

Abstract
Plant artificial micro-RNAs (amiRs) have been engineered to target viral genomes and induce their degradation. However, the exceptional evolutionary plasticity of RNA viruses threatens the durability of the resistance conferred by these amiRs. It has recently been shown that viral populations not experiencing strong selective pressure from an antiviral amiR may already contain enough genetic variability in the target sequence to escape plant resistance in an almost deterministic manner. Furthermore, it has also been shown that viral populations exposed to subinhibitory concentrations of the antiviral amiR speed up this process. In this article, we have characterized the molecular evolutionary dynamics of an amiR target sequence in a viral genome under both conditions. The use of Illumina ultradeep sequencing has allowed us to identify virus sequence variants at frequencies as low as 2 × 10−6 and to track their variation in time before and after the viral population was able of successfully infecting plants fully resistant to the ancestral virus. We found that every site in the amiR-target sequence of the viral genome presented variation and that the variant that eventually broke resistance was sampled among the many coexisting ones. In this system, viral evolution in fully susceptible plants results from an equilibrium between mutation and genetic drift, whereas evolution in partially resistant plants originates from more complex dynamics involving mutation, selection, and drift.
Keywords: artificial micro-RNAs, experimental evolution, next-generation sequencing, population genetics, resistant plants, virus evolution
Introduction
Micro-RNAs (miRNAs) are short RNAs found in eukaryotic cells that operate as post-transcriptional regulators of gene expression (He and Hannon 2004). They regulate the abundance of target mRNAs by guiding the RNA-induced silencing complex (RISC) to cleave the corresponding complementary sequence. As changes in the 21-nt-long miRNA sequence do not affect miRNA biogenesis and maturation (Vaucheret et al. 2004; Guo et al. 2005), it is possible to redesign the miRNA sequence to target different transcripts using different pre-miRNAs as backbones (Niu et al. 2006; Schwab et al. 2006; Qu et al. 2007; Warthmann et al. 2008). One application of this technology is to produce plants expressing artificial miRNAs (amiRs) targeting viral genomes, thus conferring resistance to viral infection (Niu et al. 2006; Qu et al. 2007). Niu et al. (2006) used the pre-miRNA159a precursor as backbone to construct two different amiR159 with sequences complementary to the RNA genome of turnip yellow mosaic virus (TYMV) and turnip mosaic virus (TuMV). In short, the original sequences forming the stem of the pre-miRNA159a were replaced by the appropriate viral target sequences in complementary polarities to maintain the correct stem–loop structure and subsequent processing by the DCL1 nuclease (Niu et al. 2006). Transgenic expression of these amiRs in Arabidopsis thaliana conferred high levels of specific resistance against the corresponding virus. Similarly, a gene-silencing mechanism (RNAi) has been used in in vitro assays as antiviral agent to inhibit the replication of human viruses, such as human immunodeficiency virus type 1 (HIV-1; Coburn and Cullen 2002), hepatitis C virus (Krönke et al. 2004), and influenza A virus (Ge et al. 2003). In all these experiments, a single amiR was expressed, and thus, resistance strictly depended on the match between this amiR and the corresponding viral sequence.
A major issue confronting these amiR-based antiviral strategies has been the emergence of resistant virus variants (Boden et al. 2003; Das et al. 2004; Gitlin et al. 2005; Westerhout et al. 2005; Lin et al. 2009; Lafforgue et al. 2011). These variants differ from the wild-type (WT) virus by at least one point mutation in the 21-nt target leading to imperfect matching with the corresponding amiR and hence to inefficient or ineffective processing by RISC (Westerhout et al. 2005; Sabariegos et al. 2006; von Eije et al. 2008). Although the RNAi machinery tolerates changes in some positions of the 21-nt target, it is sensitive to changes in some others, particularly at the center of the target site (Elbashir et al. 2001; Westerhout and Berkhout 2007; Lin et al. 2009). Moreover, it has been shown that the 21 TuMV genotypes resulting from introducing every single synonymous mutation in the 21-nt target that successfully infected transgenic plants expressing amiR accumulated additional changes at alternative sites within the 21-nt target, further jeopardizing the resistance of the transgenic plants (Lin et al. 2009).
