

Highlights
-
•
Metabolism and safety profile of chitosan and its derivatives on mucosal application.
-
•
Mechanisms of chitosan as potent mucosal adjuvant.
-
•
Different types and forms of chitosan in pre-clinical applications.
-
•
Clinical perspectives.
Keywords: Chitosan, Adjuvant, Mucosal immunity, Marine drug, Clinical application
Abstract
Mucosal vaccination, which is shown to elicit systemic and mucosal immune responses, serves as a non-invasive and convenient alternative to parenteral administration, with stronger capability in combatting diseases at the site of entry. The exploration of potent mucosal adjuvants is emerging as a significant area, based on the continued necessity to amplify the immune responses to a wide array of antigens that are poorly immunogenic at the mucosal sites. As one of the inspirations from the ocean, chitosan-based mucosal adjuvants have been developed with unique advantages, such as, ability of mucosal adhesion, distinct trait of opening the junctions to allow the paracellular transport of antigen, good tolerability and biocompatibility, which guaranteed the great potential in capitalizing on their application in human clinical trials. In this review, the state of art of chitosan and its derivatives as mucosal adjuvants, including thermo-sensitive chitosan system as mucosal adjuvant that were newly developed by author's group, was described, as well as the clinical application perspective. After a brief introduction of mucosal adjuvants, chitosan and its derivatives as robust immune potentiator were discussed in detail and depth, in regard to the metabolism, safety profile, mode of actions and preclinical and clinical applications, which may shed light on the massive clinical application of chitosan as mucosal adjuvant.
1. Introduction
Vaccination has arguably been the most successful and effective medical intervention for preventing infectious diseases, resulting in decreased mortality, extended life expectancy, and improved quality of life [1], [2], [3]. More than 600 million vaccine formulations are now administered by subcutaneous or intramuscular injections, according to WHO records [4].
However, some shortcomings have persisted throughout the development of the vaccine industry, such as needle-stick injuries, the potential for contamination, and the need for highly trained personnel [5]. Furthermore, most pathogens invade the body at mucosal sites. The mucosal immune system, comprising epithelial cells, mucosa-associated lymphoid tissues (MALTs), and lamina propria, contains almost 80% of the immunocytes in the human body [6] and constitutes the first line of defense against infection [7]. Unfortunately, vaccination via injection fails to achieve acceptable immune protection at mucosal sites [8].
Needle-free vaccination offers immense potential in terms of patient compliance and ease of administration, and has attracted great attention recently [3], [9], [10], [11]. The significance of mucosal immunity and its potential for improving global health has inspired research demonstrating that mucosal vaccines could be an attractive alternative to parenteral administration [6], [7].
Nonetheless, the administration of antigens via mucosal routes normally leads to poor immunogenic stimulation. To overcome this, live pathogens have been employed in commercial vaccines such as oral polio [12], [13], oral cholera [14], [15], [16], [17], oral rotavirus [18], [19], [20] and nasal influenza (FluMist™ [21], [22], [23], NASOVAC™ [24]), but these still retain some potential hazards for large-scale application. Hence, rational design of an appropriate adjuvant system with a potent capacity to stimulate immunity is essential [25]. Modern manufacturing technology is obliged to meet the following requirements: (1) stability of the antigen in an acidic, alkaline, or enzymatic environment [26], [27], [28]; (2) retention of the antigen despite constant clearance at mucosal sites [10], [29]; (3) potent induction of both systemic and mucosal immunity [30]; and (4) a balance between enhanced mucosal immunity and the possibility of immune tolerance when using high doses of antigen [10].
As a promising solution, chitosan possesses well-defined properties including biocompatibility, low cost [31], high levels of tolerance, a broad antibody response, and the capacity to stimulate robust Th1 responses [32], [33]. Chitosan has been demonstrated to have distinct mucoadhesivity, which prolongs the antigen retention time at mucosal sites [5], as well as the ability to open the tight junctions between epithelial cells to intensify the transport of antigen to sites of immune induction and effector responses [6], [7], [34], [35]. Moreover, the versatility of chitosan enables a diversity of formulations for chitosan-based delivery systems, such as solutions, powders, micro/nanoparticles, hydrogels, and microneedle patches, to meet the various needs of novel vaccination methods [9], [10], [36], [37]. Recent research has proposed several promising chitosan-based formulations that are highly suitable as mucosal adjuvants with outstanding safety profiles. This article aims to provide an up-to-date review of pre-clinical and clinical applications for these novel adjuvant systems. Although chitosan has been investigated for many years as a basic material for mucosal adjuvants, it is not yet present in any marketed vaccine formulation. The main obstacles may be a lack of understanding of its metabolism and concerns regarding safety issues. Hence, a more comprehensive evaluation of its metabolism, safety profile, modes of action, and clinical applications is also presented in the following sections.
2. Chitosan & chitosan derivatives: Metabolism and safety issues
2.1. Chitosan and its derivatives
Chitosan, a natural cationic polysaccharide, is made up of copolymers of glucosamine (β(1–4)-linked 2-amino-2-deoxy-d-glucose) and N-acetylglucosamine (2-acetamido-2-deoxy-d-glucose), and is derived by the partial deacetylation of chitin obtained from the shells of crustaceans or the mycelium of fungi [8]. A wide range of chitosan polymers is available with diverse molecular weights (MW), degrees of de-acetylation (DD), and modifications, all of which may impact on their solubility, biocompatibility, mucoadhesive behavior, and degradation, and thus influence their immunogenicity [38]. Scherließ et al. successfully showed that three commercialized chitosans with different MW and DD possessed varied levels of adjuvant activity, and the impurities (protein contaminants) in the chitosan were found to play only a minor role. They also pointed out that these diverse adjuvant effects cannot be attributed to a single factor but rather an interplay between molecular weight, degree of deacetylation, particle size, and solubility [39].
To overcome the poor solubility of chitosan at neutral pH [40], various chitosan derivatives have been developed by chemical modification of hydrophilic groups or by simply grafting on water-soluble polymers (e.g., polyethylene glycol) [41], [42], [43]. The derivatization of chitosan not only enhances its water solubility, but also improves the cationic nature of the material, which enhances its interactions with antigens or cell membranes, boosting antigen adsorption/encapsulation and cellular internalization [44], as well as facilitating the residence time at mucosal sites [45]. Among the water-soluble chitosan derivatives, N-trimethyl chitosan (TMC) and N-(2-hydroxy) propyl-3-trimethyl ammonium chitosan chloride (HTCC) are the most commonly used mucosal adjuvants. The chemical structures and main properties are summarized in Table 1 [46].
Table 1.
Currently employed chitosan derivatives as mucosal permeation enhancer.
| Type | Chemical structure | Preparation | Properties | Ref |
|---|---|---|---|---|
| TMC | 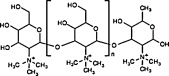 |
Reductive alkylation before quaternization with MeI; direct trimethylation of unprotected chitosan by MeI or DMS with TBDMS & KI | Mucoadhension, permeation enhancer, antimicrobial properties | [47] |
| HTCC | 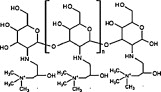 |
Direct alkylation by EPTAC | Enhanced solubility in water, stronger positive charge, mucoadhensive capacity, antibacterial and antifungal property | [48] |
Abbreviations: TMC: N-trimethyl chitosan, HTCC: N-(2-hydroxypropyl)-3-trimethylammonium chitosan chloride, MeI: methyl iodide, DMS: dimethylsulfate, KI: potassium iodide, TBDMS: tert-butyl dimethylsilyl, EPTAC: 2,3-epoxypropyl trimethyl ammonium chloride.
2.2. Degradation and distribution
A comprehensive understanding of the metabolism of a polymer is a prerequisite for its development as a novel adjuvant [49]. As a natural polysaccharide, chitosan is usually regarded as a non-toxic, biologically compatible polymer [50], [51]. In vitro studies have proven that chitosan can be degraded efficiently by lysozyme, resulting in approximately a 100% decrease in the weight of the chitosan hydrogel within 60 days [52]. Three enzymatically active forms of human chitinases have been identified in investigations of the degradation of chitosan and its derivatives [53], namely acidic mammalian chitinase (AMCase), di-N-acetylchitobiase, and chitotriosidase, which have been found in the gastrointestinal tract, liver, and plasma, respectively [54], [55], [56], [57]. Recent research has revealed that the degradation kinetics depend strongly on both the molecular weight and the degree of deacetylation [39], [58]. Typically, a higher enzyme-reaction rate was found for the degradation of high-MW chitosan. Low-DD chitosan was degraded more rapidly, and the remaining 3 to 4 acetyl groups may serve as essential contacts in the active sites of chitosanases [57]. TMC was degraded by lysozyme at a similar rate to native chitosan, but the degradation rate was independent of pH [59].
In a study of in vivo degradation, chitosan underwent partial digestion and uptake by the gastrointestinal tract, and was then deposited in tissues, or circulated in the plasma to eventually be eliminated from the body [53]. Limited data on the degradation and distribution of chitosan have been presented, because of difficulties tracking trace amounts of chitosan in the living body [60]. However, some conclusions can be deduced.
First of all, the in vivo degradation and distribution of chitosan is largely dependent on the molecular weight. FITC-labeled chitosan with a small molecular weight (3.8 kDa) was absorbed by the gastrointestinal tract to give a significant plasma concentration. However, chitosan with a MW higher than 230 kDa (with similar DD) was excreted in the feces without any uptake [61], which indicated that it was not chitosan polymers but chitosan oligosaccharides that could be adsorbed in the gut. In addition, the optimal MW of chitosan for renal clearance was estimated to be 10 kDa. If the MW of the adsorbed chitosan oligomers was larger than that, they would undergo further biodegradation [62].
The failure of intranasal formulations in clinical trials suggests that toxicity may be caused by the uptake of a degradation product by the brain and irritation of the nasal mucosa during degradation. This may lead to serious local symptoms or neurological disorders such as Bell's palsy, which is a facial nerve paralysis caused by the intranasal administration of a heat-labile enterotoxin used as an adjuvant. In the case of chitosan-based mucosal adjuvants, understanding the distribution and degradation of chitosan is vitally important for dispelling safety concerns. Recently, Smith et al. pointed out that high-DD chitosan was degraded much more slowly under the local pH (5.5–6.5) of the nasal cavity and might guarantee that the administered chitosan would be cleared by mucociliary movement and swallowed into the alimentary tract, which might protect the body from severe adverse effects and toxicological responses [5]. Chitosan with a high DD can probably serve as an ideal raw material for developing intranasal vaccines.
Most of the above results have concentrated on the metabolic behavior of chitosan polymers. On the other hand, the metabolism of chitosan powder and smaller particles may be a very different story. Although trimethyl-chitosan oligomers/DNA nanoparticles were shown to be absorbed in the gastric tract [63], there is still insufficient evidence to support the deduction that other chitosan formulations would be degraded in a similar manner.
