

Abstract
Mammalian cytochrome P450 enzymes often metabolize many pharmaceuticals and other xenobiotics, a feature that is valuable in a biotechnology setting. However, extant P450 enzymes are typically relatively unstable, with T50 values of ∼30–40 °C. Reconstructed ancestral cytochrome P450 enzymes tend to have variable substrate selectivity compared with related extant forms, but they also have higher thermostability and therefore may be excellent tools for commercial biosynthesis of important intermediates, final drug molecules, or drug metabolites. The mammalian ancestor of the cytochrome P450 1B subfamily was herein characterized structurally and functionally, revealing differences from the extant human CYP1B1 in ligand binding, metabolism, and potential molecular contributors to its thermostability. Whereas extant human CYP1B1 has one molecule of α-naphthoflavone in a closed active site, we observed that subtle amino acid substitutions outside the active site in the ancestor CYP1B enzyme yielded an open active site with four ligand copies. A structure of the ancestor with 17β-estradiol revealed only one molecule in the active site, which still had the same open conformation. Detailed comparisons between the extant and ancestor forms revealed increases in electrostatic and aromatic interactions between distinct secondary structure elements in the ancestral forms that may contribute to their thermostability. To the best of our knowledge, this represents the first structural evaluation of a reconstructed ancestral cytochrome P450, revealing key features that appear to contribute to its thermostability.
Keywords: cytochrome P450, synthetic biology, drug metabolism, enzyme structure, protein structure, membrane protein, X-ray crystallography, alpha-naphthoflavone, ancestral reconstruction, CYP1B1, estradiol, human, enzyme stability, thermostability, reconstructed ancestral protein, protein engineering, 16beta-estradiol
Introduction
Cytochrome P450 enzymes are a diverse superfamily of hemoproteins, which carry out a wide variety of important physiological functions in organisms ranging from bacteria to higher animals and plants. In humans, the cytochrome P450 1 (CYP1)2 family consists of three genes (CYP1A1, CYP1A2, and CYP1B1). The corresponding proteins each metabolize a broad but overlapping set of endogenous and environmental chemicals, including steroids, pharmaceutical compounds, and dietary pro-carcinogens. CYP1 gene expression is regulated by the aryl hydrocarbon receptor, a transcriptional regulator with roles in both development and the response to exposure to environmental toxins, such as dioxins and polycyclic aromatic hydrocarbons. Many CYP1 substrates are aryl hydrocarbon receptor agonists, inducing the up-regulation of CYP1 gene expression and thereby their own clearance (reviewed in Refs. 1–3).
CYP1B1, the most recently identified of the human forms, metabolizes a wide range of compounds including xenobiotics, such as 7-ethoxyresorufin, caffeine, and theophylline (4), as well as endobiotics, including testosterone, estradiol, retinol, arachidonic acid, and melatonin (4–9). CYP1B1 is expressed in numerous extrahepatic tissues, including eye, brain, kidney, and breast (reviewed in Ref. 10), and is overexpressed in various cancer cell types (11, 12). This distribution is of interest, given the ability of CYP1B1 to bioactivate environmental carcinogens, including polycyclic aromatic hydrocarbons and estradiol, to DNA-reactive products (13–15). CYP1B1 is essential for the immune response in mice (16, 17), and CYP1B1 polymorphisms have been associated with congenital glaucoma (18, 19), hinting at an important but as yet unidentified physiological role.
The physiological relevance of CYP1B1 is also reflected in its relatively high degree of conservation in vertebrates, compared with the CYP1A subfamily forms. The CYP1A and CYP1B lineages were created by a gene duplication event that took place in early deuterostomes, resulting in the CYP1A/CYP1D and CYP1B/CYP1C clades. The CYP1C and CYP1D subfamilies diverged from the CYP1B and CYP1A lineages, respectively, after a second duplication event in fish (20–22). CYP1C forms are present in amphibia, fish, reptiles, and some avian species (20), whereas CYP1D forms are present in fish, amphibia, reptiles, and some mammals but are pseudogenes in humans (23).
Little is known about the historical function of the CYP1B/CYP1C clade aside from what has been gleaned from observing the activities of modern-day forms. In an effort to explore the evolution of the CYP1 forms, we recently reconstructed a series of CYP1 ancestors from key nodes of the evolutionary tree.3 In line with studies on other ancestral P450s, many of the CYP1 ancestors showed increased thermal tolerance compared with extant CYP1 human enzymes. N98_CYP1B1_Mammal, the predicted ancestor of mammalian CYP1B1 forms, was closest to the extant human mammalian CYP1B1 forms, sharing 81.8% sequence identity to human CYP1B1, yet had a 60T50 (the temperature at which 50% of the protein remains folded after heating for 60 min) of 43 °C, which is 6 ºC higher than that of extant human CYP1B1.3 Whereas P450 enzymes are of interest for use in biotechnology, the extant forms are not stable enough for such applications, but engineered forms with increased thermostability are more likely candidates.
The N98_CYP1B1_Mammal ancestor also showed intriguing differences in the metabolism of representative substrates compared with the extant human CYP1B1 enzyme. For example, human CYP1B1 predominantly metabolizes estradiol to 4-hydroxyestradiol (6). However, whereas both N98_CYP1B1_Mammal and an older common ancestor of all CYP1B forms (N20_CYP1B) were less active than human CYP1B1, they produced both 4- and 2-hydroxyestradiol in similar amounts. However, these ancestors both showed slightly greater activity than human CYP1B1 toward testosterone. Both human CYP1B1 and the two ancestors produced similar, small amounts of 6β-hydroxytestosterone and 16α-hydroxytestosterone plus trace quantities of two unidentified metabolites (8).3 Thus, the reconstructed ancestors vary in both thermostability and metabolic capabilities, both important characteristics to understand for biotechnology applications.3
To initiate an analysis of how CYP1 ancestral sequences have resulted in increased thermostability and varied substrate metabolism, structures of several ancestral forms are being pursued. Crystals of the N98_CYP1B1_Mammal ancestor were successfully produced with either 17β-estradiol or the polycyclic aromatic compound α-naphthoflavone, diffracting to limiting resolutions of 3.1 and 2.95 Å, respectively (Table 1). The structure of this ancestral CYP1B1 with the small, planar α-naphthoflavone permits comparison with structures of all three extant human CYP1B1, CYP1A1, and CYP1A2 enzymes generated with this same ligand (PDB: 3PM0 (26), 4I8V (27), and 2HI4 (28), respectively). This is particularly useful because many cytochrome P450 enzymes from families 1–3 demonstrate substantial reconfiguration of the active-site dimensions with different active-site ligands. To evaluate the ligand-specific effects on N98_CYP1B1_Mammal conformation, a structure with the endogenous steroid estradiol was subsequently solved. Both represent the first reported ancestral cytochrome P450 structures and suggest structural features that may modulate catalytic activity and thermostability.
Table 1.
Data collection and refinement statistics
| N98_CYP1B1_Mammal/α-naphthoflavone (6OYU) | N98_CYP1B1_Mammal/estradiol (6OYV) | |
|---|---|---|
| Data collection | ||
| Space group | I4 | I4 |
| Cell dimensions (Å) | 164.64, 164.64, 96.30 | 162.36, 162.36, 95.69 |
| Molecules/asymmetric unit | 2 | 2 |
| Resolution (Å) | 50.00–2.95 (3.00–2.95)a | 38.27–3.10 (3.31–3.10) |
| Total reflections | 207,156 (27,530) | 154,025 (27,165) |
| Unique reflections | 27,530 (1,364) | 22,678 (4,085) |
| Redundancy | 7.5 (6.6) | 6.8 (6.6) |
| Rpim | 0.064 (0.788) | 0.047 (0.883) |
| 〈I/σ(I)〉 | 53.0 (5.0) | 10.4 (1.2) |
| CC1/2 | 0.998 (0.399) | 0.998 (0.688) |
| Completeness (%) | 100 (100) | 99.9 (97.6) |
| Refinement | ||
| Resolution (Å) | 48.15–2.95 | 38.27–3.10 |
| No. of reflections | 26,007 | 22,189 |
| R/Rfree (%) | 19.9 / 25.8 | 24.3 / 27.7 |
| Ramachandran (%) favored/allowed/outliers | 94.41/5.25/0.34 | 93.66/5.89/0.45 |
| No. of non-H atoms/B factors (Å2) | ||
| Protein | 7,244/54.11 | 7,159/111.56 |
| Ligand | 168/48.84 | 40/107.37 |
| Heme | 86/32.45 | 86/113.04 |
| Glycerol | 36/54.47 | |
| Water | 44/38.13 | |
| RMSD bond (Å) | 0.006 | 0.003 |
| RMSD angle (degrees) | 0.679 | 0.609 |
| Coordinate error (maximum likelihood--based) (Å) | 0.33 | 0.42 |
a Statistics for the highest-resolution shell are shown in parentheses.
