

Abstract
In the present study, a thorough comparison of the infectious bronchitis virus (IBV) TC07–2/GVI-1 linage was conducted by comparing the S1 gene sequences of GVI-1 viruses with those of viruses representing the established genotypes and lineages. IBV GVI-1 strains were found to be closely genetically related to each other, irrespective of where the viruses were isolated, and differed from other known IBV genotypes and lineages; thus, it was confirmed that GVI represents a novel genotype. However, the GVI-1 viruses exhibited variable antigenicity when compared to each other. Further analysis found that strains CO8089L and CO8091L, which were isolated in Colombia in 2003, were closely related to GVI-1 viruses, suggesting that GVI-1 viruses likely originated from Colombia and are prevalent in at least five countries (Colombia, China, the Republic of Korea, Japan, and Vietnam). Analysis of the complete GVI-1 virus genomes suggested that the GVI-1 strains in China may be independently derived from recombination events that occurred between GI-19 strains and CO8089L/CO8091L-like viruses following the introduction of the viruses from Colombia. Similar to the viruses isolated in the Republic of Korea, GVI-1 viruses isolated in China also showed an affinity for the respiratory tract of chickens, which differed from one of the deduced parental viruses, the GI-19 strain. This difference may be due to recombination events that occurred in the genomes of the GVI-1 viruses, resulting in the replacement of the spike gene sequences in an YX10-like strain of GI-19 lineage.
Keywords: Infectious bronchitis virus, TC07–2/GVI-1 lineage, Complete genome, Antigenicity, Pathogenicity, Tissue tropism
1. Introduction
Respiratory viral disease cases submitted to our laboratory have yielded a high frequency of infectious bronchitis virus (IBV) isolations compared to other respiratory pathogens (Xu et al., 2018). IBV is the etiological agent of infectious bronchitis (IB), which is an acute and highly contagious respiratory and urogenital viral disease that affects domestic fowl (Gallus gallus) worldwide (Cavanagh, 2007), leading to important economic losses related to mortality, severe egg drop, and poor egg quality. IBV belongs to subgenus Igacovirus of genus Gammacoronavirus in the family Coronaviridae, and its genome consists of a single positive sense RNA strand approximately 27.6 kb in length that encodes four structural proteins: 1) spike (S) glycoprotein; 2) membrane (M); 3) envelop (E); and 4) nucleocapsid (N), in addition to an RNA-dependent RNA polymerase and at least four accessory proteins (3a, 3b, 5a, and 5b) (Boursnell et al., 1987). The S protein is cleaved into the S1 and S2 subunits consisting of approximately 535 and 625 amino acids, respectively; of these, the S1 subunit is important in viral adsorption to the cellular receptor and viral entry into the host cell, as well as inducing neutralizing antibodies (Boursnell et al., 1987; Cavanagh, 2007).
Although immunosuppression, concurrent infections, and inadequate biosecurity measures have been identified to be the causes of vaccine failure in field conditions (Haghighat-Jahromi et al., 2008; Hassan et al., 2016; Tarnagda et al., 2011), field IBV isolates could still cause episodes of disease followed by a persistent infection in commercial birds, in which these viruses appear to undergo continuous genetic evolution, resulting in the generation of IBV variant progenies, further complicating the effective control of this infection. The genetic alterations found in the S1 gene and the corresponding protein of the IBV variants involved in the field outbreaks may be able to change the antigenicity, tissue tropism, and pathogenicity of these viruses when they infect birds (Cavanagh, 2007; Toro et al., 2012). These genetic changes may also influence the effectiveness of the host immune response against these viruses and favor viral persistence.
Historically, the IBV Massachusetts and Connecticut serotypes were the first isolates discovered in the 1930s and 1950s, respectively (Schalk and Hawn, 1931; Jungherr et al., 1956). Subsequently, a number of serotypes/genotypes and other variants have been identified and associated with disease outbreaks around the world (Cook et al., 2012; De Wit et al., 2011). The classification of IBV genetic types have been primarily performed without universal criteria regarding the nomenclature, methods used to compare the viral molecular data, and the precise genetic region that is to be analyzed. Consequently, groups of genetically-related viruses have been assigned as a new genotype and >50 genotypes have been reported (De Wit et al., 2011). Recently, a more definitive phylogeny-based classification system has been used and six genotypes which included 32 lineages were originally distinguished by Valastro et al. (2016). Next new lineages GI-28 and GI-29 of GI genotype and even one new genotype (GVII) were recently added (Chen et al., 2017; Jiang et al., 2017; Ma et al., 2019). This system provides reliable reference sequences and lineage prototypes to guide viral classification. Based on the criteria used in this classification system, at least 16 lineages of IBV strains, including the GI-1 (Mass) (Liu et al., 2006), GI-3 (Valastro et al., 2016), GI-4 (Valastro et al., 2016), GI-5 (Valastro et al., 2016), GI-6 (Valastro et al., 2016), GI-7 (TW I and TW II) (Ma et al., 2012; Xu et al., 2016), GI-9 (Ark) (Han et al., 2018), GI-13 (793/B, CR88, 4/91 or 1/96) (Han et al., 2011), GI-16 (ck/CH/LDL/97I or Q1) (Liu et al., 2007; Yu et al., 2001), GI-18 (Valastro et al., 2016), GI-19 (LX4 or QX) (Liu and Kong, 2004), GI-22 (ck/CH/LSC/99I) (Liu et al., 2006), GI-28 (Chen et al., 2017), GI-29 (Jiang et al., 2017), GVI-1 (TC07–2) (Li et al., 2010), and GVII-1 (Ma et al., 2019), as well as a number of recombinants have been isolated in China (Xu et al., 2018).
Although the majority of such variants cause only transitory problems, which subsequently resolve or remain confined within a specific geographical region, there are some variants that are able to persist and spread to new regions where they continue to cause disease (de Wit et al., 2011). IBV GVI-1 represents a genetically distinct lineage present in Asia (Valastro et al., 2016). Strains of the GVI-1 lineage (also known as the TC07–2 type in China) were believed to be first isolated and identified in Guangxi province in China in 2007 during an outbreak of respiratory disease and were designated as the Chinese New-Type (Li et al., 2010). Later, viruses closely related to this lineage were also isolated in the Republic of Korea in 2009, which were originally grouped in the ‘Korean New Cluster II’ (Lim et al., 2012), and in Japan in 2009 which were assigned to a group termed, JP-IV (Mase et al., 2010), by partial sequencing of the S1 gene. To date, because no sequences of JP-IV-like S1 are available, it cannot be determined whether the so-called “JP-IV strains” are included in GVI-1 or if they cluster into a separate lineage (Valastro et al., 2016). Moreover, primary pathogenicity results have revealed these strains to be of respiratory nature (Li et al., 2010; Lim et al., 2012). Recently, a GVI-1 lineage virus, KrD1515, isolated in 2015 in the Republic of Korea, was found to be a Chinese TC07–2 IBV and a non-TC07–2 Korean IBV recombinant virus that engages in respiratory tropism in chickens (Jang et al., 2018). Although originally isolated 10 years ago, the origin of the GVI-1 viruses was unknown partially because no data currently exists regarding the analysis of complete genomic sequences of this lineage.
