

Abstract
Antibody preparations have a long history of providing protection from infectious diseases. Although antibodies remain the only natural host-derived defense mechanism capable of completely preventing infection, as products, they compete against inexpensive therapeutics such as antibiotics, small molecule inhibitors and active vaccines. The continued discovery in the monoclonal antibody (mAb) field of leads with broadened cross neutralization of viruses and demonstrable synergy of antibody with antibiotics for bacterial diseases, clearly show that innovation remains. The commercial success of mAbs in chronic disease has not been paralleled in infectious diseases for several reasons. Infectious disease immunotherapeutics are limited in scope as endemic diseases necessitate active vaccine development. Also, the complexity of these small markets draws the interest of niche companies rather than big pharmaceutical corporations. Lastly, the cost of goods for mAb therapeutics is inherently high for infectious agents due to the need for antibody cocktails, which better mimic polyclonal immunoglobulin preparations and prevent antigenic escape. In cases where vaccine or convalescent populations are available, current polyclonal hyperimmune immunoglobulin preparations (pIgG), with modern and highly efficient purification technology and standardized assays for potency, can make economic sense. Recent innovations to broaden the potency of mAb therapies, while reducing cost of production, are discussed herein. On the basis of centuries of effective use of Ab treatments, and with growing immunocompromised populations, the question is not whether antibodies have a bright future for infectious agents, but rather what formats are cost effective and generate safe and efficacious treatments to satisfy regulatory approval.
Anti-infective antibodies: new opportunities for a proven technology
In the late 1890s Behring and Kitasato developed the first widely available and effective antimicrobial treatment by showing that transfer of immune sera could provide passive immunity to diphtheria [1]. In the next 50 years, serum transfer was used as a successful treatment of many infections including, pneumococcal pneumonia, meningococcal meningitis, and streptococcal infection [2]. However, due to both safety concerns and the discovery of antibiotics, serum therapy was largely abandoned by the late 1940s with the exception of a limited number of toxin- and viral-mediated diseases, which continued to rely on serum due to a lack of alternative options. The mainstream human immunoglobulin preparations used today are not recognizable as the old serum therapies. These products are highly purified, treated and filtered to ensure viral inactivation and removal, and use highly reproducible release assays to avoid the historical problems associated with polyclonal antibody (Ab) such as impurities, resident viruses, and lot variation.
Two troubling developments in infectious diseases have led to a necessary resurgence in the development of antibody-based therapeutics. First, the rampant emergence of multi-drug resistant forms of new and old pathogens; second, the recent explosion of the world's immunocompromised population has provided an unprotected population from which combinations of complex infections are emerging. New adjunctive antibody therapeutics to every major disease of infection type may be the best strategy in both treating and preventing the new wave of drug resistant infectious diseases.
Pathogenesis is linked to treatability
The pathogenesis of infectious organisms can be very complex. The co-evolution of host and pathogen over evolutionary time has resulted in many types of inflammatory response. A fine balance exists between protective versus over zealous host responses, either through deregulation or through subversion by the pathogen. Although Ab therapies are either in development or in use to many viral, bacterial, fungal, and prion-mediated infections (Table 1, Table 2, Table 3 ) these pathogens exhibit huge differences in pathology and virulence. In general, highly virulent and acute infections are more likely to require the immediate protection provided by antibody treatments. Moreover, in outbreak situations, the early implementation of public health measures may help to limit spread when no vaccine is available, as was shown with SARS 3, 4. During stretches of a dramatically increased risk of exposure, immunotherapy may provide a means of protection preferable over conventional vaccines [3]. The half-life of passive antibody therapeutics obviates the use of these types of products except when needed.
Table 1.
Passive Ab therapies to bacterial agents/toxins
| Target | Name/format | Details | Refs |
|---|---|---|---|
| Pseudomonas aeruginosa | KB001; Pegylated Fab’ against PCR-V made from humaneering murine mAb 166 | C-Kalobios/Sanofi Pasteur protects against strains with PCR-V | 92, 93 |
| Vibrio cholera | Murine mAb 72.1 LPS specific |
R – Protects against Ogawa and Inaba strains in mouse infection model | [94] |
| Yersinia pestis | Human Fab mAbs m252 (F1 specific) and m253 and m254 (V-specific) expressed as hu-IgG1 | R – Protect mice from death in a bubonic plague model | [95] |
| Escherichia coli (EHEC) Shigatoxin | Anti-Stx1 IgY; chicken egg yolk immunoglobulin (IgY) production | R – Blocks the binding of Stx1 to the Hela cells and protect BALB/c mice from toxin challenges | [96] |
| Ricinus communis Ricin – Plant Toxin | Murine mAb RAC18 and polyclonal murine antibody to A chain | R – Inhibits cell death in vitro and protects mice from inhalational ricin exposure | [97] |
| Bacillus anthracis | Purified human Polyclonal IgG (AIG) from high titer vaccinated donors (multiple epitopes/isotypes) | M – Cangene Corp; protective in several animal models | [98] |
| Bacillus anthracis – PA toxin | ABthrax (raxibacumab) Human IgG1/λ mAb to PA toxin; Anthim; Humanized deimmunized IgG1/k mAb to PA toxin; Valortim (MDX-1303) Human IgG1/k | M – Human Genome Sciences; protective in several animal models C – Elusys; protective in rabbit inhalational spore model M – Pharmathene; protective in rabbit inhalational spore model |
99, 100, 101, 102, 103 |
| Chlamydia trachomatis | Murine MP-33b (IgG3) and MP-A5d (IgA) mAbs against the major outer membrane protein delivered by hybridoma backpack | R – Reduced bacterial pathology and shedding in ascending chlamydial infection in a murine genital tract model | [104] |
| S. aureus (MRSA) + E. coli, other gram negatives | Human mAb F598 binds to poly-N-acetyl glucosamine (PNAG) | C – Alopexx/Sanofi-Aventis; target conserved epitope and protects mice from lethal infection | 105, 56 |
|
Clostridium botulinum Neurotoxin |
Despeciated blended equine hyperimmune F(ab′)2 – Heptavalent | M – Protects in vivo against death by all 7 serotypes of neurotoxin | 98, 106 |
|
Clostridium botulinum Neurotoxin |
Human scFv mAbs expressed as hu-IgG1 | R/C – Individual mAbs protect mice in vivo from one serotype of neurotoxin | 107, 108 |
|
Clostridium difficile Toxins A and B |
Human mAbs CDA-1 (to C terminus of TcdA) and CDB-1 (to C-terminus of TcdB) | C – Medarex/Merck Reduced recurrence in humans infection in phase 2 trial |
[5] |
|
Clostridium difficile Toxins A and B |
Humanized mAbs mPA-39 (to toxin A) mPA-50 (to toxin A) and mPA-41 (to N-terminus of toxin B) | C – Progenics; mAb mixtures protect hamsters with higher efficacy than Merck mAbs | [109] |
M – marketed; R – research; C – commercial, under development (excludes those which have been dropped or suspended due to clinical failure).
Table 2.