2.3. Safety of the mucosal administration of chitosan
With regard to the safety profile of chitosan, cytotoxicity is dependent on the concentration, molecular weight, and degree of de-acetylation, with a half-maximal inhibitory concentration (IC50) of 0.2–2.0 mg/mL in most cell models [64], [53], [65]. In addition, different forms of chitosan have shown similar cytotoxic effects [66], [67]. A number of studies have shown the good tolerance and safety performance of chitosan administered via oral routes. In vivo, very few adverse effects have occurred in both mouse and rat models after oral intake of chitosan in doses of 1–15 g/kg/day for 3 months [68]. In addition, no relevant clinical signs were found in healthy human volunteers after taking oral doses of up to 6.75 g/day [69]. Moreover, people with shrimp allergies have shown no allergic reactions after oral administration of shellfish-derived glucosamine [70].
Archimedes and other groups have conducted a large number of preclinical or clinical studies on chitosan administered via the intranasal route. They concluded that chitosan could be well tolerated with no adverse effects or only mild symptoms such as erythema, itchy nose, rhinorrhea, and sore throat [71]. In view of the above facts, chitosan is generally thought to be a biodegradable and biocompatible biomaterial with no or minor toxicity and offers the potential to be a safe pharmaceutical material for clinical vaccine formulations [68], [69].
Consequently, the raw material should be selected carefully with regard to the molecular weight, degree of deacetylation, and purity. Moreover, it is worth noticing that the physiochemical and biological properties of chitosan are also influenced by the original raw material, the manufacturing procedure, and even by the season and weather. Batch-to-batch consistency and stability of the raw material are other challenges when selecting chitosan-based adjuvants for clinical trials. The greatest difference between vaccine adjuvants and drug delivery systems is that vaccines are administered to healthy people, especially populations at high risk of contracting infectious diseases such as infants and the elderly, which means any side effects must be avoided [72] [73]. Nevertheless, the truth about adjuvants is that “the nearer they get to killing you, the more effective they are”. There is an appropriate level of inflammatory response that is undesirable in a drug-delivery system but obligatory for immune stimulation. Future research must manage the balance between toxicity and adjuvanticity, in order to advance the clinical application of chitosan-based mucosal adjuvants.
3. Mode of action
Chitosan-based formulations have been found to orchestrate both cellular and humoral responses, and represent the most promising candidates for adjuvant and delivery systems for mucosal vaccines [37]. Therefore, it is essential to understand the mechanisms of mucosal immunity and the mode of action of chitosan-based adjuvants, to further the rational design of a chitosan-based adjuvant system with a profound ability to stimulate immunity and a distinct safety performance [9]. Numerous studies have demonstrated the potent induction of cellular responses, activation of the innate immune system, and positive effects on mucosal adjuvanticity [39], [74], [75]. In this section, general strategies for developing chitosan into a promising mucosal adjuvant are addressed in detail with reference to recent reports.
3.1. Activation of the innate immune system
The innate immune system not only initiates immediate and consistent responses towards infection, but also directly triggers the adaptive immune system. Recognition by the innate immune system requires the activation of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs, one of the membrane-bound PRRs) and nucleotide-binding domain leucine-rich repeat containing receptors (NLRs, one of the cytoplasmic PRRs), which are specific for conserved ligands associated with cellular damage (danger associated molecular patterns, DAMPs) or invading pathogens (pathogen associated molecular patterns, PAMPs) [76].
Upon stimulation by danger signals, DCs undergo activation/maturation to an active antigen-presenting phenotype [77], with up-regulation of co-stimulatory membrane proteins such as CD80 and CD86, and secretion of proinflammatory cytokines such as TNF (tumor necrosis factor), interleukin (IL)-6, or IL-12. Recent evidence has showed that all three signals are key factors for T-lymphocyte activation [78]. Villiers et al. have demonstrated that chitosan induces the activation of DCs at the level of membrane-receptor recognition. Chitosan promoted the activation of DCs via a TLR4-dependent mechanism, with up-regulation of membrane proteins such as CD80 and CD86 [79]. However, it failed to elicit the robust cytokine production that activates T cells, which indicates that chitosan alone was unable to evoke a T-cell response. Nevertheless, when combined with antigen, chitosan and its derivatives stimulated the expression of major histocompatibility complex (MHC) class I molecules, which profoundly up-regulate the expression of co-stimulator molecules, namely CD80, CD40, and CD86, as well as the secretion of the cytokines IL-6, IL-12, MIP-1, and TNF-α. [80] This led to stronger CD8+ cells proliferation and cytotoxic T lymphocyte (CTL) responses [74].
As one of the best characterized NLR family members, the NLR family pyrin domain containing 3 (NLRP3) was shown to be associated with potent inflammatory responses, which were activated by a multi-protein complex composed of NLRP3, the adaptor apoptosis-associated speck-like protein (ASC), and the protease caspase-1 (Fig. 1 ). Currently, chitosan particles are proven triggers of a potent adaptive inflammatory response through the NLRP3 pathway, which facilitates the recruitment of neutrophils [81], [82], [83] and maturation of APCs, and creates a pro-inflammatory environment characterized by the secretion of cytokines such as IL-1β and IL-18 [84], [85].
Fig. 1.
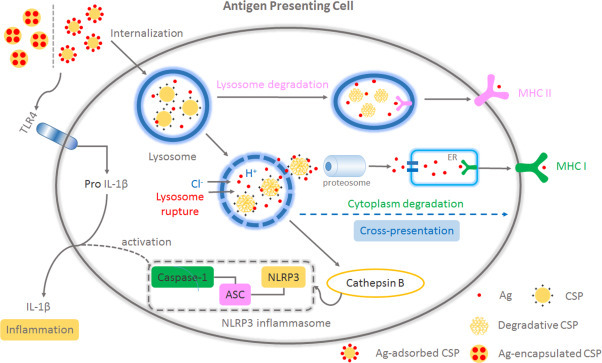
Model of CSP-induced NLRP3 inflammasome activation and cross-presentation: Ag together with CSP (via adsorption or encapsulation) was internalized by APCs. The protonation of the amino groups (“proton sponge effect”) leads to an extensive inflow of ions and water into the lysosome, which caused the osmotic swelling and deconstruction of the lysosome. The entrapped components (CSP and Ag) were released and finally presented onto MHC I, by cytoplasm degradation (with proteosome and ER involved); After the rupture, lysosome enzymes, cathepsin B, was also leaked into the cytoplasm, which induced the assembly and activation of NLRP3 complex. The capacity of TLR4 stimulation of CSP also played an important role in the intracellular synthesis of pro IL-1β and triggered the secretion of IL-1β and inflammatory responses, together with NLRP3 activation. (Ag: antigen; CSP: chitosan particle.)
As shown in Fig. 1, the disruption of lysosomes is thought to trigger the chitosan-particle-induced NLRP3 response. After rupture, the leakage of lysosomal enzymes such as cathepsin B, which can prompt the assembly and activation of NLRP3 complex, stimulates the secretion of IL-1β and subsequent inflammatory responses [86], [87], [88]. Recent research has suggested that the activation of the inflammasome by chitosan particles (CSPs) is dependent upon the physiochemical properties of CSPs, such as particle size, charge [88], [89], and surface texture [90]. Neumann et al. found that only positively charged chitosan nanoparticles and alum induced significant IL-1β secretion after pre-incubation with LPS to activate TLR4 in vitro [91].
3.2. Cross-presentation
Cellular immunity, which is caused by CD 8+ T-cell activation, is vital for the success of prophylactic and therapeutic vaccines [92]. Typically, exogenous antigens are endocytosed by professional antigen presenting cells (APCs), and are then degraded into short peptides within their endosomes. These degraded peptides are loaded onto MHC class II molecules to present antigen to CD4+ helper T cells, which leads to humoral immunity. Only some of the intracellular antigens would escape from the endosome and be presented to CD8+ T lymphocytes through the MHC class I pathway, to stimulate cellular immune responses. Traditional adjuvant systems using Alum, MF59, and AS03, failed to elicit potent cellular immunity [93]. On the other hand, chitosan particles possess a robust capacity to elicit cross-presentation of exogenous antigens and induce potent cellular immune responses.
Cross-presentation can be induced by several means such as TLR activation [94], [95], mannose receptor-mediated uptake [96], and up-regulation of the expression of heat shock proteins [97], [98]. In the case of CSPs, tuning the cellular trafficking of antigen is triggered by the “lysosome escape” pathway, which indicates that the internalized chitosan particles can disrupt the lysosome [99], [100] and escape into the cytoplasm through a “proton sponge effect” [101]. As shown in Fig. 1, the protonation of amino groups present on the chitosan chains leads to an extensive inflow of ions and water into the lysosome. When the “proton sponge effect” becomes strong enough, the entrapped components (CSP and antigen) are released by rupture of the lysosomes [102], [103]. The escaped exogenous antigen can be cross-presented through the MHC class I pathway, like endogenous antigens, to prime CD8+ T cells and evoke robust CTL responses [100], [104].
3.3. Mucosal adjuvanticity
Briefly, mucosal surfaces are constructed of a single layer of epithelial cells joined by tight junctions [11], which acts as a natural barrier to protect the body from the outside world. When pathogens frequently breach the epithelial barrier, epithelial tissues are activated and serve as sensors for detecting danger signals through pattern-recognition receptors such as TLRs [105], [106]. They respond by releasing chemokines and cytokines to recruit MALT-resident immunocytes and trigger innate and adaptive immune responses [6]. Recently, chitosan-based adjuvant systems have been proven to have mucoadhesive qualities and the unique ability to open the tight junctions between epithelial cells at natural portals, which stimulates a robust mucosal immune response by facilitating the transport of antigens via an alternative yet effective route for antigen delivery [5], [30], [39], [74], [75].
Numerous studies have confirmed the mucoadhesive properties of chitosan that prolong the residence time of formulations at mucosal sites, including the nasal mucosa [8], [10], [29], [107], gastrointestinal surfaces [108], and rectal and genital tracts [109]. Our group employed an in vivo imaging system to evaluate the ability of a thermo-sensitive HTCC hydrogel to prolong the residence time of Cy5 labeled H5N1 antigen in the nasal cavity. As shown in Fig. 2a, no fluorescent signal was detected in the PBS/H5N1 group 1 h post administration. However, strong fluorescence intensity could be observed in the HTCC/H5N1 group. More than 54% of the administered antigens persisted in the nasal cavity after 2 h (Fig. 2b), which shows the ability of chitosan to offer a sustained antigen-mucosal interaction without being cleared away [45].
Fig. 2.
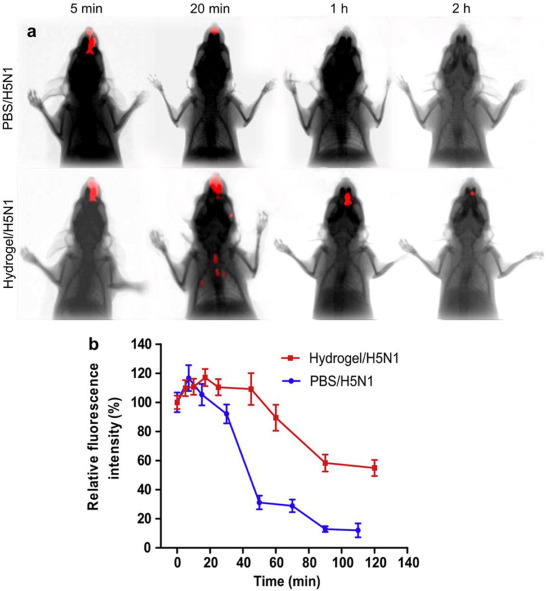
(a) Residence time of Cy5 labeled H5N1 antigen in nasal cavity was determined by In vivo Imaging System. (b) Relative fluorescence intensity in the nasal cavity over time. (Relative fluorescence intensity was calculated from absolute fluorescence of the initial fluorescence in the nasal cavity) [44].