Results and discussion
Comparison of overall structures: Ancestral CYP1B1 versus extant human CYP1B1
Structures of the ancestral reconstructed mammalian CYP1B1 enzyme, N98_CYP1B1_Mammal, with α-naphthoflavone as the bound ligand, revealed that this enzyme maintains a canonical cytochrome P450 fold (Fig. 1A). Structural comparison with the extant human CYP1B1 (PDB: 3PM0) reveals that the mammalian ancestor has similar Cα backbone positioning. The Cα root mean square deviation (RMSD) between the two structures with α-naphthoflavone is 1.43 Å (Fig. 1A). Whereas most of the boundaries for helices and β-strand regions are also conserved, significant differences are observed in the conformation of the F helix (Fig. 1A). First, the ancestral CYP1B1 has its F helix extended by eight residues compared with the extant human CYP1B1 (Fig. 1B). Second, whereas the extant human CYP1B1 and all of the other human CYP1 family enzymes have an unusual break near the middle of the F helix (26–28), the ancestral CYP1B1 F helix is intact throughout these residues (Fig. 1B). The net result is that the C terminus of the F helix is much farther away from the heme in the ancestral CYP1B1 structure than in the extant human structure, and this substantially changes the active-site accessibility and ligand binding (see below).
Figure 1.

Comparisons of the mammalian ancestral and human CYP1B1 cytochrome P450 enzymes. A, the global structure of the extant human CYP1B1 (green) compared with that of the ancestral CYP1B1 (blue). Brighter blue in the ancestral CYP1B1 structure indicates the positions of differences in the amino acid sequences. Both were crystallized with α-naphthoflavone (not shown for clarity). The heme is shown as black sticks with the iron as an orange sphere. B, aligned amino acid sequences for both enzymes are annotated with the nonconserved amino acids (in red text) and secondary structure features (helices highlighted in blue; β strands highlighted in green; named above the sequence). Amino acid numbering corresponds to the full-length human CYP1B1 sequence. N98 1B1_M is the N98_CYP1B1_Mammal sequence. Note that the extant human CYP1B1 structure (PDB: 3PM0) was determined from a version of the protein that has a naturally occurring SNP resulting in an A119S substitution.
The residues that form the immediate central active-site cavity are largely conserved, as well as regions important for heme binding and proximal cysteine ligation, such as the L helix. Differences in the primary sequences of the CYP1B1 forms are distributed throughout the tertiary structure and generally occur farther from the heme and active site (Fig. 1A, bright blue). Examination of the amino acid differences in the linear alignment (Fig. 1B, red text) reveals some notable groupings of divergent amino acids compared with the human enzyme. Portions of the H helix and H/I loop have multiple substitutions, but there is considerable disorder in this region (Fig. 1A) for both the human structure (amino acids 308–311 of the H/I loop disordered) and the ancestral CYP1B1 structure (amino acids 300–311 of the H/I loop disordered), which precludes structural comparisons.
Ancestral CYP1B1 structure with α-naphthoflavone
Co-crystallization of the mammalian ancestral CYP1B1 with the flavonoid α-naphthoflavone was initially pursued to facilitate comparison with the human CYP1B1 structure containing the same ligand. Surprising substantial differences were first observed in ligand binding. Whereas the human CYP1B1 enzyme has a single copy of α-naphthoflavone (Fig. 2A) within a relatively small, planar, enclosed active-site cavity, the ancestral CYP1B1 structure displays clear evidence for binding four copies of α-naphthoflavone within a substantially expanded active site (Fig. 2B), largely generated by reconfigurations in helix F discussed below.
Figure 2.

Comparison of CYP1B1/ligand interactions. A, the extant human CYP1B1 crystal structure (PDB: 3PM0, ribbons) with α-naphthoflavone (green sticks) shown from the same perspective as the ancestral CYP1B1 structures in B and C. B, the mammalian ancestral CYP1B1 structure (ribbons) with α-naphthoflavone (blue sticks) clearly indicates four copies of this ligand in the active site. All four are located higher up in the active site, whereas a putative water network (red spheres) is closer to the heme (black sticks). C, the mammalian ancestral CYP1B1 structure (ribbons) with estradiol (purple sticks) has the same overall structure as when α-naphthoflavone is bound, but only one copy of estradiol is present, located near the heme and oriented with C6, C7, and C15 closest to the heme (black sticks) iron (orange sphere). In both B and C, blue mesh represents electron density for the respective ligands and heme using a σA-weighted composite omit map at 1σ. D, absolute spectra of titration of the mammalian ancestral CYP1B1 with α-naphthoflavone. The initial, ligand-free spectrum (main plot, black line) is consistent with water coordination of the heme iron (λmax = 417 nm). As α-naphthoflavone is titrated in, the 417-nm peak decreases in intensity, and a shoulder at 394 nm, consistent with water displacement by the ligand, increases to approximately equal intensity (gray lines) until 1.6 μm (blue line). However, upon further addition of α-naphthoflavone, the 394 peak progressively decreases, reforming only the peak at 417 nm (red lines). E, in the same titration observed in difference mode (inset), a type I spectral shift is observed at lower α-naphthoflavone concentrations (gray lines) until 1.3 μm (blue line), but on further addition of α-naphthoflavone, the spectrum returns to baseline in the region of the trough (red lines) except for background absorbance due to the ligand below ∼375 nm. A fit of the change in absorbance between 0 and 1.3 μm (main panel, red line) reveals cooperative binding with sigmoidal fit generating a Hill coefficient of 1.6. These ligand concentration–dependent spectral changes suggest that α-naphthoflavone is positioned near the heme at low ligand concentrations but relocates to allow water interaction with the heme at high ligand concentrations. The structure in B is most consistent with the high concentrations of ligand employed in crystallization. F, in contrast, titration of estradiol into the mammalian ancestral CYP1B1 reveals a progressive decrease in the 417-nm peak with concomitant increases at 396 nm associated with ligand disruption of water coordination to the heme iron. G, in the same estradiol titration observed in difference mode (inset), a type I spectral shift is observed that can be fit (main panel, red line) with a single-site binding equation. For clarity, not all intermediate spectra are shown for D–G.
In the ancestral CYP1B1 structure, the benzochromenone core of the α-naphthoflavone closest to the heme essentially occupies the same space as it does in the human structure, tilted toward the central I helix. However, the ligand is flipped by 180° such that the phenyl ring is directed toward the heme in the human structure and away from the heme in the ancestral CYP1B1 complex. As a result, in the ancestral CYP1B1 complex α-naphthoflavone, atoms C8 and C9 on the fused ring are closest to the heme iron, but with distances of 7.4 and 7.2 Å, respectively. This orientation is consistent with these atoms as likely sites of metabolism, as opposed to the orientation found in the human CYP1B1 structure. However, this α-naphthoflavone molecule would need to move closer to the heme (to 4–5 Å) for metabolism. In both α-naphthoflavone complexes, this ligand forms a π-π stacking interaction with the conserved F helix Phe-231 (Fig. 2, A and B). This interaction was also key in α-naphthoflavone binding within human CYP1A1 (27) and CYP1A2 (28). In all three human structures, the positioning of this Phe helped to define the narrow and planar active-site architecture. Whereas this interaction is maintained in the ancestral enzyme, repositioning of the end of the F helix 5–8 Å toward the distal side means that Phe-231 in the F helix is located higher up in the active site, consistent with the ligand location further from the heme. An additional interaction is formed with this same copy of α-naphthoflavone in the N98_CYP1B1_Mammal structure. The F helix His-227 is a hydrogen bond donor to the α-naphthoflavone carbonyl oxygen. This interaction is not observed in the human CYP1B1 structure because the F helix break and repositioning of the C-terminal end of the F helix result in direction of this residue away from the active-site cavity.
In the ancestral CYP1B1 complex, the next two copies of α-naphthoflavone pack parallel with each other via π stacking. They are mostly confined by van der Waals and/or hydrophobic interactions between the F helix and sequences immediately following the K helix. Whereas the K helix side is largely unperturbed compared with the human structure, the ancestral CYP1B1 F helix repositioning 5–8 Å toward the distal side opens up the active site to bulk solvent. The fourth α-naphthoflavone ligand is at the mouth of this cleft or channel. It is possible that the captured positioning of the α-naphthoflavone molecules in the ancestral structure represents a substrate entry and/or egress pathway.
The active site of the ancestral CYP1B1 enzyme also had residual electron density between the heme and one of the paired, mid-channel α-naphthoflavone molecules. This density was not consistent with α-naphthoflavone or any of the crystallization components, but rather with a water network (Fig. 2B). Hydrogen bonding appears to occur among these waters, as well as with atoms on the backbone of I helix residues Ala-330 and Asp-333, Leu-509 of the β4-loop, and a carbonyl of one of the α-naphthoflavone molecules. The presence of water within the active site is not unexpected, considering that the N98_CYP1B1_Mammal active site is ultimately open to bulk solvent (Fig. 3A).
Figure 3.
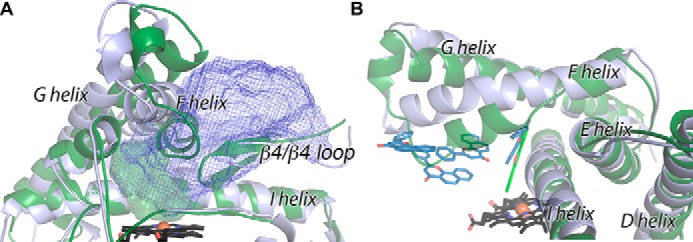
Comparison of ancestral CYP1B1 (blue) and extant human CYP1B1 (green) enzymes highlighting structural differences in helices F through G, the β4 loop, and helix I. Shown are the protein backbone (ribbons) and heme (black sticks). A, the active site for the extant human CYP1B1 (solid green) is much smaller than the active site and channel observed for the ancestral CYP1B1 (blue mesh). B, α-naphthoflavone ligands (colored sticks).
