

Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is one of the most common malignancy worldwide. Accumulating evidences have highlighted the importance of transcriptome data during HNSCC tumorigenesis. The aim of this study was to identify significant genes as effective biomarkers for HNSCC and constructed miRNA-mRNA regulatory network for a more comprehensive understanding of the underlying molecular mechanisms.
Methods
A total of four independent microarrays conducted on HNSCC samples were downloaded from the Gene Expression Omnibus (GEO) and analyzed through R software. FunRich was applied to predict potential transcription factors and targeted genes of miRNAs. Protein-protein interaction (PPI) network and miRNA-mRNA regulatory network were constructed in Cytoscape. Additionally, the database for annotation, visualization, and integrated discovery (DAVID) was utilized to perform GO and KEGG pathway enrichment analyses. Validation of gene expression levels was conducted by online databases and qPCR experiments.
Results
A total of 35 and 193 differentially expressed miRNAs (DEMs) and mRNAs (DEGs) were screened out by the limma package in R. The interactive network of the overlapping DEGs presented three significant modules and ten hub genes (FN1, MMP3, SPP1, STAT1, LOX, CXCL5, CXCL11, ISG15, IFIT3, and RSAD2). Predicted target genes of DEMs were visualized in Cytoscape and six miRNA-mRNA regulatory pairs were identified. Further validation demonstrated the upregulation of SLC16A1 and COL4A1 in HNSCC.
Conclusions
We performed an integrated and comprehensive bioinformatics analysis of miRNAs and mRNAs in HNSCC, contributing to explore the underlying regulatory mechanisms and to identify genetic biomarkers and therapeutic targets for HNSCC.
Keywords: Biomarker, Gene Expression Omnibus (GEO), head and neck squamous cell carcinoma (HNSCC), miRNA-mRNA network
Introduction
Head and neck squamous cell carcinoma (HNSCC) is listed as one of the most frequent malignancies all over the world and it was reported that approximately 500,000 new cases are diagnosed annually. In spite of the substantial efforts devoted to developing innovative and effective treatment methods for HNSCC, the survival rate of HNSCC patients stayed at 50% in the past 40 years (1). Therefore, in depth investigation of precise biomarkers for diagnosis and effective targets for treatment is especially valuable.
According to the newly displayed modern technologies such as microarray and next-generation deep sequencing, human genome codes have primarily been divided into two groups, namely messenger RNAs and non-coding RNAs. Non-coding RNAs represent novel regulatory layers in the transcriptional and post-transcriptional gene regulation. They influence gene regulation through multiple aspects, such as epigenetic regulation, transcription, and mRNA splicing (2). The high proportion and complexity of non-coding RNAs in the genome made them significant factors in a good array of malignancies (3,4). MicroRNAs (miRNAs), single-stranded non-coding RNAs with around 20 nucleotides in length, were reported to possess the ability of regulating gene expression. In most circumstances, miRNAs perform their functions by binding with the mRNAs of target genes post-transcriptionally (5). A multiple of studies have demonstrated the vital roles of miRNAs in various aspects of biological phenotypes, which may play indispensable roles in carcinogenesis (6). Furthermore, the tissue-specific property of miRNAs renders them comparatively accurate and effective biomarkers in the diagnosis, treatment, and prognosis of cancers. Recently, a series of tumor-promoting or tumor-suppressive miRNAs have been implicated in HNSCC progression. For example, miR-300 was demonstrated to inhibit the EMT transition process by regulating twist in HNSCC (7). Also, miR-125b-1 dysregulation was verified to stimulate the pathogenesis and deterioration of HNSCC (8). In addition, some kinds of miRNAs were upregulated in HNSCC and acted as stimulators for tumor progression, such as miR-21 (9), miR-134 (10), and miR-196a (11), mainly by facilitating cell growth and promoting cell motility.
The miRNA-mRNA network is an innovative model for presenting gene expression regulation and representing complicated interactions between coding and noncoding RNAs. The ability of miRNA binding to mRNA constitutes an indispensable part in the underlying molecular mechanisms involving various physiological processes. Therefore, the integration and analysis of differentially expressed miRNAs (DEMs) and mRNAs (DEGs) through microarray data-based analysis is conductive to the discovery of diagnostic biomarkers and therapeutic targets. For example, by analyzing the interactive relationship of miRNA and mRNA in breast cancer, the study identified essential molecular markers that may facilitate the prognosis evaluation of breast cancer patients (12). Another study found out candidate miRNA-mRNA network that could be utilized to predict chemoresistance response in osteosarcoma (13). Moreover, a study provided a meaningful characterization of miRNA-mRNA co-expression network in chondrosarcoma and identified crucial miRNAs and mRNAs involved in chondrosarcoma carcinogenesis (14).
Although prior studies have detected some kinds of aberrantly expressed miRNAs that may mediate the tumorigenesis process of HNSCC, a majority of them were dependent on a relatively small sample size that may result in biased and unreliable results. In the present study, we applied multiple microarray datasets of HNSCC from Gene Expression Omnibus (GEO) and made a series of functional analyses. Investigating the expression pattern of miRNAs and mRNAs and identifying candidate miRNA-mRNA regulatory pairs could be helpful for understanding the underlying regulatory mechanisms in HNSCC.
Methods
Overall design of the study
Firstly, an integrated analysis of three individual GEO datasets was performed by R software and identified DEGs. Then, selected DEGs underwent a series of further analyses including enrichment analysis, PPI network analysis and public database validation. Subsequently, DEMs were screened out and miRNA target genes were predicted by FunRich software. Significant miRNA-mRNA regulatory network was finally established and potential molecular regulatory mechanism was detected. More importantly, selected hub genes were verified by qPCR experiment conducted on HNSCC cell lines and clinical samples.