Taken together, these results show that some changes in the 21-nt target sequence may generate virus escape variants. However, the relevance of these escape variants in natural viral populations has not been clarified. In other words, to evaluate the viability of antiviral therapies based on the transgenic expression of amiRs in plants, it is essential to understand how likely are viral populations to contain escape variants, which may be subsequently transmitted to immunized plants. Moreover, it is also crucial to evaluate whether variation in the expression of amiR transgenes in different tissues or at different stages of plant development, especially at subinhibitory concentrations, might affect the accumulation and evolution of viral escape mutants. More specifically, we are interested in addressing the following issues: 1) What is the likelihood of virus escape mutations arising and accumulating in a WT host population? 2) Does partial resistance favor the accumulation of escape mutants? 3) At what frequencies are these mutant viruses maintained in susceptible hosts? and 4) What sites in the 21-nt target are more critical for escaping from amiR surveillance?
To address the first of these issues, in a previous study, we experimentally evolved 25 independent lineages of TuMV (family Potyviridae) in susceptible WT A. thaliana plants. At each passage until resistance was broken (Lafforgue et al. 2011), we evaluated the infectivity of the evolving populations in transgenic A. thaliana plants (line 12-4) that were fully resistant to the ancestral virus due to high-level expression of amiR159-HCPro, an engineered variant of miRNA159 that is complementary to 21 nt within the TuMV cistron that encodes for the multifunctional protein HC-Pro (Niu et al. 2006). In the same work, we addressed the second issue, by evolving in the same way 25 additional TuMV lineages in A. thaliana transgenic plants (line 10-4) expressing the amiR159-HCPro at subinhibitory concentrations. Our results showed that TuMV populations replicating in both susceptible hosts (WT and 10-4) accumulated resistance-breaking alleles, resulting in overcoming the resistance of 12-4 plants. The rate at which resistance was broken was significantly faster for TuMV populations experiencing subinhibitory concentrations of the antiviral amiR159-HCPro during their evolution, thus suggesting that TuMV escape alleles were at higher frequencies in the partially resistant plants, possibly because of a selective advantage. This previous study mainly focused on making quantitative inferences about the likelihood of resistance breaking. We confirmed that resistance breaking had a genetic basis by characterizing the consensus sequence of the TuMV population isolated from the 12-4-resistant plants that first showed infection symptoms. However, we did not determine whether escape mutants were already present in the evolving populations before being inoculated in the 12-4 plants nor the level of polymorphism in the population replicating in these plants (especially whether they contained a fraction of nonresistant TuMV genotypes).
To address these issues, classical Sanger dideoxynucleotide sequencing methods are not appropriate, as they only allow the detection of viral variants present in the population at frequencies around 2 × 10−1 (e.g., Zagordi et al. 2011). On the other hand, next-generation sequencing (NGS) techniques can generate massive amounts of genetic data, which can be used to detect variants at much lower frequencies (Wang et al. 2007; Eriksson et al. 2008; Solmone et al. 2009; Cordey et al. 2010; Eckerle et al. 2010; Murcia et al. 2010; Zagordi et al. 2010; Willerth et al. 2010; Bunnik et al. 2011; Guo et al. 2011; Wrigth et al. 2011). In this study, we have generated tens of millions of short reads with Illumina sequencing to obtain an unprecedented ultradeep coverage of amiR targets in the TuMV genome, thus characterizing in great detail the genetic composition of TuMV lineages evolving in WT and 10-4 plants at different time points during the experimental evolution process and right after the first successful infection of 12-4-resistant plants. Although in general the limited length of Illumina reads makes it difficult to assess linkage among mutations, in our particular case this was not an issue because the 21-nt sequences corresponding to the amiR159-HC-Pro target were completely covered by the 76-nt-long reads. The validity of the Illumina technology for assessing virus diversity has been proved with studies of the severe acute respiratory syndrome coronavirus (Eckerle et al. 2010), human rhinovirus (Cordey et al. 2010), HIV-1 (Willerth et al. 2010), and foot-and-mouth disease virus (Wrigth et al. 2011).
Materials and Methods
Plant Material and Growth Conditions
Two homozygous T4 transgenic A. thaliana Col-0 lines expressing amiR159-HCPro were used in this study: 10-4 and 12-4 (Niu et al. 2006; Lafforgue et al. 2011). Plants were maintained in a growth chamber under 16 h light 25°C/8 h darkness 22°C; 12-4 plants were fully resistant to infection with the ancestral TuMV clone, whereas 10-4 plants showed incomplete penetrance and variable expressivity of the resistance character (Lafforgue et al. 2011).