The striking differences in the conformations of the extant and ancestral CYP1B1 enzymes with α-naphthoflavone are the result of significant alterations in positioning of structural elements forming parts of the active-site roof. The most significant structural change is the conformation of the F helix (Fig. 3A). The RMSD for Cα in this region is 3.2 Å, compared with 1.43 Å overall. As described previously, the extant human CYP1B1 structure (and those of CYP1A1 and CYP1A2 with α-naphthoflavone) has a notable break in the F helix (Fig. 3B, green), which is not typically observed in other family 2 P450 enzymes and which contains a conserved Phe that projects into the active site to π-stack with the planar elements of ligands (26–28). In the ancestral CYP1B1 structure, this break is not observed, and the F helix maintains its α-helical character as it passes above the active site (Fig. 3B). This longer, intact helix and a slight change in the tilt angle of the F helix overall mean that the C-terminal portion of the F helix is substantially farther away from the active site (Fig. 3A). Repositioning of the F helix is accommodated by corresponding shifts in the G′- and G-helices, but it is the F helix shift that is the major change opening up the channel from the active site proper to the protein surface (Fig. 3A). In the extant human CYP1B1 structure, the N-terminal half of the F helix interacts with the loop at the tip of the β4 system to close off this channel, but in the ancestral CYP1B1 structure, this region is retracted in the opposite direction from the F helix shift and also contributes to the open channel, resulting in an ∼16-fold increase in volume (Fig. 3A).
Another region of significant structural difference between the extant and ancestral enzyme structures occurs in the middle of the I helix as it crosses the heme and forms one wall of the active site. Whereas the overall backbone and side-chain positions of the I helix are highly conserved, in the ancestral enzyme, a single helical turn (residues 329–333) has a significant bulge such that Ala-330 protrudes into the lower part of the active-site cavity (Fig. 3B). This potentially restricts the ligand α-naphthoflavone from obtaining closer proximity to the heme in the ancestral structure.
Ancestral CYP1B1 titrations with α-naphthoflavone
To further probe α-naphthoflavone binding and interactions near the heme, the effects of ligand binding on the heme coordination were monitored by changes in the Soret heme absorbance. The as-isolated ancestral CYP1B1 protein has a Soret peak absorbance maximum at 417 nm (Fig. 2D, black spectrum), consistent with water coordination to the heme iron, as is typical for isolated P450 enzymes in the oxidized state. Titration of this enzyme with α-naphthoflavone to concentrations up to 1.6 μm produced partial high-spin character with absorbance increases at 394 nm and decreases at 417 nm (Fig. 2D, gray spectra). This response is consistent with α-naphthoflavone binding in the active site and partially displacing coordinated water from the heme. Indeed, difference spectra showing these initial titrations are characteristic of these “type I” spectral changes (Fig. 2E, gray spectra). However, when higher concentrations of α-naphthoflavone were added, the high-spin component at 394 nm began to decrease, eventually returning to a Soret maximum at absorbance at 417 nm, indicating water binding to the heme (Fig. 2D, red spectra). This reversal in binding mode was also reflected in the difference spectra, as the trough returned to baseline at higher concentrations (Fig. 2E, red lines). The absorbance peak is overshadowed by intrinsic ligand absorbance at below ∼375 nm at these high concentrations. With the reversal in binding mode halfway through the titration, the typical plot for the absorbance change versus ligand concentration cannot be fit to a simple two-state model. Even using only the data at the low ligand concentrations corresponding to the initial type I spectral shift reveals a sigmoidal trend (Fig. 2E, main panel), which is best fit using the Hill equation, resulting in a Hill coefficient of 1.6 (95% CI: 0.24–0.30) and S50 of 0.26 μm (95% CI: 1.4–1.9). Human CYP1B1 shows a “reverse type I” spectral change upon binding α-naphthoflavone (29), which is likely the result of ligand binding in such a way as to promote water ligation. Overall, the spectral changes observed for the ancestral CYP1B1 enzyme are consistent with α-naphthoflavone binding near the heme iron at lower concentrations where the ligand:P450 stoichiometry is 1:1 or lower and reverting to water-coordinated heme when additional molecules pack into the protein, as observed in the crystal structure.
Ancestral CYP1B1 metabolism of α-naphthoflavone
Both the human CYP1B1 and N98_CYP1B1_Mammal enzymes convert α-naphthoflavone to three monooxygenated products (Fig. 4, M1, M2, and M3). M1 was the major product in both cases (12 ± 4 and 20 ± 6% relative yield for human CYP1B1 and N98_CYP1B1_Mammal, respectively), followed by M2 (∼1%) and M3 (<0.05%). Comparison with products formed by hCYP1A1 and hCYP1A2 run in the same system suggested that M1 was likely to be the 5,6-epoxide and M2 was the 7-hydroxy metabolite. The identity of M3 could not be clarified (possibly the 7,8-dihydrodiol, 5,6-dihydrodiol, 7,8-epoxide, or 6-hydroxy metabolite). Application of a two-tailed, unpaired Student's t test suggested that there was no significant difference between the ratio of M1/M2 produced by the two P450 enzymes (however, N98_CYP1B1_Mammal had a slightly higher ratio of M2/M3 compared with human CYP1B1 (15:1 versus 13:1, p < 0.01)).
Figure 4.

Metabolism of α-naphthoflavone by ancestral and extant CYP1B1 forms. The percentages of α-naphthoflavone converted to each of three metabolites (M1, M2, and M3) by the extant human CYP1B1 and the ancestral N98_CYP1B1_Mammal are compared with a negative control (bacterial membranes containing human NADPH-cytochrome P450 reductase but no P450). All three metabolites compared in A, B, and C show the production of M2 and M3 at expanded scales. The amount of substrate remaining at the end of the assays was 70 ± 20% for N98_CYP1B1_Mammal, 76 ± 9% for hCYP1B1, and 90 ± 10% for the hCPR control. From comparison with incubations of hCYP1A1 and hCYP1A2, M1 is likely to be the 5,6-epoxide and M2 is the 7-hydroxy metabolite, but the identity of M3 is unclear. Reactions were carried out at 37 °C for 1 h, with 0.5 μm P450 and 25 μm α-naphthoflavone. α-Naphthoflavone and its metabolites were analyzed by LC-MS/MS and quantified by the relative size of MS/MS fragment 103, which is present in the mass spectra of both the substrate and products. Data represent the mean ± S.D. (error bars) of n = 3 reactions. Results that are significantly different from the negative control (hCPR only; unpaired two-tailed Student's t test) are indicated by p values.
Human CYP1B1 has previously been shown to metabolize α-naphthoflavone to more polar, unidentified metabolites (30), but α-naphthoflavone is also a potent inhibitor of human CYP1B1 (4). In the current studies, each of the three products produced by both ancestral and human enzymes had a mass of 289, consistent with the addition of a single oxygen. Whereas these metabolites could not be identified unambiguously by LC-MS alone, and the quantities produced precluded identification by NMR, the fragmentation patterns of M1 and M2 are consistent with an oxide or hydroxyl group added to one of the terminal rings, in positions 5–10 (Fig. S1). The substrate specificity of the human CYP1 enzymes frequently overlaps, so M1, the major metabolite for both human CYP1B1 and N98_CYP1B1_Mammal, may be the 5,6-oxide reported for human CYP1A1 and CYP1A2 (Fig. S1) (31). Similar reasoning suggests that M2 may result from 7-hydroxylation and M3 from 6- or 9-hydroxylation, as observed for rat CYP1A1 (Fig. S2).
Ancestral CYP1B1 structure with 17β-estradiol
One of the key questions that arises from the ancestral CYP1B1 crystal structure with α-naphthoflavone is whether the open conformation of the protein was induced by the stacking of multiple ligand molecules within the active-site channel or whether this is an intrinsic feature of the ancestral CYP1B1 protein. To potentially answer this question, attempts were made to co-crystallize with several other ligands. 17β-Estradiol could be co-crystallized with the ancestral CYP1B1 by employing crystallization conditions similar to those yielding the α-naphthoflavone structure. The global structure of the mammalian ancestral 1B1 bound to 17β-estradiol was highly similar to the structure bound to α-naphthoflavone, with an RMSD of 0.57 Å. Thus, the 17β-estradiol–bound structure also had a large channel from the active site proper extending between the F helix and β4-loop to the protein surface. However, only one copy of 17β-estradiol was present, located directly adjacent to the heme in the active site proper (Fig. 2C). Despite the open conformation, density was not observed for ordered water molecules in the 17β-estradiol structure. From this, we can conclude that the overall open protein conformation observed is not dependent on the specific ligand or number of copies of the ligand. Thus, it is likely that the additional three α-naphthoflavone molecules take advantage of the open channel to bind fortuitously. As always, however, one cannot exclude the possibility that multiple protein conformations occur and that the specific conformation selected depends on its facility to pack into crystals for structure determination.