Passive Ab therapy to viral agents
| Target | Format | Details | Refs |
|---|---|---|---|
| Respiratory syncytial virus (RSV) | Respigam; Human polyclonal immune globulin to RSV | M – Medimmune, Astra Zeneca; prophylactic treatment of infants; | 110, 111 |
| Varicella Zoster Virus (chicken pox) | VariZIG Human polyclonal immune globulin with titer to Varicella zoster | M – Cangene Corp; purified human polyclonal antibody | 98, 112 |
| Vaccinia | VIG; Human polyclonal immune globulin to vaccinia virus from immunized people | M – VIGIV Cangene and VIGIV Dynport; purified human polyclonal antibody from donors with high titer; for treatment of smallpox vaccine side effects | [113] |
| Hepatitis B virus (HBV) | HepaGamB; Human polyclonal immune globulin to HBV | M – Cangene Cop.; used to prevent re-infection with hepatitis B disease in HBV Ag positive liver transplant patients. | [98] |
| Hepatitis C virus (HCV) | Civacir; Human polyclonal immune globulin to HCV | C – Nabi; prevent re-infection with hepatitis C disease in HCV Ag positive liver transplant patients. | [114] |
| Hepatitis C | mAbs to Ara C epitope on Hepatitis C E2 protein | R – Scripps; used Fab display library made from chronically infected donor and showed broad protection in a novel chimeric liver mouse model | [84] |
| Respiratory syncytial virus (RSV) | Synagis; a humanized IgG1/k mAb derived from murine mAb 1129 | M – Medimmune Astra Zeneca; NSO cell line expressed mAb for prophylactic treatment of infants | 22, 115 |
| Dengue virus (DENV) | Murine mAbs DENV1-E105, and DENV1-E106 to the E protein | C – Macrogenics licensed several of the mAbs; mAbs exhibited therapeutic activity even four days after infection with heterologous virus | 116, 117 |
| HIV | Hu mAbs IgGb12, 2F5, 4E10, 2G12 | R – provides protection against mucosal SHIV challenge in macaques even at low titers | 118, 119, 120 |
| Ebola | Hu mAb KZ52, murine mAbs 13F6 13C6 6D8, Can9G1a | R – Isolated from survivors and provide protection in rodents | 121, 122 |
| Nipah/Hendrah | Human mAb m102.4 optimised from naïve library | Binds to the surface glycoprotein and cross protects both Nipah and related hendrah virus | [123] |
| Influenza A infection | Several new Cross reactive mAbs | C – Kirin – M2e C – Cytos M2e R-C Harvard, Crossreactive mAbs to HA |
124, 125, 63 |
| General Infection | Normal Human polyclonal immune globulin (IVIG) | M – Talecris, Bayer, CSL, Baxter others; used to reduce/prevent infection in post exposure prophylaxis of some viral disease and for individuals with immunodeficiency | 126, 127, 128, 129 |
M – marketed; R – research; C – commercial, under development.
JDB manuscript in preparation.
Table 3.
Passive Ab therapy to fungi and yeast
| Target | Format | Details | Refs |
|---|---|---|---|
| Paracoccidioides brasiliensis | Murine mAb E3 against the gp43 protein | R – mAbs show protection in vivo via reduction in fungal burden and less inflammation in murine models; gp43 is a protective antigen | [130] |
| Cryptococcus neoformans | Murine mAb 2G8 anti β-glucan | R – MAb caused a reduction in the fungal burden in the brains and livers of mice systemically infected with a highly virulent, encapsulated C. neoformans strain | [60] |
| Cryptococcus neoformans | Murine 3E5 MAb anti-capsular ps | R – IgG1, IgG2a, and IgG2b switch variants prolong the survival of lethally infected mice, whereas the IgG3 MAb does not; all protect in c’ deficient mice. | [131] |
| Asperigillius fumigatus | Murine MAb A9 anti-cell wall surface of hyphae | R – murine model of invasive aspergillosis | [132] |
| Candida spp. | Efungumab (Mycograb) is a hu-mAb against HSP 90 | C – Novartis; prevents a conformational change needed for fungal viability; shows efficacy in humans and lowered mortality; abandoned in 2010 | [133] |
| Pneumocystis carnii | Murine anti-kex1 mAb 4F11 and its F(ab′)2 derivative | R – reduced infectious burden in an intranasal immunoprophylaxis model | [134] |
M – marketed; R – research; C – commercial, under development.
Not all infections are suitable for antibody therapies. The development of a therapeutic can cost hundreds of millions of dollars and take years to find its way through the various stages of research and development, clinical development, and the government regulatory approval processes. At the end of this process the manufacturing costs factor into the cost per dose of the final product (Table 4 ). Cheaper to manufacture antibiotics have an advantage in the post development and FDA approval processes. Until antibody products produced via novel recombinant expression systems gain regulatory approval, monoclonal antibody (mAb) manufacturing remains limited cell culture. Currently every FDA approved mAb has been manufactured using classical mammalian cell culture expression systems.
Table 4.
Therapeutics antibodies, antibiotics and small molecules
| Criteria | Poly Ig | mAbs | RNAi (antivirals) | Antibiotics (bacterial treatments |
|---|---|---|---|---|
| Ease of Delivery | High (IV, IM, SC) | High (IV, IM, SC) | ? Low–High (Oral, IV) | Low–High (Oral, IV) |
| Regulatory Approval Cost | High | High | High | High |
| Manufacturing Cost | Medium | High | ? | Low |
| Cost per dose | Medium (100s) | High (1000s) | ? | Low (1s) |
| Damage to Microbiome | None | None | ? | Yes |
| Specific Activity | Yes | Yes | ? | No |
| Toxicity | Low | Low | ? | Low |
| QA effort required | High to prevent lot variation | Low | ? | Low |
| Source | Humans, animals | Tissue culture | ? | Bacterial Fermentation |
| History of regulatory approval | Yes | Yes | No | Yes |
To be considered viable for the development of an antibody therapy, an infectious disease pathogen needs very specific attributes. The antibody must show efficacy in vivo; such as help clear the infection, reduce the infectious burden, reduce the time to heal, or prevent infection/intoxication from occurring. However, before addressing the biology of the antibody, numerous issues around the target market drive commercial interests. The continued development of antibody therapeutics, due to their inherent narrow specificity and high cost of production, is largely dependent on competing antimicrobial therapies and vaccines. For example, despite significant investment in the development of protective mAbs against Clostridium difficile toxins [5], of which several are in clinical trials, a novel toxoid vaccine from Sanofi Pasteur may convert the relatively large and growing market for C. difficile treatment to niche market for breakthrough infections, unimmunized, or the immunocompromised. By contrast, the advent of a C. difficile specific antibiotic such as fidaxomicin (under development by Optimer Pharmaceuticals), which purportedly leaves the normal gut flora intact while clearing C. difficile, would probably leave the anti-toxin therapeutic market intact as the immunotherapeutic could still be administered as an adjunctive therapy to specifically remove the effects of the toxin. A summary of the characteristics that determine the pros and cons of the various classes of competing antimicrobial drugs is shown in Table 4.
One target, numerous biological effects
Monoclonal antibody is a unique class of drugs in that numerous biological effects, depending upon the isotype, can be induced through binding to a single microbial epitope (Fig. 1 ). Many good reviews exist which discuss contemporary mAb development [6] and more specifically for infectious agents [7]. Many of these effects are dependent on other components of the immune system; including antibody dependent cellular cytotoxicity (ADCC), complement dependent cytotoxicity (CDC), opsonization, and immunomodulation. Virus neutralization can be achieved by interfering with one of the various methods used by viruses to enter host cells. In the case of human immunodeficiency virus (HIV), Abs can prevent viral attachment to the host cell by binding to either the envelope protein of the virus, or a host receptor such as CD4 or CCR5 8, 3. Antibodies may also neutralize the influenza A virus by preventing the low pH induced conformational change required in hemagglutinin [9] or by preventing the release of progeny virions from infected cells [10]. Interestingly, antibodies can also be directly antimicrobial, as has been described in Candida albicans [11], and Cryptococcus neoformans [12]. In the latter, IgG1 and IgM mAbs induce fungal gene expression and metabolic changes in an animal model of cryptococcus [12].
Figure 1.

Antibody effector mechanisms used to combat microorganisms mediated by the various isotypes of human immunoglobulin.
Anti-infective antibodies: monoclonal or polyclonal?
Immunoglobulin from high titer plasma of immune individuals (poly Ig) and bulk intravenous immune globulin (IVIG) remain predominant therapeutics used today in passive antibody therapy for treating infectious diseases. Unlike the modest antigenic differences between normal and tumor cells which warrant a monospecific approach to treating cancer, infectious agents are generally foreign to the immune system allowing the less specific and more readily available polyclonal approach to become the norm in treating infectious disease. IVIG treatment of disease has been reviewed recently and is recommended in the treatment of Kawasaki's disease, cytomegalovirus (CMV)-induced pneumonitis, neonatal sepsis, rotaviral enterocolitis, and staphylococcal toxic shock syndrome 13, 14. As a high titer alternative, poly Ig is used clinically in the prevention of infections caused by CMV, hepatitis A and B viruses (HBV), rabies, varicella, and measles virus while anti-tetanus Ig is used to treat tetanus [14]; rabies hepatitis B and tetanus donors are usually vaccinated, and CMV, measles, varicella are typically screened for high titer. In this regard, stimulation of human donors with vaccines has been used to develop effective polyclonal anthrax immune globulin (AIG) for passive therapy [15]. Poly Ig is also used to prevent mother-to-child transmission of HBV and varicella-zoster virus [16]. Conversely, only a single mAb to RSV is currently licensed for the prophylactic treatment of an infectious disease [17]. Both polyclonal and monoclonal IgG preparations have been successfully used for infectious disease prevention with the majority of experience being with purified polyclonal IgG.