The distinguishing trait of chitosan to mimic pathogen entry by opening tight junctions between epithelial cells has also been investigated in recent research [73], [110]. Artursson's group suggested that the ability to open junctions was due to the positively charged amino group on the C-2 position of chitosan, which could interact with the negatively charged mucosal surface [111]. In another study, they compared differences in the transport of [14C]-mannitol, among three chitosan-derivative solutions, including chitosan hydrochloride, chitosan glutamate, and TMC, all with a similar degree of de-acetylation. In the intestinal epithelial cell line Caco-2, the transport of [14C]-mannitol was increased 34-fold by chitosan hydrochloride, 25-fold by chitosan glutamate, and 11-fold by TMC at 0.25% w/v concentrations in vitro, compared with the administration of mannitol alone [112]. By determining the change in transepithelial electrical resistance to indicate the tightness of the junctions and using confocal laser scanning microscopy in vitro, they further demonstrated the interactions between chitosan and zona occludens (ZO)-1 protein, which is regarded as the target of chitosan and its derivatives [113], [114], [115]. Hsu et al. elucidated the signaling mechanism underlying the opening of tight junctions by chitosan in Caco-2 cell monolayers. As shown in Fig. 3 , the cascade of tight-junction opening is accompanied by the clustering of integrins (αvβ3) along the cell periphery, which led to the phosphorylation of focal adhesion kinase (FAK, also known as protein tyrosine kinase 2, PTK2) and Src (a family of non-specific tyrosine kinases). This also resulted in the regulation of tight-junction permeability via the redistribution of CLDN4 (a tight-junction protein) from the cell membrane to the cytosol and its eventual degradation in lysosomes [116], [117].
Fig. 3.
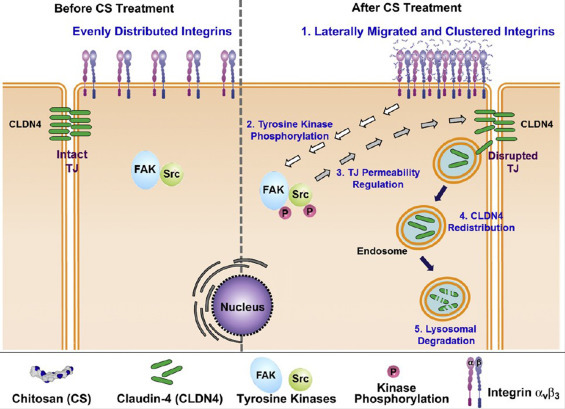
The signaling mechanism of CS-mediated TJ opening [83].
4. Chitosan employed as immune potentiator in pre-clinical research
In pre-clinical and clinical research, chitosan and its derivatives have demonstrated low-toxicity, mucoadhesive capabilities, the ability to enhance the internalization of antigens by APCs, and immune-stimulatory properties, which makes them promising candidates for effective mucosal adjuvants [5]. The current evidence supporting chitosan as a potent mucosal adjuvant is summarized and recapitulated in Table 2 , in a multitudinous array of vaccine formulations, different chitosan types, dosage forms, model animals, and routes of administration. In the following section, applications for adjuvant systems based on chitosan and its derivatives are reviewed with regard to their routes of administration.
Table 2.
Pre-clinical application of chitosan-based adjuvant systems.
| Antigen | Chitosan type | Dosage form | Other adjuvant | Model animal | Routes | Results | References |
|---|---|---|---|---|---|---|---|
| Chitosan particles | |||||||
| Tetanus toxoid (TT) | CS (23–38 kDa, DD = 87%) | NP (150–450 nm); Encapsulation | – | BALB/c | i.n | Increasing and long-lasting humoral immune response (IgG & IgA levels) | [118] |
| Diphtheria toxoid (DT) | CS & CS-PEG (100 kDa, DD = 86%) | PEG-modified NP (100–500 nm), encapsulation | – | BALB/c | i.n | Profound IgA & IgG level; But CS-PEG is more significant in adjuvant performance, which may due to the controlled releasing of antigen | [119] |
| H3N2 subunit antigen | TMC (40–177 kDa, DD = 26%) | NP (350 nm); encapsulation | – | C57Bl/6 | i.n | Higher IgG & IgA levels than i.m administration of antigen alone; balanced cellular and humoral immunity | [120] |
| Dermonecrotoxin (BBD) | MCM (mannosylated chitosan microspheres) | MP (3–5 μm); adsorption | – | BALB/c | i.n | In vitro, target on macrophages, in vivo significantly higher IgA responses, IL-6 and TNF-α concentration | [121] |
| DT | TMC (177 kDa, DD = 93%) |
MP (1–6 μm), encapsulation | – | Guinea pigs | pulmonary | High IgA and IgG titer compared with Alum s.c group | [122] |
| pDNA(SARS-CoV) | CHS | Biotin and bfFp modified NP (200 nm); encapsulation | – | BALB/c | i.n/i.m | Increasing IgG1, IgG2a, IgG2b titers and IFN-γlevels in both i.n and i.m groups. But i.n group is lower than i.m group | [123] |
| OVA | TMC (120 kDa, DD = 92%) CS (120 kDa, DD = 92%) |
NP (300 nm); encapsulation | – | BALB/c | Intraduodenal | Boosting M-cell dependent uptake of antigen; activation on DCs; High IgG response | [124] |
| Chitosan hybrid particles | |||||||
| HBsAg | CHS (Seacure 210, DD = 83%); PLGA (40 kDa–75 kDa) | CS modified PLGA MP (1–10 μm); encapsulation | – | BALB/c | i.n | Achieve the comparable IgG levels with alum groups; potent secretion of IgA and cytokines and IFN-γ in spleen homogenates | [125] |
| HBsAg | CS (DD = 95%) | NP (300-600 nm); encapsulation | CpG | BALB/c | i.n | Robust IgG, sIgA and IFN-γ production, ThI response; but The generation of Th1-biased antigen-specific systemic antibodies was observed only when HbsAg loaded NP were applied together with Class B CpG ODN | [126] |
| Recombinant Influenza A virus H1N1 | CS (190 kDa, DD > 75%); PCL (14.8 kDa) | NP (125 nm); encapsulation | – | BALB/c | i.n/i.m | Humoral (both systemic and mucosal) and cellular immune responses upon i.n administration; comparable results between i.n and i.m administration | [127] |
| Urease/BSA | TMC, CS (200 kDa, DD = 95%) alginate | NP (180–330 nm), encapsulation | – | Kunming mice | oral | Good performance on transepithelial electrical resistance (TEER) value; High IgG titer and IgA titer | [128] |
| OVA | TMC-SH (43 kDa, DD = 83%, DQ = 30%, 50%); HA-SH (15 kDa) | NP (300 nm); mixture | – | BALB/c | i.n/i.d | High stability; thiol sensitivity; superior immunogenicity compared to non-stabilized particles (IgG, IgG1, IgG2, IgA) | [129] |
| rHBsAg | CS (hydrochloride salt, 125 kDa, DD = 86%); Miglyol 813; soybean lecithin | Core-corona NP (200 nm); adsorption | imiquimod (TLR 7 agonist) | BALB/c | i.n | Ability to enter macrophages evoked more secretion of pro-inflammatory cytokines (IL-1α, IL-10, IL-6 and TNF-α); increasing IgG levels over time, elicited both cellular and humoral immunity | [130] |
| HBsAg | CS (110–150 kDa); GC (250 kDa); PLGA (40–50 kDa, 50:50); | NP (175 nm); encapsulation | – | BALB/c | i.n | GC-PLGA exhibited the best mucus adhesion ability and induced highest IgG & IgA titer as well as IL-4, IL-2, INF-γ | [131] |
| HBsAg | CS (DD = 95%); alginate (18 kDa) | NP (1 μm); adsorption | CPG | BALB/c | oral | High IgG titer with more secretion of IL-2; Activation of CD4+ and CD8+ T cells in the spleen | [132] |
| Other delivery system based on chitosan | |||||||
| Anthrax rPA | CG (Protasan UPG 213) | Dry powder, mixture | MPL-A | NWR | i.n | Significant anti-rPA IgG, IgA titers. distinct increace in survivor proportions to a mean aerosol challenge, nine weeks after immunization | [133] |
| OVA | CS (viscosity < 200 mPa) | Microneedle; encapsulation | – | SD rat | i.d | CS microneedle can remain in the epidermis and dermis (approximately 600 μm) for 14 days to sustained delivery of antigen. Antigen depot effect. Significant higher antibody level lasts for 6 weeks via single vaccination than intramusclar administration | [134] |
| H5N1 split | HTCC | Hydrogel; mixture | – | BALB/c | i.n | Gelate at body temperature on the surface of nasal mucosa in a short time; Enhanced antigen uptake; robust serum and mucosal antibody levels, secretion of cytokines and potent response of antigen-specific memory CD8+ T cells | [45] |
| OVA | CS | Powder encapsulation | – | C57Bl/6 | i.n | The local cellular immune response in the cervical lymph nodes was modest and only for the CSMP and the agarose NP was there a significant difference compared to s.c. injection of ovalbumin in alum | [135] |
| Adenovirus | HTCC | Hydrogel; mixture | – | BALB/c | i.n | Simultaneously induced systemic and humoral immunity; upgrade the secretion of cytokines (IL-12, IFN-γ) | [136] |
Abbreviations: CG: chitosan glutamate, CHS: chitosan hydrochloride salt, HA: hyaluronic acid, MP: microparticle, NP: nanoparticle, SIV: swine influenza virus, SARS-CoV: severe acute respiratory syndrome coronavirus, rPA: recombinant protective antigen, NWR: new zealand rabbit, i.n: intranasal, s.c: subcutaneous, i.m: intramuscular, i.d: intradermal, i.t: intratumoral, bfFp: functionalized with biotin and bifunctional fusion protein, LHRH: luteinizing hormonereleasing hormone, DD: degree of deacetylation, GC: glycol chitosan, TNF-α: tumor necrosis factor-α, IL-2: interleukin-2, IFN-γ: interferon-γ, HTCC: N-[(2-hydroxy-3-trimethylammonium) propyl] chitosan chloride, TNF: tumour necrosis factor.
4.1. Oral delivery
Since the adoption and widespread administration of the sabin oral polio vaccine (OPV) [13], oral delivery has been considered the preferred route of administration for vaccines, especially for children [8]. Recently, chitosan has been extensively studied as a promising candidate for oral administration to promote the internalization of antigen and activate both systemic and mucosal immunity [110].