Microarray datasets
Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) is a web-accessible database with a collection of gene expression datasets and platform records. In this study, four independent HNSCC microarrays were downloaded from GEO for differential expression analysis. The detailed information of each microarray was summarized in Table 1.
Table 1. Details of the GEO datasets.
Identification of DEMs and DEGs from multiple datasets
DEMs and DEGs were selected by the limma package in R language with different criterions. P value <0.01 and |logFC| >1.5 were set as the threshold for DEGs. While those met the criteria of P value <0.05 and |logFC| >1.0 were screened out as DEMs. Overlapping DEGs were identified through the intersection of the results from three individual GEO microarrays.
GO and KEGG pathway enrichment analysis
Gene ontology (GO, http://www.geneontology.org/) is a universally used bioinformatics tool for investigating functional relationships between gene products and predicting three main aspects: biological process (BP), cellular component (CC), and molecular function (MF). Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/) is a database for analyzing enriched pathways of the selected genes in order to further understand gene functions. DAVID is an online database (https://david.ncifcrf.gov/) which was frequently utilized to conduct GO and KEGG pathway analysis.
Protein-protein interaction (PPI) network and module analysis
STRING database (https://string-db.org) is an online software which could provide an integrated analysis of the direct or indirect associations among selected genes. The overlapping DEGs were mapped to STRING to create an interactive network and then visualized through the Cytoscape software (www.cytoscape.org). Subsequently, the Molecular Complex Detection (MCODE) plugin was applied to screen out significant gene modules. Meanwhile, the cytoHubba plugin was utilized to identify the key mRNAs in this complicated network. The top 10 genes with highest scores were determined as hub genes and subjected to further analyses.
Identification of potential transcription factors of DEMs
FunRich (http://www.funrich.org) is a public accessible software that has the ability of identifying the enriched transcription factors of uploaded miRNAs. In the current study, DEMs were subjected to FunRich for analysis and the top 10 essential transcription factors that may regulate the DEMs were finally identified.
Prediction of miRNA target genes and miRNA-mRNA regulatory network
It was suggested that the functional roles of miRNAs mainly reside in the regulation of target genes. Therefore, target gene prediction is especially important, which could help understand the biological functions and enriched pathways of miRNAs indirectly (15). The miRNA enrichment function in FunRich was adopted to implement miRNA target prediction. Moreover, the intersection of predicted target genes of DEMs and DEGs was determined as significantly differentially expressed target genes. Later, these significant differentially expressed target genes and the corresponding miRNAs were put to the Cytoscape software to establish the miRNA-mRNA regulatory network.
Validation of gene expression levels in multiple online databases
GEPIA (http://gepia.cancer-pku.cn/) is a newly established platform which recollects TCGA and GTEx data and presents differential expression analysis of human genes. In the present study, we overviewed the mRNA level of selected genes with |log2FC| =1.0 and P value =0.05 set as the cutoff.
The human protein atlas (HPA) (https://www.proteinatlas.org/) is a database that offers transcription and translation data on human normal and pathological tissues. In this study, the distribution and protein level of differentially expressed target genes was investigated in HPA by immunohistochemistry analysis.
Cell culture
The detailed resource and culture conditions of the cells used in this study were as described before (16). Specifically, SCC-4, SCC-25 and CAL 27 cells were purchased from the American Type Culture Collection (ATCC, U.S.A.), and the human HNSCC cell lines HN4, HN6 and HN30 were kindly provided by the University of Maryland Dental School. SCC-4 and SCC-25 cells were cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (GIBCO-BRL, U.S.A.) supplemented with 10% fetal bovine serum (GIBCO-BRL, U.S.A.) and 1% penicillin and streptomycin. The other cells were cultured in DMEM medium with the same additives. Furthermore, normal oral epithelial cells which were obtained from primary culture were used as normal counterpart and they were cultured in keratinocyte serum-free medium (GIBCO-BRL, USA) containing 0.2 ng/mL recombinant epidermal growth factor (Invitrogen, USA). All the cells were maintained at 37 °C in a humidified atmosphere.
Tissue specimens
HNSCC samples were obtained from the Department of Oral and Maxillofacial-Head and Neck Oncology, Ninth People’s Hospital, Shanghai Jiao Tong University of Medicine (Shanghai, China). In total, 33 pairs of HNSCC tissues and adjacent non-tumor tissues were collected from patients and then quickly frozen in liquid nitrogen and stored at −80°C until use. The protocols were approved by Review Board of the Medical Ethics Committee of the Ninth People’s Hospital. This study received written informed consent from all the patients involved. Additionally, tissue samples used in this study were pathologically confirmed as HNSCC and tumor pathological differentiation and clinical stage were defined according to World Health Organization Classification of Tumors and the TNM classification system of the International Union Against Cancer (1988), respectively.
RNA extraction and quantitative real-time PCR
Total RNA of cells and tissues was extracted by TRIzol reagent (Takara, Japan) and reverse-transcribed into cDNA using a PrimeScript™ RT reagent kit (Takara, Japan). Real-time PCR was performed using SYBR Premix Ex Taq Reagent Kit (Takara, Japan) with ABI StepOne real-time PCR system (Applied Biosystems, USA). The reaction conditions were performed according to the manufactures’ instructions. The detailed sequences of the primers used in the experiment are presented as follows. SLC16A1 (forward primer: 5'-AGGTCCAGTTGGATACACCCC-3' and reverse primer: 5'-GCATAAGAGAAGCCGATGGAAAT-3'), COL4A1 (forward primer: 5'-GGACTACCTGGAACAAAAGGG-3' and reverse primer: 5'-GCCAAGTATCTCACCTGGATCA-3'), and GAPDH (forward primer: 5'-ACAACTTTGGTATCGTGGAAGG-3' and reverse primer: 5'-GCCATCACGCCACAGTTTC-3'). mRNA levels of genes were normalized against GAPDH and 2−ΔΔCt method was adopted to assess the relative expression level.