In this study, the complete genomes of six viruses belonging to the GVI-1 lineage were determined and analyzed to elucidate the origin and evolution of the viruses. Additionally, the pathogenicity, tissue tropism, and serological relatedness were also examined to elucidate the disease potential of this lineage.
2. Materials and methods
2.1. Viruses
Six IBV GVI-1 viruses, ck/CH/LHB/110615 (LHB/110615), ck/CH/LHB/110617 (LHB/110617), I0916/16, I1209/16, I0221/17, and I0725/17, isolated in 2011, 2016, and 2017 in Hebei, Hunan, Guangxi, Henan, and Fujian provinces in China, respectively, were used in this study (Xu et al., 2018). All of the viruses originated from respiratory disease in layers and broilers occurring at 25, 67, 34, and 65 days of age. All IBV strains were propagated in 9-day-old specific pathogen-free (SPF) embryonated chicken eggs to prepare the viral stocks before titration. The viral titers were determined using standard methods (Reed and Muench, 1938) in 9-day-old SPF eggs.
2.2. Cloning and sequencing of the complete GVI-1 viral genomes
Viral genomic RNA from each of the six GVI-1 viruses was extracted from the allantoic fluid with RNAiso Plus reagent (Takara Bio, Shiga, Japan) and RT-PCR was performed using a One-step RT-PCR Kit (Takara Bio, Shiga, Japan) following the manufacturer's protocol. The primers previously used for amplifying the IBV genome (Liu et al., 2013) were used in this study to amplify the entire genomes of the IBV GVI-1 lineage. The reaction program was as follows: reverse transcription at 50 °C for 30 min, 95 °C for 2 min; 30 cycles of 94 °C for 30 s, 55 °C–65 °C for 30 s, 72 °C for 1 min; followed by 72 °C for 10 min. The 3′ and 5′ ends of the viral genomes were confirmed by rapid amplification of cDNA ends (RACE) using a 3′/5’ RACE kit (Takara Bio, Shiga, Japan) as previously described (Liu et al., 2013). Each of the resulting PCR products representing the different fragments was purified by agarose gel electrophoresis and a QIA quick Gel Extraction Kit (Qiagen, Valencia, California, USA), and was sequenced directly or cloned into a pMD 18-T vector (Takara Bio, Shiga, Japan). Sanger sequencing was performed (Big Dye Terminator) and 3–5 clones of each PCR product were sequenced to determine a consensus sequence for any given genomic region. Open reading frame (ORF) predictions were carried out using the ORF-finder program (https://www.ncbi.nim.nih.gov/orffinder/), and the ORFs were compared with that of the GVI-1 virus, CK/CH/SD09/005.
2.3. Genotyping and analysis of the GVI-1 virus S1 gene/fragments
Phylogenetic trees were constructed using a dataset, including our six IBV strains and 70 reference strains (including the TC07–2 strain) representing the established genotypes and lineages (Chen et al., 2017; Jiang et al., 2017; Ma et al., 2019; Valastro et al., 2016) (Supplementary Table 1) using Mega version 6.0 software (http://www.megasoftware.net/). The maximum likelihood method with the Tamura-Nei substitution model and 1000 bootstrap replicates was used to assess the robustness of the branches. An additional 44 complete S1 gene nucleotide sequences of the IBV strains were selected to construct phylogenetic trees with our six strains and the 70 reference strains (Supplementary Table 1). The S1 gene sequences of the 44 viruses were available in the GenBank database and closely related to our six IBV strains (>92%) by BLAST analysis using the complete S1 gene sequence of our IBV strains. In total, 45 TC07–2-like sequences, including 37 from China, 7 from the Republic of Korea, and 1 from Vietnam were included in this study. The percentage identity between the TC07–2-like strains, and between the TC07–2-like strains and the representative strains from each lineage based on the phylogenetic trees were calculated at both the nucleotide and amino acid level.
In addition, based on the results of the BLAST analysis in the GenBank database using the S1 gene of our six GVI-1 viruses, it was revealed that the IBV strains CO8089L and CO8091L from Colombia shared a 96% amino acid identity with our six GVI-1 IBV strains. Hence, the available 100 amino acid sequences at the N terminus of the S1 subunit of the spike protein of strains CO8089L and CO8091L were selected to perform a pairwise comparison with those of the 45 TC07–2-like strains using Multiple Alignment with Fast Fourier Transformation (MAFFT) version v6 (http://mafft.cbrc.jp/alignment/software/).
2.4. Analysis of GVI-1 virus complete genomes
To thoroughly compare the complete genomes of our six GVI-1 viruses, phylogenetic trees were constructed using a dataset, including the complete genomes of our six GVI-1 strains and those of five reference viruses of the GVI-1 lineage and 99 reference strains of other lineages (Supplementary Table 1) (Chen et al., 2017; Jiang et al., 2017; Ma et al., 2019; Valastro et al., 2016) from GenBank (www.ncbi.nlm.nih.gov/genbank/). The complete genomic sequences of our six IBV strains and the 104 reference viruses were aligned using Clustal W, and phylogenetic trees were constructed using both Mega version 6.0 software (http://www.megasoftware.net/) using the maximum likelihood method and the Clustal V method using DNAStar software (Liu et al., 2006), respectively; the topology of the two methods was similar. In addition, the complete genomic sequences of 11 GVI-1 viruses, including the six in this study and five from the GenBank database were analyzed using MAFFT version 6. The percentage identities between the 11 GVI-1 viruses were calculated.
2.5. Recombination analysis
Based on the results from the phylogenetic analysis, the complete genomic sequence of strain YX10 (GI-19) were selected to compare with those of 11 GVI-1 viruses to analyze the possible recombination events, the probable parental viruses of the GVI-1 strains, and the likely recombination breakpoints using SimPlot version 3.5.1 (http://sray.med.som.jhmi.edu/SCRopftware/simplot/) (Lole et al., 1999). The IBV strain GX-YL5 was used as a query virus. The window width and step size were set to 1000 bp and 100 bp, respectively. To confirm the recombination events that occurred in the genomes of the GVI-1 viruses, three phylogenetic trees were constructed based on the results of the SimPlot analysis for the nucleotide fragments 1–20,196 (5′ untranslated region (UTR) to 3′ end of Gene 1); 20, 197–24,450 (3′ end of Gene 1 to 5′ end of Gene 3); and 24, 451–27,673 (5′ end of Gene 3 to 3’ UTR), from six IBV strains, including three viruses (YX10, DY07, and P100) in the GI-19 lineage, and three Mass strains (B17, H52 and H120) as an outgroup.