Population Passages and Evaluation of Pathogenicity in A. thaliana 12-4 Plants
Details of the evolution experiments and evaluation of the pathogenicity of evolving populations were described elsewhere (Lafforgue et al. 2011). In short, a large stock of infectious sap was obtained from TuMV-infected Nicotiana benthamiana inoculated with a plasmid containing a TuMV cDNA (GenBank accession no. AF530055.2). Saps were obtained by grinding-infected tissues with 20 volumes of grinding buffer (50 mM potassium phosphate pH 7.0, 3% polyethylene glycol 6000). Aliquots of 5 µl 10% carborundum were applied to three different A. thaliana leaves, and inoculation was done mechanically by gentle rubbing with a cotton swab soaked with infectious sap. Twenty-five WT and 25 transgenic 10-4 A. thaliana plants were inoculated. Each plant represented the starting point for an independent evolutionary lineage. Fourteen days postinoculation (dpi), symptomatic tissue was collected for each lineage and ground in a mortar with liquid N2 and stored at −80°C. A portion of the ground tissue was extracted with grinding buffer and used to inoculate the next set of plants. A second portion was used for the pathogenicity tests in 12-4 plants. A third portion was used to purify RNA for reverse-transcription followed by polymerase chain reaction (RT-PCR) amplification of TuMV cDNA.
For the pathogenicity tests, 20 plants from the 12-4 line were inoculated as described earlier. Infection was determined as 14 dpi and the frequency of infected plants, that is pathogenicity, recorded. These challenge experiments were performed after every passage for each of the 50 evolving lineages.
RNA Preparation and RT-PCR Amplification of a TuMV cDNA
TuMV-infected tissue from A. thaliana plants (0.2 g) was ground with mortar and pestle in the presence of liquid N2 and RNA purified by chromatography using silica gel spin columns (Zymo Research). RNA was finally eluted from the columns with 12 µl 20 mM Tris–HCl, pH 8.5 and quantified by spectrophotometry (Nanodrop, Thermo Scientific).
RNA aliquots from TuMV-infected A. thaliana plants (100 ng) were subjected to reverse transcription with 50 U M-MuLV reverse transcriptase (Fermentas) in the presence of 5 pmol primer PI (5′-CAAGCAGAAGACGGCATACGAGCCTGATTCTGTTGTGACAC-3′, sequence complementary to TuMV-AF530055.2 positions 2,095–2,115 underlined) in 10 µl reactions for 45 min at 42°C, 10 min at 50°C, and 5 min at 60°C. Before the reaction, the primer was allowed to anneal to the RNA by incubation for 1.5 min at 98°C and snap cooling on ice. Reverse transcription reactions were stopped by heating at 72°C for 15 min. PCR amplifications were performed in 20 µl with 0.4 U of the high-fidelity Phusion DNA polymerase (Finnzymes) in HF buffer (Finnzymes) and 3% dimethyl sulfoxide, 0.2 mM dNTPs, and 1 µl of the previous reverse transcription reaction. Reactions also contained 0.5 µM primer PI and 0.5 µM of a particular version of primer PII. For multiplex deep sequencing, a series of 45 different PII primers was designed including a constant 5′ sequence required for the deep sequencing protocol, a variable 4-nt sequence (XXXX) for bar coding and a constant 3′ sequence (underlined) homologous to TuMV-AF530055.2 positions 2,011–2,031: 5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGACGATCXXXXGAAAGGCGAACCGGTAGAAC-3′. The reactions were incubated for 30 s at 98°C followed by 20 cycles of 10 s at 98°C, 30 s at 55°C, and 30 s at 72°C and a final extension of 10 min at 72°C. PCR products were separated by electrophoresis in 2% agarose gels in buffer TAE (40 mM Tris, 20 mM sodium acetate, and 1 mM ethylenediaminetetraacetic acid, pH 7.2) and stained with ethidium bromide. The TuMV cDNA amplification products (177 bp) from the different reactions were eluted from the gel using silica gel spin columns (Zymo Research) and quantified by spectrophotometry.