Statistical analysis
All the statistical analyses were performed by the SPSS 23.0 software. Student's two-tailed t test was applied to make comparisons between two groups. P<0.05 was considered statistically significant throughout this study.
Results
Identification of DEMs and DEGs in the GEO datasets
Three mRNA microarray datasets (GSE23558, GSE31056, GSE30784) and one miRNA microarray dataset (GSE34496) were obtained from GEO database and analyzed through R language. A total of 35 DEMs and 193 DEGs were screened out from the collected data of HNSCC samples. Volcano plots were generated to manifest upregulated (red) and downregulated (blue) genes between cancer and normal controls from three studies, respectively (Figure 1A,B,C). Then, 193 overlapping DEGs were identified through the intersection of three independent datasets (Figure 1D).
Figure 1.
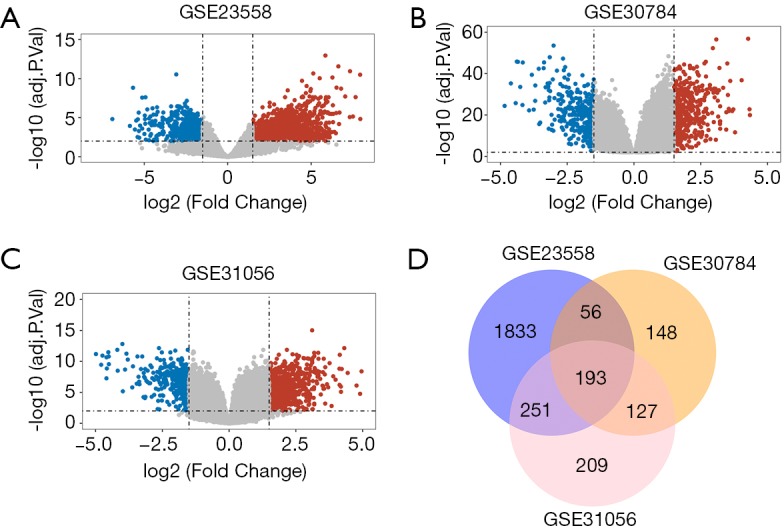
Differentially expressed genes (DEGs) identification by bioinformatics analysis. (A) Volcano plot of DEGs in GSE23558. (B) Volcano plot of DEGs in GSE30784. (C) Volcano plot of DEGs in GSE31056. Red, blue and gray color indicates relatively high, low and equal expression of genes, respectively. P<0.01 and |log(FC)| >1.5 were set as the threshold. (D) Overlapping DEGs commonly screened out from three GEO datasets.
GO and KEGG analysis of the overlapping DEGs
To have a more profound understanding of selected DEGs, GO and KEGG pathway enrichment analysis was performed in David. The top 10 biological processes that these DEGs enriched in were presented in Figure 2A, including cell adhesion, extracellular matrix organization, and inflammatory response. For cellular component, the results showed enrichment in extracellular regions, such as extracellular matrix, extracellular space, and extracellular exosome (Figure 2B). The importance of extracellular components in cancer progression has been gradually recognized as more and more findings illustrated the intimate communications between tumor environment and extracellular matrix. Therefore, our findings highlighted the critical roles of selected DEGs in HNSCC progression. Regarding molecular function classification, the DEGs particularly participated in the following functions: oxygen and heparin binding, peptidase activity, and cytokine activity (Figure 2C). More importantly, KEGG analysis suggested the overlapping DEGs were primarily enriched in cancer-related pathways, such as focal adhesion, ECM-receptor interaction, and cytokine-cytokine receptor interaction (Figure 2D).
Figure 2.

GO and KEGG pathway enrichment analysis of the overlapping DEGs in HNSCC. (A) Top10 of biological process. (B) Top10 of cellular component. (C) Top10 of molecular function. (D) Top10 of KEGG pathway.
PPI network of overlapping DEGs and hub gene identification
A PPI network was established by the STRING database. The three most outstanding modules were subsequently identified by the MCODE application based on the connective degrees (Figure 3A,B,C). In the meantime, cytoHubba plugin was applied to screen out hub genes, defined as having the highest degree of connectivity within the PPI network (Figure 3D). It turned out that FN1 was the most significant gene with the connectivity degree of 32, followed by STAT1 (degree =15) and MMP3 (degree =13).
Figure 3.

Protein-protein interactions (PPI) network, module analysis, and hub gene identification. (A,B,C) The most significant modules screened out by using Molecular Complex Detection (MCODE) plugin in Cytoscape software. (D) The top ten hub genes identified by the CytoHubba plugin in Cytoscape.
Determination of hub gene expression in GEPIA database
To demonstrate the reliability and accuracy of the results of bioinformatics analyses, we examined the transcriptional level of hub genes in GEPIA database, a platform from which TCGA data for multiple cancers could be obtained. We set the cutoff as |log2FC| =1.0 and P value =0.05. It was shown in Figure 4 that all hub genes were remarkably upregulated in HNSCC, implying their possible oncogenic functions.
Figure 4.

Validation of hub gene expression level in GEPIA database. The threshold was set as P value=0.05 and |log2(FC)| =1. Red indicates HNSCC samples and grey indicates normal samples. *, P<0.05.