The complete genomic sequences of the six GVI-1 viruses, LHB/110615, LHB/110617, I0916/16, I1209/16, I0221/17, and I0725/17, were deposited in GenBank under the accession numbers, MK574042, MK574043, MK217374, MK217375, MK217372, and MK217373, respectively.
2.6. Antigenic characterization of GVI-1 IBV strains by cross-virus neutralization
Ethical approval to perform this study was obtained from the Ethical and Animal Welfare Committee of Heilongjiang province, China (License no. HSY-IACUC-2019-91). Five GVI-1 viruses, including LHB/110615, I0916/16, I1209/16, I0221/17, and I0725/17, were used in the cross-virus neutralization (VN) tests. The antiserum against each of the viruses was prepared. Briefly, 25 SPF chickens were divided into five groups. Five chickens in each group were maintained in separate positive-pressure isolators and inoculated via the ocular-nasal route with 103.5/0.1 mL median embryo infective doses (EID50) of each of the virus at three weeks of age to produce monospecific antiserum against each of the viruses used in this study. Three weeks later, the chickens in each group were inoculated with 104.0 EID50 of the virus by the intratracheal and intravenous routes. Blood was collected 3 weeks after the last inoculation, the serum was harvested, pooled for each virus, inactivated at 56 °C for 30 min, and stored at −70 °C for use in VN tests.
The VN tests were performed between the GVI-1 viruses using the β procedure (constant virus diluted serum method). Each of the 100 EID50 of the virus were mixed with equal volumes of two-fold dilutions of the antisera for 1 h and inoculated via the allantoic cavity route into five 9-day-old SPF chicken eggs for one dilution. Neutralization indices were used to calculate the antigenic relatedness value (r) between the viruses using the Archetti and Horsfall (1950).
2.7. Pathogenicity of the GVI-1 viruses
Ethical approval to perform this study was obtained from the Ethical and Animal Welfare Committee of Heilongjiang province, China (License no. HSY-IACUC-2019-103). Six groups of 15 1-day-old SPF chickens were used in this study. Chickens in groups 1 to 5 were individually inoculated with strains LHB/110615, I0916/16, I1209/16, I0221/17, and I0725/17, respectively, via the ocular and nasal routes with 200 μL of the virus, each containing approximately 104.0 EID50. Group 6 served as an uninfected control. Respiratory rales, including tracheal or/and nasal, were assessed by listening closely to each individual bird in each group at 5 and 8 days post infection (dpi), and scored as: 0 (absent); 1 (mild); 2 (moderate); or 3 (severe) as described previously (Han et al., 2018). At 5 dpi, five chickens from each group were humanely killed and necropsied. Trachea and kidney samples were collected and used for viral titration and detection of the IBV antigen by immunohistopathology (IHC) (Gao et al., 2016), respectively. The remaining birds in each group were investigated for another 20 days and the morbidity and mortality were recorded. The blood samples were collected at 4, 8, 12, 16, 20, and 24 dpi from the remaining chickens in each group. IBV-specific IgG in the sera was detected using a commercial IBV enzyme-linked immunosorbent assay (ELISA) kit (IDEXX Lab. Inc., Westbrook, Maine, USA) following the manufacture's protocol.
2.8. Viral titration for the trachea and kidney tissues
Tissues from the trachea and kidneys were individually homogenized in 0.01 M PBS (pH 7.2) and clarified by centrifugation at 8000 ×g for 5 min at 4 °C, filtered through 0.45 μm membranes as described previously (Han et al., 2018). The IBVs were titrated in 9-day-old embryonated SPF chicken eggs, and the EID50 was determined using the Reed and Muench (1938).
2.9. Statistical analysis
Data are expressed as mean ± standard deviation. The viral titers were analyzed with a Student's t-test using GraphPad Prism for Windows version 5 (GraphPad Software, La Jolla, CA, USA). Differences were considered significant if the p value was <0.05 (⁎ p < .05; ⁎⁎ p < .01; ⁎⁎⁎ p < .001).
3. Results
3.1. Genetic characteristics of the six GVI-1 viruses
The complete sequences of the S1 genes for the six GVI-1 viruses were compared with each other and with the reference representatives of specific lineages and genotypes available from GenBank. A phylogenetic analysis revealed that our six IBV strains were clustered together with strain TC07–2, which was isolated in 2007 in Guangxi province (Li et al., 2010), 30 IBV strains from other provinces in China, seven strains from the Republic of Korea, and one from Vietnam (Fig. 1 ). Moreover, at both the nucleotide and deduced amino acid levels, the GVI-1 viruses were most closely related to each other (> 92%), irrespective of where the viruses were isolated. However, the viruses were more distinct to representatives of other IBV lineages/genotypes (< 75%), suggesting that IBV strains of the GVI-1 lineage may have originated from the same ancestor and exhibited the greatest evolutionary distance from all other lineages. Substitutions were also found across the S1 gene of GVI-1 viruses when they were compared between each other (Supplementary Fig. 1). Viruses of the GVI-1 lineage were isolated in at least in nine provinces in China, the majority of which were isolated in Southeast China where the first GVI-1 strain, TC07–2, was isolated in 2007 (Li et al., 2010) (Supplementary Fig. 2). In contrast, the GVI-1 viruses isolated in the Republic of Korea were classified together in the phylogenetic tree and exhibited a close relationship compared to the viruses isolated in other countries.
Fig. 1.

Maximum-likelihood phylogeny of our six GVI-1 strains, other 39 GVI-1 strains, and 70 IBV reference strains based on the complete nucleotide sequences of S1 genes. Our six IBV strains are highlighted with a ★. The countries where the IBV strains were isolated, the GenBank accession numbers for each of the viruses, and the years when the IBV strains were isolated are indicated in parentheses.
In this study, we found that the GVI-1 viruses showed a high sequence similarity with two IBV strains, CO8089L and CO8091L, which were isolated in 2003 in Colombia (Alvarado et al., 2005) by using the nucleotide BLAST search tool in the GenBank database with the S1 amino acid sequences from our six IBV strains. By further comparing the available 100 amino acid sequences of the S1 subunit for strains CO8089L and CO8091L, we found that only one amino acid residue differed between CO8091L and strains TC07–2, LHB/110615, and LHB/110617, which were located at the HVR1 (Fig. 2 ). In addition, four amino acid residues differed between CO8089L and strains TC07–2, LHB/110615, and LHB/110617, which were located at the HVR1 of the S1 subunit and one of which was an insertion compared with those of the other GVI-1 strains (Fig. 2).
Fig. 2.