NGS
The amplified TuMV cDNAs from the evolution experiments in WT and 10-4 plants were mixed in separate pools. Each pool contained normalized amounts of the different cDNAs at 18 ng/µl. The two cDNA pools were subjected to NGS using the Illumina HiSeq 2000 sequencer at the Rockefeller University Genomics Resource Center.
Data Filtering and Analysis of Sequence Diversity
To exclude the considering sequencing or experimental miscalls as true mutations, we carefully considered the nucleotide qualities provided by the Illumina sequencing platform. Each nucleotide in each read has a quality score Q, which can be translated to measure of the probability, P, of it being an erroneous call with the expression 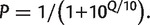 The average error probability increased with nucleotide position along the reads and reached 1% after nucleotide 65: these levels are incompatible with the reliable identification of low-frequency polymorphisms. Typically, this poor average performance is due to a restricted number of very low-quality reads, which suffered some problems during the base-calling process. Therefore, we removed all the reads from the analysis with an average sequencing error per nucleotide higher than 0.1% (corresponding to 5–15% of the sample total). The coverage of the samples obtained with the filtered reads ranged between 3.5 × 105 and 8.5 × 105-fold. Reads were then aligned to the reference sequence with a simple score-assigning routine, and new, sample-specific consensus sequences were obtained.
The average error probability increased with nucleotide position along the reads and reached 1% after nucleotide 65: these levels are incompatible with the reliable identification of low-frequency polymorphisms. Typically, this poor average performance is due to a restricted number of very low-quality reads, which suffered some problems during the base-calling process. Therefore, we removed all the reads from the analysis with an average sequencing error per nucleotide higher than 0.1% (corresponding to 5–15% of the sample total). The coverage of the samples obtained with the filtered reads ranged between 3.5 × 105 and 8.5 × 105-fold. Reads were then aligned to the reference sequence with a simple score-assigning routine, and new, sample-specific consensus sequences were obtained.
Subsequently, we estimated the frequency of site-specific polymorphisms from the frequency of mismatches to the reference genome found in the aligned reads. A proportion of these mismatches are expected to be artifacts, arising from base miscalling. To discriminate between real variation and sequencing artifacts, we used a variation of the method developed by Wrigth et al. (2011): we considered the probability of a sequencing error appearing at genome position i in read j, Pij, and we averaged over all the reads to obtain 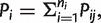 where ni is the coverage of site i. Values of Pi ∼0.03% corresponded to Q = 35. If all the erroneous callings are independent, the probability of a mutation appearing in x reads at site i to be a sequencing error follows a binomial distribution B(xi|Pi/3, ni), where ni is the coverage of site i, and Pi/3 is the average probability of observing a specific base calling error. Pi is averaged over all the nucleotides aligned at site i bearing a mutation with respect to the consensus. Furthermore, we assumed that all possible sequencing errors occur with the same probability (e.g., A can be mistaken for C, G, or U with no preferences).
where ni is the coverage of site i. Values of Pi ∼0.03% corresponded to Q = 35. If all the erroneous callings are independent, the probability of a mutation appearing in x reads at site i to be a sequencing error follows a binomial distribution B(xi|Pi/3, ni), where ni is the coverage of site i, and Pi/3 is the average probability of observing a specific base calling error. Pi is averaged over all the nucleotides aligned at site i bearing a mutation with respect to the consensus. Furthermore, we assumed that all possible sequencing errors occur with the same probability (e.g., A can be mistaken for C, G, or U with no preferences).
At each site, we ranked the frequencies of the nucleotides observed in the reads and assigned a score to each variant, defined as sik = B(xi,k|Pi/3, ni), where xik is the number of reads aligned at site i bearing nucleotide k ∈ {A, C, G, U}. sik increases if a mutation is observed in a large number of reads, making it unlikely to be a sequencing error: when sik is small, we can reject the hypothesis “the observed mutation is generated by sequencing error.” We considered only mutations where sik < q, with q being a threshold chosen to be 0.05. All these analyses were performed with custom-made C scripts.
Finally, we estimated the probability of resequencing the same fragment more than once. Let M be the initial number of fragment present in the sample and N the coverage of a site: the probability of sampling a fragment l times is distributed according to the binomial law B(l|1/M, N), where 1/M is the probability of choosing a particular fragment among the M available. Therefore, the probability of resequencing the same fragment is 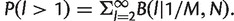 For the typical values N = 5 × 105 and M = 1.2 × 1011 (De la Iglesia et al. 2012), P(l > 1) ≈ 8 × 10−12, which is sufficiently small to be ignored.