Within the active site, the positioning of side chains is also largely conserved compared with the same ancestral enzyme bound to α-naphthoflavone. 17β-Estradiol extends across the heme, with its flat, unsubstituted planar α-surface approximately orthogonal to the heme plane and the steroidal A and B rings packing against the against the I helix Gly-329–Ala-330 peptide bond (Fig. 2C). Because 17β-estradiol is positioned closer to the heme than α-naphthoflavone, it does not interact with Phe-231 or other F helix residues, but 17β-estradiol does appear to make two hydrogen bonds with active-site residues. The 17β-estradiol C3 hydroxyl is a hydrogen bond donor to the backbone carbonyl of Leu-509 in the β4 loop, whereas the C17 hydroxyl hydrogen bonds with the side chain of Asp-326 in the I helix. The overall result is that 17β-estradiol carbons 7 (4.2 Å), 15 (4.9 Å), and 6 (5.1 Å) are closest to the heme iron (Fig. 2C).
Ancestral CYP1B1 titrations with 17β-estradiol
Titration of the ancestral CYP1B1 protein with 17β-estradiol resulted in decreases in the Soret band at 417 nm associated with iron-water coordination and concomitant increases in absorbance at 396 nm corresponding to the high-spin state (Fig. 2F). Corresponding titrations done in difference mode indicate that 17β-estradiol binds with a classic type 1 binding mode (Fig. 2G, inset) with a hyperbolic response (Fig. 2G, main panel) best fit to a single-site binding equation with a Kd of 2.75 μm (95% CI: 2.55–2.96). This is an order of magnitude larger than the α-naphthoflavone S50 and with a 2-fold reduction in the maximal absorbance change (ΔAmax = 0.03 for 17β-estradiol versus 0.07 for α-naphthoflavone). Overall, these observations correspond to positioning of 17β-estradiol in the ancestral CYP1B1 structure.
Ancestral CYP1B1 metabolism of 17β-estradiol
It has been observed previously that the ancestral CYP1B1 enzyme is ∼9-fold less active toward 17β-estradiol than human CYP1B13 but also shows differences in the regioselectivity. Extant human CYP1B1 produces 4-hydroxy-17β-estradiol as the major metabolite, with smaller amounts of 2-hydroxyestradiol and trace amounts of a third hydroxy-17β-estradiol metabolite likely to be 15α-hydroxy-17β-estradiol. The ancestral CYP1B1 enzyme produces these same three metabolites but in approximately equimolar amounts.3 The orientation of 17β-estradiol observed in the crystallographic structure is consistent with the formation of 15α-hydroxy-17β-estradiol, but to form the 2- and 4-hydroxymetabolites, 17β-estradiol would need to reorient in the active site so that C2 and C4 were presented to the iron for catalysis at these positions.
The regioselectivity of extant CYP1B1 forms for 4- or 2-hydroxylation is known to vary among species; human, dog, and monkey isoforms preferentially produce 4-hydroxy-17β-estradiol, whereas rodent isoforms produce more 2-hydroxy-17β-estradiol (32). This preference has been attributed to the residue at position 395 (32). Forms containing Val at this position produced more 4-hydroxy-17β-estradiol, whereas those containing Leu produced more 2-hydroxy-17β-estradiol, and exchanging these two residues reversed the regioselectivity. The current results suggest that additional factors influence regioselectivity because whereas the ancestral CYP1B1 has Val at position 395, it does not exhibit a strong preference for either 2- or 4-hydroxylation. In the ancestral CYP1B1 structure with 17β-estradiol, the terminal methyl of Val-395 is 3.6 Å from C16.
Structural changes to the redox partner binding surface
One final notable structural difference between the extant and ancestral CYP1B1 structures occurs on the opposite side of the heme from the active site, on the proximal surface of the protein comprising part of the surface that binds the redox partner NADPH-cytochrome P450 reductase required for catalysis. In particular, the loop between the C and D helices contains three positively charged Arg residues that have been implicated in electrostatic interactions with the negatively charged surface of the reductase. This loop has different conformations in all three structures (Fig. 5A). In the extant human CYP1B1 structure, the C/D loop is positioned closer to the H helix (Fig. 5A, green), whereas in the ancestral CYP1B1 structure with α-naphthoflavone, it is displaced in the opposite direction, toward the L helix (Fig. 5A, blue). In the ancestral CYP1B1 structure with 17β-estradiol, the C/D loop adopts an intermediate conformation (Fig. 5A, purple). In the ancestral CYP1B1 structures, the C/D loop conformations are coincident with similar shifting of the H helix on one side and the loop preceding the J′ helix on the opposite side. As a result, the placement of the positively charged C/D loop guanidine groups is different for all three structures. These differences are reflected in the surface shape and exposed charge for this region (Fig. 5, B–D), in which the ancestral CYP1B1 structures display a less prominent concavity and positive charge. The C/D loop region in P450 enzymes is thought to be a critical binding surface for reductase, so it is possible that changes in the charged features in this region affect reductase binding for the ancestral CYP1B1 enzyme. The ancestral CYP1B1 has similar catalytic activity for α-naphthoflavone as the human CYP1B1 enzyme, suggesting that its C/D-loop conformation is compatible with reductase binding. However, when the ancestral CYP1B1 enzyme is bound to 17β-estradiol, the enzyme has much lower activity toward production of all three metabolites than human CYP1B1. One possible explanation for this is that the conformation of the CD loop observed in this structure prevents effective reductase binding and/or electron transfer.
Figure 5.

Comparison of the proximal surface of extant human CYP1B1 and ancestral CYP1B1. A, significant conformational differences are observed in the region of the C-D loop region of the human CYP1B1 (green) versus the ancestral CYP1B1 crystallized with α-naphthoflavone (blue) versus the ancestral CYP1B1 crystallized with estradiol (purple). As a result, three positively charged Arg residues (sticks) are repositioned, some of which have been implicated in binding the negatively charged surface of its redox partner NADPH-cytochrome P450 reductase. B–D, electrostatic surfaces (blue, positive; red, negative) for the proximal, reductase-binding surfaces of extant human CYP1B1 crystallized with α-naphthoflavone (B), the ancestral CYP1B1 crystallized with α-naphthoflavone (C), and the ancestral CYP1B1 crystallized with estradiol (D).
Structural elements and interactions proposed to confer thermostability
A key consideration in engineering P450 enzymes for biocatalysis is thermostability. In the previous literature, several structural features have been proposed to contribute to thermostability, especially when comparing mesophilic and thermophilic P450s. This information is based on a limited data set of four thermophilic P450 enzymes for which structures are available (33–36). A recent meta-analysis of these structures compared with their mesophilic P450 relatives (37) concluded that cytochrome P450 thermostability may correlate with 1) smaller, more compact enzymes, especially due to N-terminal truncation and shorter loops; 2) a preference for charged and hydrophobic residues versus noncharged, polar, and Ala residues; 3) increased salt bridges, particularly complex, extended salt-bridged networks; and perhaps 4) extended clusters of aromatic side chains. These enzymes were all soluble, microbial cytochrome P450 enzymes.
The advantages of using bacterial cytochrome P450 enzymes for biocatalysis often include better stability, but many of them are highly specific for their substrates and the products generated. If a particular soluble P450 can be identified that performs a commercially useful transformation (38) or can be engineered to do so (39–42), then this is may be the most advantageous route. However, many of the less stable mammalian cytochrome P450 enzymes have evolved to intrinsically bind and metabolize a wide diversity of foreign compounds and may be more commercially useful if one could engineer in thermostability.
Although the difference in thermostability of the ancestral mammalian CYP1B1 enzyme compared with extant human CYP1B1 is not as dramatic as thermophilic versus mesophilic bacterial P450 enzymes, it does provide one of the first real opportunities to determine whether some of the same four concepts can confer increased thermostability in eukaryotic microsomal P450s.
First, the overall protein size and lengths of loops and N and C termini were compared. The ancestral CYP1B1 and extant human CYP1B1 structures reveal loop regions of similar length (Fig. 1, A and B). Only small differences are observed in a few of the loops, and they are not always in the anticipated direction. The D/E loop is three residues longer in the ancestral CYP1B1 enzyme (Fig. 1B). Likewise, the H/I loop appears to be six residues longer in the ancestral enzyme, a region that is disordered in both enzymes. A three-residue helical segment occurs in the ancestral CYP1B1 enzyme corresponding to the human K″/L loop, whereas a short, two-strand β-sheet (the β-4 sheet) occurs in the human enzyme (Fig. 1B). Although both enzymes were truncated to remove the N-terminal transmembrane helix for crystallization, there are obvious differences in the remaining N termini. In the ancestral CYP1B1 enzyme, residues 48–60 could be modeled but not residues 61–67 (due to insufficient density to trace the backbone). By comparison, the human CYP1B1 structure could only be modeled beginning at residue 68 where the A helix begins. Because they are present in the construct crystallized, the absence of electron density for residues 48–60 in the human 1B1 structure must be due to high flexibility. It is not obvious from the primary sequence why this part of the ancestor would be more stable, but this region forms a small β-strand (Phe-55–Trp-57) that interacts with β-sheet 1. Additionally Trp-57 forms a potential face-to-edge aromatic interaction with Phe-74 (A helix). At the C-terminal end of the protein, the ancestor is 16 residues shorter, reducing the overall size by 2 kDa. In human CYP1B1, only six residues in the extended C terminus could be modeled. However, these were packing against symmetry-related molecules (26) and may be more conformationally flexible in solution. This shorter C terminus in the ancestral CYP1B1 enzyme is perhaps comparable with the shorter N termini observed in soluble thermophilic P450 enzymes and may reduce conformational flexibility.