Advances in mAb technology necessitate a movement towards the more defined but expensive to manufacture mAb-based therapy. The ability to reproduce mAb therapeutics in scalable quantities to meet demand represents a significant difference from the harvesting processes for poly Ig preparations. Furthermore, although human IgG preparations used today have the highest safety record of all biologics and a clear pathway with the FDA, there are still some lingering fears regarding the chance of human poly Ig to transmit an infection, even if exceedingly low [18]. For example, UK citizens are still not allowed to donate their blood in the USA or the UK as a fall out of the prion incidents in the early 1990s, despite the fact that resins and complexing agents which remove prions, are well entrenched in poly IgG purification protocols from the plasma supply [19]. Although those concerns could be reduced through the use of mAbs produced via tissue culture or microbial expression systems, the cost per dose of mAbs is significantly higher and may not warrant development for many niche market products (Table 4). Indeed, it is not always obvious what models are relevant in qualifying a treatment or which antigens are going to be protective, so in many cases poly Ig proceeds as a cheaper alternative to mAbs. In such cases, where a vaccine is not at least through phase 1 and available, harvesting poly Ig from endemic regions may alleviate concerns of the low titer of some IVIG reagents. For instance IVIG with a titer to West Nile virus is collected from selected Israeli donors to treat infection [13]. Furthermore, mAbs typically are limited in both specificity and functionality, are fragile macromolecules which are expensive to produce, and can take considerably longer to isolate and optimize than poly Ig or IVIG. The current status of research aimed at expanding the limits of mAbs in terms of specificity and functionality, in particular to deal with infectious agents, is discussed below.
One isotype, one function
Inherent in the use of a single designed antibody to a protective target with a chosen isotype is that the effector functions mediated by the single isotype must alone combat the infection. A polyclonal IgG preparation is relatively resistant to antigenic variation through targeting a range of epitopes. It also has widened biological effector functions as it consists of multiple classes of antibody (which comprises typically IgG1 > IgG2 > IgG3 > IgG4). The ability to induce ADCC or CDC is dependent on both the isotype and glycosylation status of the antibody [20], thus the variability inherent in polyclonal preparations should in many cases provide added benefit and a protective advantage. Each isotype has characteristics that make it more or less desirable during an immune response [21] and many new recombinant Fc scaffolds have been designed in recent years [22]. Among IgG isotypes, IgG1 has long been the isotype of choice for mAb cancer therapy due to its long half life, partiality for protein antigens, ability to activate complement, and affinity for Fc receptors (FcγI, II, and IIIa/b) [21]. Synagis®, a mAb for the prevention of severe RSV in infants, remains the sole mAb for infectious agents marketed for use in humans and is an IgG1/κ, and no other mAb isotype has been approved for infectious diseases to date [2]. However, although IgG1 is effective at inducing ADCC by natural killer (NK) cells, it is a considerably less potent activator of polymorphonuclear cells (PMN) 23, 24. Furthermore, the activation of inflammatory pathways by IgG1 can be detrimental depending on the immune status of the individual [25]. The IgG2 class has relatively inert effector functions; however, it is generated in response to stimulation by inherently inflammatory bacterial cell wall polysaccharides making it important in protection against bacterial infection [26]. Moreover, two IgG2 formatted mAbs have been approved for use in human chronic diseases and at least eight more are currently in clinical development [21] owing to the longest half-life, fewest allotypes, and inability to activate complement. The IgG3 class has similar biological functions as IgG1 but has received little use to date as a recombinant mAb due to a considerably shorter half-life and extensive polymorphism [27]. The IgG4 class is unique in its inability to cross-link antigens in vitro, and is generated in response to persistent antigen stimulation, making it important for chronic viral and bacterial infections [28]. At least two IgG4 class mAbs are on the market with at least another nine in different phases of research and development for chronic disease indications [21]. In natural infections, the IgM class antibodies are generally produced the earliest in response to T-dependent antigens, and can persist in response to T-cell-independent antigens, such as lipopolysaccharides (LPS). The pentameric form of IgM makes it highly avid, although atypical to commercially purify, and the most potent inducer of CDC [29]. A human IgM mAb from Kenta Biotech, KBPA101, has been under development for some time and targets the surface O-polysaccharide LPS of Pseudomonas aeruginosa [30]. Provided expression of the native pentamer can be achieved at commercial scale, as has been described recently in human PER.C6® cells [31], polymeric IgM may prove to be the isotype of choice for the treatment of many bacteria, particularly for local infections at mucosal surfaces. Human IgA1 has unique O-linked glycoforms but the relatively short half-life of both IgA1 and IgA2 may also be best suited to be used locally. Indeed, intranasal administration of an IgA mAb pre- or post-infection protected mice from a sublethal challenge of H5N1 influenza virus [32]. Even IgD binds respiratory Gram-negative bacteria Moraxella catarrhalis and Haemophilus influenza, mediates internalization by mast cells and basophiles, and triggers innate antimicrobial responses [33].
Clearly the choice of mAb isotype is a crucial decision that will ultimately determine its function in vivo. Other factors such as dose, route of administration, half-life, and site of infection also require attention. The choice of isotype, however, is often not crystal clear as illustrated by the recent observation that despite the potent CDC and ADCC inducing ability of IgG1 in C. neoformans mouse models, only human IgG2 and IgG4 were able to protect mice from infection when antibodies carrying the same variable region but different constant regions were passively administered [34]. Also, passively administered serum IgG has shown protection in pig-tailed macaques from rotavirus infection in a mucosal gut model [35]. These findings point out the still empirical nature of designing therapeutic antibody to treat infectious diseases.
The inherent flexibility of recombinant mAb approaches permits the modification of immunoglobulin to fit specific needs. To address the problem posed by the use of a single isotype, it is possible to engineer the Fc region to increase its biological function. Targeted amino acid substitutions increase the affinity of the Fc region for FcγRIIIa and increase ADCC by natural killer cells 2 fold [36] and the glycosylation patterns of the Fc region can be further altered to increase ADCC [37]. A different approach is to engineer a bispecific mAb where one half of the Fab binds to the microbe, and the other recognizes a host receptor. Examples include mAbs specific for either P. aeruginosa [38], or bacteriophage [39] that also bind a host complement receptor through the other Fab domain. Other groups have taken lessons learned from clinical trials involving immunotoxin based therapies in oncology, and applied them successfully to infectious disease. MAbs conjugated to radioisotopes have been used to treat experimental infections by C. neoformans [40], Histoplasma capsulatum [41], Streptococcus pneumoniae [42], and HIV [43].
Monoclonal specificity: pro or con?
The exquisite specificity of mAbs may be both its biggest advantage, and biggest disadvantage. The high specificity of mAb therapy results in little cross reactivity with both host cells and normal flora, which is important considering the link between chronic diseases and certain types of cancer with the use of broad spectrum antibiotics (Table 4) 2, 44, 45. Furthermore, mAb therapy is unlikely to select for drug resistance in extraneous microbes due to the lack of intra-microbe cross reactivity [2]. Unlike poly IgG, which carries many irrelevant specificities, mAbs can be developed which target specific microbial epitopes important in pathogenesis, thereby theoretically increasing the potency of the specific biological activity per dose, in particular if the target epitope is highly protective. Polyclonals also can have isoagglutinins (anti A and anti B) which can be undesirable in some cases. Nevertheless, the high specificity also implies that as a narrow-spectrum antimicrobial drug, a quick and accurate diagnosis is required before therapy. The resulting small market for anti-infective mAbs may limit the economical feasibility of a mAb based approach in certain instances [14] and can be overshadowed by the breadth of reactivity of poly IgG which comes from targeting multiple epitopes with multiple IgG isotypes.