Screening of potential transcription factors and target genes of DEMs
Analysis result of GSE34496 identified 35 DEMs and a volcano plot showing the distribution of these DEMs was presented in Figure 5A. Afterwards, we used the heatmap2 package in R to plot a heatmap based on the expression levels of DEMs in GSE34496 (Figure 5B). Each row represented an individual sample and each column represented a specific miRNA. Since transcription factors were demonstrated to act as essential factors in miRNA, the top 10 enriched transcription factors were investigated by FunRich software, namely EGR1, SP1, SP4, POU2F1, NFIC, RREB1, RORA, ZFP161, FOXD1 and HOXD8 (Figure 5C). Subsequently, the target genes predicted by FunRich were subjected to GO/KEGG analyses and detailed outcomes were shown in Figure 5D,E,F,G.
Figure 5.

Screening of potential transcription factors and target genes of DEMs. (A) Volcano plot of DEMs in GSE34496. Red, blue and gray color represents relatively high, low and equal expression of genes, respectively. P<0.05 and |log(FC)| >1.0 were set as the criteria. (B) The heatmap showing the DEMs expression profiles in HNSCC and compared normal samples in GSE34496. Red represents upregulation and green represents downregulation. (C) Identification of the potential transcription factors of DEMs by FunRich software. (D,E,F) The top 10 of biological process (D), cellular component (E), and molecular function (F) of the target genes of DEMs. (G) The top 10 enriched KEGG pathways of the target genes of DEMs.
miRNA-mRNA regulatory network
For a more intuitional visualization of target genes composition and correlation, Cytoscape was utilized to make a comprehensive target gene network. To clarify the complicated correlations between miRNAs and mRNAs, essential miRNA-mRNA pairs were identified based on the expression profiles. The significantly differentially expressed target genes were screened out by the intersection of predicted target genes of DEMs and overlapping DEGs (Figure 6A). Finally, six essential miRNA-mRNA pairs were identified, implying the crucial effect of them on HNSCC (Figure 6B).
Figure 6.

miRNA-mRNA regulatory network in HNSCC. (A) Venn diagram of the overlapping DEGs and the target genes of DEMs. (B) A total of six miRNA-mRNA pairs were identified underlying HNSCC progression.
Detection of the differentially expressed target genes in multiple online databases
The transcriptional expression of the differentially expressed target genes between HNSCC tissues and normal tissues were determined in GEPIA. The mRNA levels of ITGA6, COL4A1, and SLC16A1 were shown to be significantly higher in HNSCC, suggesting their potential oncogenic effect (Figure 7A). Meanwhile, the relative underexpression of PPM1L, GREM2, and ID4 implied the tumor suppressor role of them in cancer development. The corresponding protein level of the target genes was further explored through HPA database, with similar expression pattern suggested in Figure 7B.
Figure 7.

Detection of the differentially expressed target genes in multiple online databases. (A) Relative mRNA level of differentially expressed target genes was investigated in GEPIA database. The threshold was set as |Log2FC| =1 and P=0.05; *, P<0.05. (B) Investigation of differentially expressed target genes protein level in HPA database (magnification: 200×).
Investigation of SLC16A1 and COL4A1 expression in HNSCC cell lines and clinical samples
To lay a solid groundwork for further research, we chose to verify the expression level of SLC16A1 and COL4A1 in clinical samples. The results of qPCR experiment showed a remarkable higher level of SLC16A1 and COL4A1 in HNSCC cell lines and tissues when compared to normal controls (Figure 8A,B). Further analysis of the relationship of SLC16A1 and COL4A1 with clinicopathological parameters found that the upregulation of COL4A1 in HNSCC was positively correlated with advanced tumor stage and lymph node metastasis (Figure 8C,D; Tables 2,3). Moreover, statistical analysis suggested that the expression level of SLC16A1was closely related with smoking and drinking behaviors, with higher levels in patients with smoking or drinking history (Figure 8E,F). However, there are three highly expressers of SLC16A1 that may skew the correlations of smoking and alcohol consumption analysis. From our perspective, the statistical significance may not firmly come to the conclusion that SLC16A1 was intimately associated with tobacco or alcohol. We need to collect more HNSCC samples in the future to get a more reliable analysis.
Figure 8.

Validation of SLC16A1 and COL4A1 in HNSCC cell lines and tissues. (A) The relative higher level of SLC16A1 and COL4A1 in HNSCC cell lines when compared to normal oral epithelial cells. (B) SLC16A1 and COL4A1 mRNA levels were determined by qPCR in 33 pairs of HNSCC tumor tissues and adjacent normal tissues. (C) The upregulation of COL4A1 was correlated with advanced TNM stage in HNSCC patients. (D) The relative higher expression level of COL4A1 was associated with lymph node metastasis in HNSCC patients. (E,F) A significant higher level of SLC16A1 was observed in HNSCC patients with smoking or drinking history. *, P<0.05.
Table 2. Relationship between SLC16A1 level and clinicopathologic features (N=33).