Several studies have demonstrated that associating the antigens with CSPs can enhance their uptake by M cells and reduce the risk of vaccine degradation. Van der Lubben et al. reported that the size of microparticles (MPs) should be 10 μm for efficient uptake by M cells [34]. They also found that fluorescently labeled chitosan MPs can be taken up by M cells in Peyer's patches at intestinal surfaces [137]. In later research, van der Lubben et al. further showed that antigen was released from MPs after its uptake by M cells [138]. Moreover, a dose-dependent immune reaction was revealed for mice vaccinated with different doses of antigen associated with chitosan MPs [139], which proved the ability of chitosan to promote the internalization of vaccines.
With the aid of an enteric coating, chitosan delivery systems can be protected from stomach acid and released from within the enteric coating after reaching the intestine [140]. Hori et al. developed ovalbumin (OVA)-containing chitosan MPs (Chi-OVA), and coated them with Eudragit L100 (ER-Chi-OVA). Compared to an uncoated Chi-OVA delivery system, significantly higher intestinal mucosal IgA and systemic IgG responses were detected after oral administration of coated Chi-OVA. [141]. In another study, Cho et al. presented a mucoadhesive and pH-sensitive thiolated Eudragit-coated chitosan microparticle, with an enhanced capacity for retaining the structural integrity of the encapsulated protein [142]. Recently, HBsAg-loaded chitosan microparticles were formulated. To overcome the limitations imposed by enzymatic degradation and permeation barriers, bacitracin was introduced as a protease inhibitor, which resulted in better protective levels of immunity after oral administration compared with aprotinin as a protease inhibitor. Moreover, the MPs remained stable at room temperature for up to four months [143].
4.2. Nasal delivery
The nasal route is an excellent alternative to conventional forms of vaccination, and is effective at inducing systemic and mucosal immunity in not only the upper and lower respiratory tracts, but also the gastric and genital tracts via cross-protection [25], [29]. As mentioned above, the chitosan-based adjuvant system can provide effective immune stimulation through potent enhancement of antigen retention, mucosal permeability, cellular uptake, and protection of the antigen [144], as well as a good safety profile [145].
The past few years have witnessed the wide application of chitosan-based adjuvants in pre-clinical research on intranasal vaccines. Vila et al. prepared tetanus toxoid-encapsulated chitosan nanoparticles as an intranasal antigen delivery system, which resulted in an increasing and long-lasting humoral immune response (IgG and IgA levels) [118]. The controlled release of antigen by chitosan particles is another important factor contributing to adjuvanticity. Mokarram et al. has demonstrated the effect of PEG (polyethylene glycol) modifications on the immune-stimulating performance of chitosan nanoparticles, using diphtheria toxoid-loaded chitosan nanoparticles with or without PEG modifications (CS-PEG and CS). In the study, CS-PEG and CS nanoparticles both elicited profoundly higher IgA and IgG levels compared with fluid antigen. In addition, the performance of CS-PEG as an adjuvant was significantly better, which may be due to the controlled releasing of antigen [119]. Scherließ et al. proposed a novel spray-dried and β-irradiated sterilized vaccine with outstanding stability for intranasal administration against influenza A(H1N1)pdm09 in non-human primates, and employed chitosan and a stabilizing and protecting solution (SPS). The chitosan in the formulation not only served as an inducer of immunity, but also stabilized the antigen, especially in combination with SPS. The seroconversion results revealed that the effectiveness of the pilot formulation was comparable to Pandemrix®. Moreover, the hemagglutination-inhibition titer of the group administered sterilized vaccine showed no difference from the group administered unsterilized vaccine [146].
Chitosan derivatives, such as TMC [120], CG [133], and chitosan hydrochloride salt [123], have also been employed in the intranasal delivery of antigen. Our group has developed a thermo-sensitive hydrogel formulated from the chitosan derivative HTCC and α,β-glycerophosphate (α,β-GP) as an intranasal vaccine delivery system for the H5N1 split antigen [107]. This hydrogel was designed to gelate in a short time at body temperature on the surface of the nasal mucosa, which significantly reduced mucociliary clearance and prolonged the antigen residence time in the nasal cavity. Using confocal laser scanning microscopy and scanning electron microscopy, we observed the stained nasal epithelia after intranasal administration of chitosan hydrogel, and have provided significant evidence for the disorganization of ZO-1 protein in nasal epithelial tissue. The thermo-sensitive hydrogel facilitated the transportation of antigen into the sub-mucosa via the paracellular pathway rather than the endocytosis pathway (Fig. 4 ). When employed in the intranasal delivery of H5N1 split antigen [45] and adenovirus antigen [136], the hydrogel system enhanced robust serum and mucosal antibody levels, the secretion of cytokines, and a potent response by antigen-specific memory CD8+ T cells.
Fig. 4.
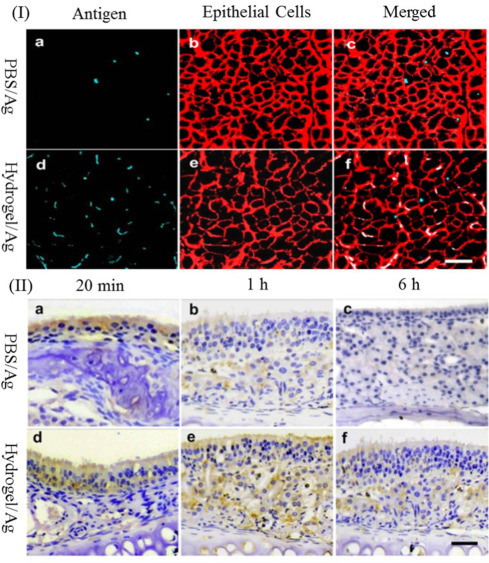
(I) (a–c) CLSM images of nasal epithelia following administration of NHS-Cy5-Ag in PBS; (d–f) NHS-Cy5-Ag loaded hydrogel formulation; (II) (a–f) Antigen distribution examinations by immunohistochemistry method [44].
When chitosan and its derivatives were added to a hybrid system, an increase in the adjuvanticity of other biodegradable polymers was observed. Jaganathan et al. reported that intranasal administration of chitosan hydrochloride-modified PLGA MPs led to significantly higher anti-HbsAg IgG and IgA titers and levels of cytokines (IL-2 and IFN-γ) in spleen homogenates than unmodified PLGA MPs [125]. In another study, Pawar et al. prepared uncoated PLGA nanoparticles, chitosan-modified PLGA NP (CS-PLGA), and Glycol chitosan-modified PLGA NP (GC-PLGA) for encapsulation of the antigen, and administered these via the nasal tract. The results showed that GC-PLGA exhibited the highest level of mucus adhesion and elicited the highest IgG and IgA titers, as well as the secretion of IL-4, IL-2, and INF-γ [131]. Vicente et al. developed a polymer/oil-based core-corona nanocapsule, formulated with an oily core of Miglyol® 812, a neutral oil composed of triglycerides of medium chain fatty acids, and a positively charged shell of chitosan [75]. Through co-delivery of rHBsAg antigen and the TLR7 agonist Imiquimod, the vaccine orchestrated greater secretion of pro-inflammatory cytokines (IL-1α, IL-6, and TNF-α), increasing IgG levels over time and boosting both cellular and humoral immunity [147].
4.3. Other routes of administration
Chitosan-based adjuvant systems have also been employed in mucosal vaccination via other routes. Chen et al. developed a novel transdermal delivery system consisting of embeddable chitosan microneedles with a supporting array composed of a polylactic acid (PLA) polymer, for the sustained delivery of encapsulated antigen, which in this case was OVA. The chitosan microneedle remained in the epidermis and dermis (at a depth of approximately 600 μm) for 14 days as the chitosan degraded (Fig. 5 ). The antibody levels were significantly higher in the microneedle group than the intramuscular-administration group [134].
Fig. 5.
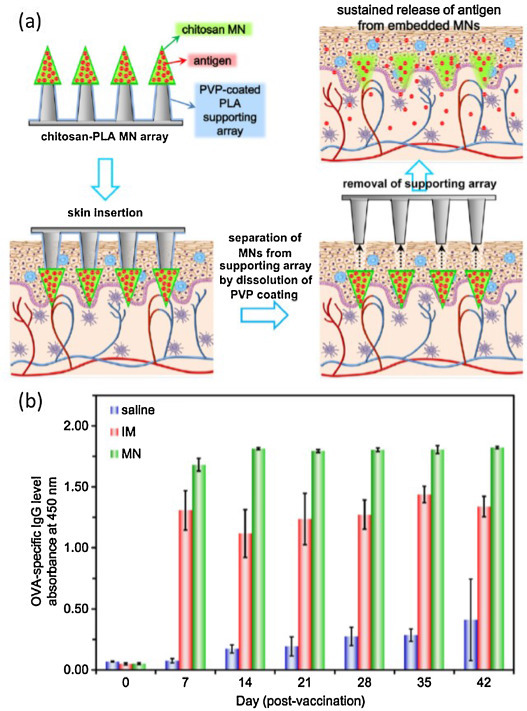
(a) Scheme of the sustain release of the antigen using chitosan-PLA microneedle array. (b) OVA-specific IgG levels of rats after a single dose of antigen: saline (non-immunized group), IM (intramuscular administration) and MN (microneedle administration) [106].
For pulmonary administration, microparticles containing diphtheria toxoid (DT) and chitosan were prepared using a spray-drying technique with a supercritical mixture of carbon dioxide (CO2) and ethanol. After immunization, a strong immune response was observed in the group receiving chitosan-DT, with higher IgM, IgG, IgG1, and IgG2 titers than the group receiving alum-absorbed DT, which supports the potential of chitosan as a potent delivery system for pulmonary vaccines [122].
5. Clinical applications of chitosan-based systems
In the food industry, chitosan has been designated generally-recognized-as-safe (GRAS) in the US, Japan, and Italy [148]. However, chitosan has still not been utilized in any marketed vaccine formulations. Only a few studies have been published on the clinical applications of chitosan as an adjuvant.
Mills et al. have reported the safety and immune-stimulatory capacity of a chitosan-glutamate intranasal delivery system for the diphtheria toxoid antigen CRM197, among healthy volunteers. In Phase A of the study, three groups of subjects were administered intranasal diphtheria vaccine (50 μg of CRM197 with 9.5 mg of chitosan glutamate 213, and 2.5 mg of mannitol), antigen alone (on Day 0 and 28), or alum-adsorbed diphtheria toxoid vaccine via intramuscular injection (single administration on Day 0). Serum IgG and IgA from nasal wash were collected on Days 27 and 42. The results suggested that the chitosan intranasal formulation was well tolerated with only transient mild-to-moderate adverse effects (AEs), such as nasal discharge, blockage, and discomfort. There were no significant differences in the circulating antibody titers of the groups after first administration, but the antibody levels of the group receiving CRM197 and chitosan intranasally were boosted after a second vaccination [149].
Norovirus infection is the most common cause of viral gastroenteritis in humans [150], [151]. Parra et al. developed a norovirus virus-like particle (VLP) vaccine, which orchestrated distinct immune responses in the rabbit after intranasal vaccination [152]. The vaccine (NV-VLP) was formulated as spray-dried powder, which was composed of chitosan (ChiSys™) and norovirus VLP antigen, with MPL (monophosphoryl lipid) as an immune enhancer. ChiSys™ is a drug delivery technology based on chitosan, which is provided by Archimedes Development Limited.
