200. doi: 10.1099/0022-1317-75-5-1195. [DOI] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocherhans R., Bridgen A., Ackermann M., Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Porcine epidemic diarrhea virus: an emerging and re-emerging epizootic swine virus. Virol. J. 2015;12:193. doi: 10.1186/s12985-015-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-K., Park C.-K., Kim S.-H., Lee C. Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Res. 2010;149:175–182. doi: 10.1016/j.virusres.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Park G.-S., Shin J.-H., Lee C. Full-genome sequence analysis of a variant strain of porcine epidemic diarrhea virus in South Korea. Genome Announc. 2014;2 doi: 10.1128/genomeA.01116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Li H., Liu Y., Pan Y., Deng F., Song Y., Tang X., He Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012;18:1350–1353. doi: 10.3201/eid1808.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.N., Chung W.B., Chang S.W., Wen C.C., Liu H., Chien C.H., Chiou M.T. US-like strain of porcine epidemic diarrhea virus outbreaks in Taiwan, 2013–2014. J. Vet. Med. Sci. 2014;76:1297–1299. doi: 10.1292/jvms.14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Fuentes S., Elizondo-Quiroga D., Farfan-Ale J.A., Loroño-Pino M.A., Garcia-Rejon J., Gomez-Carro S., Lira-Zumbardo V., Najera-Vazquez R., Fernandez-Salas I., Calderon-Martinez J., Dominguez-Galera M., Mis-Avila P., Morris N., Coleman M., Moore C.G., Beaty B.J., Eisen L. Use of Google Earth™ to strengthen public health capacity and facilitate management of vector-borne diseases in resource-poor environments. Bull. World Health Organ. 2008;86:718–725. doi: 10.2471/BLT.07.045880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.P., Murrell B., Golden M., Khoosal A., Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1 doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T., Murakami S., Takahashi O., Miyazaki A., Ohashi S., Yamasato H., Suzuki T. New porcine epidemic diarrhoea virus variant with a large deletion in the spike gene identified in domestic pigs. Arch. Virol. 2015;160:2565–2568. doi: 10.1007/s00705-015-2522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojkic D., Hazlett M., Fairles J., Marom A., Slavic D., Maxie G., Alexandersen S., Pasick J., Alsop J., Burlatschenko S. The first case of porcine epidemic diarrhea in Canada. Can. Vet. J.: La revue veterinaire canadienne. 2015;56:149–152. [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Moon H.J., Luo Y., Kim H.K., Kim E.M., Yang J.S., Song D.S., Kang B.K., Lee C.S., Park B.K. Cloning and further sequence analysis of the ORF3 gene of wild- and attenuated-type porcine epidemic diarrhea viruses. Virus Genes. 2008;36:95–104. doi: 10.1007/s11262-007-0164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Kim H.K., Song D.S., Moon H.J., Park B.K. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Arch. Virol. 2011;156:577–585. doi: 10.1007/s00705-010-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensaert M., De Bouck P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978;58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J. 2013. Tracer 1.6 [Computer Program] [Google Scholar]
- Rambaut A., Drummond A.J. 2014. FigTree 1.4.2 [Computer Program] [Google Scholar]
- Sun D.B., Feng L., Shi H.Y., Chen J.F., Liu S.W., Chen H.Y., Wang Y.F. Spike protein region (aa 636789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol. 2007;51:149–156. [PubMed] [Google Scholar]
- Sun D., Feng L., Shi H., Chen J., Cui X., Chen H., Liu S., Tong Y., Wang Y., Tong G. Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet. Microbiol. 2008;131:73–81. doi: 10.1016/j.vetmic.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R.Q., Cai R.J., Chen Y.Q., Liang P.S., Chen D.K., Song C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012;18:161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Ma J., Wang Y., Wang M., Song W., Zhang W., Lu C., Yao H. Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. J. Clin. Microbiol. 2015;53:1484–1492. doi: 10.1128/JCM.02898-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013 doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temeeyasen G., Srijangwad A., Tripipat T., Tipsombatboon P., Piriyapongsa J., Phoolcharoen W., Chuanasa T., Tantituvanont A., Nilubol D. Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infect. Genet. Evol. 2014;21:205–213. doi: 10.1016/j.meegid.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Theuns S., Conceição-Neto N., Christiaens I., Zeller M., Desmarets L.M.B., Roukaerts I.D.M., Acar D.D., Heylen E., Matthijnssens J., Nauwynck H.J. Complete genome sequence of a porcine epidemic diarrhea virus from a novel outbreak in Belgium, January 2015. Genome Announc. 2015;3:e00506–e00515. doi: 10.1128/genomeA.00506-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vui D.T., Thanh T.L., Tung N., Srijangwad A., Tripipat T., Chuanasa T., Nilubol D. Complete genome characterization of porcine epidemic diarrhea virus in Vietnam. Arch. Virol. 2015;160:1931–1938. doi: 10.1007/s00705-015-2463-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequences of TH1–6 represent sequences and their most closely related strain.
Amino acids sequences of TH1–6 represent sequences and their most closely related strain.
The phylogenetic tree conducted neighbor-joining using Tamura-Nei parameter, pairwise deletion, and, 1000 bootstraps. Grey colored label the strains that closest to each TH1–6 represent sequences.
Sequences using in this study for phylogenetic analysis provided with country, strain name, accession number and their genogroup in this study compare with other study.
Sequences of US samples using to compare their evolutionary analysis with Thai sequences in this study.
Evolutionary rate of each dataset and clock model performed in this study.
KML file containing the Google maps of the most important areas described in this article.