Multiple alignments of the amino acid sequences at the N-terminus of the S1 subunit of the spike protein in 47 viruses of the GVI-1 lineage. The 47 GVI-1 strains included strains CO8089L and CO8091L which were isolated from Colombia and 45 GVI-1 strains included in Fig. 1. The 45 GVI-1 viruses included 37 isolated in China, seven in the Republic of Korea, and one from Vietnam. The amino acid sequences of TC07–2 are listed and only the amino acids that differed from those of TC07–2 are shown. The sequences that contained the hypervariable region 1 are indicated in bold. The deleted nucleotides are represented as “–”. The sequences of CK/CH/2014/QL1403, LHB/110615, LHB/110617, CK/CH/HN10/05, CK/CH/GD/KP10, and ck/CH/IBTZ/2012 share an identical sequence with that of TC07–2; hence, only the TC07–2 sequence is listed in the alignment. The CK/CH/GX/YL1501–2, CK/CH/HuB/HC1402–1, CK/CH/GX/NN16–11, GX-QZ130064, and GX-QZ130065 sequences are identical and only the sequence for CK/CH/GX/YL1501–2 is shown. IBV strains K/CH/GX/NN11–4, GX-NN120079, CK CH GD YX1101, and GX-NN09032 had the same sequences and the sequence of K/CH/GX/NN11–4 is presented. The sequences of VNUA11, I0221/17, and CK/CH/GD/XX16–9 were identical and the sequence of VNUA11 is shown. The sequence pairs of CK CH HB HC1104 and CK/CH/HB/2016, I0916/16 and CK/CH/GX/GL16, GX-NN120084 and GX-NN120089, CK/CH/GX/NN16–2 and GXNanningHuangdajiu2016.8, as well as K23/10 and K46/10 are identical, and only each sequence of the former viruses are listed, respectively, in this alignment. The GenBank accession numbers for the S1 gene sequences of the viruses are the same as those presented in Fig. 1.
3.2. GVI-1 viruses were possibly mosaics, displaying extensive genetic diversity across the genome
As determined and compared with those of the five reference GVI-1 strains, the full-length genomes ranged in size for the different strains of the same lineage (Supplementary Table 2). Moreover, deletions or insertions were found in different genes or gene fragments in the genomes of 11 GVI-1 viruses when they were compared with each other. Interestingly, a 57-bp deletion was detected in the 3a gene of strain I0221/17 (Fig. 3A), resulting in a frameshift event and a C-terminally truncated protein with 33 amino acids (Fig. 3B). However, all six strains included in this study and five reference GVI-1 viruses exhibited similar genome organization with at least 10 ORFs: 5’-UTR-1a-1b-S (S1 and S2)-3a-3b-3c (E)-M-5a-5b-N-3’-UTR (Supplementary Table 2), which is typical of IBV strains.
Fig. 3.

The alignments for the nucleotide (A) and amino acid (B) sequences of ORFs 3a and flanking sequences of our six GVI-1 strains in the present study. The sequences of strain LHB/110615 are listed, and only nucleotide sequences/amino acid residues differing from those of LHB/110615 are depicted. The start codons of the 3a and 3b genes are highlighted in bold and the stop codons of the spike and 3a genes are highlighted in italics. A deletion (represented as “–”) in the ORF3a of I0221/17 resulted in a truncated 3a protein.
Further identification by phylogenetic analysis showed that our six GVI-1 strains were clustered into the same genetic group with five other GVI-1 reference strains and separated from other distinct genetic groups (Supplementary Fig. 3). Viruses in the GVI-1 lineage showed an identity ranging from 94.0% (LHB/110615 and I1209/16) to 99.7% (LHB/110615 and LHB/110617) between each other. Compared to the S and Gene 3 regions, the mutations were found to be more extensive in other regions across the genome (Fig. 4A, Supplementary Fig. 4). In general, the complete genomic sequences of 11 GVI-1 viruses exhibited a close genetic relationship with those of viruses in the GI-19, GI-28, GI-29, and GVII-1 lineages in the phylogenetic tree. Comparatively, viruses in the GVI-1 lineage displayed a somewhat higher identity with that of GI-19, which ranged from 90.5% to 93.4%, compared with those of the GI-28, GI-29, and GVII-1 lineages.
Fig. 4.

Recombination events in the genome of GVI-1 strains. A Simplot analysis was performed to detect recombination within the genomes (A). The complete genomic sequence of the GX-YL5 strain was used as the query sequence in the SimPlot analysis. The window width and step size were set to 1000 bp and 100 bp, respectively. The grey region shows the spike gene and Gene 3 sequences of the GVI-1 viruses. Phylogenetic trees were constructed using different nucleotide fragments based on the results of the SimPlot analysis (B). The GenBank accession numbers for the S1 gene sequences of the viruses are the same as those presented in Supplementary Fig. 3.
In a Simplot analysis, viruses in the GVI-1 lineage were closely related to the viruses in the GI-19 lineage on fragments I and III, which encompassed the 5’-UTR, nearly the entire Gene 1 sequence, and the sequences from the M gene to the 3’-UTR, respectively, but shared a low sequence similarity throughout the complete S and Gene 3 (Fig. 4A). A number of phylogenetic trees were generated to describe the relationship of the IBV strains based on the different gene fragments (Fig. 4B). The phylogenetic trees revealed that viruses in the GVI-1 and GI-19 lineages belonged to the same clades based on fragments I and III; however, the GVI-1 viruses differed from those of the GI-19 lineage based on fragment II (Fig. 4B). These data suggested that the GVI-1 viruses emerged as a result of a replacement of the spike gene and Gene 3 sequences in an YX10-like strain of GI-19 lineage through recombination. The YX10-like strain is one of the parental viruses. Another is an unidentified virus from which the spike gene and Gene 3 sequences were derived.
Strains LHB/110615 and LHB/110617 showed high sequence identities based on both S1 gene and the complete genome, thus only LHB/110615 was used in the following VN and pathogenicity tests.
3.3. Antigenic characterization of the GVI-1 viruses using cross-virus neutralization tests
We achieved titers of 104.5/0.1 mL EID50 for strain LHB/110615, 104.2/0.1 mL EID50 for strain I0916/16, 104.6/0.1 mL EID50 for strain I1209/16, 104.4/0.1 mL EID50 for strain I0221/17 and 105.4/0.1 mL EID50 for strain I0725/17. VN tests were performed to evaluate the antigenic characterization and differences between our five GVI-1 viruses. The VN titers between virus pairs of the GVI-1 lineage are listed in Table 1 . The antigenic relatedness value calculated using the Archetti and Horsfall (1950) formula indicated that the antigenic relatedness value was 94.9% between LHB/110615 and I0221/17, indicating that the two strains were the same serotype. Other strains shared 9.4%–37.5% antigenic relatedness values between each other, which was <50%, implicating distinct antigenicity.
Table 1.