Second, because thermophilic bacterial P450 enzymes seemed to have more charged and hydrophobic residues and fewer noncharged, polar, and Ala residues than related mesophilic P450 enzymes, the amino acid compositions of the ancestral CYP1B1 and extant human CYP1B1 were compared. The charged amino acids and noncharged but polar amino acids were very similar (≤0.5% different). The more thermostable ancestral CYP1B1 has 2% fewer hydrophobic amino acids, which is opposite to the trends in soluble P450 enzymes. Conversely, the ancestral enzyme also had 1% fewer Ala residues, which is consistent with the thermostable bacterial P450 trends. Of the 12 times that Ala in human CYP1B1 was replaced by another amino acid in the ancestral CYP1B1, 10 of these substitutions were to nonconserved amino acids (Pro (five), Glu (three), Arg (one), or Ser (one)).
Third, salt-bridge pairs and networks were compared between the two structures. In the extant human CYP1B1 structure, 13 two-residue salt bridges were found, whereas in the ancestor CYP1B1 structure, 14 were present. Evaluation of salt-bridge networks involving three or more residues revealed that human CYP1B1 had six, and the ancestral CYP1B1 had eight (Table S1). It is useful to examine two-residue salt bridges and networks that are unique to only one of the CYP1B1 enzymes (Fig. 6, A and B, spheres). In the extant human CYP1B1, five of the seven unique two-residue interactions and the one unique network occur within the same structural element, generally with residues found in close proximity within the primary sequence (Fig. 6A (black text) and Table S1 (red text but not boldface)). Two unique two-residue salt bridges occur between secondary structural elements (Fig. 6A (colored text) and Table S1 (red and boldface text)). In the mammalian ancestor, of the six unique two-residue salt-bridge interactions (Fig. 6B, gray spheres), three of them occur between different secondary structure elements (Fig. 6B (spheres with colored text) and Table S1 (red boldface text)). Additionally, of the three unique networks in the ancestor (Fig. 6B, black spheres), all three occur between secondary structure elements (Fig. 6B (colored text) and Table S1 (red, boldface text)). Such interactions likely help stabilize multiple regions of the protein and could be a contributor to the thermostability of the ancestor.
Figure 6.

Analysis of unique salt bridges and aromatic interactions in human CYP1B1 and the ancestral CYP1B1 structures, both co-crystallized with α-naphthoflavone. A and B, extant human CYP1B1 (green ribbons) (A) and ancestral CYP1B1 (blue ribbons) (B) showing unique two-residue salt bridges (gray spheres) and more extended networks (black spheres). Not shown (for clarity) are instances where one or two new amino acids interact with a previously existing network, referred to as partials; two partials are present in human CYP1B1 and two partials in the ancestor (see Table S1). C and D, extant human CYP1B1 (green ribbons) (C) and ancestral CYP1B1 (blue ribbons) (D) showing unique aromatic and π-cation stacking interactions (gray sticks). Partials in the stacking interactions are not shown; only one partial is found in extant CYP1B1 (see Table S2). In all four images, the text indicates the secondary structure element in which the indicated residues belong, with black text indicating that the residues are in the same secondary structure element and colored text indicating the pairings of residues that are in distinct secondary structure elements.
Finally, soluble thermophilic cytochrome P450 enzymes, such as CYP119A1 (33) and CYP119A2 (34) from archaea, have extended networks of aromatic residues. Analysis of aromatic and/or cation-π stacking interactions in the human and ancestral CYP1B1 proteins indicated some differences (Table S2). Human CYP1B1 contained six two-residue pairs, three of which had stacking interactions with a third aromatic residue. By comparison, the ancestral CYP1B1 structure had eight two-residue aromatic or π-cation stacking pairs, with only one stacking pair interacting with a third aromatic residue. Unique aromatic and π-cation stacking interactions between the two proteins are shown in Fig. 6 (C and D). Human CYP1B1 contains two unique two-residue aromatic interactions (Fig. 6C (gray sticks) and Table S2 (red)) and one three-residue network (Fig. 6C (black sticks) and Table S2 (red)). Two of the aromatic/π-cation stacking two-residue pairs are composed of residues that are more distant in the primary sequence and likely facilitate interactions between different parts of the tertiary protein structures (Fig. 6C (colored text labels) and Table S2 (red boldface)). By comparison, the ancestral structure has five unique two-residue aromatic interactions (Fig. 6D). Four of these interaction pairs occur between residues found in distinct secondary structure elements and thus are more likely to contribute to global protein structure.
Conclusions
The first structural evaluation of a reconstructed ancestral cytochrome P450 has reinforced several existing concepts about enzyme functionality and stability but also suggested some new ones. The ancestral CYP1B1 enzyme retains most of the active-site residues of the extant human CYP1B1 enzyme and retains some of the metabolic capabilities and metabolite stoichiometries for some substrates (in this case α-naphthoflavone) while showing very different metabolic capacity and metabolite ratios for others (e.g. 17β-estradiol). Subtle amino acid substitutions outside the active site seem to control overall protein conformation, in this case restoring the helical nature of the entire F helix and opening up a broad channel from the active site to the protein surface. Whereas ideas about shorter loops contributing to thermostability did not appear to apply here, a C-terminal truncation may be relevant. The somewhat more thermostable ancestral enzyme did show a few more electrostatic and aromatic interactions that could contribute to thermostability, but the locations of such interactions between distinct secondary structure elements, often far apart in the primary sequence, may be more critical. Additional structures will be required to determine whether these trends continue in even older reconstructions that have higher stability.
Experimental procedures
Materials
pCW plasmids containing CYP1B1, N98_CYP1B1_Mammal and human NADPH-cytochrome P450 reductase (hCPR) were constructed in previous studies (7, 26).3 Catalytic assays with human CYP1B1 used construct #3 from Ref. 7. All binding, catalytic assays, and crystallography for the ancestral CYP1B1 were done with the N-terminally truncated, C-terminally His-tagged version of N98_CYP1B1_Mammal,3 with the sequence shown in Fig. 1B. The chaperone co-expression plasmid, pGro7, was obtained from Prof. K. Nishihara (HSP Research Institute, Kyoto, Japan) (43) and from Takara Bio Inc. DH5αF′IQ Escherichia coli cells were purchased from Thermo Fisher Scientific (Scoresby, Australia).
Bactotryptone, bactopeptone, and yeast extract were supplied by Becton Dickinson Pty. Ltd. (North Ryde, Australia). 17β-Estradiol metabolites were purchased from Steraloids Inc. (Newport, RI) and kindly made available for this work by Prof. F. Peter Guengerich (Vanderbilt University). Emulgen 913 and 911 were obtained from Kao Chemicals (Tokyo, Japan). All other materials, including α-naphthoflavone and 17β-estradiol, were purchased at the highest available grade from Sigma-Aldrich-Merck (Castle Hill, Australia) where possible or from a local supplier.
Preparation of bacterial membranes for activity assays
P450s and hCPR were expressed in E. coli and isolated in bacterial membranes as described previously (44, 45). P450 content was quantified by Fe(II).CO versus Fe(II) difference spectroscopy (46, 47). NADPH-cytochrome P450 reductase was quantified by the reduction of the surrogate electron acceptor, cytochrome c, as described previously (48).
Metabolic assays
Metabolic incubations were carried out as described previously with modifications (49). Bacterial membranes containing 0.5 μm P450 were incubated for 60 min at 37 °C in 100 mm potassium phosphate, pH 7.4, with 25 μm α-naphthoflavone in the presence and absence of an NADPH-generating system (100 mm glucose 6-phosphate, 250 μm NADP+, and 0.5 units/ml glucose-6-phosphate dehydrogenase). Progesterone (5 μm) was added as an internal standard for α-naphthoflavone assays, prior to quenching and extraction with 1 ml of ethyl acetate.