In Phase I clinical studies, two randomized, double-blinded, controlled studies of NV-VLP vaccine have been performed. Healthy volunteers (18–49 years old) were randomized to receive two intranasal administrations of NV-VLP or act as controls (with a interval of 21 days). In study 1, 5 μg, 15 μg, and 50 μg dosages of Norwalk antigen and chitosan alone were evaluated. In study 2, four groups of healthy subjects were administered MPL/chitosan (50 or 100 μg VLP per dose, as a powder), chitosan only, or placebo (puff of air). The symptoms were recorded for 7 days after vaccination, and the safety evaluation was followed up for 180 days. Samples of serum and peripheral blood mononuclear cells were also collected. The results indicated that the most common symptoms were nasal stuffiness and discharge, and sneezing. No vaccine-related serious adverse effects (SAEs) were observed. The most significant immune response was found for the 100-μg dose, with IgG and IgA titers increased by 4.8 and 9.1-fold, respectively. All subjects who received the vaccine dose of 50 or 100 μg produced IgA antibody-secreting cells (ASCs), with surface molecules that are associated with homing to mucosal and peripheral lymphoid tissues (CD19+, CD27+, CD62L−, integrin α4/β7+, CD19+, CD27+, CD62L+, and integrin α4/β7+). The results indicated that intranasal administration of NV-VLP may evoke IgA secretion by intestinal mucosal tissues (the entry site of norovirus) [153].
In other studies, healthy adults (18–50 years of age) were randomized and received primary and booster injections of either vaccine (100 μg VLP per dose) or placebo, administered 3 weeks apart. Three weeks after the second injection, the subjects were challenged with Norovirus. Serology data were collected and the clinical efficacy was assessed by the occurrence of infection, the onset severity, and the duration of the illness caused by Norovirus. AEs of nasal stuffiness, nasal discharge, and sneezing were also reported in this trial, and occurred with similar frequency among vaccine and placebo recipients. A significant IgA response (a 4-fold increase) was detected in 70% of vaccine recipients. Moreover, the occurrence of norovirus-induced gastroenteritis (69% of placebo recipients vs. 37% of vaccine recipients, P = 0.006) and virus infection (82% of placebo recipients vs. 61% of vaccine recipients, P = 0.05) was significantly reduced by NV-VLP vaccination [154], [155].
6. Conclusion and perspectives
Chitosan-based adjuvant systems have been developed with versatility in formulation, excellent tolerance, distinct mucoadhesivity, and a robust capacity to stimulate cellular and humoral immunity in both clinical and pre-clinical studies. Application of these adjuvant systems would significantly improve vaccine development to meet the needs of reduced cost, increased stability, and easier administration. However, only a few chitosan adjuvant systems have been approved for clinical trials (15 on-going clinical experiments), and none of them have yet been included in a licensed vaccine product. Numerous challenges are still hindering the development of chitosan-based mucosal adjuvant systems in humans.
Diverse types of chitosan at different concentrations with various DD and MW are now being studied by separate research groups using a diverse range of antigens (Table 2), which could also lead to controversy and confusion regarding its potential application in human medicine. Future research is needed to investigate the mode of action of chitosan, based on its different aforementioned properties, and establish criteria for advancing chitosan development.
We envisage a booming future for chitosan as an adjuvant in novel mucosal vaccine formulations. Although there are hurdles that still need to be overcome, it is reasonable to expect that it will not be very long before chitosan-based delivery systems for vaccines are released onto the market.
Acknowledgments
We thank the financial support provided by National Key Basic Research Program of China (973 Program): 2013CB531505, National Science and Technology Major Project (No. 2014ZX09102045) and National High Technology Research and Development Program of China (No. 2014AA093604).
Contributor Information
Jie Wu, Email: wujie@ipe.ac.cn.
Guanghui Ma, Email: ghma@ipe.ac.cn.
References
- 1.Roush S.W., Murphy T.V. Vaccine preventable Dis T historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA J Am Med Assoc. 2007;298:2155–2163. doi: 10.1001/jama.298.18.2155. [DOI] [PubMed] [Google Scholar]
- 2.Rappuoli R., Miller H.I., Falkow S. The intangible value of vaccination. Science. 2002;297:937–939. doi: 10.1126/science.1075173. [DOI] [PubMed] [Google Scholar]
- 3.Reed S.G., Bertholet S., Coler R.N., Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30:23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Perrie Y., Mohammed A.R., Kirby D.J., McNeil S.E., Bramwell V.W. Vaccine adjuvant systems: enhancing the efficacy of sub-unit protein antigens. Int J Pharm. 2008;364:272–280. doi: 10.1016/j.ijpharm.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 5.Smith A., Perelman M., Hinchcliffe M. Chitosan: a promising safe and immune-enhancing adjuvant for intranasal vaccines. Hum Vaccines Immunother. 2014;10:797–807. doi: 10.4161/hv.27449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmgren J., Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11:S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 7.Neutra M.R., Kozlowski P.A. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 8.Islam M.A., Firdous J., Choi Y.J., Yun C.H., Cho C.S. Design and application of chitosan microspheres as oral and nasal vaccine carriers: an updated review. Int J Nanomed. 2012;7:6077–6093. doi: 10.2147/IJN.S38330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAllister L., Anderson J., Werth K., Cho I., Copeland K., Le Cam Bouveret N. Needle-free jet injection for administration of influenza vaccine: a randomised non-inferiority trial. Lancet. 2014;384:674–681. doi: 10.1016/S0140-6736(14)60524-9. [DOI] [PubMed] [Google Scholar]
- 10.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nat Rev Immunol. 2012;12:592–605. doi: 10.1038/nri3251. [DOI] [PubMed] [Google Scholar]
- 11.Amorij J.-P., Hinrichs W.L.J., Frijlink H.W., Wilschut J.C., Huckriede A. Needle-free influenza vaccination. Lancet Infect Dis. 2010;10:699–711. doi: 10.1016/S1473-3099(10)70157-2. [DOI] [PubMed] [Google Scholar]
- 12.Pasetti M.F., Simon J.K., Sztein M.B., Levine M.M. Immunology of gut mucosal vaccines. Immunol Rev. 2011;239:125–148. doi: 10.1111/j.1600-065X.2010.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawken J., Troy S.B. Adjuvants and inactivated polio vaccine: a systematic review. Vaccine. 2012;30:6971–6979. doi: 10.1016/j.vaccine.2012.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker R.I. Considerations for development of whole cell bacterial vaccines to prevent diarrheal diseases in children in developing countries. Vaccine. 2005;23:3369–3385. doi: 10.1016/j.vaccine.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 15.Shin S., Desai S.N., Sah B.K., Clemens J.D. Oral vaccines against cholera. Clin Infect Dis. 2011;52:1343–1349. doi: 10.1093/cid/cir141. (An Official Publication of the Infectious Diseases Society of America) [DOI] [PubMed] [Google Scholar]
- 16.Levine M.M., Dougan G. Optimism over vaccines administered via mucosal surfaces. Lancet. 1998;351:1375–1376. doi: 10.1016/S0140-6736(05)79439-3. [DOI] [PubMed] [Google Scholar]
- 17.Czerkinsky C., Holmgren J. Enteric vaccines for the developing world: a challenge for mucosal immunology. Mucosal Immunol. 2009;2:284–287. doi: 10.1038/mi.2009.22. [DOI] [PubMed] [Google Scholar]
- 18.Madhi S.A., Cunliffe N.A., Steele D., Witte D., Kirsten M., Louw C. Effect of human rotavirus vaccine on severe diarrhea in African infants. N Engl J Med. 2010;362:289–298. doi: 10.1056/NEJMoa0904797. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg H.B., Estes M.K. Rotaviruses: from pathogenesis to vaccination. Gastroenterology. 2009;136:1939–1951. doi: 10.1053/j.gastro.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vesikari T., Matson D.O., Dennehy P., Van Damme P., Santosham M., Rodriguez Z. Safety and efficacy of a pentavalent human–bovine (WC3) reassortant rotavirus vaccine. N Engl J Med. 2006;354:23–33. doi: 10.1056/NEJMoa052664. [DOI] [PubMed] [Google Scholar]
- 21.Carter N., Curran M. Live attenuated influenza vaccine (FluMist®; Fluenz™) Drugs. 2011;71:1591–1622. doi: 10.2165/11206860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 22.Dormitzer P.R., Galli G., Castellino F., Golding H., Khurana S., Del Giudice G. Influenza vaccine immunology. Immunol Rev. 2011;239:167–177. doi: 10.1111/j.1600-065X.2010.00974.x. [DOI] [PubMed] [Google Scholar]
- 23.Heinonen S., Silvennoinen H., Lehtinen P., Vainionpää R., Ziegler T., Heikkinen T. Effectiveness of inactivated influenza vaccine in children aged 9 months to 3 years: an observational cohort study. Lancet Infect Dis. 2011;11:23–29. doi: 10.1016/S1473-3099(10)70255-3. [DOI] [PubMed] [Google Scholar]
- 24.Dhere R., Yeolekar L., Kulkarni P., Menon R., Vaidya V., Ganguly M. A pandemic influenza vaccine in India: from strain to sale within 12 months. Vaccine. 2011;29(Suppl 1):A16–A21. doi: 10.1016/j.vaccine.2011.04.119. [DOI] [PubMed] [Google Scholar]
- 25.Mitragotri S. Immunization without needles. Nat Rev Immunol. 2005;5:905–916. doi: 10.1038/nri1728. [DOI] [PubMed] [Google Scholar]
- 26.Hejazi R., Amiji M. Chitosan-based gastrointestinal delivery systems. J Control Release. 2003;89:151–165. doi: 10.1016/s0168-3659(03)00126-3. [DOI] [PubMed] [Google Scholar]
- 27.Merkus F.W.H.M., Verhoef J.C., Marttin E., Romeijn S.G., van der Kuy P.H.M., Hermens W.A.J.J. Cyclodextrins in nasal drug delivery. Adv Drug Delivery Rev. 1999;36:41–57. doi: 10.1016/s0169-409x(98)00054-4. [DOI] [PubMed] [Google Scholar]
- 28.Chaturvedi K., Ganguly K., Nadagouda M.N., Aminabhavi T.M. Polymeric hydrogels for oral insulin delivery. J Controlled Release. 2013;165:129–138. doi: 10.1016/j.jconrel.2012.11.005. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 29.Jabbal-Gill I. Nasal vaccine innovation. J Drug Target. 2010;18:771–786. doi: 10.3109/1061186X.2010.523790. [DOI] [PubMed] [Google Scholar]