Many cutting edge strategies are being developed to address the narrow specificity of mAb therapy, such as broadening the utility and pathogen range and are summarized in Fig. 2 . Typically the selection of Ab to protective antigens is the key criterion in the development of an effective passive immunization strategy. Highly conserved antigens are the most valuable targets for passive immunotherapeutics as they reduce both the number of mAbs needed to make an effective cocktail and the chance of escape from antibody. These can generally be placed into several key themes.
Figure 2.
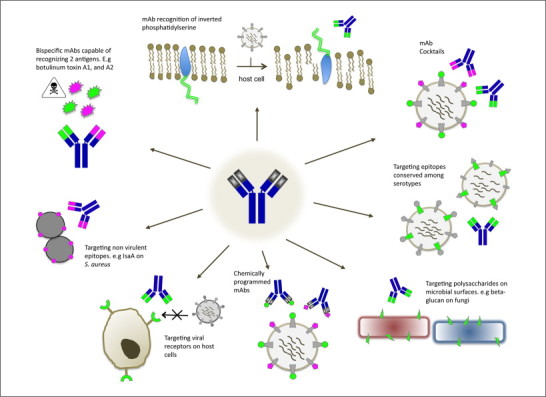
Exploiting recombinant technology to address monospecificity issues surrounding mAb therapy for the treatment/prevention of infectious diseases.
The creation of antibody cocktails to target groups of infectious agents
Certain types of infection consistently involve a predictable cadre of infectious agents. These may be associated with a particular injury (e.g. post burn infections), environmental exposure (e.g. contaminated food) or intentional biothreat release (e.g. hemorrhagic viruses). Chemically programmed mAbs, in which the unlimited chemical diversity of synthetic molecules is coupled with the effector functions of antibodies provides a means in which a single mAb may can have specificity for multiple epitopes 47, 48. In this fashion, effector functions can be brought in by pre-immunized circulating antibody to “tags” on the small molecule adapters. These small molecule [48], or single chain variable fragment (scFv) [49] adapters can be expressed cheaply in Escherichia coli and are significantly less expensive to manufacture than full-length mAbs.
Targeting virulence factor associated antigens/epitopes
Current trends for bacterial immunotherapeutics target virulence associated factors and are promising candidates for several reasons: First, they prevent direct damage to the host caused by large bacterial exotoxins and LPS through cytolysis or inflammation; second, virulence factors lend resistance to harsh immunological conditions as seen with capsules, spores, or antigenic variation; Third, virulence factors allow adhesion and or entry into host cell environments leading to evasion and chronic infection. Targeting a single virulence factor can be very effective where disease symptoms and pathogenesis are interrupted. For example, in gastro-intestinal tract infections with C. difficile, two large cytotoxins, TcdA and Tcd B cause the associated disease and diarrhea. Recently, Merck published clinical data showing that a cocktail of two mAbs (one to TcdA another to TcdB) at 10 mg/kg can greatly reduce reoccurrence rates compared to controls, probably due to reduced damage to the intestinal lining [5]. The concept of targeting virulence factors is confounded in some cases where a bacterium expresses a plethora of virulence factors which collectively contribute to disease. For example, Staphylococcus aureas produces a broad range of virulence factors and toxins, which may have temporal roles in specific types of infection. Other approaches target antigenically variable capsules [50] clumping factor (ClfA) 51, 52, 53, alpha hemolysin and finally Panton-Valentine leukocidin (PVL) [54]. In these cases, removal of one virulence factor seems to be compensated by another. When this occurs, targeting surface structures or housekeeping proteins such as lsaA [55], as discussed below, may be more appropriate.
Protection mediated by mAbs to indirect virulence factors
A related strategy is to target bacterial proteins that have an indirect role in pathogenesis; hence are subject to less selective pressure for antigenic variation. Broadly protective antibodies are not common treatments for many bacteria, including one of the most disconcerting modern infections, methicillin-resistant S. aureus (MRSA). Recently Dr. Gerald Pier and Alopexx developed a human mAb against the carbohydrate backbone of poly N-acetylglucosamine (PNAG) in S. aureus [56]. The mAb was effective initially in models of MRSA by inducing opsonization and CDC. These mAbs are a promising immunotherapy for both prophylaxis and adjunctive treatment of MRSA. In a related study, mAbs targeting the housekeeping protein lsaA, a suggested lytic transglycosylase in S. aureus provided protection in a mouse model by promoting opsonization and inducing highly reactive oxygen species [55]. MAbs against quorum sensing auto-inducers protect mice from lethal challenge from S. aureus, albeit using a clinically irrelevant dosage [57], and mAbs specific for type III secretion system components from gram-negative bacteria Yersinia species [58], P. aeruginosa [59], and Aeromonas salmonicida [46] have all shown protection in animal models as well. MAbs have also been used against multi-drug efflux pumps of C. difficile, and Enterococcus species as part of a combination therapy with small molecule pump inhibitors [46]. Fungal infections may be a more promising area for mAb therapy due to the presence of conserved epitopes between species. Therapy with an IgG2 mAb specific for laminarin (beta-glucan) was effective at inducing fungal phagocytosis by monocytes and preventing infection from C. neoformans, C. albicans, and Aspergillis spp. 60, 61.
Antibody to conserved viral targets and host receptors
The incredible variability of viruses poses a significant challenge to mAb therapy. One way this can be countered is by using recombinant technology to derive an antibody specific for an epitope conserved among different viral strains [62]. Using phage display, Sui et al. selected hemagglutinin-5 specific mAbs capable of neutralizing both H5N1, and H1N1 [63]. A similar approach has been used to select cross protective mAbs against SARS coronavirus 64, 65 and Dengue virus type 1 [66]. Although potentially advantageous under outbreak conditions in which diagnoses are delayed, treatment of viruses capable of great antigenic variation using a single mAb is not ideal for the reasons discussed below.
As an alternative to targeting the viral proteome, several mAbs have been developed that target host proteins that double as virus receptors and co-receptors. These mAbs inhibit entry of a diverse range of virus subtypes including New World arenaviruses through anti-transferrin receptor 1 mAbs [67], HCV through anti-claudin-1 mAbs [68], and several R tropic viruses through anti-CCR5 mAbs [3]. While showing potential, mAbs against host proteins are a long way from clinical use, largely due to complications inherent in blocking host receptors. For example, blocking the CCR5 receptor may impair the host's ability fight off other viral infections, such as West Nile virus, as CCR5 is involved in lymphocyte traffic to the brain 3, 69. Lastly, one particularly promising area of research is the use of an anti-phospholipid mAb capable of identifying virally infected cells and inducing ADCC [70]. The mAb designated Bavituximab (Peregrine Pharmaceuticals) functions by recognizing phosphatidylserine, which is normally inaccessible to serum antibodies. Upon virus-induced cell activation, phosphatidylserine is exposed on the cell surface due to a loss in lipid symmetry, allowing the mAb to bind 70, 46. This mAb has prevented CMV and Pichinde arenavirus infection in mice, and clinical trials are currently underway for the treatment of HCV and HIV.
Design of mAb cocktails to reduce antigenic escape
The potent neutralizing ability of mAbs can inadvertently result in increased antigenic escape in experimental infection with highly variant RNA viruses. Indeed, treatment with a single mAb has culminated in escape variants in the treatment of HIV, hepatitis C virus (HCV), and influenza A 71, 72, 73, 74. To counter this, scientists have devised mixtures of two or more mAbs specific for distinct protective epitopes that can greatly prevent the rise of escape mutants in models of infection [74]. Indeed, development of such cocktails is being explored for H5N1 influenza A [75], SARS coronavirus [76], lymphocytic choriomeningitis virus [77], HCV [78], HBV [79], and rabies virus 80, 81, 82. These so called “cocktails” of mAbs address two major problems posed by targeting a single epitope. First, by targeting at minimum two neutralizing epitopes, escape mutants generated from each mAb are cleared by the complementing antibody [75]. Second, by increasing the antibody density (valency) on the viral surface, which may be the most important factor in virus neutralization [83], the neutralizing ability of the antibody is amplified considerably [3].