| Characteristics | No. of patients | PYGM △Cta, mean ± SD | Non-parametric test value | P value | |
|---|---|---|---|---|---|
| No. | % | ||||
| Age (years) | |||||
| <60 | 10 | 30.3 | 60.96±86.49 | Z =−0.838 | 0.402 |
| ≥60 | 23 | 69.7 | 44.32±100.29 | ||
| Gender | |||||
| Female | 10 | 30.3 | 49.21±88.17 | Z =−0.627 | 0.531 |
| Male | 23 | 69.7 | 52.32±97.82 | ||
| Smoking history | |||||
| Nonsmoker | 21 | 63.6 | 22.68±15.68 | Z =−2.330 | 0.026 |
| Smoker | 12 | 36.4 | 95.53±139.66 | ||
| Alcohol history | |||||
| Nondrinker | 23 | 69.7 | 26.42±18.62 | Z =−2.136 | 0.041 |
| Drinker | 10 | 30.3 | 95.06±146.97 | ||
| Tumor size (cm) | |||||
| ≤4 | 23 | 69.7 | 45.42±69.25 | Z =−0.353 | 0.724 |
| >4 | 10 | 30.3 | 65.09±138.38 | ||
| Lymph node metastasis | |||||
| pN0 | 23 | 69.7 | 43.44±71.20 | Z =−0.726 | 0.468 |
| pN1 to pN2 | 10 | 30.3 | 67.26±130.36 | ||
| TNM stage | |||||
| I-II | 17 | 51.5 | 53.47±81.80 | Z =−0.108 | 0.914 |
| III-IV | 16 | 48.5 | 49.41±106.07 | ||
| Pathological differentiation | |||||
| Well | 30 | 90.9 | 40.34±61.77 | Z =−2.277 | 0.030 |
| Moderately/poorly | 3 | 9.1 | 161.75±254.64 | ||
| Disease site | |||||
| Tongue | 11 | 33.3 | 25.09±18.19 | H =1.871 | 0.143 |
| Gingival | 8 | 24.2 | 24.23±10.43 | ||
| Cheek | 8 | 24.2 | 10.83±5.65 | ||
| Floor of mouth | 2 | 3.0 | 39.83±24.57 | ||
| Oropharynx | 4 | 12.1 | 15.61±14.93 | ||
| Recurrence | |||||
| No | 26 | 78.8 | 60.17±103.95 | Z =−1.321 | 0.186 |
| Yes | 7 | 21.2 | 18.74±12.53 | ||
SD, standard deviation; pN, pathological lymph node status; TNM stage, tumor-lymph node-metastasis stage. a△Ct indicates the difference in the cycle number at which a sample’s fluorescent signal passes a given threshold above baseline (Ct) derived from a specific gene compared with that of β-actin in tumor tissues.
Table 3. Relationship between COL4A1 level and clinicopathologic features (N=33).
| Characteristics | No. of Patients | PYGM △Cta, mean ± SD | Non-parametric test value | P value | |
|---|---|---|---|---|---|
| No. | % | ||||
| Age (years) | |||||
| <60 | 10 | 30.3 | 28.61±19.99 | Z =−1.449 | 0.147 |
| ≥60 | 23 | 69.7 | 19.58±13.11 | ||
| Gender | |||||
| Female | 10 | 30.3 | 18.34±9.70 | Z =−0.588 | 0.557 |
| Male | 23 | 69.7 | 24.05±17.68 | ||
| Smoking history | |||||
| Nonsmoker | 21 | 63.6 | 19.92±13.03 | Z =−1.160 | 0.246 |
| Smoker | 12 | 36.4 | 26.51±19.59 | ||
| Alcohol history | |||||
| Nondrinker | 23 | 69.7 | 21.43±18.15 | Z =−1.528 | 0.127 |
| Drinker | 10 | 30.3 | 24.36±8.43 | ||
| Tumor size (cm) | |||||
| ≤4 | 23 | 69.7 | 20.04±12.79 | Z =−0.666 | 0.505 |
| >4 | 10 | 30.3 | 26.72±21.28 | ||
| Lymph node metastasis | |||||
| pN0 | 23 | 69.7 | 17.32±9.11 | Z =−2.350 | 0.019 |
| pN1 to pN2 | 10 | 30.3 | 33.82±21.65 | ||
| TNM stage | |||||
| I-II | 17 | 51.5 | 16.04±8.21 | Z =−2.089 | 0.037 |
| III-IV | 16 | 48.5 | 28.99±19.13 | ||
| Pathological differentiation | |||||
| Well | 30 | 90.9 | 22.84±16.13 | Z =−0.679 | 0.497 |
| Moderately/poorly | 3 | 9.1 | 14.24±0.81 | ||
| Disease site | |||||
| Tongue | 11 | 33.3 | 29.37±19.32 | H =0.826 | 0.520 |
| Gingival | 8 | 24.2 | 18.50±12.90 | ||
| Cheek | 8 | 24.2 | 18.24±16.80 | ||
| Floor of mouth | 2 | 3.0 | 22.46±9.62 | ||
| Oropharynx | 4 | 12.1 | 19.67±4.82 | ||
| Recurrence | |||||
| No | 26 | 78.8 | 22.37±17.03 | Z =−0.264 | 0.792 |
| Yes | 7 | 21.2 | 22.15±10.81 | ||
SD, standard deviation; pN, pathological lymph node status; TNM stage, tumor-lymph node-metastasis stage. a△Ct indicates the difference in the cycle number at which a sample’s fluorescent signal passes a given threshold above baseline (Ct) derived from a specific gene compared with that of β-actin in tumor tissues.
Discussion
HNSCC is a tumor type that determined as one of the most prevalent malignancies worldwide. Although enormous work has been devoted to exploring molecular pathology and investigating targeted therapies for HNSCC, there still lacks effective biomarkers or efficient treatment methods to screen and treat HNSCC patients (17). To develop innovative approach to cancer treatment, clarifying the mechanisms involved in the pathogenesis of HNSCC has spawned great interest from researchers.