The antigenic relatedness values (r) of the virus neutralization tests between viruses in the GVI-I lineage (serum dilution using a constant amount of virus).
| Virus |
Serum |
||||
|---|---|---|---|---|---|
| LHB110615 | I0916/16 | I1209/16 | I0221/17 | I0725/17 | |
| LHB110615 | 100% | 9.4% | 11.4% | 94.9% | 27.7% |
| I0916/16 | 100% | 10.7% | 15.6% | 14.7% | |
| I1209/19 | 100% | 37.5% | 29.7% | ||
| I0221/17 | 100% | 26.0% | |||
| I0725/17 | 100% | ||||
3.4. GVI-1 viruses exhibited comparable pathogenicity in one-day-old SPF chickens
No mortality was recorded among the trials. Mild to moderate respiratory signs were detected in chickens inoculated with the five GVI-1 viruses during the 25 days of monitoring (Fig. 5 ). The clinical signs appeared three days after infection. All chickens challenged with the five GVI-1 strains exhibited similar clinical signs (e.g., depression and ruffled feathers; tracheal rales, sneezing, and gasping) in some of the birds in each group, whereas the control chickens remained healthy throughout the experiment. The severity of the clinical signs was comparable between the challenge groups, although the clinical signs in the group infected with I0725/17 were slightly more severe compared to those in the other GVI-1 virus-infected groups. The clinical signs disappeared 12 days after challenge.
Fig. 5.

Respiratory symptoms were detected at 5 and 8 dpi in chickens infected with five GVI-1 viruses. The mean score was calculated from individual scores based on the severity of the respiratory symptoms detected in individual chickens.
Nearly all the five chickens infected with each of the five GVI-1 lineage viruses developed mild, serous, or catarrhal exudates in the trachea at 5 dpi, as observed in the chickens infected with most of the IBV strains (Cavanagh, 2007). None of the birds in the negative control group exhibited obvious tracheal lesions at 5 dpi. No obvious gross lesions were observed in the kidneys of the infected chickens, and they were comparable to those of the negative control birds.
The antibody response induced by infection with the five GVI-1 strains was detectable by an ELISA in only two or three birds at 16 dpi (Table 2 ). At 24 dpi, antibodies were detectable in the majority of the infected birds. The chickens in the negative control group had no detectable antibody response at any of the observed timepoints.
Table 2.
Overview of the results of the challenge tests using our four IBV strains.
| Group | Morbidity | Mortality |
Antibody response |
|||||
|---|---|---|---|---|---|---|---|---|
| 4 da | 8 d | 12 d | 16 d | 20 d | 24 d | |||
| LHB/110615 | 8/10 | 0/10 | 0/10b | 0/10 | 0/10 | 2/10 | 5/10 | 8/10 |
| I0916/16 | 10/10 | 0/10 | 0/10 | 0/10 | 1/10 | 4/10 | 9/10 | 9/10 |
| I1209/16 | 8/10 | 0/10 | 0/10 | 0/10 | 1/10 | 1/10 | 3/10 | 5/10 |
| I0221/17 | 9/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 3/10 | 5/10 |
| I0725/17 | 9/10 | 0/10 | 0/10 | 0/10 | 3/10 | 5/10 | 8/10 | 9/10 |
| Negative control | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 |
Days after challenge.
Number of chickens that seroconverted/the number of chickens tested.
3.5. GVI-1 viruses displayed an affinity for the respiratory tract
In the trachea, the average viral titers were comparable for the five GVI-1 viruses at 5 dpi, which varied from 2 to 3 log10 in the present study (Fig. 6 ). The titer of the I0221/17 strain in the trachea of infected chickens was moderately higher compared to those of the other GVI-1 viruses. Although replication in the kidneys was observed in some of the chickens infected with strains LHB/110615, I1209/16, I0221/17, and I0725/17, the average titers were <0.25 log10. Moreover, IBV replication was not detected in the kidneys of chickens infected with strain I0916/16. No virus was detected in any of the tracheas or kidneys of the negative control birds.
Fig. 6.
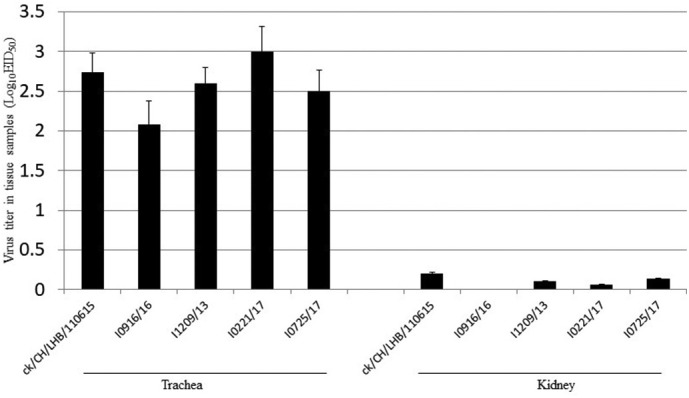
Replication of five GVI-1 strains in the trachea and kidney of chickens at 5 dpi. Viral titration was performed by inoculating 9-day-old embryonated, specific pathogen-free eggs through the allantoic route. Data are expressed as the mean ± standard deviation.
IBV antigen expression was detected in mucosal epithelial cells in the tracheas of all five of the GVI-1-infected chickens at 5 dpi (Fig. 7 ); however, no viral antigens were detected in the kidneys of the corresponding chickens (data not shown). In comparison, antigen-positive epithelial cells were more intensely labeled in the chickens infected with strain I0221/17. No viral antigens were detected in the tracheas or kidneys of the negative control chickens. Taken together, these results suggested that the GVI-1 viruses in this study showed an affinity for the respiratory tract and were determined to be non-nephropathogenic strains.
Fig. 7.

A detection of the IBV antigen by immunohistochemical analysis in the trachea of chickens at 5 dpi following an infection with the IBV GVI-1 strains (A and B for I0221/17, C and D for I0725/17, E and F for I0916/16, G and H for I1209/16, and I and J for LHB/110615).
4. Discussion
The IBV GVI-1 lineage was considered to be detected for the first time in China in 2007 (Li et al., 2010) and was previously believed to be an indigenous Asian lineage because the viruses in this lineage were strictly geographically confined to Asia (Valastro et al., 2016). A deep molecular investigation of the GVI-1 viruses isolated in different parts of the world has led to speculation regarding the origin of this lineage, as two IBV isolates (CO8089L and CO8091L) were detected in Colombia in 2003 (Alvarado et al., 2005). Moreover, this discovery was made 4 years before TC07–2 was isolated and was found to be closely related to the sequences of GVI-1 viruses isolated in other countries, suggesting that strains CO8089L and CO8091L may be ancestors of the GVI-1 lineage. Despite the fact that only a small part of the S1 gene is currently available for the CO8089L and CO8091L strains, the sequence analysis of the S1 gene fragment alone was not sufficient to confirm or deny this hypothesis. This finding was very similar to the case of the GI-16 lineage (ck/CH/LDL/97I or Q1 genotype), which was believed to have originated in China in 1996, after which it subsequently spread worldwide (Valastro et al., 2016). However, recent studies have revealed a close relationship between the sequences of the GI-16 lineage and IBV strain 624I that had circulated in Italy in the early 1990s (Laconi et al., 2019). Further studies have demonstrated that after its emergence at the beginning of the XX century, GI-16 was able to persist until today in Italy (Franzo et al., 2018). In the late 1980s, GI-16 migrated to Asia, which became the primary nucleus for further spread to the Middle East, Europe, African countries, and especially South America, likely through multiple introduction events (Franzo et al., 2018). For the GVI-1 lineage, further studies that monitor, isolate, and sequence more complete genomes of the GVI-1 strains from different countries may provide insight into the origin and evolution of the GVI-1 viruses. However, irrespective of where and when it originated, the GVI-1 lineage has spread worldwide (detected in at least five countries including Colombia, China, Korea, Japan, and Vietnam).