A promising recent development concerns antibody to the hepatitis C virus (HCV), the leading cause of liver disease leading to transplantation in the U.S.A. Antigenic variation of HCV remains a serious challenge in antibody therapy and active vaccine design. Poly Ig for HCV is one means by which this might be dealt with, as antibody would be prepared from immunizations with distinct HCV virus vaccines. To date there has been no active vaccine available for HCV so this has not been a possible route for a therapeutic. Law et al. instead identified human mAbs that neutralize genetically diverse HCV isolates using phage display [84]. The mAbs were isolated using recombinant HCV E2A glycoprotein from a Fab phage library developed from the bone marrow RNA of a donor chronically infected with HCV. These mAbs were elegantly shown to bind discontinuous epitopes that cluster in three distinct antigenic regions in the E2A protein; one of which (antigenic region 3) appears to be highly conserved among HCV genotypes. Furthermore, the mAbs show protection against heterologous HCV quasispecies challenge in a human liver–chimeric mouse model. This represents a major advancement as the lack of a suitable model has for years prevented advancement on this field. The results provide strong evidence that broadly neutralizing antibodies to HCV can protect against heterologous HCV virus infection and furthermore, suggest that a prophylactic vaccine against HCV may be achievable and may avoid any potential antibody enhancement of infectivity effects which may be due to irrelevant antibody in a poly IgG preparation. Advent of an approved phase 1 vaccine would lead rapidly to potential donor stimulation program and the development of a human polyclonal therapeutic for testing.
Immunotherapy is also one of the more promising approaches for the treatment of prion diseases. White et al. were the first to show that mAbs could both inhibit prion replication and delay disease development [85]. More recently, a camelid antibody with specificity for both PrPC and PrPSc demonstrated the ability to cross the blood brain barrier, reduce prion replication in vivo, and eliminate prion replication in N2a neuroblastoma cells in vitro [86].
Forthcoming developments
It is becoming evident that treatment of an infectious disease with multiple mAbs is most probably required to provide optimal protection. While mAb cocktails are being developed to address this problem, another approach may be to couple antibody therapy with existing antimicrobials. Protocols for clinical development of new antibody therapies for infectious diseases will require clinicians to include the current standard of care which in many cases are antibiotics or antivirals. Antibody acts in synergy with antibiotics and with antivirals to provide increased protection against infection (Table 5 ). Indeed, naturally resistant bacteria can be rendered susceptible to antibiotics by mAb therapy, and antivirals can increase the efficacy of anti-viral mAbs by inhibiting viral replication, thereby decreasing the likelihood of escape mutants [14].
Table 5.
Antibody – drug synergy in the treatment of infections
| Infectious agents | Disease | Treatment | Synergy | Refs |
|---|---|---|---|---|
| Pseudomonas aeruginosa | Human – Colonized Cystic fibrosis patients (chronic pulmonary infection) | Conventional antibiotic treatment plus Ps-ivIGa and conventional IVIG | Yes – transient but improved clinical scores | [135] |
| Mixed infection | Human – trauma infection | Penicillin and IVIG versus albumin in controls | Yes – reduced septic complications and improved serum bactericidal activity | [136] |
| Staphylococcus aureus (MRSA) and E. coli and Proteus spp. | Rat – experimental models for both gram positive and gram negative organisms | Mezlocillin and IVIG for gram negatives E coli and Proteus spp.; Cephalothin or Cephamandole with IVIG for S. aureus | Yes – IVIG together rendered Cephalothin as effective as oxacillin in beta-lactamase postive S. aureus and made cefamandole immediately bactericidal | [137] |
| P. aeruginosa; Klebsiella pneumonia, Seratia. marcescens, Proteus mirabilis | Experimental – murine burn model | Piperacillin plus human anti-LPS IgG and conventional IVIG | Yes – however strain specific (P. aeruginosa; not K. pneumonia, S. marcescens, P. mirabilis) | [138] |
|
Escherichia coli Proteus spp. |
Experimental – rat granuloma pouch model | Mezlocillin plus human IgG and conventional IVIG | Yes – for betalactamase + E. coli and Proteus spp. no protection by individual treatments | [139] |
| Pseudomonas aeruginosa | Experimental – murine thigh infection | Ceftazidime plus murine mAb Ld3-2F2 to LPS. | Yes – significant reduction in bacteria recovered | [140] |
| Pseudomonas aeruginosa (antibiotic resistant) | Experimental – murine burn model | Ceftazidime plus murine mAb Ld3-2F2 to LPS. | Yes – compared to no survival with individual treatments | [141] |
| Klebsiella pneumonia | Experimental – murine burn model | Ceftazidime (suboptimal dose) plus locally (sc) delivered human IgG | Yes –reduction in burn bacterial burden compared to either monotherapies | [142] |
| Bacillus anthracis | Experimental – inhalational anthrax | Ciprofloxacin plus Human mAb AVP-21D9 to PA toxinb plus | Yes – 100% protection in mice, guinea pigs; mAb protected rabbits without antibiotics | [143] |
| Clostridium difficile | Human – gastrointestinal infection | Metronidazole/vancomycin plus two human mAbs CDA-1 and CDB-1 to toxins A and B respectively | Yes – reduced recurrence rates significantly | [5] |
| Staphylococcus aureus (MRSA) | Experimental – clearance of bacteremia in rabbits | Vancomycin and human SA-IVIGc | Yes – accelerated clearance | [53] |
| Human immunodeficiency virus (HIV-1) | Human – treatment/control of infection | Anti-CCR5 antibody plus small molecule ccr5 inhibitors | Yes – blocking host receptor | [144] |
| Hepatitis B virus (HBV) | Human – prevents post liver transplant recurrence | HBIG (hyperimmune immunoglobulin) plus lamivudine (Nabi) | Yes – combination with nucleoside analogue allows reduced dose of HBIG | [145] |
Ps-ivIG is a purified human Ig preparation from pooled plasma with elevated titer to Pseudomonas aeruginosa (pH 4.25, from Cutter Biological).
PA toxin – anthrax protective antigen.
SA-IVIG – is a purified human Ig preparation from plasma donors with elevated titers of anti-ClfA antibody selected from the general donor population (Massachusetts Public Health Biological Laboratories).
Niche markets prevail in antibody therapy for infectious diseases as disease pathogens with large potential markets are generally approached from the vaccine or antibiotic standpoint. The development of immunotherapies for the prevention of severe RSV infections in young children exemplifies the complexity of developing treatments for infectious diseases which tend to have very specific target populations. Originally a poly Ig product Respigam®, was developed by MedImmune and approved for prophylactic use in infants. While efficacious, a humanized IgG1 mAb product called Synagis® (palivizumab) was designed to replace Respigam®. Synagis® remains the only mAb approved for infectious disease in humans [17], the cost effectiveness relative to Respigam® is still under analysis [87]; however, it is a more defined product and can be produced in tissue culture infinitely. Despite focused efforts by MedImmune/AstraZeneca to gain regulatory approval for a third generation fully human mAb treatment, Numax (motavizumab) a higher affinity fully human replacement for Synagis®, has long been underway. However, AstraZeneca recently announced it was discontinuing the development of Numax, despite the $445 million dollar investment, due to unforeseen complications that arose during the clinical testing and regulatory approval process. Thus, niche markets require a very different approach to biotechnology and the investment of time, expenses, and research opportunity costs. Few niche market companies in infectious disease biotechnology could endure this risk and many might have instead simply continued to manufacture more doses of the earlier generation Respigam® or Synagis®, rather than boldly attempt to bring forward a new product with seemingly incremental improvement. In summary, recent developments in broadening both the utility and pathogen range of mAb therapeutics, multi-drug resistance, and the expected future efficiencies in mAb production methods 88, 89, 90, 91, combined with rational use of proven polyclonal Ig methods, enable us to predict that Ab therapy for infectious pathogens will continue to be a leading treatment for emerging, difficult to contain infectious diseases.