Microarray technology and integrated molecular analysis could provide innovative insights into the molecular mechanisms underlying tumor formation (18,19). Moreover, the combined application of molecular biology and bioinformatics technique contributes to the discovery of relative accurate therapy targets and reliable biomarkers for cancer diagnosis and prognosis. Computational analyses predict that almost 30% of all human genes are regulated more or less by miRNAs (20). To be more specific, each miRNA can target multiple genes, and one specific gene may be regulated by a diversity of miRNAs. Therefore, miRNAs and their potential target genes could form a complicated and comprehensive miRNA-mRNA regulatory network (15). In order to establish miRNA-target gene regulatory network in HNSCC and elucidate the underlying mechanisms involved in tumor progression, we made an integrated bioinformatics-based analysis. Since the integration of multiple microarray datasets could lead to more reliable and accurate results (21), we utilized four independent HNSCC datasets in this study. Through bioinformatics analysis, 35 DEMs and 193 DEGs were screened out and GO analysis showed that the candidate genes mainly participate in cell adhesion, extracellular matrix organization and disassembly, and inflammatory response, all are indispensable biological processes involved in carcinogenesis (22). In the meantime, a majority of genes were enriched in extracellular region including matrix and exosome and it was explicitly illustrated that there exist complex communications between tumor cells and the ECM (23). Moreover, KEGG analysis suggested these genes showed enrichment in focal adhesion, ECM-receptor interaction and cytokine-cytokine receptor interaction, which were intimately involved in HNSCC pathogenesis (24,25). KEGG pathway enrichment analyses provide us with a more in-depth understanding of the HNSCC pathogenesis and enable us to explore the potential molecular mechanisms of DEGs.
Networks offer a clear and straightforward representation of intricate interactions between gene nodes. Many domains have utilized network-based analysis, such as gene coexpression network (26), protein-protein interaction network (27,28), and cell-cell interaction network (29). In the present study, PPI network based on overlapping DEGs was constructed and hub genes were identified. Our results showed that FN1 was the most significant gene with the highest connectivity degree, followed by STAT1 and MMP3. FN1, a composition part of the extracellular matrix, was described to be elevated in different malignancies including hepatocellular carcinoma, gastric cancer, and head and neck cancer (30). It could mediate interactions between cells and extracellular matrix so as to influence cell differentiation, proliferation, migration, and adhesion. Signal transducer and activator of transcription (STAT) proteins are transcription factors downstream of cytokines and growth factors. Previous studies indicated STAT1 may be an effective prognostic indicator and a specific biomarker for evaluating the response to drug treatment in HNSCC patients (31,32). Matrix metalloproteinases (MMPs), traditionally recognized to function at matrix remolding, were suggested to mediate several biological process in cancer, such as tumor invasion, angiogenesis, and metastasis. In this study, MMP3 was identified to be a hub gene in HNSCC and a published study showing increased MMP3 expression in neoplasms supported our findings (33).
Bioinformatics predictions of miRNA target genes could facilitate the investigation of miRNA functions and help to illuminate the gene regulation network of miRNAs. All the predicted target genes of DEMs underwent enrichment analyses and the results emphasized their potential functions in cancer. Specifically, it turned out that these genes were mainly enriched in intrinsic apoptotic signaling pathway and transcription regulation, both were closely related to the biology and phenotypes of tumor cells. KEGG pathway enrichment results further suggested their intimate associations with tumor progression, such as TGF-β signaling pathway, Notch signaling pathway, and cAMP signaling pathway. It implied the target genes of miRNAs may mediate HNSCC by a variety of signaling pathways. Also, the enrichment category of ‘microRNAs in cancer’ enhanced the reliability of our analysis. Finally, we identified 6 miRNA-mRNA regulatory pairs involving three kinds of miRNAs. Several studies have reported the tumor suppressive role of miR-140-3p, which was validated to influence lung cancer progression by targeting BRD9 and ATP8A1 directly (34,35). miR-342-3p, a newly developed cancer-related miRNA, was reported to be involved in a number of physiological and pathological processes, serving as a promising anticancer therapy target (36,37). miR-34c-5p, which belongs to the miR-34 family, consists of three main members, namely miR-34a, miR-34b, and miR-34c. The hypermethylation of miR-34c-5p promoter led to its repression and its tumor suppressive functions on multiple human cancers (38). It could also affect cancer progression, drug resistance, and patient prognosis (39). Meanwhile, six key genes (ID4, PPM1L, GREM2, ITGA6, COL4A1, SLC16A1) were further investigated in our research. ID4, a member of the inhibitor of DNA binding (ID) family proteins, was demonstrated to be correlated with tumor differentiation and patient prognosis and it hinted that promoter hypermethylation may account for the silencing of ID4 in cancers (40). PPM1L, originally discovered as a potent regulator of stress-activated protein kinase signaling, has been investigated its candidate tumor suppressor roles in recent years (41). GREM2 is a gene located in chromosome 1q43 and belongs to the cystine knot superfamily. A study discovered many differentially expressed genes in tumor clusters including GREM2, laying the foundation of its possible role in tumorigenesis (42). Another study demonstrated that GREM2 could biologically mediate the proliferation, apoptosis, migration, and invasion of cancer cells through mediating the JNK signaling pathway, making GREM2-mediated JNK signaling pathway a promising therapeutic strategy (43). It is obvious that tumor microenvironment has increasingly attracted more and more attention and acknowledged as an essential factor in tumor context. Integrins not only regulate interactions within the extracellular matrix (ECM), but also mediate intracellular signaling events that communicate from the tumor microenvironment to the inside of tumor cells. ITGA6, a vital component of integrins, was suggested to contribute to the aggressive phenotypes of cancer directly (44). Therefore, future studies concentrated on the genetic modulation of ITGA6 may be valuable in exploring therapeutic modalities for tumors. COL4A1, which belongs to the collagen family, constitutes a major component of the basement membrane that surrounds tumor vasculature, leading to an increased angiogenesis activity in cancers. Moreover, it has been verified to be tightly associated with cell viability, cell cycle arrest, and cell adhesion. SLC16A1 is a transcription factor which encodes MCT1 protein, a member of monocarboxylate transporter (MCT) family. Its primary function is to transport lactate into and out of tumor cells, serving as a key mediator in maintaining tumor microenvironment homeostasis. A number of tumors presented elevated levels of SLC16A1 and targeting MCT1 has been certified to inhibit tumor progression and enhance chemotherapeutic drugs sensitivity (45). More in depth investigation suggested the involvement of NF-κB pathway underlying MCT1-mediated cancer progression (46). From what has been discussed above, we came to the conclusion that our selected genes were associated with different cancers, either act as regulators in biological processes or serve as efficient biomarkers for diagnosis and prognosis.