α-Naphthoflavone and its metabolites were identified using a Shimadzu Nexera Ultraperformance LC system coupled to an Orbitrap Elite mass spectrometer. Chromatography was performed on an AccucoreTM C30 column (150 × 2.1 mm, 2.6 μm) at 0.4 ml/min at 50 °C. Buffer A comprised 1% acetonitrile, 0.1% formic acid, and buffer B comprised 90% acetonitrile, 0.1% formic acid. The gradient was developed as follows: 0–0.5 min, 50% B; 0.5–3 min, linear gradient to 98% B; 3–4.5 min, 98% B; 4.5–4.6 min, linear gradient to 50% B; 4.6–5 min, 50% B. The mass spectrometer was operated in positive mode. Source parameters included S lens 60; ion spray voltage 4.5 kV; capillary temperature 380 °C; heater 400 °C; sheath gas and auxiliary gases 50 and 16, respectively. Data-dependent acquisition was performed across 50–1500 m/z, and the top five precursors were selected. Higher-energy collisional dissociation fragmentation was optimized to use a normalized collision energy of 60. Data were processed using XcaliburTM software. Spectra were searched against publicly available databases. α-Naphthoflavone metabolites were quantified using UPLC-APCI-MRM with the same chromatography conditions, but with a SCIEX 5500 QTRAP mass spectrometer operating in positive multiple-reaction-monitoring (MRM) mode. Source parameters included a nebulizer current of 3 μA, temperature at 500 °C, nebulizing gas (GS1) at 45, declustering potential at 120, and collision cell exit potential of 12. For each compound, the collision energy, entrance potential, and cell exit potential were optimized for maximum sensitivity by manual infusion. Data were processed using both AnalystTM 1.7 and MultiquantTM 3.0 (SCIEX) software. MRM transitions used for detection are listed in Table S3, and the recovery of α-naphthoflavone and its metabolites were estimated based on the relative intensity of MS/MS fragment 103.
Protein expression and purification for ligand-binding assays and crystallization
Expression and purification of the N98_CYP1B1_Mammal for crystallization were based on methods used for expression of human CYP1A1 for crystallography (50) but modified extensively, so a detailed expression protocol is described below for clarity. The expression plasmid pCW_N98_1B1_Mammal was transformed into E. coli DH5α chemically competent cells already containing the plasmid pGro7 for coexpression of the molecular chaperones GroEl/GroES. Transformed cells were plated on lysogeny broth (LB)-agar supplemented with 100 μg/ml carbenicillin for selection of the P450 plasmid and 20 μg/ml chloramphenicol for selection of the pGro7 plasmid and grown for ∼18 h at 37 °C. A single colony from this plate was inoculated into a 5-ml LB starter culture, supplemented with antibiotics described above, and grown for ∼7 h at 37 °C and shaking at 250 rpm. This starter culture (50 μl) was then used to inoculate a 200-ml LB culture, supplemented with the above antibiotics, grown for ∼16 h at 37 °C with shaking at 250 rpm. Finally, this overnight culture (10 ml) was used to inoculate 250-ml cultures of Terrific Broth in 1-liter Erlenmeyer flasks containing the above antibiotics. The culture volume grown for a single purification typically totaled 2 liters or eight flasks. These expression cultures were grown at 37 °C with 250 rpm shaking until they reached an optical density at 600 nm (OD600) of 0.3. At this point, chaperone expression was induced by the addition of 2 mg/ml arabinose, and δ-aminolevulinic acid was added to 1 mm as a heme precursor. The temperature and shaking of the cultures were then reduced to 25 °C and 225 rpm, respectively. At OD600 of 0.6, N98_CYP1B1_Mammal expression was induced by adding 1 mm isopropyl 1-thio-β-d-galactopyranoside. Cultures were grown for 48 h after induction.
Isolation of ancestral N98_CYP1B1_Mammal was performed using a purification method that was developed for human CYP1A1 engineered for crystallization as described (50) with only slight changes. Modifications included depletion of the detergent CHAPS during ion-exchange chromatography by using a carboxymethyl Sepharose wash buffer (10 mm potassium phosphate, 100 mm NaCl, 20% (v/v) glycerol, 1 mm EDTA, pH 7.4) for the wash step and carboxymethyl elution buffer (50 mm potassium phosphate, 500 mm NaCl, 20% (v/v) glycerol, 1 mm EDTA, pH 7.4) to elute the bound protein. Additionally, the final protein buffer used during gel filtration was a lower ionic size-exclusion chromatography buffer (20 mm potassium phosphate, 20% glycerol, 0.1 m NaCl, pH 7.4). Under these conditions, the main N98_CYP1B1_Mammal peak eluted from the size-exclusion column as a trimer, with a shoulder corresponding to a dimeric species. The major trimeric peak was pooled and used in ligand binding and crystallization experiments. The final purity of the protein was assessed by SDS-PAGE and the ratio of the Soret peak to the 280-nm peak. The protein appeared to be a single band on the SDS-polyacrylamide gel and typically had A416 nm/A280 nm = 1.2. The protein concentration was determined using the absorbance of the Soret band and an extinction coefficient of 100 mm−1 cm−1 in the size-exclusion chromatography buffer listed above. Finally, a reduced carbon monoxide-difference spectrum (47) revealed a peak exclusively at 450 nm, consistent with active enzyme, and no peak or shoulder at 420 nm.
Ligand-binding assays
α-Naphthoflavone and 17β-estradiol binding to N98_CYP1B1_Mammal were monitored spectrophotometrically. Initial titrations in the absolute spectrum mode employed 1 μm P450 in buffer (50 mm potassium phosphate, 500 mm NaCl, 20% glycerol, 0.5% (w/v) CHAPS, pH 7.4) in 1-cm quartz cuvettes, with additions of ligand dissolved in DMSO being added to the sample cuvette and equal volumes of ligand added to the reference cuvette only containing buffer. Subsequent titrations accomplished in difference mode to focus on ligand-induced changes used 1 μm protein in 1-cm quartz cuvettes in the same buffer. Data were fit to a simple one-site hyperbolic binding equation for estradiol and to the Hill equation for α-naphthoflavone over the concentration ranges shown in the figures.
Crystallization, data collection, structure determination, and analysis
Purified N98_CYP1B1_Mammal was first saturated with either α-naphthoflavone or 17β-estradiol by the addition of 100 μm ligand during final concentration using centrifugal ultrafiltration (Amicon Ultra-15 filters with a molecular mass cut-off of 50 kDa), once for α-naphthoflavone and three times for estradiol. Crystals of the ancestral CYP1B1 protein with α-naphthoflavone were then grown at 20 °C using the sitting-drop vapor diffusion method in 96-well plates by mixing 0.5 μl of N98_CYP1B1_Mammal/α-naphthoflavone (protein at 290 μm as determined by absolute spectrum) with 0.5 μl of crystallization solution (900 mm sodium citrate tribasic, 90 mm Tris base/HCl, pH 7.0, 180 mm NaCl, 10% glycerol) and equilibrating against a 50-μl reservoir of the same crystallization solution. Cocrystallization with estradiol was performed at 20 °C in sitting-drop 24-well plates by mixing 0.9 μl of N98_CYP1B1_Mammal/estradiol (290 μm) with 1.1 μl of crystallization solution (1 m sodium citrate tribasic, 0.1 m Tris/HCl, pH 7.0, 0.2 m NaCl) and equilibrating against 250 μl of the same crystallization solution. Cubic crystals grew in 2–3 days. Harvested crystals were cryoprotected using the respective crystallization solution supplemented with either additional 20% glycerol (α-naphthoflavone) or 22% glycerol (estradiol) before being flash-cooled in liquid nitrogen. The N98_CYP1B1_Mammal/α-naphthoflavone diffraction data were collected on a single crystal at the Advanced Photon Source LS-CAT beamline 21-ID-G. The N98-_CYP1B1_Mammal/estradiol diffraction data were collected on a single crystal on beamline 12-2 at the Stanford Synchrotron Radiation Lightsource.
Data processing for the N98_CYP1B1_Mammal/α-naphthoflavone data set was performed with HKL2000 (52). The N98_CYP1B1_Mammal/estradiol data set was processed in XDS (53) with scaling in AIMLESS (54). A molecular replacement solution for N98_CYP1B1_Mammal/α-naphthoflavone was initially obtained using MolRep (55) using the CYP1B1/α-naphthoflavone crystal structure (PDB: 3PM0) as a search model (contrast score of 9.9). Inspection of packing and electron density as well as initial statistics after one round of refinement (R = 0.34 and Rfree = 0.43) indicated a correct solution with two molecules in the asymmetric unit. A molecular replacement solution for the N98_CYP1B1_Mammal/estradiol structure was obtained using Phaser (56) and the N98_CYP1B1_Mammal/α-naphthoflavone (PDB: 6OYU) structure as a search model. The molecular replacement solution was successful with a log likelihood of 3,702 and translation function Z score of 58. For both structures, multiple rounds of model building and refinement were performed using COOT (57) and PHENIX (58). Torsion angle noncrystallographic symmetry (NCS) with automatic group NCS group determination was employed in Phenix for the slightly lower-resolution 17β-estradiol structure. Coordinates and restraints for α-naphthoflavone and estradiol were obtained from the CCD (Chemical ID: BHF and EST, respectively) and minimized through PHENIX eLBOW (59) using AM1 geometry optimization.