Seven genotypes which together comprise 35 distinct IBV lineages are recognized worldwide (Chen et al., 2017; Jiang et al., 2017; Ma et al., 2019; Valastro et al., 2016). The constant emergence of new IBV types was believed to be the result of insertions, deletions, point mutations, and recombination events in the IBV genome, especially in the S1 subunit, since specific IBV serotypes and neutralization epitopes have been mapped in the S1 subunit (Kant et al., 1992; Koch et al., 1990). Moreover, extensive amino acid sequence variability of the spike protein S1 subunit was found and the S1 subunit shows a high sequence diversity (up to 50% among different genotypes) among IBV strains (Kusters et al., 1987, Kusters et al., 1989; Toro et al., 2012). Interestingly, the S1 gene sequences were somewhat conserved among the viruses in the GVI-1 lineages, compared to those of other genes or regions in the genomes. The findings of the present study demonstrated that the GVI-1 viruses emerged as a result of a replacement of the spike gene and Gene 3 sequences in an YX10-like strain of GI-19 lineage through recombination. Therefore, it is likely that not all the viruses in the GVI-1 lineage were derived from a common ancestor that originated from a recombination event between an YX10-like strain of GI-19 lineage and an unidentified strain. In contrast, recombination may occur in separate events between unidentified viruses and different GI-19 strains with variable genomic sequences. It is important to note that the potential parent, an IBV strain of GI-19 lineage, represented the most prevalent lineage, believed to have initially originated in China, and has not been detected in Colombia to date (Cook et al., 2012). Hence, any recombination events likely occurred in China between strains of GI-19 lineage and CO8089L/CO8091L-like viruses following the introduction of CO8089L/CO8091L-like viruses from Colombia by unknown routes. Consequently, the unidentified viruses should be Colombian CO8089L/CO8091L-like viruses, which were derived in Colombia and subsequently introduced into China. Similarly, the IBV strain, KrD1515, was isolated from Korea in 2015, found to be of the TC07–2/GVI-I lineage, and considered to have originated from a recombination event (Jang et al., 2018); the authors did not analyze the sequences of the CO8089L and CO8091L strains and concluded that the KrD1515 virus was a recombinant virus between a Chinese TC07–2 and a non-TC07–2 Korean IBV. These results suggested that the recombination events of the GVI-1 viruses in China and Korea occurred independently. Therefore, we can predict that the CO8089L/CO8091L-like strains were independently introduced from Colombia to China and Korea, and they were most likely the common parental viruses of the GVI-1 viruses isolated from both countries. Whether this is also the case for the emergence and evolution of GVI-1 viruses in other Asian countries requires further confirmation. This hypothesis can be confirmed by the availability and analysis of the complete genomic sequences of the CO8089L/CO8091L-like strains in Colombia and GVI-1 viruses in Asian countries. Increasing evidence shows that the circulation of many “novel” genotypes and lineages often predate their first identification (Franzo et al., 2017, Franzo et al., 2018). This is likely also the case of the GVI-1 lineage. Thus, we speculate that some of the so called “emerging” genotypes may actually be ancient genetic groups that have not been correctly identified or classified (Franzo et al., 2018).
Based on the VN results, strains LHB/110615 and I0221/17 were found to be of the same serotype and shared 94.9% antigenic relatedness; however, despite the comparative sequence conservation of the S1 subunit for the GVI-1 viruses in this study, obvious antigenic diversity was also found. This has also been reported in other viruses which were found to belong to the same lineage but displayed different antigenicity (Gelb Jr et al., 1997; Lin et al., 2016; Nix et al., 2000). As the main antigenic viral protein containing epitopes for neutralization (Cavanagh and Davis, 1986; Cavanagh et al., 1986b, Cavanagh et al., 1986c; Kant et al., 1992; Koch et al., 1990), only a few changes in the amino acid composition in the S1 portion of the virus spike protein, especially within the hypervariable regions, may contribute to the altered antigenicity of the viruses (Cavanagh et al., 1992); however, the precise locations of these changes are difficult to access. In the present study, because amino acid substitutions were observed in the S1 subunits of the GVI-1 viruses at both the HVRs and sequences encoding the neutralizing epitopes, these substitutions may account for such antigenic changes.
Despite the different sites of recombination in the genomes of the IBV strains that have been reported, the recombination events that occurred in the IBV gene encoding the spike protein may potentially cause antigenic, pathogenic, and tissue tropism changes, possibly even a host shift event, which could lead to the emergence of a novel IBV genotype or avian coronaviruses. It has been proposed that the emergence of coronaviruses in turkeys (Hughes, 2011; Jackwood et al., 2010) and guinea fowl (Brown et al., 2016) resulted from recombination events involving IBVs and an unidentified coronavirus that donated a spike gene that encoded a protein of low amino acid identity to those of IBVs. These recombinations have resulted in a host shift from chickens to turkeys and guinea fowl, respectively. For turkey coronaviruses, such recombination has also resulted in an altered tissue tropism of the virus from the upper respiratory tract to the intestine. Recently, a novel genotype (GVII-1) (Ma et al., 2019) and lineages (GI-28 and GI-29) (Chen et al., 2017; Jiang et al., 2017) of IBVs have been found to have emerged in China, which were derived from recombination events that led to the replacement of the complete spike gene between IBVs. In some cases, recombination events have resulted in changes in tissue tropism from nephropathogenicity to an affinity for the respiratory tract of chickens (Ma et al., 2019), compared to the deduced parental virus. This was also the case for viruses of the GVI-1 lineage, which displayed respiratory tropism in chickens in this study and for virus isolated in the Republic of Korea (Jang et al., 2018); however, the deduced parental viruses exhibited obvious nephropathogenesis in chickens (Ma et al., 2019). Interestingly, the commercial broilers where the CO8089L and CO8091L strains were isolated only exhibited the presence of respiratory disease (Alvarado et al., 2005). In China, nephropathogenic IBV strains (Chen et al., 2017; Han et al., 2011; Gao et al., 2016; Jiang et al., 2017) of different serotypes have been reported to represent the predominant health problems affecting poultry. However, the wide prevalence of recombinant GVI-1 viruses which display an affinity for the respiratory tract complicated the epidemiology of IBV in China.
The following are the supplementary data related to this article.
Supplementary Fig. 1.