In this study, we validated the upregulation of SLC16A1 and COL4A1 in HNSCC tissues and emphasized their potential oncogenic roles in HNSCC progression. However, there also exist some deficiencies in this study. Firstly, the sample size for validation was relatively small. Secondly, there is lack of in-depth in vitro experiments. Last but not the least, HPV status was not taken into account in our analysis. To get a more comprehensive analysis, we intend to determine the HPV status of the clinical samples we will collect in the future and make corresponding investigation. What is more, a recent review on non-coding RNAs discovered a number of long non-coding RNAs (lncRNAs) whose expression level could be utilized as biomarkers for head and neck tumors (2). More importantly, some of the lncRNAs function by sponging miRNAs or affecting miRNA stability. Therefore, more comprehensive studies focused on lncRNA-miRNA-mRNA network may be conducted in the future.
Conclusions
In conclusion, our research applied integrated bioinformatics methods to systematically analyze miRNAs and mRNAs in HNSCC. Ten hub genes were investigated as potent markers and six key miRNA-mRNA regulatory pairs were screened out as essential mediators in HNSCC. These findings could remarkably improve the overall understanding of the potential molecular interactions and the intricate mechanisms of HNSCC, facilitating the discovery of effective biomarkers for diagnosis and prognosis for HNSCC patients.
Acknowledgments
Funding: This work was supported by the National Key R&D Program of China (grant numbers 2017YFC 0840100, 2017YFC 0840110).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The protocols were approved by Review Board of the Medical Ethics Committee of the Ninth People’s Hospital. This study received written informed consent from the patients involved.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell 2004;5:311-6. 10.1016/S1535-6108(04)00090-X [DOI] [PubMed] [Google Scholar]
- 2.Cossu AM, Mosca L, Zappavigna S, et al. Long Non-coding RNAs as Important Biomarkers in Laryngeal Cancer and Other Head and Neck Tumours. Int J Mol Sci 2019;20. doi: . 10.3390/ijms20143444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun M, Gadad SS, Kim DS, et al. Discovery, Annotation, and Functional Analysis of Long Noncoding RNAs Controlling Cell-Cycle Gene Expression and Proliferation in Breast Cancer Cells. Mol Cell 2015;59:698-711. 10.1016/j.molcel.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014;25:666-81. 10.1016/j.ccr.2014.03.010 [DOI] [PubMed] [Google Scholar]
- 5.Beermann J, Piccoli MT, Viereck J, et al. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol Rev 2016;96:1297-325. 10.1152/physrev.00041.2015 [DOI] [PubMed] [Google Scholar]
- 6.Oliveira-Carvalho V, Carvalho VO, Silva MM, et al. MicroRNAs: a new paradigm in the treatment and diagnosis of heart failure? Arq Bras Cardiol 2012;98:362-9. 10.1590/S0066-782X2012000400011 [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Xie F, Bao X, et al. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol Cancer 2014;13:121. 10.1186/1476-4598-13-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakanishi H, Taccioli C, Palatini J, et al. Loss of miR-125b-1 contributes to head and neck cancer development by dysregulating TACSTD2 and MAPK pathway. Oncogene 2014;33:702-12. 10.1038/onc.2013.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mydlarz W, Uemura M, Ahn S, et al. Clusterin is a gene-specific target of microRNA-21 in head and neck squamous cell carcinoma. Clin Cancer Res 2014;20:868-77. 10.1158/1078-0432.CCR-13-2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CJ, Shen WG, Peng SY, et al. miR-134 induces oncogenicity and metastasis in head and neck carcinoma through targeting WWOX gene. Int J Cancer 2014;134:811-21. 10.1002/ijc.28358 [DOI] [PubMed] [Google Scholar]
- 11.Qin X, Guo H, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol 2019;20:12. 10.1186/s13059-018-1604-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao Y, Liu R, Gao C, et al. Identification of prognostic biomarkers for breast cancer based on miRNA and mRNA co-expression network. J Cell Biochem 2019;120:15378-88. 10.1002/jcb.28805 [DOI] [PubMed] [Google Scholar]
- 13.Dai P, He Y, Luo G, et al. Screening candidate microRNA-mRNA network for predicting the response to chemoresistance in osteosarcoma by bioinformatics analysis. J Cell Biochem 2019;120:16798-810. 10.1002/jcb.28938 [DOI] [PubMed] [Google Scholar]
- 14.Sui J, Liu Q, Zhang H, et al. Deep integrative analysis of microRNA-mRNA regulatory networks for biomarker and target discovery in chondrosarcoma. J Cell Biochem 2019;120:9631-8. 10.1002/jcb.28241 [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215-33. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin X, Yan M, Wang X, et al. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics 2018;8:921-40. 10.7150/thno.22182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Argiris A, Karamouzis MV, Raben D, et al. Head and neck cancer. Lancet 2008;371:1695-709. 10.1016/S0140-6736(08)60728-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajendran V, Gopalakrishnan C, Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. J Cell Biochem 2018;119:918-25. 10.1002/jcb.26257 [DOI] [PubMed] [Google Scholar]
- 19.Rajendran V. Structural analysis of oncogenic mutation of isocitrate dehydrogenase 1. Mol Biosyst 2016;12:2276-87. 10.1039/C6MB00182C [DOI] [PubMed] [Google Scholar]