Structural comparisons with extant human P450 enzymes were executed using molecule B of N98_CYP1B1_Mammal complexed with α-naphthoflavone. Whereas the two molecules in the asymmetric unit of each structure with both ligands are all similar, the rationale was that the α-naphthoflavone structure had higher resolution than the estradiol structure, that all of the human CYP1 structures were obtained with α-naphthoflavone as the ligand, and that chain B of the α-naphthoflavone structure is most complete. By comparison, chain A of the α-ancestral CYP1B1 naphthoflavone structure has an additional disordered gap for amino acids 448–452 comprising the K′-K″ loop. Calculation of the active-site void volumes for both N98_CYP1B1_Mammal/α-naphthoflavone and human CYP1B1 (PDB: 3PM0) (26) was performed using VOIDOO (60) with a probe radius of 1.4 Å and grid spacing of 1.0. For the N98_CYP1B1_Mammal/α-naphthoflavone structure, it was necessary to add a network of water molecules to close the large cleft on the surface of the protein for the purposes of active-site volume calculation. Structural alignments were performed using the super command in PyMOL (25). Sequence alignment was performed using CLC Genomics Workbench (61). Analysis of salt bridges was performed in COOT (57) using a definition of ≤4 Å between the oxygen of Glu or Asp side chains and the nitrogen group(s) of Arg, His, or Lys (36, 37). Aromatic and π-cation stacking interactions were defined by selection of aromatic residues with ring-to-ring or ring-to-arginine guanidinium group distances of ≤4 Å using Schrödinger Maestro (Schrödinger, LLC, New York). All salt-bridge and stacking interactions were visually inspected to ensure that residue positions were well-defined in the electron density. Structure figures were prepared using PyMOL (25) with electrostatic potential surfaces generated using APBS (24).
Data availability
The atomic coordinates and structure factors for the ancestral CYP1B1 structures have been deposited in the Protein Data Bank under accession codes 6OYU (complex with α-naphthoflavone) and 6OYV (complex with estradiol). For titrations in Fig. 2, not all intervening scans are shown, but they are available from the corresponding author. All other data are included in the article.
Author contributions
A. G. B., K. L. H., E. M. J. G., and E. E. S. conceptualization; A. G. B., K. L. H., E. M. J. G., and E. E. S. formal analysis; A. G. B., K. L. H., E. M. J. G., and E. E. S. supervision; A. G. B., K. L. H., E. M. J. G., and E. E. S. funding acquisition; A. G. B., K. L. H., E. M. J. G., and E. E. S. investigation; A. G. B., K. L. H., E. M. J. G., and E. E. S. visualization; A. G. B., K. L. H., E. M. J. G., and E. E. S. writing-original draft; A. G. B., K. L. H., E. M. J. G., and E. E. S. project administration; A. G. B., K. L. H., E. M. J. G., and E. E. S. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to Prof. F. Peter Guengerich for the gift of the estradiol metabolites, to Peter Josh for assistance with the LC-MS/MS analysis of the α-naphthoflavone metabolites, to Raine Thomson for clarification of the steroid metabolite chromatography, and to Drs. Shalini Andersson and Ulrik Jurva for helpful discussions and support with the metabolic assays. Crystallization employed instrumentation in the University of Michigan Center for Structural Biology (CSB), which receives support from the U-M Life Sciences Institute, the U-M Rogel Cancer Center, and the U-M Medical School Endowment for the Basic Sciences and grants from the National Institutes of Health. X-ray diffraction data were collected at both Advanced Photon Source and the Stanford Synchrotron Radiation Lightsource. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the DOE, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by NIGMS, National Institutes of Health.
This work was supported by National Institutes of Health Grant R37 GM076343 (to E. E. S.) and by the 2018 M. G. and R. A. Plowman Scholarship in Inorganic Chemistry (School of Chemistry and Molecular Biosciences at the University of Queensland) (to K. L. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3 and Figs. S1 and S2.
K. L. Harris, Y. Gumulya, G. Foley, P. Syed, T. Janosik, A. Sandinge, S. Andersson, U. Jurva, M. Bodén, and E. M. J. Gillam, unpublished observations.
- CYP
- cytochrome P450
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- CI
- confidence interval
- hCPR
- human NADPH-cytochrome P450 reductase
- MRM
- multiple-reaction-monitoring
- LB
- lysogeny broth
- OD
- optical density
- NCS
- noncrystallographic symmetry.
References
- 1. Nebert D. W., Roe A. L., Dieter M. Z., Solis W. A., Yang Y., and Dalton T. P. (2000) Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem. Pharmacol. 59, 65–85 10.1016/S0006-2952(99)00310-X [DOI] [PubMed] [Google Scholar]
- 2. Noakes R. (2015) The aryl hydrocarbon receptor: a review of its role in the physiology and pathology of the integument and its relationship to the tryptophan metabolism. Int. J. Tryptophan Res. 8, 7–18 10.4137/IJTR.S19985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hankinson O. (2016) The role of AHR-inducible cytochrome P450s in metabolism of polyunsaturated fatty acids. Drug Metab. Rev. 48, 342–350 10.1080/03602532.2016.1197240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimada T., Gillam E. M. J., Sutter T. R., Strickland P. T., Guengerich F. P., and Yamazaki H. (1997) Oxidation of xenobiotics by recombinant human cytochrome P450 1B1. Drug Metab. Dispos. 25, 617–622 [PubMed] [Google Scholar]
- 5. Choudhary D., Jansson I., Stoilov I., Sarfarazi M., and Schenkman J. B. (2004) Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1B1. Drug Metab. Dispos. 32, 840–847 10.1124/dmd.32.8.840 [DOI] [PubMed] [Google Scholar]
- 6. Hayes C. L., Spink D. C., Spink B. C., Cao J. Q., Walker N. J., and Sutter T. R. (1996) 17β-Estradiol hydroxylation catalysed by human cytochrome P450 1B1. Proc. Natl. Acad. Sci. U.S.A. 93, 9776–9781 10.1073/pnas.93.18.9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shimada T., Wunsch R. M., Hanna I. H., Sutter T. R., Guengerich F. P., and Gillam E. M. J. (1998) Recombinant human cytochrome P450 1B1 expression in Escherichia coli. Arch. Biochem. Biophys. 357, 111–120 10.1006/abbi.1998.0808 [DOI] [PubMed] [Google Scholar]
- 8. Crespi C. L., Penman B. W., Steimel D. T., Smith T., Yang C. S., and Sutter T. R. (1997) Development of a human lymphoblastoid cell line constitutively expressing human CYP1B1 cDNA: substrate specificity with model substrates and promutagens. Mutagenesis 12, 83–89 10.1093/mutage/12.2.83 [DOI] [PubMed] [Google Scholar]
- 9. Ma X., Idle J. R., Krausz K. W., and Gonzalez F. J. (2005) Metabolism of melatonin by human cytochromes P450. Drug Metab. Dispos. 33, 489–494 10.1124/dmd.104.002410 [DOI] [PubMed] [Google Scholar]
- 10. Murray G. I., Melvin W. T., Greenlee W. F., and Burke M. D. (2001) Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. Toxicol. 41, 297–316 10.1146/annurev.pharmtox.41.1.297 [DOI] [PubMed] [Google Scholar]
- 11. Murray G. I., Taylor M. C., McFadyen M. C. E., McKay J. A., Greenlee W. F., Burke M. D., and Melvin W. T. (1997) Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 57, 3026–3031 [PubMed] [Google Scholar]
- 12. McKay J. A., Melvin W. T., Ah-See A. K., Ewen S. W., Greenlee W. F., Marcus C. B., Burke M. D., and Murray G. I. (1995) Expression of cytochrome P450 CYP1B1 in breast cancer. FEBS Lett. 374, 270–272 10.1016/0014-5793(95)01126-Y [DOI] [PubMed] [Google Scholar]
- 13. Shimada T., Hayes C. L., Yamazaki H., Amin S., Hecht S. S., Guengerich F. P., and Sutter T. R. (1996) Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 56, 2979–2984 [PubMed] [Google Scholar]
- 14. Shimada T., Gillam E. M. J., Oda Y., Tsumura F., Sutter T. R., Guengerich F. P., and Inoue K. (1999) Metabolism of benzo[a]pyrene to trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene by recombinant human cytochrome P4501B1 and purified liver epoxide hydrolase. Chem. Res. Toxicol. 12, 623–629 10.1021/tx990028s [DOI] [PubMed] [Google Scholar]
- 15. Oda Y., Aryal P., Terashita T., Gillam E. M. J., Guengerich F. P., and Shimada T. (2001) Metabolic activation of heterocyclic amines and other procarcinogens in Salmonella typhimurium umu tester strains expressing human cytochrome P4501A1, 1A2, 1B1, 2C9, 2D6, 2E1, and 3A4 and human NADPH-P450 reductase and bacterial O-acetyltransferase. Mutat. Res. 492, 81–90 10.1016/S1383-5718(01)00154-1 [DOI] [PubMed] [Google Scholar]
- 16. Dragin N., Shi Z., Madan R., Karp C. L., Sartor M. A., Chen C., Gonzalez F. J., and Nebert D. W. (2008) Phenotype of the Cyp1a1/1a2/1b1−/− triple-knockout mouse. Mol. Pharmacol. 73, 1844–1856 10.1124/mol.108.045658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ward J. M., Nikolov N. P., Tschetter J. R., Kopp J. B., Gonzalez F. J., Kimura S., and Siegel R. M. (2004) Progressive glomerulonephritis and histiocytic sarcoma associated with macrophage functional defects in CYP1B1-deficient mice. Toxicol. Pathol. 32, 710–718 10.1080/01926230490885706 [DOI] [PubMed] [Google Scholar]