References
- 1.Brock T.D., editor. Milestones in Microbiology: 1556 to 1940. ASM Press; 1998. [Google Scholar]
- 2.Saylor C. Monoclonal antibody-based therapies for microbial diseases. Vaccine. 2009;27S:G38–G46. doi: 10.1016/j.vaccine.2009.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marasco W.A., Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat. Biotechnol. 2007;25:1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean A.R., editor. SARS a Case Study in Emerging Infections. Oxford University Press; New York: 2005. [Google Scholar]
- 5.Lowy I. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 6.Beerli R.R., Rader C. Mining human antibody repertoires. MAbs. 2010;2:365–378. doi: 10.4161/mabs.2.4.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry J.D. Rational monoclonal antibody development to emerging pathogens, biothreat agents and foreign animal disease: the antigen scale. Vet. J. 2005;170:193–211. doi: 10.1016/j.tvjl.2004.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burton D.R. A model for neutralization of viruses based on antibody coating of the virion surface. Curr. Top. Microbiol. Immunol. 2001;260:109–143. doi: 10.1007/978-3-662-05783-4_7. [DOI] [PubMed] [Google Scholar]
- 9.Imai M. Fusion of influenza virus with the endosomal membrane is inhibited by monoclonal antibodies to defined epitopes on the hemagglutinin. Virus Res. 1998;53:129–139. doi: 10.1016/s0168-1702(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 10.Webster R.G., Laver W.G. Preparation and properties of antibody directed specifically against the neuraminidase of influenza virus. J. Immunol. 1967;99:49–55. [PubMed] [Google Scholar]
- 11.Moragues M.D. A monoclonal antibody directed against a Candida albicans cell wall mannoprotein exerts three anti-C. albicans activities. Infect. Immun. 2003;71:5273–5279. doi: 10.1128/IAI.71.9.5273-5279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClelland E.E. Ab binding alters gene expression in Cryptococcus neoformans and directly modulates fungal metabolism. J. Clin. Invest. 2010;120:1355–1361. doi: 10.1172/JCI38322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orange J.S. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the primary immunodeficiency committee of the American academy of allergy, asthma and immunology. J. Allergy Clin. Immunol. 2006;117:S524–S553. doi: 10.1016/j.jaci.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 14.ter Meulen J. Monoclonal antibodies for prophylaxis and therapy of infectious diseases. Expert Opin. Emerg. Drugs. 2007;12:525–540. doi: 10.1517/14728214.12.4.525. [DOI] [PubMed] [Google Scholar]
- 15.Plotkin S. Countering anthrax: vaccines and immunoglobulins. Clin. Infect. Dis. 2008;46:129–136. doi: 10.1086/523578. [DOI] [PubMed] [Google Scholar]
- 16.Couderc T. Prophylaxis and therapy for Chikungunya virus infection. J Infect Dis. 2009;200:516–523. doi: 10.1086/600381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu H. Immunoprophylaxis of RSV infection: advancing from RSV-IVIG to palivizumab and motavizumab. Curr. Top. Microbiol. Immunol. 2008;317:103–123. doi: 10.1007/978-3-540-72146-8_4. [DOI] [PubMed] [Google Scholar]
- 18.Casadevall A., Scharff M.D. Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995;21:150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burdick M.D. Clearance of prions during plasma protein manufacture. Transfus Med. Rev. 2006;20:57–62. doi: 10.1016/j.tmrv.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Progress. 2005;21:11–16. doi: 10.1021/bp040016j. [DOI] [PubMed] [Google Scholar]
- 21.Salfeld J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007;25:1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 22.Johnson S. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 2010;176:1215–1224. doi: 10.1086/514115. [DOI] [PubMed] [Google Scholar]
- 23.Beck A. Therapeutic antibodies and related products: choosing the right structure for success. Med. Sci. (Paris) 2009;12:1024–1032. doi: 10.1051/medsci/200925121024. [DOI] [PubMed] [Google Scholar]
- 24.Dechant M. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood. 2002;100:4574–4580. doi: 10.1182/blood-2002-03-0687. [DOI] [PubMed] [Google Scholar]
- 25.Lendvai N. Mechanism for the isotype dependence of antibody-mediated toxicity in Cryptococcus neoformans-infected mice. J. Immunol. 2000;164:4367–4374. doi: 10.4049/jimmunol.164.8.4367. [DOI] [PubMed] [Google Scholar]
- 26.Sarvas H. Effect of Gm allotypes on IgG2 antibody responses and IgG2 concentrations in children and adults. Infect. Immunol. 1990;2:317–332. doi: 10.1093/intimm/2.4.317. [DOI] [PubMed] [Google Scholar]
- 27.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin. Biol. Ther. 2007;7:1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 28.van der Zee J.S. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J. Immunol. 1986;137:3566–3571. [PubMed] [Google Scholar]
- 29.Spiegelberg H.L. Biological role of different antibody classes. Int. Arch. Allergy Appl. Immunol. 1989;90:22–57. doi: 10.1159/000235071. [DOI] [PubMed] [Google Scholar]
- 30.Horn M.P. Preclinical in vitro and in vivo characterization of the fully human monoclonal IgM antibody KBPA101 specific for Pseudomonas aeruginosa serotype IATS-O11. Antimicrob. Agents Chemother. 2010;54:2338–2344. doi: 10.1128/AAC.01142-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tchoudakova A. High level expression of functional human IgMs in human PER.C6® cells. MAbs. 2009;1:1–9. doi: 10.4161/mabs.1.2.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye J. Intranasal delivery of an IgA monoclonal antibody effective against sublethal H5N1 influenza virus infection in mice. Clin. Vaccine Immunol. 2010;17:1363–1370. doi: 10.1128/CVI.00002-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol. 2009;10:889–898. doi: 10.1038/ni.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beenhouwer D.O. Human immunoglobulin G2 (IgG2) and IgG4, but not IgG1 or IgG3 protect mice against Cryptococcus neoformans. Infect. Immun. 2007;75:1424–1435. doi: 10.1128/IAI.01161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westerman F.E. Serum IgG mediates mucosal immunity against rotavirus infection. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7268–7273. doi: 10.1073/pnas.0502437102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazar G.A. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 38.Taylor R.P. Bispecific monoclonal antibody complexes facilitate erythrocyte binding and liver clearance of a prototype particulate pathogen in a monkey model. J. Immunol. 1997;159:4035–4044. [PubMed] [Google Scholar]
- 39.Lindorfer M.A. Targeting of Pseudomonas aeruginosa in the bloodstream with bispecific monoclonal antibodies. J. Immunol. 2001;167:2240–2249. doi: 10.4049/jimmunol.167.4.2240. [DOI] [PubMed] [Google Scholar]
- 40.Dadachova E. Ionizing radiation delivered by specific antibody is therapeutic against a fungal infection. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10942–10947. doi: 10.1073/pnas.1731272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dadachova E. Susceptibility of the human pathogenic fungi Cryptococcus neoformans and Histoplasma capsulatum to gamma-radiation versus radioimmunotherapy with alpha- and beta-emitting radioisotopes. J. Nucl. Med. 2004;45:313–320. [PubMed] [Google Scholar]
- 42.Dadachova E. Feasibility of radioimmunotherapy of experimental pneumococcal infection. Antimicrob. Agents Chemother. 2004;48:1624–1629. doi: 10.1128/AAC.48.5.1624-1629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Little S.J. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 2002;347:385–394. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- 44.Velicer C.M. Antibiotic use in relation to the risk of breast cancer. JAMA. 2004;291:827–835. doi: 10.1001/jama.291.7.827. [DOI] [PubMed] [Google Scholar]
- 45.Kozyrsky A.J. Increased risk of childhood asthma from antibiotic use in early life. Chest. 2007;131:1753–1759. doi: 10.1378/chest.06-3008. [DOI] [PubMed] [Google Scholar]