- 20.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15-20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 21.Ma T, Liang F, Oesterreich S, et al. A Joint Bayesian Model for Integrating Microarray and RNA Sequencing Transcriptomic Data. J Comput Biol 2017;24:647-62. 10.1089/cmb.2017.0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonastre E, Brambilla E, Sanchez-Cespedes M. Cell adhesion and polarity in squamous cell carcinoma of the lung. J Pathol 2016;238:606-16. 10.1002/path.4686 [DOI] [PubMed] [Google Scholar]
- 23.Oudin MJ, Jonas O, Kosciuk T, et al. Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov 2016;6:516-31. 10.1158/2159-8290.CD-15-1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eke I, Cordes N. Focal adhesion signaling and therapy resistance in cancer. Semin Cancer Biol 2015;31:65-75. 10.1016/j.semcancer.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 25.Liu Q, Li A, Tian Y, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev 2016;31:61-71. 10.1016/j.cytogfr.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn R, Gupta R, Lai K, et al. Network analysis of psoriasis reveals biological pathways and roles for coding and long non-coding RNAs. BMC Genomics 2016;17:841. 10.1186/s12864-016-3188-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia J, Liu Z, Xiao X, et al. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition. J Biomol Struct Dyn 2016;34:1946-61. 10.1080/07391102.2015.1095116 [DOI] [PubMed] [Google Scholar]
- 28.Li BQ, Huang T, Liu L, et al. Identification of colorectal cancer related genes with mRMR and shortest path in protein-protein interaction network. PLoS One 2012;7:e33393. 10.1371/journal.pone.0033393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartwell LH, Hopfield JJ, Leibler S, et al. From molecular to modular cell biology. Nature 1999;402:C47-52. 10.1038/35011540 [DOI] [PubMed] [Google Scholar]
- 30.Kim H, Park J, Kim Y, et al. Serum fibronectin distinguishes the early stages of hepatocellular carcinoma. Sci Rep 2017;7:9449. 10.1038/s41598-017-09691-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laimer K, Spizzo G, Obrist P, et al. STAT1 activation in squamous cell cancer of the oral cavity: a potential predictive marker of response to adjuvant chemotherapy. Cancer 2007;110:326-33. 10.1002/cncr.22813 [DOI] [PubMed] [Google Scholar]
- 32.Schmitt NC, Trivedi S, Ferris RL. STAT1 Activation Is Enhanced by Cisplatin and Variably Affected by EGFR Inhibition in HNSCC Cells. Mol Cancer Ther 2015;14:2103-11. 10.1158/1535-7163.MCT-15-0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005;436:123-7. 10.1038/nature03688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong W, Yao C, Teng X, et al. MiR-140-3p suppressed cell growth and invasion by downregulating the expression of ATP8A1 in non-small cell lung cancer. Tumour Biol 2016;37:2973-85. 10.1007/s13277-015-3452-9 [DOI] [PubMed] [Google Scholar]
- 35.Huang H, Wang Y, Li Q, et al. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett 2019;446:81-9. 10.1016/j.canlet.2019.01.007 [DOI] [PubMed] [Google Scholar]
- 36.Xue X, Fei X, Hou W, et al. miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett 2018;412:170-8. 10.1016/j.canlet.2017.10.024 [DOI] [PubMed] [Google Scholar]
- 37.Li XR, Chu HJ, Lv T, et al. miR-342-3p suppresses proliferation, migration and invasion by targeting FOXM1 in human cervical cancer. FEBS Lett 2014;588:3298-307. 10.1016/j.febslet.2014.07.020 [DOI] [PubMed] [Google Scholar]
- 38.Gu J, Wang G, Liu H, et al. SATB2 targeted by methylated miR-34c-5p suppresses proliferation and metastasis attenuating the epithelial-mesenchymal transition in colorectal cancer. Cell Prolif 2018;51:e12455. 10.1111/cpr.12455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tung SL, Huang WC, Hsu FC, et al. miRNA-34c-5p inhibits amphiregulin-induced ovarian cancer stemness and drug resistance via downregulation of the AREG-EGFR-ERK pathway. Oncogenesis 2017;6:e326. 10.1038/oncsis.2017.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L, Liu C, Vandeusen J, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet 2005;37:265-74. 10.1038/ng1521 [DOI] [PubMed] [Google Scholar]
- 41.Thean LF, Loi C, Ho KS, et al. Genome-wide scan identifies a copy number variable region at 3q26 that regulates PPM1L in APC mutation-negative familial colorectal cancer patients. Genes Chromosomes Cancer 2010;49:99-106. [DOI] [PubMed] [Google Scholar]
- 42.Edfeldt K, Bjorklund P, Akerstrom G, et al. Different gene expression profiles in metastasizing midgut carcinoid tumors. Endocr Relat Cancer 2011;18:479-89. 10.1530/ERC-10-0256 [DOI] [PubMed] [Google Scholar]
- 43.Ran A, Guan L, Wang J, et al. GREM2 maintains stem cell-like phenotypes in gastric cancer cells by regulating the JNK signaling pathway. Cell Cycle 2019;18:2414-31. 10.1080/15384101.2019.1646561 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Yang B, Chen Z, Huang Y, et al. Identification of potential biomarkers and analysis of prognostic values in head and neck squamous cell carcinoma by bioinformatics analysis. Onco Targets Ther 2017;10:2315-21. 10.2147/OTT.S135514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li KK, Pang JC, Ching AK, et al. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum Pathol 2009;40:1234-43. 10.1016/j.humpath.2009.02.003 [DOI] [PubMed] [Google Scholar]
- 46.Zhao Z, Wu MS, Zou C, et al. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF-kappaB pathway. Cancer Lett 2014;342:150-8. 10.1016/j.canlet.2013.08.042 [DOI] [PubMed] [Google Scholar]