Supplementary material
Acknowledgments
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2017YFD0500105), the China Agriculture Research Systerm (No. CARS-40-K18), National “Twelfth Five-Year” Plan for Science & Technology Support (2015BAD12B03) and the Provincial Supported Science Foundation of Heilongjiang Province for The National Key Technology R&D Program (GX16B003).The authors declare that they have no competing interests.
Competing interests
The authors declare that they have no competing interests.
References
- Alvarado I.R., Villegas P., Mossos N., Jackwood M.W. Molecular characterization of avian infectious bronchitis virus strains isolated in Colombia during 2003. Avian Dis. 2005;49:494–499. doi: 10.1637/7202-050304R.1. [DOI] [PubMed] [Google Scholar]
- Archetti I., Horsfall F.L. Persistent antigenic variation of influenza a viruses after incomplete neutralization in ovo with heterologous immune serum. J. Exp. Med. 1950;92:441–462. doi: 10.1084/jem.92.5.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursnell M.E., Brown T.D., Foulds I.J., Green P.F., Tomley F.M., Binns M.M. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J. Gen. Virol. 1987;68:57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- Brown P.A., Touzain F., Briand F.X., Gouilh A.M., Courtillon C., Allée C., Lemaitre E., De Boisséson C., Blanchard Y., Eterradossi N. First complete genome sequence of European Turkey coronavirus suggests complex recombination history related with US turkey and Guinea fowl coronaviruses. J. Gen.Virol. 2016;97:110–120. doi: 10.1099/jgv.0.000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J. Coronavirus IBV: removal of spike glycopolypeptide S1 by urea abolishes infectivity and haemagglutination but not attachment to cells. J. Gen. Virol. 1986;67:1443–1448. doi: 10.1099/0022-1317-67-7-1443. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Darbyshire J.H., Peters R.W. Coronavirus IBV: virus retaining spike glycopolypeptide S2 but not S1 is unable to induce virus-neutralizing or haemagglutination-inhibiting antibody, or induce chicken tracheal protection. J. Gen. Virol. 1986;67:1435–1442. doi: 10.1099/0022-1317-67-7-1435. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Pappin D.J. Coronavirus IBV glycopolypeptides: locational studies using proteases and saponin, a membrane permeabilizer. Virus Res. 1986;4:145–156. doi: 10.1016/0168-1702(86)90038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Cook J.K., Li D., Kant A., Koch G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992;21:33–43. doi: 10.1080/03079459208418816. [DOI] [PubMed] [Google Scholar]
- Chen Y., Jiang L., Zhao W., Liu L., Zhao Y., Shao Y., Li H., Han Z., Liu S. Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI-28) in China. Vet. Microbiol. 2017;198:108–115. doi: 10.1016/j.vetmic.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook J.K., Jackwood M., Jones R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012;41:239–250. doi: 10.1080/03079457.2012.680432. [DOI] [PubMed] [Google Scholar]
- de Wit J.J., Cook J.K., van der Heijden H.M. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol. 2011;40:223–235. doi: 10.1080/03079457.2011.566260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo G., Massi P., Tucciarone C.M., Barbieri I., Tosi G., Fiorentini L., Ciccozzi M., Lavazza A., Cecchinato M., Moreno A. Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS One. 2017;12 doi: 10.1371/journal.pone.0184401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo G., Cecchinato M., Tosi G., Fiorentini L., Faccin F., Tucciarone C.M., Trogu T., Barbieri I., Massi P., Moreno A. GI-16 lineage (624/I or Q1), there and back again: the history of one of the major threats for poultry farming of our era. PLoS One. 2018;13 doi: 10.1371/journal.pone.0203513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Wang Q., Zhao W., Chen Y., Zhang T., Han Z., Xu Q., Kong X., Liu S. Serotype, antigenicity, and pathogenicity of a naturally recombinant TW I genotype infectious bronchitis coronavirus in China. Vet. Microbiol. 2016;191:1–8. doi: 10.1016/j.vetmic.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb J., Jr., Keeler C.L., Nix W.A., Rosenberger J.K., Cloud S.S. Antigenic and S-1 genomic characterization of the Delaware variant serotype of infectious bronchitis virus. Avian Dis. 1997;41:661–669. [PubMed] [Google Scholar]
- Haghighat-Jahromi M., Asasi K., Nili H., Dadras H., Shooshtari A.H. Coinfection of avian influenza virus (H9N2 subtype) with infectious bronchitis live vaccine. Arch.Virol. 2008;153:651–655. doi: 10.1007/s00705-008-0033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Sun C., Yan B., Zhang X., Wang Y., Li C., Zhang Q., Ma Y., Shao Y., Liu Q., Kong X., Liu S. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011;11:190–200. doi: 10.1016/j.meegid.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Jiang L., Zhao W., Chen Y., Xu L., Sun J., Zhao Y., Liu S. Isolation and characteristics of the Arkansas-type infectious bronchitis virus in China. Avian Dis. 2018;62:18–27. doi: 10.1637/11719-072517-Reg.1. [DOI] [PubMed] [Google Scholar]
- Hassan K.E., Shany S.A., Ali A., Dahshan A.H., El-Sawah A.A., El-Kady M.F. Prevalence of avian respiratory viruses in broiler flocks in Egypt. Poult. Sci. 2016;95:1271–1280. doi: 10.3382/ps/pew068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A.L. Recombinational histories of avian infectious bronchitis virus and Turkey coronavirus. Arch.Virol. 2011;156:1823–1829. doi: 10.1007/s00705-011-1061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackwood M.W., Boynton T.O., Hilt D.A., McKinley E.T., Kissinger J.C., Paterson A.H., Robertson J., Lemke C., McCall A.W., Williams S.M., Jackwood J.W., Byrd L.A. Emergence of a group 3 coronavirus through recombination. Virology. 2010;398:98–108. doi: 10.1016/j.virol.2009.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang I., Lee H.J., Bae Y.C., Park S.C., Lee H.S., Choi K.S. Genetic and pathologic characterization of a novel recombinant TC07-2-type avian infectious bronchitis virus. Avian Dis. 2018;62:109–113. doi: 10.1637/11764-103017-ResNote.1. [DOI] [PubMed] [Google Scholar]
- Jiang L., Zhao W., Han Z., Chen Y., Zhao Y., Sun J., Li H., Shao Y., Liu L., Liu S. Genome characterization, antigenicity and pathogenicity of a novel infectious bronchitis virus type isolated from South China. Infect. Genet. Evol. 2017;54:437–446. doi: 10.1016/j.meegid.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungherr E.I., Chomiak T.W., Luginbuhi R.E. Proceedings 60th Annual Meeting US Livestock Sanitary Association. 1956. Immunological differences in strains of infectious bronchitis virus; pp. 203–209. (Chicago, IL) [Google Scholar]