- 46.Pai J.C. Progress towards recombinant anti-infective antibodies. Recent Pat. Antiinfect. Drug Discov. 2009;4:1–17. doi: 10.2174/157489109787236319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo F. Breaking the one antibody-one target axiom. Proc. Natl. Acad. Sci. U.S.A. 2006;29:11009–11014. doi: 10.1073/pnas.0603822103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popkov M. Instant immunity through chemically programmable vaccination and covalent self-assembly. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4378–4383. doi: 10.1073/pnas.0900147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sepulveda J. Efficient serum clearance of botulinum neurotoxin achieved using a pool of small antitoxin binding agents. Infect. Immun. 2010;78:756–763. doi: 10.1128/IAI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fattom A.S. Safety and immunogenicity of a booster dose of Staphylococcus aureus types 5 and 8 capsular polysaccharide conjugate vaccine (StaphVAX) in a hemodialysis patient. Vaccine. 2004;23:656–663. doi: 10.1016/j.vaccine.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 51.Kaufman D. Veronate (Inhibitex) Curr. Opin. Invest. Drugs. 2006;7:172–179. [PubMed] [Google Scholar]
- 52.Hall A.E. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 2003;71:6864–6870. doi: 10.1128/IAI.71.12.6864-6870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vernachio J. Anti-clumping factor A immunoglobulin reduces the duration of methicillin-resistant Staphylococcus aureus bacteremia in an experimental model of infective endocarditis. Antimicrob. Agents Chemother. 2003;47:3400–3406. doi: 10.1128/AAC.47.11.3400-3406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bubeck Wardenburg J. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 2007;13:1405–1406. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 55.Lorenz U. Functional antibodies targeting IsaA of Staphylococcus aureus augment host immune response and open new perspectives for antibacterial therapy. Antimicrob. Agents Chemother. 2011;55:165–173. doi: 10.1128/AAC.01144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly-Quintos C. Characterization of the opsonic and protective activity against Staphylococcus aureus of fully human monoclonal antibodies specific for the bacterial surface polysaccharide poly-N-acetylglucosamine. Infect. Immun. 2006;74:2742–2750. doi: 10.1128/IAI.74.5.2742-2750.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park J. Infection control by antibody disruption of bacterial quorum sensing signalling. Chem. Biol. 2008;14:1119–1127. doi: 10.1016/j.chembiol.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neely A.N. Passive anti-pcrv treatment protects burned mice against Pseudomonas aeruginosa challenge. Burns. 2005;31:153–158. doi: 10.1016/j.burns.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 59.Imamura Y. Effect of anti-pcrv antibody in a murine chronic airway Pseudomonas aeruginosa infection model. Eur. Respir. J. 2007;29:965–968. doi: 10.1183/09031936.00147406. [DOI] [PubMed] [Google Scholar]
- 60.Rachini A. An anti-beta-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 2007;75:5085–5094. doi: 10.1128/IAI.00278-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torosantucci A. A novel glyco-conjugate vaccine against fungal pathogens. J. Exp. Med. 2005;202:597–606. doi: 10.1084/jem.20050749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Throsby M. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;4:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sui J. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sui J. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog. 2008;4:e1000197. doi: 10.1371/journal.ppat.1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rockx B. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J. Virol. 2008;82:3220–3235. doi: 10.1128/JVI.02377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shrestha B. The development of therapeutic antibodies that neuralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog. 2010;6:e1000823. doi: 10.1371/journal.ppat.1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Radoshitzky S.R. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans M.J. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 69.Glass W.G. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soares M.M. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 2008;14:1357–1362. doi: 10.1038/nm.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKeating J.A. Characterization of the HIV-1 neutralization escape mutants. AIDS. 1989;3:777–784. doi: 10.1097/00002030-198912000-00001. [DOI] [PubMed] [Google Scholar]
- 72.Keck Z.Y. Mutations in hepatitis C virus E2 located outsie the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J. Virol. 2009;83:6149–6160. doi: 10.1128/JVI.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zharikova D. Influenza type A virus escape mutants emerge in vivo in the presence of antibodies to the ectodomain of matrix protein 2. J. Virol. 2005;79:6644–6654. doi: 10.1128/JVI.79.11.6644-6654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Logtenberg T. Antibody cocktails: next-generation biopharmaceuticals with improved potency. Trends Biotechnol. 2007;25:390–394. doi: 10.1016/j.tibtech.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 75.Prabakaran M. Combination therapy using chimeric monoclonal antibodies protects mice from lethal H5N1 infection and prevents formation of escape mutants. PLoS One. 2009;4:e5672. doi: 10.1371/journal.pone.0005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meulen J.T. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3:e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seiler P. In vivo selection of neutralization-resistant virus variants but no evidence of B cell tolerance in lymphocyttic choriomeningitis virus carrier mice expressing a transgenic virus-neutralizing antibody. J. Immunol. 2009;162:4536–4541. [PubMed] [Google Scholar]
- 78.Study of XTL6865 in Patients With Chronic Hepatitis C Virus Infection. March 6, 2007. http://www.clinicaltrials.gov/show/NCT00300807
- 79.Bregenholt S. Recombinant human polyclonal antibodies: a new class of therapeutic antibodies against viral infections. Curr. Pharm. Des. 2007;12:2007–2015. doi: 10.2174/138161206777442173. [DOI] [PubMed] [Google Scholar]
- 80.Bakker A.B.H. Novel human monoclonal antibody combination effectively neutralizing natural rabies virus variants and individual in vitro escape mutants. J. Virol. 2005;79:9062–9068. doi: 10.1128/JVI.79.14.9062-9068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Kruif J. A human monoclonal antibody cocktail as a novel component of rabies postexposure prophylaxis. Annu. Rev. Med. 2007;58:359–368. doi: 10.1146/annurev.med.58.061705.145053. [DOI] [PubMed] [Google Scholar]
- 82.Goudsmit J. Comparison of an anti-rabies human monoclonal antibody combination with human polyclonal anti-rabies immune globulin. J. Infect. Dis. 2006;193:796–801. doi: 10.1086/500470. [DOI] [PubMed] [Google Scholar]
- 83.Burton D.R. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 84.Law M. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008;14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 85.White A.R. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 86.Rhys Jones D. A camelid anti-PrP antibody abrogates PrPSc replication in prion-permissive neuroblastoma cell lines. PLoS One. 2010;5:e9804. doi: 10.1371/journal.pone.0009804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prescott W.A., Jr. Cost effectiveness of respiratory syncytial virus prophylaxis: a critical and systematic review. Pharmacoeconomics. 2010;28:279–293. doi: 10.2165/11531860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 88.Nielsen L.S. Single-batch production of recombinant human polyclonal antibodies. Mol. Biotechnol. 2010;45:257–266. doi: 10.1007/s12033-010-9270-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pogue G.P. Production of pharmaceutical-grade recombinant aprotinin and a monoclonal antibody product using plant-based transient expression systems. Plant Biotechnol. J. 2010;8:638–654. doi: 10.1111/j.1467-7652.2009.00495.x. [DOI] [PubMed] [Google Scholar]
- 90.Kuroiwa Y. Antigen-specific human polyclonal antibodies from hyperimmunized cattle. Nat. Biotechnol. 2009;27:173–181. doi: 10.1038/nbt.1521. [DOI] [PubMed] [Google Scholar]
- 91.Kuczewski M. A single-use purification process for the production of a monoclonal antibody produced in a PER.C6 human cell line. Biotechnol. J. 2011;6:56–65. doi: 10.1002/biot.201000292. [DOI] [PubMed] [Google Scholar]
- 92.Baer M. An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 2009;77:1083–1090. doi: 10.1128/IAI.00815-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frank D.W. Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J. Infect. Dis. 2002;186:64–73. doi: 10.1086/341069. [DOI] [PubMed] [Google Scholar]