- 30.Mutsch M., Zhou W.G., Rhodes P., Bopp M., Chen R.T., Linder T. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N Engl J Med. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez J.A., Rodriguez-Amado I., Montemayor M.I., Fraguas J., Gonzalez Mdel P., Murado M.A. Chondroitin sulfate, hyaluronic acid and chitin/chitosan production using marine waste sources: characteristics, applications and eco-friendly processes: a review. Marine Drugs. 2013;11:747–774. doi: 10.3390/md11030747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu L., Sun Y., Wu Y. Advances in chitosan-based drug delivery vehicles. Nanoscale. 2013;5:3103–3111. doi: 10.1039/c3nr00338h. [DOI] [PubMed] [Google Scholar]
- 33.Muzzarelli R.A. Chitins and chitosans as immunoadjuvants and non-allergenic drug carriers. Marine Drugs. 2010;8:292–312. doi: 10.3390/md8020292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Lubben I.M., Verhoef J.C., Borchard G., Junginger H.E. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci. 2001;14:201–207. doi: 10.1016/s0928-0987(01)00172-5. [DOI] [PubMed] [Google Scholar]
- 35.Arca H.Ç., Günbeyaz M., Şenel S. Chitosan-based systems for the delivery of vaccine antigens. Expert Rev Vaccines. 2009;8:937–953. doi: 10.1586/erv.09.47. [DOI] [PubMed] [Google Scholar]
- 36.Kammona O., Kiparissides C. Recent advances in nanocarrier-based mucosal delivery of biomolecules. J Controlled Release. 2012;161:781–794. doi: 10.1016/j.jconrel.2012.05.040. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 37.Amidi M., Mastrobattista E., Jiskoot W., Hennink W.E. Chitosan-based delivery systems for protein therapeutics and antigens. Adv Drug Delivery Rev. 2010;62:59–82. doi: 10.1016/j.addr.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Aranaz I., Mengíbar M., Harris R., Paños I., Miralles B., Acosta N. Functional characterization of chitin and chitosan. Curr Chem Biol. 2009;3:203–230. [Google Scholar]
- 39.Scherließ R., Buske S., Young K., Weber B., Rades T., Hook S. In vivo evaluation of chitosan as an adjuvant in subcutaneous vaccine formulations. Vaccine. 2013;31:4812–4819. doi: 10.1016/j.vaccine.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 40.Jabbal-Gill I., Watts P., Smith A. Chitosan-based delivery systems for mucosal vaccines. Expert Opin Drug Deliv. 2012;9:1051–1067. doi: 10.1517/17425247.2012.697455. [DOI] [PubMed] [Google Scholar]
- 41.Sashiwa H., Kawasaki N., Nakayama A., Muraki E., Yajima H., Yamamori N. Chemical modification of chitosan Part 15: Synthesis of novel chitosan derivatives by substitution of hydrophilic amine using N-carboxyethylchitosan ethyl ester as an intermediate. Carbohydr Res. 2003;338:557–561. doi: 10.1016/s0008-6215(02)00492-5. [DOI] [PubMed] [Google Scholar]
- 42.Benediktsdottir B.E., Baldursson O., Masson M. Challenges in evaluation of chitosan and trimethylated chitosan (TMC) as mucosal permeation enhancers: from synthesis to in vitro application. J Controlled Release. 2014;173:18–31. [PubMed] [Google Scholar]
- 43.Jimtaisong A., Saewan N. Utilization of carboxymethyl chitosan in cosmetics. Int J Cosmet Sci. 2014;36:12–21. doi: 10.1111/ics.12102. [DOI] [PubMed] [Google Scholar]
- 44.Sayin B., Somavarapu S., Li X.W., Thanou M., Sesardic D., Alpar H.O. Mono-N-carboxymethyl chitosan (MCC) and N-trimethyl chitosan (TMC) nanoparticles for non-invasive vaccine delivery. Int J Pharm. 2008;363:139–148. doi: 10.1016/j.ijpharm.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 45.Wu Y., Wei W., Zhou M., Wang Y., Wu J., Ma G. Thermal-sensitive hydrogel as adjuvant-free vaccine delivery system for H5N1 intranasal immunization. Biomaterials. 2012;33:2351–2360. doi: 10.1016/j.biomaterials.2011.11.068. [DOI] [PubMed] [Google Scholar]
- 46.Kurita K. Chitin and chitosan: functional biopolymers from marine crustaceans. Marine Biotechnol. 2006;8:203–226. doi: 10.1007/s10126-005-0097-5. [DOI] [PubMed] [Google Scholar]
- 47.Benediktsdóttir B.E., Baldursson Ó., Másson M. Challenges in evaluation of chitosan and trimethylated chitosan (TMC) as mucosal permeation enhancers: from synthesis to in vitro application. J Controlled Release. 2014;173:18–31. [PubMed] [Google Scholar]
- 48.Kaminski K., Szczubialka K., Zazakowny K., Lach R., Nowakowska M. Chitosan derivatives as novel potential heparin reversal agents. J Med Chem. 2010;53:4141–4147. doi: 10.1021/jm1001666. [DOI] [PubMed] [Google Scholar]
- 49.Gupta R.K., Relyveld E.H., Lindblad E.B., Bizzini B., Benefraim S., Gupta C.K. Adjuvants—a balance between toxicity and adjuvanticity. Vaccine. 1993;11:293–306. doi: 10.1016/0264-410x(93)90190-9. [DOI] [PubMed] [Google Scholar]
- 50.Hutmacher D.W., Goh J.C.H., Teoh S.H. An introduction to biodegradable materials for tissue engineering applications. Ann Acad Med Singapore. 2001;30:183–191. [PubMed] [Google Scholar]
- 51.Thanou M., Verhoef J.C., Junginger H.E. Oral drug absorption enhancement by chitosan and its derivatives. Adv Drug Delivery Rev. 2001;52:117–126. doi: 10.1016/s0169-409x(01)00231-9. [DOI] [PubMed] [Google Scholar]
- 52.Ma Z., Yang C., Song W., Wang Q., Kjems J., Gao S. Chitosan hydrogel as siRNA vector for prolonged gene silencing. J Nanobiotechnol. 2014;12:23–31. doi: 10.1186/1477-3155-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kean T., Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Delivery Rev. 2010;62:3–11. doi: 10.1016/j.addr.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Hartl D., He C.H., Koller B., Da Silva C.A., Homer R., Lee C.G. Acidic mammalian chitinase is secreted via an ADAM17/epidermal growth factor receptor-dependent pathway and stimulates chemokine production by pulmonary epithelial cells. J Biol Chem. 2008;283:33472–33482. doi: 10.1074/jbc.M805574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu Z., Zheng T., Homer R.J., Kim Y.-K., Chen N.Y., Cohn L. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 56.Malaguarnera L. Chitotriosidase: the yin and yang. Cell Mol Life Sci: CMLS. 2006;63:3018–3029. doi: 10.1007/s00018-006-6269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huet J., Rucktooa P., Clantin B., Azarkan M., Looze Y., Villeret V. X-ray Structure of papaya chitinase reveals the substrate binding mode of glycosyl hydrolase family 19 chitinases. Biochemistry. 2008;47:8283–8291. doi: 10.1021/bi800655u. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H., Neau S.H. In vitro degradation of chitosan by bacterial enzymes from rat cecal and colonic contents. Biomaterials. 2002;23:2761–2766. doi: 10.1016/s0142-9612(02)00011-x. [DOI] [PubMed] [Google Scholar]
- 59.Verheul R.J., Amidi M., van Steenbergen M.J., van Riet E., Jiskoot W., Hennink W.E. Influence of the degree of acetylation on the enzymatic degradation and in vitro biological properties of trimethylated chitosans. Biomaterials. 2009;30:3129–3135. doi: 10.1016/j.biomaterials.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 60.Ebihara K., Schneeman B.O. Interaction of bile-acids, phospholipids, cholesterol and triglyceride with dietary-fibers in the small-intestine of rats. J Nutr. 1989;119:1100–1106. doi: 10.1093/jn/119.8.1100. [DOI] [PubMed] [Google Scholar]
- 61.Chae S.Y., Jang M.K., Nah J.W. Influence of molecular weight on oral absorption of water soluble chitosans. J Controlled Release. 2005;102:383–394. doi: 10.1016/j.jconrel.2004.10.012. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 62.Onishi H., Machida Y. Biodegradation and distribution of water-soluble chitosan in mice. Biomaterials. 1999;20:175–182. doi: 10.1016/s0142-9612(98)00159-8. [DOI] [PubMed] [Google Scholar]
- 63.Zheng F., Shi X.W., Yang G.F., Gong L.L., Yuan H.Y., Cui Y.J. Chitosan nanoparticle as gene therapy vector via gastrointestinal mucosa administration: results of an in vitro and in vivo study. Life Sci. 2007;80:388–396. doi: 10.1016/j.lfs.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 64.Opanasopit P., Aumklad P., Kowapradit J., Ngawhiranpat T., Apirakaramwong A., Rojanarata T. Effect of salt forms and molecular weight of chitosans on in vitro permeability enhancement in intestinal epithelial cells (Caco-2) Pharm Dev Technol. 2007;12:447–455. doi: 10.1080/10837450701555901. [DOI] [PubMed] [Google Scholar]
- 65.Schipper N.M., Vårum K., Artursson P. Chitosans as absorption enhancers for poorly absorbable drugs: influence of molecular weight and degree of acetylation on drug transport across human intestinal epithelial (Caco-2) cells. Pharm Res. 1996;13:1686–1692. doi: 10.1023/a:1016444808000. [DOI] [PubMed] [Google Scholar]
- 66.Prego C., Garcia M., Torres D., Alonso M.J. Transmucosal macromolecular drug delivery. J Controlled Release. 2005;101:151–162. doi: 10.1016/j.jconrel.2004.07.030. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 67.Garcia-Fuentes M., Prego C., Torres D., Alonso M.J. A comparative study of the potential of solid triglyceride nanostructures coated with chitosan or poly(ethylene glycol) as carriers for oral calcitonin delivery. Eur J Pharm Sci. 2005;25:133–143. doi: 10.1016/j.ejps.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 68.Baldrick P. The safety of chitosan as a pharmaceutical excipient. Regul Toxicol Pharmacol: RTP. 2010;56:290–299. doi: 10.1016/j.yrtph.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 69.Garcia-Fuentes M., Alonso M.J. Chitosan-based drug nanocarriers: where do we stand? J Controlled Release. 2012;161:496–504. doi: 10.1016/j.jconrel.2012.03.017. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 70.Villacis J., Rice T.R., Bucci L.R., El-Dahr J.M., Wild L., DeMerell D. Do shrimp-allergic individuals tolerate shrimp-derived glucosamine. Clin Exp Allergy. 2006;36:1457–1461. doi: 10.1111/j.1365-2222.2006.02590.x. [DOI] [PubMed] [Google Scholar]
- 71.Smith A., Perelman M., Hinchcliffe M. Chitosan A promising safe and immune-enhancing adjuvant for intranasal vaccines. Hum Vaccines Immunother. 2014;10:797–807. doi: 10.4161/hv.27449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi T., Fukushima K., Sannan T., Saito N., Takiguchi Y., Sato Y. Evaluation of the effectiveness and safety of chitosan derivatives as adjuvants for intranasal vaccines. Viral Immunol. 2013;26:133–142. doi: 10.1089/vim.2012.0057. [DOI] [PubMed] [Google Scholar]
- 73.Benediktsdottir B.E., Baldursson O., Masson M. Challenges in evaluation of chitosan and trimethylated chitosan (TMC) as mucosal permeation enhancers: from synthesis to in vitro application. J Controlled Release. 2013 (Official Journal of the Controlled Release Society) [PubMed] [Google Scholar]