- Kant A., Koch G., van Roozelaar D.J., Kusters J.G., Poelwijk F.A., van der Zeijst B.A. Location of antigenic sites defined by neutralizing monoclonal antibodies on the S1 avian infectious bronchitis virus glycopolypeptide. J. Gen. Virol. 1992;73:591–596. doi: 10.1099/0022-1317-73-3-591. [DOI] [PubMed] [Google Scholar]
- Koch G., Hartog L., Kant A., van Roozelaar D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: correlation with biological functions. J. Gen. Virol. 1990;71:1929–1935. doi: 10.1099/0022-1317-71-9-1929. [DOI] [PubMed] [Google Scholar]
- Kusters J.G., Niesters H.G., Bleumink-Pluym N.M., Davelaar F.G., Horzinek M.C., Van der Zeijst B.A. Molecular epidemiology of infectious bronchitis virus in the Netherlands. J. Gen. Virol. 1987;68:343–352. doi: 10.1099/0022-1317-68-2-343. [DOI] [PubMed] [Google Scholar]
- Kusters J.G., Jager E.J., Lenstra J.A., Koch G., Posthumus W.P., Meloen R.H., van der Zeijst B.A. Analysis of an immunodominant region of infectious bronchitis virus. J. Immunol. 1989;143:2692–2698. [PubMed] [Google Scholar]
- Laconi A., Listorti V., Franzo G., Cecchinato M., Naylor C., Lupini C., Catelli E. Molecular characterization of whole genome sequence of infectious bronchitis virus 624I genotype confirms the close relationship with Q1 genotype. Transbound. Emerg. Dis. 2019;66:207–216. doi: 10.1111/tbed.13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Xue C., Chen F., Qin J., Xie Q., Bi Y., Cao Y. Isolation and genetic analysis revealed no predominant new strains of avian infectious bronchitis virus circulating in South China during 2004-2008. Vet. Microbiol. 2010;143:145–154. doi: 10.1016/j.vetmic.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim T.H., Kim M.S., Jang J.H., Lee D.H., Park J.K., Youn H.N., Lee J.B., Park S.Y., Choi I.S., Song C.S. Live attenuated nephropathogenic infectious bronchitis virus vaccine provides broad cross protection against new variant strains. Poult. Sci. 2012;91:89–94. doi: 10.3382/ps.2011-01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.Y., Li Y.T., Chen Y.T., Chen T.C., Hu C.J., Chen H.W. Identification of an infectious bronchitis coronavirus strain exhibiting a classical genotype but altered antigenicity, pathogenicity, and innate immunity profile. Sci. Rep. 2016;23 doi: 10.1038/srep37725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Kong X. A new genotype of nephropathogenic infectious bronchitis virus circulating in vaccinated and non-vaccinated flocks in China. Avian Pathol. 2004;33:321–327. doi: 10.1080/0307945042000220697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.W., Zhang Q.X., Chen J.D., Han Z.X., Liu X., Feng L., Shao Y.H., Rong J.G., Kong X.G., Tong G.Z. Genetic diversity of avian infectious bronchitis coronavirus strains isolated in China between 1995 and 2004. Arch.Virol. 2006;151:1133–1148. doi: 10.1007/s00705-005-0695-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Han Z., Chen J., Liu X., Shao Y., Kong X., Tong G., Rong J. S1 gene sequence heterogeneity of a pathogenic infectious bronchitis virus strain and its embryo-passaged, attenuated derivatives. Avian Pathol. 2007;36:231–234. doi: 10.1080/03079450701338730. [DOI] [PubMed] [Google Scholar]
- Liu X., Shao Y., Ma H., Sun C., Zhang X., Li C., Han Z., Yan B., Kong X., Liu S. Comparative analysis of four Massachusetts type infectious bronchitis coronavirus genomes reveals a novel Massachusetts type strain and evidence of natural recombination in the genome. Infect. Genet. Evol. 2013;14:29–38. doi: 10.1016/j.meegid.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G., Ingersoll R., Sheppard H.W., Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Shao Y., Sun C., Han Z., Liu X., Guo H., Liu X., Kong X., Liu S. Genetic diversity of avian infectious bronchitis coronavirus in recent years in China. Avian Dis. 2012;56:15–28. doi: 10.1637/9804-052011-Reg.1. [DOI] [PubMed] [Google Scholar]
- Ma T., Xu L., Ren M., Shen J., Han Z., Sun J., Zhao Y., Liu S. Novel genotype of infectious bronchitis virus isolated in China. Vet. Microbiol. 2019;230:178–186. doi: 10.1016/j.vetmic.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mase M., Kawanishi N., Ootani Y., Murayama K., Karino A., Inoue T., Kawakami J. A novel genotype of avian infectious bronchitis virus isolated in Japan in 2009. J. Vet. Med. Sci. 2010;72:1265–1268. doi: 10.1292/jvms.10-0080. [DOI] [PubMed] [Google Scholar]
- Nix W.A., Troeber D.S., Kingham B.F., Keeler C.L., Jr., Gelb J., Jr. Emergence of subtype strains of the Arkansas serotype of infectious bronchitis virus in Delmarva broiler chickens. Avian Dis. 2000;44:568–581. [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent end points. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
- Schalk A.E., Hawn M.C. An apparently new respiratory disease of baby chicks. J. Am. Vet. Med. Assoc. 1931;78:413–422. [Google Scholar]
- Tarnagda Z., Yougbare I., Kam A., Tahita M.C., Ouedraogo J.B. Prevalence of infectious bronchitis and Newcastle disease virus among domestic and wild birds in H5N1 outbreaks areas. J. Infect. Dev. Ctries. 2011;5:565–570. doi: 10.3855/jidc.1441. [DOI] [PubMed] [Google Scholar]
- Toro H., van Santen V.L., Jackwood M.W. Genetic diversity and selection regulates evolution of infectious bronchitis virus. Avian Dis. 2012;56:449–455. doi: 10.1637/10072-020212-Review.1. [DOI] [PubMed] [Google Scholar]
- Valastro V., Holmes E.C., Britton P., Fusaro A., Jackwood M.W., Cattoli G., Monne I. S1 gene-based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect. Genet. Evol. 2016;39:349–364. doi: 10.1016/j.meegid.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Han Z., Wang Q., Zhang T., Gao M., Zhao Y., Shao Y., Li H., Kong X., Liu S. Emergence of novel nephropathogenic infectious bronchitis viruses currently circulating in Chinese chicken flocks. Avian Pathol. 2016;45:54–65. doi: 10.1080/03079457.2015.1118435. [DOI] [PubMed] [Google Scholar]
- Xu L., Han Z., Jiang L., Sun J., Zhao Y., Liu S. Genetic diversity of avian infectious bronchitis virus in China in recent years. Infect. Genet. Evol. 2018;66:82–94. doi: 10.1016/j.meegid.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Jiang Y., Low S., Wang Z., Nam S.J., Liu W., Kwangac J. Characterization of three infectious bronchitis virus isolates from China associated with proventriculus in vaccinated chickens. Avian Dis. 2001;45:416–424. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material