- 94.Dharmasena M.N. Characterization of a novel protective monoclonal antibody that recognizes an epitope common to Vibrio cholerae Ogawa and Inaba serotypes. Microbiology. 2009;155:2353–2364. doi: 10.1099/mic.0.025726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xiao X. Human anti-plague monoclonal antibodies protect mice from Yersinia pestis in a bubonic plague model. PLoS One. 2010;5:e13047. doi: 10.1371/journal.pone.0013047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Q. Passive protection of purified yolk immunoglobulin administered against Shiga toxin 1 in mouse models. Can. J. Microbiol. 2010;56:1003–1010. doi: 10.1139/W10-087. [DOI] [PubMed] [Google Scholar]
- 97.Pratt T.S. Oropharyngeal aspiration of ricin as a lung challenge model for evaluation of the therapeutic index of antibodies against ricin A-chain for post-exposure treatment. Exp. Lung Res. 2007;33:459–481. doi: 10.1080/01902140701731805. [DOI] [PubMed] [Google Scholar]
- 98.http://www.cangene.com
- 99.http://www.hgsi.com
- 100.Migone T.S. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009;361:135–144. doi: 10.1056/NEJMoa0810603. [DOI] [PubMed] [Google Scholar]
- 101.Mohamed N. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect. Immun. 2005;73:795–802. doi: 10.1128/IAI.73.2.795-802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.http://www.pharmathene.com
- 103.Vitale L. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 2006;74:5840–5847. doi: 10.1128/IAI.00712-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cotter T.W. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect. Immun. 1995;63:4704–4714. doi: 10.1128/iai.63.12.4704-4714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.http://www.alopexx.com
- 106.Centers for Disease Control and Prevention (CDC) Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin. Morb. Mortal Wkly. Rep. 2010;59:299. [PubMed] [Google Scholar]
- 107.Garcia-Rodriguez C. Neutralizing human monoclonal antibodies binding multiple serotypes of botulinum neurotoxin. Protein Eng. Des. Sel. 2010;13:1–11. doi: 10.1093/protein/gzq111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.http://www.xoma.com
- 109.http://www.progenics.com
- 110.http://www.medimmune.ca
- 111.Morris S.K. A meta-analysis of the effect of antibody therapy for the prevention of severe respiratory syncytial virus infection. BMC Infect. Dis. 2009;9:106. doi: 10.1186/1471-2334-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Centers for Disease Control and Prevention (CDC) A new product (VariZIG) for postexposure prophylaxis of varicella available under an investigational new drug application expanded access protocol. Morb. Mortal Wkly. Rep. 2006;55:209–210. [PubMed] [Google Scholar]
- 113.Wittek R. Vaccinia immune globulin: current policies, preparedness, and product safety and efficacy. Int. J. Infect. Dis. 2006;10:193–201. doi: 10.1016/j.ijid.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 114.http://www.nabi.com
- 115.Beeler J.A., van Wyke Coelingh K. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: effect of mutation upon fusion function. J. Virol. 2009;63:2941–2950. doi: 10.1128/jvi.63.7.2941-2950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rajamanonmani R. On a mouse monoclonal antibody that neutralizes all four dengue virus serotypes. J. Gen. Virol. 2009;90:799–809. doi: 10.1099/vir.0.006874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shrestha B. The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog. 2010;6:e1000823. doi: 10.1371/journal.ppat.1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hessell A.J. Broadly neutralizing monoclonal antibodies 2F5 and 4E10, directed against the human immunodeficiency virus type 1 (HIV-1) gp41 membrane proximal external region (MPER), protect against SHIVBa-L mucosal challenge. J. Virol. 2009;84:1302–1313. doi: 10.1128/JVI.01272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hessell A.J. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hessell A.J. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 2009;15:951–956. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilson J.A. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–1666. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- 122.Shedlock D.J. Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology. 2010;401:228–235. doi: 10.1016/j.virol.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu Z. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J. Infect. Dis. 2008;197:846–853. doi: 10.1086/528801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang R. Therapeutic potential of a fully human monoclonal antibody against influenza A virus M2 protein. Antiviral Res. 2008;80:168–177. doi: 10.1016/j.antiviral.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 125.Beerli R.R. Prophylactic and therapeutic activity of fully human monoclonal antibodies directed against influenza A M2 protein. Virol. J. 2009;6:224. doi: 10.1186/1743-422X-6-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.http://www.talecris.com
- 127.http://www.cslplasma.com
- 128.http://www.bayer.com
- 129.http://www.baxter.com
- 130.Buissa-Filho R. The monoclonal antibody against the major diagnostic antigen of Paracoccidioides brasiliensis mediates immune protection in infected BALB/c mice challenged intratracheally with the fungus. Infect. Immun. 2008;76:3321–3328. doi: 10.1128/IAI.00349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shapiro S. Immunoglobulin G monoclonal antibodies to Cryptococcus neoformans protect mice deficient in complement component C3. Infect. Immun. 2002;70:2598–2604. doi: 10.1128/IAI.70.5.2598-2604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chaturvedi A.K. Monoclonal immunoglobulin G1 directed against Aspergillus fumigatus cell wall glycoprotein protects against experimental murine aspergillosis. Clin. Diagn. Lab. Immunol. 2005;12:1063–1068. doi: 10.1128/CDLI.12.9.1063-1068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cabezas J. Potential of anti-Candida antibodies in immunoprophylaxis. Immunotherapy. 2010;2:171–183. doi: 10.2217/imt.09.76. [DOI] [PubMed] [Google Scholar]
- 134.Gigliotti F. Passive intranasal monoclonal antibody prophylaxis against murine pneumocystis carinii pneumonia. Infect. Immun. 2009;70:1069–1074. doi: 10.1128/IAI.70.3.1069-1074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Van Wye J.E. Pseudomonas hyperimmune globulin passive immunotherapy for pulmonary exacerbations in cystic fibrosis. Pediatr. Pulmonol. 1990;9:7–18. doi: 10.1002/ppul.1950090104. [DOI] [PubMed] [Google Scholar]
- 136.Douzinas E.E. Prevention of infection in multiple trauma patients by high-dose intravenous immunoglobulins. Crit. Care Med. 2000;28:8–15. doi: 10.1097/00003246-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 137.Dalhoff A. Synergy between acylureidopenicillins and immunoglobulin G in experimental animals. Am. J. Med. 1984;76:91–100. doi: 10.1016/0002-9343(84)90326-7. [DOI] [PubMed] [Google Scholar]
- 138.Fomsgaard A., Holder I.A. Effect of a human IgG preparation rich in antibodies to a wide range of lipopolysaccharides on gram-negative bacterial sepsis in burned mice. APMIS. 1993;101:229–234. doi: 10.1111/j.1699-0463.1993.tb00105.x. [DOI] [PubMed] [Google Scholar]
- 139.Dalhoff A., Brunner H. Mode of interaction between immunoglobulin G and mezlocillin against beta-lactamase producing bacteria. Arzneimittelforschung. 1983;33:1666–1671. [PubMed] [Google Scholar]
- 140.Akiyama M. Antibacterial properties of Pseudomonas aeruginosa immunotype 1 lipopolysaccharide-specific monoclonal antibody (MAb) in a murine thigh infection model: combined effects of MAb and ceftazidime. Microbiol. Immunol. 2000;44:629–635. doi: 10.1111/j.1348-0421.2000.tb02543.x. [DOI] [PubMed] [Google Scholar]
- 141.Felts A.G. Locally delivered antibodies combined with systemic antibiotics confer synergistic protection against antibiotic-resistant burn wound infection. J. Trauma. 2000;49:873–878. doi: 10.1097/00005373-200011000-00014. [DOI] [PubMed] [Google Scholar]
- 142.Barekzi N.A. Locally delivered polyclonal antibodies potentiate intravenous antibiotic efficacy against gram-negative infections. Pharm. Res. 2000;19:1801–1807. doi: 10.1023/a:1021481122011. [DOI] [PubMed] [Google Scholar]
- 143.Peterson J.W. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect. Immun. 2006;74:1016–1024. doi: 10.1128/IAI.74.2.1016-1024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murga J.D. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2006;50:3289–3296. doi: 10.1128/AAC.00699-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dickson R.C. Protective antibody levels and dose requirements for IV 5% Nabi Hepatitis B immune globulin combined with lamivudine in liver transplantation for hepatitis B-induced end stage liver disease. Liver Transpl. 2006;12:124–133. doi: 10.1002/lt.20582. [DOI] [PubMed] [Google Scholar]