For the typical values N = 5 × 105 and M = 1.2 × 1011 (De la Iglesia et al. 2012), P(l > 1) ≈ 8 × 10−12, which is sufficiently small to be ignored.
Population genetic analyses were performed with ARLEQUIN 3.1 (Excoffier et al. 2005). The average nucleotide diversity per site (π) and the average number of pairwise nucleotide differences (k) were computed for each population. Using these two estimates, we computed Tajima’s D statistics (Tajima 1989) to evaluate whether the observed patterns of variability were compatible with the neutral expectation (D = 0), with the action of directional selection/population growth (D < 0) or with balancing selection/population subdivision (D > 0). Patterns of nucleotide substitutions in the amiR159-HCPro target in TuMV genome were analyzed using MEGA5 (Tamura et al. 2011). Nucleotide substitution matrices and ratios of transitions to transversion rates (κ) were estimated by maximum likelihood for each evolutionary lineage under the general time reversible model with uniform rates among sites and using a neighbor-joining phylogenetic tree (Saitou and Nei 1987). Substitution rates per synonymous (dS) and nonsynonymous (dN) sites were estimated using Nei-Gojobori’s modified method and bootstrap SEM (1,000 pseudoreplicates). IBM SPSS version 19 was used for all additional statistical analyses reported.
Results and Discussion
To study the population dynamics of evolving TuMV sequence variants and the escape mutants able to break resistance in fully resistant 12-4 plants by NGS, we chose four evolutionary lineages from our previous work (Lafforgue et al. 2011). The first lineage, labeled as L20.Col-0, consisted of 19 passages in WT. At each passage, a pathogenicity test was performed (see Materials and Methods section). All tests were negative until passage 19. The TuMV population replicating in the infected 12-4 plants was analyzed by NGS and are referred in figure 2A as passage 20 (highlighted in light green). In this case, the escape allele identified by Sanger sequencing as the most abundant one contained the mutation C11U (numbering referred to the 21 nt of the amiR159-HCPro target), a synonymous mutation that appeared in 7 of the 25 independent lineages evolved in this host. For the other three lineages, evolution took place in partially resistant 10-4 plants and occurred faster. Lineage L11.10-4 also contained the C11U escape allele, and it arose after only two passages in 10-4 plants followed by a successful pathogenicity test in 12-4 plants (labeled as passage 3 and highlighted in green in fig. 3A). Finally, lineages L1.10-4 and L10.10-4 consisted, respectively, of three and four evolutionary passages in 10-4 plants plus the subsequent positive pathogenicity tests in 12-4 plants (labeled as passages 4 and 5 and highlighted in green in fig. 3B and C, respectively). Consensus sequencing of escape alleles for these two lineages showed mutations A19C (K to T amino acid replacement) and G12A (V to M, a conservative amino acid change) (Lafforgue et al. 2011).
Fig. 2.

Diversity evolution in one TuMV lineage evolved in fully susceptible A. thaliana plants (WT). (A) Evolution of frequency for the 50 more abundant haplotypes detected for the amiR159-HCPro target sequence along the evolution experiment. The red line corresponds to the ancestral TuMV haplotype. At passage 20, we show the haplotypic composition found after resistance breaking in the 12-4 plants (shadow in light green): the numerically dominant haplotypes carry mutations in the target. However, the ancestral TuMV haplotype was still present at a detectable frequency in the population. (B) Observed diversity per each of the 21 nt in the amiR159-HCPro target along the 20 serial passages. In both panels, abscises axes are in decimal log scale.
Fig. 3.

Diversity evolution in three TuMV lineages evolved in partially resistant A. thaliana 10-4 plants. (A–C) Evolution of frequency for up to the 50 more abundant haplotypes detected for the amiR target sequence along the evolution experiments. At the final positive pathogenicity test passages (3, 4, and 5), we show the haplotypic composition (shadow in light green). (D–F) Observed diversity per each of the 21 nt in the amiR target at each passage. Panels (A and D) correspond to lineage L1.10-4, panels (B and E) to lineage L10.10-4, and panels (C and F) to lineage L11.10-4. Diversity at the last indicated passage corresponds to that observed in the population that broke the 12-4 resistance (shadow in light green). Abscises axes are in decimal log scale.