- 74.Wang Y.-Q., Wu J., Fan Q.-Z., Zhou M., Yue Z.-G., Ma G.-H. Novel vaccine delivery system induces robust humoral and cellular immune responses based on multiple mechanisms. Adv Healthcare Mater. 2013;3:670–681. doi: 10.1002/adhm.201300335. [DOI] [PubMed] [Google Scholar]
- 75.Vicente S., Diaz-Freitas B., Peleteiro M., Sanchez A., Pascual D.W., Gonzalez-Fernandez A. A polymer/oil based nanovaccine as a single-dose immunization approach. PLoS ONE. 2013;8:62407. doi: 10.1371/journal.pone.0062500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dang Y., Li S., Wang W., Wang S., Zou M., Guo Y. The effects of chitosan oligosaccharide on the activation of murine spleen CD11c+ dendritic cells via Toll-like receptor 4. Carbohydr Polym. 2011;83:1075–1081. [Google Scholar]
- 77.Underhill D.M., Ozinsky A. Toll-like receptors: key mediators of microbe detection. Curr Opin Immunol. 2002;14:103–110. doi: 10.1016/s0952-7915(01)00304-1. [DOI] [PubMed] [Google Scholar]
- 78.Curtsinger J.M., Johnson C.M., Mescher M.F. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 79.Villiers C., Chevallet M., Diemer H., Couderc R., Freitas H., Van Dorsselaer A. From secretome analysis to immunology: chitosan induces major alterations in the activation of dendritic cells via a TLR4-dependent mechanism. Mol Cell Proteomics. 2009;8:1252–1264. doi: 10.1074/mcp.M800589-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koppolu B., Zaharoff D.A. The effect of antigen encapsulation in chitosan particles on uptake, activation and presentation by antigen presenting cells. Biomaterials. 2013;34:2359–2369. doi: 10.1016/j.biomaterials.2012.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kafka D., Ling E., Feldman G., Benharroch D., Voronov E., Givon-Lavi N. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol. 2008;20:1139–1146. doi: 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 82.Stokes C.A., Ismail S., Dick E.P., Bennett J.A., Johnston S.L., Edwards M.R. Role of interleukin-1 and MyD88-dependent signaling in rhinovirus infection. J Virol. 2011;85:7912–7921. doi: 10.1128/JVI.02649-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mijares L.A., Wangdi T., Sokol C., Homer R., Medzhitov R., Kazmierczak B.I. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J Immunol. 2011;186:7080–7088. doi: 10.4049/jimmunol.1003687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H., Willingham S.B., Ting J.P.Y., Re F. Cutting edge inflammasome activation by Alum and Alum's adjuvant effect are mediated by NLRP3. J Immunol (Baltimore, MD: 1950) 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang H., Ko H.-J., Yang J.-Y., Kim J.-J., Seo S.-U., Park S.G. Interleukin-1 promotes coagulation, which is necessary for protective immunity in the lung against Streptococcus pneumoniae infection. J Infect Dis. 2013;207:50–60. doi: 10.1093/infdis/jis651. [DOI] [PubMed] [Google Scholar]
- 86.Cassel S.L., Joly S., Sutterwala F.S. The NLRP3 inflammasome: a sensor of immune danger signals. Sem Immunol. 2009;21:194–198. doi: 10.1016/j.smim.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharp F.A., Ruane D., Claass B., Creagh E., Harris J., Malyala P. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Nat Acad Sci USA. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bueter C.L., Lee C.K., Rathinam V.A., Healy G.J., Taron C.H., Specht C.A. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J Biol Chem. 2011;286:35447–35455. doi: 10.1074/jbc.M111.274936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li R., Wang X., Ji Z., Sun B., Zhang H., Chang C.H. Surface charge and cellular processing of covalently functionalized multiwall carbon nanotubes determine pulmonary toxicity. ACS Nano. 2013;7:2352–2368. doi: 10.1021/nn305567s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vaine C.A., Patel M.K., Zhu J.T., Lee E., Finberg R.W., Hayward R.C. Tuning innate immune activation by surface texturing of polymer microparticles: the role of shape in inflammasome activation. J Immunol. 2013;190:3525–3532. doi: 10.4049/jimmunol.1200492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neumann S., Burkert K., Kemp R., Rades T., Dunbar P.R., Hook S. Activation of the NLRP3 inflammasome is not a feature of all particulate vaccine adjuvants. Immunol Cell Biol. 2014;92:535–542. doi: 10.1038/icb.2014.21. [DOI] [PubMed] [Google Scholar]
- 92.Reed S.G., Orr M.T., Fox C.B. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 93.Brito L.A., Malyala P., O’Hagan D.T. Vaccine adjuvant formulations: a pharmaceutical perspective. Sem Immunol. 2013;25:130–145. doi: 10.1016/j.smim.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 94.Wilson N.S., Behrens G.M.N., Lundie R.J., Smith C.M., Waithman J., Young L. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 95.Amigorena S., Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. 2010;22:109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 96.Burgdorf S., Kautz A., Böhnert V., Knolle P.A., Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316:612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 97.Imai T., Kato Y., Kajiwara C., Mizukami S., Ishige I., Ichiyanagi T. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Nat Acad Sci USA. 2011;108:16363–16368. doi: 10.1073/pnas.1108372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pockley A.G. Heat shock proteins as regulators of the immune response. Lancet. 2003;362:469–476. doi: 10.1016/S0140-6736(03)14075-5. [DOI] [PubMed] [Google Scholar]
- 99.Irvine D.J., Swartz M.A., Szeto G.L. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hirosue S., Vokali E., Raghavan V.R., Rincon-Restrepo M., Lund A.W., Corthesy-Henrioud P. Steady-state antigen scavenging, cross-presentation, and CD8(+) T cell priming: a new role for lymphatic endothelial cells. J Immunol. 2014;192:5002–5011. doi: 10.4049/jimmunol.1302492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lin C., Engbersen J.F. Effect of chemical functionalities in poly(amido amine)s for non-viral gene transfection. J Controlled Release. 2008;132:267–272. doi: 10.1016/j.jconrel.2008.06.022. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 102.De Koker S., Lambrecht B.N., Willart M.A., van Kooyk Y., Grooten J., Vervaet C. Designing polymeric particles for antigen delivery. Chem Soc Rev. 2011;40:320–339. doi: 10.1039/b914943k. [DOI] [PubMed] [Google Scholar]
- 103.Moreira C., Oliveira H., Pires L.R., Simoes S., Barbosa M.A., Pego A.P. Improving chitosan-mediated gene transfer by the introduction of intracellular buffering moieties into the chitosan backbone. Acta Biomater. 2009;5:2995–3006. doi: 10.1016/j.actbio.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 104.Varkouhi A.K., Scholte M., Storm G., Haisma H.J. Endosomal escape pathways for delivery of biologicals. J Controlled Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 105.Kagnoff M.F., Eckmann L. Epithelial cells as sensors for microbial infection. J Clin Invest. 1997;100:S51–S55. doi: 10.1172/JCI119522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.MacDonald T.T., Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–1925. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 107.Wu J., Wei W., Wang L.Y., Su Z.G., Ma G.H. A thermosensitive hydrogel based on quaternized chitosan and poly(ethylene glycol) for nasal drug delivery system. Biomaterials. 2007;28:2220–2232. doi: 10.1016/j.biomaterials.2006.12.024. [DOI] [PubMed] [Google Scholar]
- 108.Porporatto C., Bianco I.D., Correa S.G. Local and systemic activity of the polysaccharide chitosan at lymphoid tissues after oral administration. J Leukoc Biol. 2005;78:62–69. doi: 10.1189/jlb.0904541. [DOI] [PubMed] [Google Scholar]
- 109.Jøraholmen M.W., Vanić Ž, Tho I., Škalko-Basnet N. Chitosan-coated liposomes for topical vaginal therapy: assuring localized drug effect. Int J Pharm. 2014;472:94–101. doi: 10.1016/j.ijpharm.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 110.Lam J.K., Xu Y., Worsley A., Wong I.C. Oral transmucosal drug delivery for pediatric use. Adv Drug Delivery Rev. 2014;73:50–62. doi: 10.1016/j.addr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 111.Kotze A.F., Lueen H.L., de Leeuw B.J., de Boer B.G., Verhoef J.C. Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2) Pharm Res. 1994;11:1358–1361. doi: 10.1023/a:1018967116988. [DOI] [PubMed] [Google Scholar]
- 112.Kotze A.F., Luessen H.L., de Leeuw B.J., de Boer B.G., Verhoef J.C., Junginger H.E. Comparison of the effect of different chitosan salts and N-trimethyl chitosan chloride on the permeability of intestinal epithelial cells (Caco-2) J Controlled Release. 1998;51:35–46. doi: 10.1016/s0168-3659(97)00154-5. [DOI] [PubMed] [Google Scholar]
- 113.Yee S. In vitro permeability across Caco-2 Cells (Colonic) can predict in vivo (small intestinal) absorption in man—fact or myth. Pharm Res. 1997;14:763–766. doi: 10.1023/a:1012102522787. [DOI] [PubMed] [Google Scholar]
- 114.Lu S., Gough A.W., Bobrowski W.F., Stewart B.H. Transport properties are not altered across Caco-2 cells with heightened TEER despite underlying physiological and ultrastructural changes. J Pharm Sci. 1996;85:270–273. doi: 10.1021/js950269u. [DOI] [PubMed] [Google Scholar]
- 115.Mi F.-L., Wu Y.-Y., Lin Y.-H., Sonaje K., Ho Y.-C., Chen C.-T. Oral delivery of peptide drugs using nanoparticles self-assembled by poly(gamma-glutamic acid) and a chitosan derivative functionalized by trimethylation. Bioconjugate Chem. 2008;19:1248–1255. doi: 10.1021/bc800076n. [DOI] [PubMed] [Google Scholar]
- 116.Yeh T.-H., Hsu L.-W., Tseng M.T., Lee P.-L., Sonjae K., Ho Y.-C. Mechanism and consequence of chitosan-mediated reversible epithelial tight junction opening. Biomaterials. 2011;32:6164–6173. doi: 10.1016/j.biomaterials.2011.03.056. [DOI] [PubMed] [Google Scholar]
- 117.Hsu L.W., Lee P.L., Chen C.T., Mi F.L., Juang J.H., Hwang S.M. Elucidating the signaling mechanism of an epithelial tight-junction opening induced by chitosan. Biomaterials. 2012;33:6254–6263. doi: 10.1016/j.biomaterials.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 118.Vila A., Sánchez A., Janes K., Behrens I., Kissel T., Jato J.L.V. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57:123–131. doi: 10.1016/j.ejpb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 119.Rezaei Mokarram A., Alonso M.J. Preparation and evaluation of chitosan nanoparticles containing Diphtheria toxoid as new carriers for nasal vaccine delivery in mice. Arch Razi Inst. 2007;61:13–25. [Google Scholar]