- 18. Stoilov I., Akarsu A. N., and Sarfarazi M. (1997) Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 6, 641–647 10.1093/hmg/6.4.641 [DOI] [PubMed] [Google Scholar]
- 19. Vasiliou V., and Gonzalez F. J. (2008) Role of CYP1B1 in glaucoma. Annu. Rev. Pharmacol. Toxicol. 48, 333–358 10.1146/annurev.pharmtox.48.061807.154729 [DOI] [PubMed] [Google Scholar]
- 20. Jönsson M. E., Woodin B. R., Stegeman J. J., and Brunström B. (2011) Cytochrome P450 1 genes in birds: evolutionary relationships and transcription profiles in chicken and Japanese quail embryos. PLoS ONE 6, e28257 10.1371/journal.pone.0028257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldstone J. V., Goldstone H. M. H., Morrison A. M., Tarrant A., Kern S. E., Woodin B. R., and Stegeman J. J. (2007) Cytochrome P450 1 genes in early deuterostomes (tunicates and sea urchins) and vertebrates (chicken and frog): origin and diversification of the CYP1 gene family. Mol. Biol. Evol. 24, 2619–2631 10.1093/molbev/msm200 [DOI] [PubMed] [Google Scholar]
- 22. Goldstone H. M. H., and Stegeman J. J. (2006) A revised evolutionary history of the CYP1A subfamily: gene duplication, gene conversion, and positive selection. J. Mol. Evol. 62, 708–717 10.1007/s00239-005-0134-z [DOI] [PubMed] [Google Scholar]
- 23. Kawai Y. K., Ikenaka Y., Fujita S., and Ishizuka M. (2010) The CYP1D subfamily of genes in mammals and other vertebrates. Mamm. Genome 21, 320–329 10.1007/s00335-010-9263-9 [DOI] [PubMed] [Google Scholar]
- 24. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeLano W. L. (2019) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 26. Wang A., Savas U., Stout C. D., and Johnson E. F. (2011) Structural characterization of the complex between α-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 286, 5736–5743 10.1074/jbc.M110.204420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walsh A. A., Szklarz G. D., and Scott E. E. (2013) Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 288, 12932–12943 10.1074/jbc.M113.452953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sansen S., Yano J. K., Reynald R. L., Schoch G. A., Griffin K. J., Stout C. D., and Johnson E. F. (2007) Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 282, 14348–14355 10.1074/jbc.M611692200 [DOI] [PubMed] [Google Scholar]
- 29. Shimada T., Tanaka K., Takenaka S., Foroozesh M. K., Murayama N., Yamazaki H., Guengerich F. P., and Komori M. (2009) Reverse type I binding spectra of human cytochrome P450 1B1 induced by flavonoid, stilbene, pyrene, naphthalene, phenanthrene, and biphenyl derivatives that inhibit catalytic activity: a structure-function relationship study. Chem. Res. Toxicol. 22, 1325–1333 10.1021/tx900127s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shimada T., Yamazaki H., Foroozesh M., Hopkins N. E., Alworth W. L., and Guengerich F. P. (1998) Selectivity of polycyclic inhibitors for human cytochrome P450s 1A1, 1A2, and 1B1. Chem. Res. Toxicol. 11, 1048–1056 10.1021/tx980090+ [DOI] [PubMed] [Google Scholar]
- 31. Bauer E., Guo Z., Ueng Y. F., Bell L. C., Zeldin D., and Guengerich F. P. (1995) Oxidation of benzo[a]pyrene by recombinant human cytochrome P450 enzymes. Chem. Res. Toxicol. 8, 136–142 10.1021/tx00043a018 [DOI] [PubMed] [Google Scholar]
- 32. Nishida C. R., Everett S., and Ortiz de Montellano P. R. (2013) Specificity determinants of CYP1B1 estradiol hydroxylation. Mol. Pharmacol. 84, 451–458 10.1124/mol.113.087700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yano J. K., Koo L. S., Schuller D. J., Li H., Ortiz de Montellano P. R., and Poulos T. L. (2000) Crystal structure of a thermophilic cytochrome P450 from the archaeon Sulfolobus solfataricus. J. Biol. Chem. 275, 31086–31092 10.1074/jbc.M004281200 [DOI] [PubMed] [Google Scholar]
- 34. Oku Y., Ohtaki A., Kamitori S., Nakamura N., Yohda M., Ohno H., and Kawarabayasi Y. (2004) Structure and direct electrochemistry of cytochrome P450 from the thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. J. Inorg. Biochem. 98, 1194–1199 10.1016/j.jinorgbio.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 35. Ho W. W., Li H., Nishida C. R., de Montellano P. R., and Poulos T. L. (2008) Crystal structure and properties of CYP231A2 from the thermoacidophilic archaeon Picrophilus torridus. Biochemistry 47, 2071–2079 10.1021/bi702240k [DOI] [PubMed] [Google Scholar]
- 36. Yano J. K., Blasco F., Li H., Schmid R. D., Henne A., and Poulos T. L. (2003) Preliminary characterization and crystal structure of a thermostable cytochrome P450 from Thermus thermophilus. J. Biol. Chem. 278, 608–616 10.1074/jbc.M206568200 [DOI] [PubMed] [Google Scholar]
- 37. Harris K. L., Thomson R. E. S., Strohmaier S. J., Gumulya Y., and Gillam E. M. J. (2018) Determinants of thermostability in the cytochrome P450 fold. Biochim. Biophys. Acta Proteins Proteom. 1866, 97–115 10.1016/j.bbapap.2017.08.003 [DOI] [PubMed] [Google Scholar]
- 38. Serizawa N., and Matsuoka T. (1991) A two component-type cytochrome P-450 monooxygenase system in a prokaryote that catalyzes hydroxylation of ML-236B to pravastatin, a tissue-selective inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Biochim. Biophys. Acta 1084, 35–40 10.1016/0005-2760(91)90052-J [DOI] [PubMed] [Google Scholar]
- 39. Fasan R., Chen M. M., Crook N. C., and Arnold F. H. (2007) Engineered alkane-hydroxylating cytochrome P450(BM3) exhibiting nativelike catalytic properties. Angew. Chem. Int. Ed. Engl. 46, 8414–8418 10.1002/anie.200702616 [DOI] [PubMed] [Google Scholar]
- 40. Glieder A., Farinas E. T., and Arnold F. H. (2002) Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20, 1135–1139 10.1038/nbt744 [DOI] [PubMed] [Google Scholar]
- 41. Jung S. T., Lauchli R., and Arnold F. H. (2011) Cytochrome P450: taming a wild type enzyme. Curr. Opin. Biotechnol. 22, 809–817 10.1016/j.copbio.2011.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lewis J. C., and Arnold F. H. (2009) Catalysts on demand: selective oxidations by laboratory-evolved cytochrome P450 BM3. Chimia 63, 309–312 10.2533/chimia.2009.309 [DOI] [Google Scholar]
- 43. Nishihara K., Kanemori M., Kitagawa M., Yanagi H., and Yura T. (1998) Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 64, 1694–1699 10.1128/AEM.64.5.1694-1699.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Notley L. M., De Wolf C. J. F., Wunsch R. M., Lancaster R. G., and Gillam E. M. J. (2002) Bioactivation of tamoxifen by recombinant human cytochrome P450 enzymes. Chem. Res. Toxicol. 15, 614–622 10.1021/tx0100439 [DOI] [PubMed] [Google Scholar]
- 45. Gillam E. M., Baba T., Kim B. R., Ohmori S., and Guengerich F. P. (1993) Expression of modified human cytochrome P450 3A4 in Escherichia coli and purification and reconstitution of the enzyme. Arch. Biochem. Biophys. 305, 123–131 10.1006/abbi.1993.1401 [DOI] [PubMed] [Google Scholar]
- 46. Johnston W. A., Huang W., De Voss J. J., Hayes M. A., and Gillam E. M. (2008) Quantitative whole-cell cytochrome P450 measurement suitable for high-throughput application. J. Biomol. Screen. 13, 135–141 10.1177/1087057107312780 [DOI] [PubMed] [Google Scholar]
- 47. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 48. Guengerich F. P. (2008) Analysis and characterization of enzymes and nucleic acids. in Principles and Methods in Toxicology (Hayes A. W., ed) 5th Ed., pp. 1973–2040, Raven Press, New York [Google Scholar]
- 49. Hunter D. J. B., Behrendorff J. B. Y. H., Johnston W. A., Hayes P. Y., Huang W., Bonn B., Hayes M. A., De Voss J. J., and Gillam E. M. J. (2011) Facile production of minor metabolites for drug development using a CYP3A shuffled library. Metab. Eng. 13, 682–693 10.1016/j.ymben.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 50. Bart A. G., and Scott E. E. (2018) Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 293, 19201–19210 10.1074/jbc.RA118.005588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deleted in proof.
- 52. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 53. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vagin A., and Teplyakove A. (2010) Molecular replacement with MOLREP. Acta. Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 56. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D. (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 10.1107/S0907444909029436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kleywegt G. J., and Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 10.1107/S0907444993011333 [DOI] [PubMed] [Google Scholar]
- 61. CLC Genomics Workbench, version 11.0, Qiagen, Redwood City, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors for the ancestral CYP1B1 structures have been deposited in the Protein Data Bank under accession codes 6OYU (complex with α-naphthoflavone) and 6OYV (complex with estradiol). For titrations in Fig. 2, not all intervening scans are shown, but they are available from the corresponding author. All other data are included in the article.