Description and Filtering of Illumina Data
Samples from all the evolution passages for the four lineages described earlier were sent out for Illumina sequencing, and the resulting data were subjected to the quality analyses described in the corresponding section of the Materials and Methods section. Figure 1A illustrates the quality scores Qi of the sequence reads translated into error probabilities Pi. The Qi associated with each nucleotide decreased toward the end of the reads, as the reliability of the sequencing process decreases with the number of cycles in the Illumina sequencing platform; therefore, Pi increased along the reads. After discarding reads with an average P > 0.2%, the error profile became flatter (dashed line in fig. 1A). The number of valid reads varied widely among lineages and among samples within each lineage. For L20.Col-0, the number of valid reads ranged from 291,348 for passage 9 to 858,037 for passage 15 or to 598,784 for the 20th passage, when the resistance of 12-4 plants was broken, with a median value of 485,318. For lineage L1.10-4, the number of valid reads ranged 609,097–678,729, with a median of 620,168 reads. For lineage L10.10-4, the range went from 477,218 to 1,222,462 valid reads (median: 729,630). Finally, for lineage L11.10-4, the number of valid reads runs within the interval 502,642–637,476, with a median value of 531,402. Only nucleotides with Q > 30 were used to determine haplotypes.
Fig. 1.

(A) Average errors per nucleotide in the 76-nt reads, computed with base qualities. The average error increased greatly toward the ends of the reads (solid line). The dashed line shows the average error after filtering. Positions 37–57 within the reads correspond to the 21-nt amiR159-HCPro target. (B) Relationship between the number of valid reads (R) per sample and the number of different haplotypes (H) detected in a sample. The solid line represents the fit to the power model H = 0.391 R0.567 (R2 = 0.962, F1,30 = 749.420, P < 0.001). The dashed lines correspond to the 95% confidence interval of the model.
Figure 1 B shows the log–log relationship between the number of reads and the number of detected haplotypes. The power law relationship between the number of valid reads and of detected haplotypes (R2 = 0.962, F1,30 = 749.42, P < 0.001) imposes a certain degree of uncertainty about the number of different haplotypes existing in each viral population sampled: as the power law has no asymptotic value, the more reads analyzed the more new haplotypes detected. In other words, an infinite number of valid reads would be necessary to estimate the exact number of haplotypes contained in a viral population. This being said, we can still make some valid inferences from our data. For instance, we can ask whether the number of haplotypes differed significantly between samples and/or lineages. For lineage L20.Col-0, the number of haplotypes per passage ranged between 19 (for passage 8) to 287 (for passage 20), with a median value of 28. Interestingly, the median number of haplotypes was larger for the three lineages evolved in the partially resistant 10-4 plants: for lineage L1.10-4, the number spanned between 44 and 256 (median 114), for lineage L10.10-4 between 38 and 255 (median 41), and for lineage L11.10-4 between 29 and 129 (median 57). An analysis of covariance (ANCOVA) on the log number of haplotypes using lineage as random factor and the log number of valid reads as covariable revealed a significant difference in the median number of haplotypes between the lineage evolved in WT and the three lineages evolved in 10-4 plants (F4,24 = 9.072, P < 0.001). In other words, the expression of subinhibitory amounts of an amiR by the host plant facilitates the accumulation of genetic variability in the virus amiR target. Furthermore, the analysis also detected a significant effect of the random factor on the slope of the regression line (F3,24 = 5.595, P = 0.005), which suggests that the underlying power law relationship may be different for each experiment. In this study, we are not exploring further whether this difference in slopes among lineages reflects some underlying biological process, or, in contrast, it may result from some more trivial reasons (e.g., the fact that the number of reads is different for each lineage).
For the sake of simplicity, in the following sections, we will concentrate our discussion only on the 50 most abundant haplotypes detected in each evolutionary lineage.