- 120.Amidi M., Romeijn S.G., Verhoef J.C., Junginger H.E., Bungener L., Huckriede A. N-Trimethyl chitosan (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: biological properties and immunogenicity in a mouse model. Vaccine. 2007;25:144–153. doi: 10.1016/j.vaccine.2006.06.086. [DOI] [PubMed] [Google Scholar]
- 121.Jiang H.L., Kang M.L., Quan J.S., Kang S.G., Akaike T., Yoo H.S. The potential of mannosylated chitosan microspheres to target macrophage mannose receptors in an adjuvant-delivery system for intranasal immunization. Biomaterials. 2008;29:1931–1939. doi: 10.1016/j.biomaterials.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 122.Amidi M., Pellikaan H.C., Hirschberg H., de Boer A.H., Crommelin D.J., Hennink W.E. Diphtheria toxoid-containing microparticulate powder formulations for pulmonary vaccination: preparation, characterization and evaluation in guinea pigs. Vaccine. 2007;25:6818–6829. doi: 10.1016/j.vaccine.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 123.Raghuwanshi D., Mishra V., Das D., Kaur K., Suresh M.R. Dendritic cell targeted chitosan nanoparticles for nasal DNA immunization against SARS CoV nucleocapsid protein. Mol Pharmaceutics. 2012;9:946–956. doi: 10.1021/mp200553x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Slutter B., Plapied L., Fievez V., Sande M.A., des Rieux A., Schneider Y.J. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J Controlled Release. 2009;138:113–121. doi: 10.1016/j.jconrel.2009.05.011. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 125.Jaganathan K.S., Vyas S.P. Strong systemic and mucosal immune responses to surface-modified PLGA microspheres containing recombinant hepatitis B antigen administered intranasally. Vaccine. 2006;24:4201–4211. doi: 10.1016/j.vaccine.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 126.Borges O., Cordeiro-da-Silva A., Tavares J., Santarem N., de Sousa A., Borchard G. Immune response by nasal delivery of hepatitis B surface antigen and codelivery of a CpG ODN in alginate coated chitosan nanoparticles. Eur J Pharm Biopharm. 2008;69:405–416. doi: 10.1016/j.ejpb.2008.01.019. (Official Journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV) [DOI] [PubMed] [Google Scholar]
- 127.Gupta N.K., Tomar P., Sharma V., Dixit V.K. Development and characterization of chitosan coated poly-(varepsilon-caprolactone) nanoparticulate system for effective immunization against influenza. Vaccine. 2011;29:9026–9037. doi: 10.1016/j.vaccine.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 128.Chen F., Zhang Z.R., Yuan F., Qin X., Wang M., Huang Y. In vitro and in vivo study of N-trimethyl chitosan nanoparticles for oral protein delivery. Int J Pharm. 2008;349:226–233. doi: 10.1016/j.ijpharm.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 129.Verheul R.J., Slutter B., Bal S.M., Bouwstra J.A., Jiskoot W., Hennink W.E. Covalently stabilized trimethyl chitosan-hyaluronic acid nanoparticles for nasal and intradermal vaccination. J Controlled Release. 2011;156:46–52. doi: 10.1016/j.jconrel.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 130.Vicente S., Peleteiro M., Diaz-Freitas B., Sanchez A., Gonzalez-Fernandez A., Alonso M.J. Co-delivery of viral proteins and a TLR7 agonist from polysaccharide nanocapsules: a needle-free vaccination strategy. J Controlled Release. 2013;172:773–781. doi: 10.1016/j.jconrel.2013.09.012. (Official Journal of the Controlled Release Society) [DOI] [PubMed] [Google Scholar]
- 131.Pawar D., Mangal S., Goswami R., Jaganathan K.S. Development and characterization of surface modified PLGA nanoparticles for nasal vaccine delivery: effect of mucoadhesive coating on antigen uptake and immune adjuvant activity. Eur J Pharm Biopharm. 2013;85:550–559. doi: 10.1016/j.ejpb.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 132.Borges O., Tavares J., de Sousa A., Borchard G., Junginger H.E., Cordeiro-da-Silva A. Evaluation of the immune response following a short oral vaccination schedule with hepatitis B antigen encapsulated into alginate-coated chitosan nanoparticles. Eur J Pharm Sci. 2007;32:278–290. doi: 10.1016/j.ejps.2007.08.005. (Official Journal of the European Federation for Pharmaceutical Sciences) [DOI] [PubMed] [Google Scholar]
- 133.Klas S.D., Petrie C.R., Warwood S.J., Williams M.S., Olds C.L., Stenz J.P. A single immunization with a dry powder anthrax vaccine protects rabbits against lethal aerosol challenge. Vaccine. 2008;26:5494–5502. doi: 10.1016/j.vaccine.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen M.C., Huang S.F., Lai K.Y., Ling M.H. Fully embeddable chitosan microneedles as a sustained release depot for intradermal vaccination. Biomaterials. 2013;34:3077–3086. doi: 10.1016/j.biomaterials.2012.12.041. [DOI] [PubMed] [Google Scholar]
- 135.Scherließ R., Monckedieck M., Young K., Trows S., Buske S., Hook S. First in vivo evaluation of particulate nasal dry powder vaccine formulations containing ovalbumin in mice. Int J Pharm. 2015;479:408–415. doi: 10.1016/j.ijpharm.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 136.Wu Y., Wu S., Hou L., Wei W., Zhou M., Su Z. Novel thermal-sensitive hydrogel enhances both humoral and cell-mediated immune responses by intranasal vaccine delivery. Eur J Pharm Biopharm. 2012;81:486–497. doi: 10.1016/j.ejpb.2012.03.021. (Official Journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV) [DOI] [PubMed] [Google Scholar]
- 137.van der Lubben I.M., Verhoef J.C., van Aelst A.C., Borchard G., Junginger H.E. Chitosan microparticles for oral vaccination: preparation, characterization and preliminary in vivo uptake studies in murine Peyer's patches. Biomaterials. 2001;22:687–694. doi: 10.1016/s0142-9612(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 138.van der Lubben I.M., van Opdorp F.A.C., Hengeveld M.R., Onderwater J.J.M., Koerten H.K., Verhoef J.C. Transport of chitosan microparticles for mucosal vaccine delivery in a human intestinal M-cell model. J Drug Target. 2002;10:449–456. doi: 10.1080/1061186021000038319. [DOI] [PubMed] [Google Scholar]
- 139.van der Lubben I.M., Kersten G., Fretz M.M., Beuvery C., Coos Verhoef J., Junginger H.E. Chitosan microparticles for mucosal vaccination against diphtheria: oral and nasal efficacy studies in mice. Vaccine. 2003;21:1400–1408. doi: 10.1016/s0264-410x(02)00686-2. [DOI] [PubMed] [Google Scholar]
- 140.Kumar M.N.V.R., Muzzarelli R.A.A., Muzzarelli C., Sashiwa H., Domb A.J. Chitosan chemistry and pharmaceutical perspectives. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 141.Hori M., Onishi H., Machida Y. Evaluation of Eudragit-coated chitosan microparticles as an oral immune delivery system. Int J Pharm. 2005;297:223–234. doi: 10.1016/j.ijpharm.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 142.Quan J.-S., Jiang H.-L., Kim E.-M., Jeong H.-J., Choi Y.-J., Guo D.-D. pH-sensitive and mucoadhesive thiolated Eudragit-coated chitosan microspheres. Int J Pharm. 2008;359:205–210. doi: 10.1016/j.ijpharm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 143.Premaletha K., Licy C.D., Jose S., Saraladevi A., Shirwaikar A., Shirwaikar A. Formulation, characterization and optimization of Hepatitis B surface antigen (HBsAg)-loaded chitosan microspheres for oral delivery. Pharm Dev Technol. 2012;17:251–258. doi: 10.3109/10837450.2010.535824. [DOI] [PubMed] [Google Scholar]
- 144.Lee J.W., Park J.H., Robinson J.R. Bioadhesive-based dosage forms: the next generation. J Pharm Sci. 2000;89:850–866. doi: 10.1002/1520-6017(200007)89:7<850::AID-JPS2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 145.Illum L., Jabbal-Gill I., Hinchcliffe M., Fisher A.N., Davis S.S. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Delivery Rev. 2001;51:81–96. doi: 10.1016/s0169-409x(01)00171-5. [DOI] [PubMed] [Google Scholar]
- 146.Scherließ R., Ajmera A., Dennis M., Carroll M.W., Altrichter J., Silman N.J. Induction of protective immunity against H1N1 influenza A(H1N1)pdm09 with spray-dried and electron-beam sterilised vaccines in non-human primates. Vaccine. 2014;32:2231–2240. doi: 10.1016/j.vaccine.2014.01.077. [DOI] [PubMed] [Google Scholar]
- 147.Vicente S., Goins B.A., Sanchez A., Alonso M.J., Phillips W.T. Biodistribution and lymph node retention of polysaccharide-based immunostimulating nanocapsules. Vaccine. 2014;32:1685–1692. doi: 10.1016/j.vaccine.2014.01.059. [DOI] [PubMed] [Google Scholar]
- 148.Kitozyme S.A. 2011. Chitosan GRAS notice. 〈http://www.accessdata.fda.gov/scripts/fcn/gras_notices/GRN000397.pdf〉 (accessed 11 September 2012) The reference can be obtained from wedsite. [Google Scholar]
- 149.Mills K.H.G., Cosgrove C., McNeela E.A., Sexton A., Giemza R., Jabbal-Gill I. Protective levels of diphtheria-neutralizing antibody induced in healthy volunteers by unilateral priming–boosting intranasal immunization associated with restricted ipsilateral mucosal secretory immunoglobulin A. Infect Immun. 2003;71:726–732. doi: 10.1128/IAI.71.2.726-732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lindesmith L., Moe C., Marionneau S., Ruvoen N., Jiang X., Lindblad L. Human susceptibility and resistance to Norwalk virus infection. Nat Med. 2003;9:548–553. doi: 10.1038/nm860. [DOI] [PubMed] [Google Scholar]
- 151.Allen D.J., Noad R., Samuel D., Gray J.J., Roy P., Iturriza-Gomara M. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol J. 2009;6:150. doi: 10.1186/1743-422X-6-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parra G.I., Bok K., Taylor R., Haynes J.R., Sosnovtsev S.V., Richardson C. Immunogenicity and specificity of norovirus consensus GII.4 virus-like particles in monovalent and bivalent vaccine formulations. Vaccine. 2012;30:3580–3586. doi: 10.1016/j.vaccine.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.El-Kamary S.S., Pasetti M.F., Mendelman P.M., Frey S.E., Bernstein D.I., Treanor J.J. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis. 2010;202:1649–1658. doi: 10.1086/657087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Atmar R.L., Bernstein D.I., Harro C.D., Al-Ibrahim M.S., Chen W.H., Ferreira J. Norovirus vaccine against experimental human Norwalk virus illness. N Engl J Med. 2011;365:2178–2187. doi: 10.1056/NEJMoa1101245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ramirez K., Wahid R., Richardson C., Bargatze R.F., El-Kamary S.S., Sztein M.B. Intranasal vaccination with an adjuvanted Norwalk virus-like particle vaccine elicits antigen-specific B memory responses in human adult volunteers. Clin Immunol. 2012;144:98–108. doi: 10.1016/j.clim.2012.05.006. [DOI] [PubMed] [Google Scholar]