Dynamics of Molecular Evolution in the TuMV Target of amiR159-HCPro: Passages in WT Plants
Figure 2 A shows the change in relative abundance for the 50 most common haplotypes that arose in the TuMV lineage evolved in fully susceptible WT plants (e.g., lineage L20.Col-0). The last passage represented, the 20th, corresponds to the infectivity test in which lineage L20.Col-0 broke the amiR159-HCPro-mediated resistance of 12-4 plants for the first time. Several striking conclusions can be drawn from these results. First, and most notably, no less than 21 potential escape alleles were present in the evolving population from the very beginning of the evolution experiment. The median frequency of haplotypes carrying potential escape mutations across passages was 0.02%. Indeed, the haplotype containing the escape mutation C11U that turned out to be the most abundant one in the population infecting the 12-4 plants at passage 20 (62.88%) also had the highest population frequency during most intermediate passages (ranging from 0.02% at passage 18 to 0.09% at passage 11; median 0.06%) except in passages 9 and 10. The second most abundant haplotype (17.73%) found in the 12-4 plants had mutation G12A; four other haplotypes had frequencies in the range 4.82–1.23%. Interestingly, when the diversity indexes k and π were computed pooling data from consecutive passages, we found that k was 13.3-fold larger in the comparison between passages 19 and 20 (k = 0.096) than in any other of the previous 19 pairs (average k = [7.215 ± 0.162] × 10−3; z-score, P < 0.001). Similarly, π was 25.6-fold larger (z-score, P < 0.001) in the comparison between passages 19 and 20 (π = 4.575 × 10−3) than across all the other pairs (average π = [3.435 ± 0.077] × 10−4). These results suggest that the viral population composition changed substantially after the TuMV population was moved from WT to 12-4 hosts.
The temporal persistence of these potential escape mutations suggests that the event of successfully infecting 12-4-resistant plants after passage 19 was not dependent on a steady increase of its frequency or on the accumulation of different escape alleles until some critical value was reached. Rather, the TuMV genotype that finally overcame the resistance of 12-4 plants was randomly chosen among the many coexisting ones. This can be explained by the transmission bottlenecks followed by strong selection of resistance alleles in presence of the amiR159-HCPro. Indeed, that breaking resistance does not depend only on the previous presence of escape mutation is supported by two additional sources of evidence. First, there is no correlation between the frequency of the different haplotypes in the 12-4 plant (the 20th passage) and their median frequency across the different passages in WT plants (Spearman’s rS = −0.195, 48 df, P = 0.176). Second, Tajima’s D statistic was significantly smaller than zero in every passage (supplementary fig. S1, Supplementary Material online), thus suggesting that the observed excess of low-frequency polymorphisms can be simply explained by the population bottlenecks associated with each transmission event. These results support the hypothesis of Lafforgue et al. (2011) that TuMV populations evolving in WT plants were at the mutation–drift balance.
A second observation is that 28 of the haplotypes identified in the 12-4 plant (passage 20) did not qualify between the 50 most abundant in any of the previous passages in WT. Interestingly, 25 of these haplotypes carried two mutations in the amiR159-HCPro target, in comparison with all the 21 pre-existing haplotypes, all of which had only a single-nucleotide substitution, being the difference in the number of mutations per haplotype between both groups statistically significant (Fisher’s exact test P < 0.001). Eighteen of these double-mutant haplotypes contained the most abundant mutation C11U, four of them contained the second most abundant mutation G12A, and one contained both mutations. The other three double-mutant haplotypes had mutations G16C/A18G, G14A/A18G, and C17U/A19C. Mutation A18G was already detected in most passages in WT plants. In contrast, mutation A19C was the third most common single-mutation haplotype found in the 12-4 plant (passage 20), but it was not observed in any of the previous passages in WT plants. Therefore, we can conclude that most of these double-mutant haplotypes arose when resistance was broken and resulted from imperfect replication of pre-existing single-mutation haplotypes that were strongly selected on inoculation in 12-4 plants. This result is in good agreement with the observation by Lin et al. (2009) that additional mutations in the amiR target arise and facilitate escape.
A third, very interesting observation was that the TuMV ancestral sequence was still found in 12-4 plants at a frequency of 0.69%, ranking the 25th in this population. The presence of the ancestral TuMV sequence in 12-4 plants can be explained by three nonmutually exclusive hypotheses. First, the ancestral TuMV genome is capable of evading amiR resistance only when it represents a minor fraction of the population, suggesting that the efficiency of the RNA silencing machinery in detecting allelic variation in the target depends on a threshold concentration. Second, the escape mutants that dominate the population express the silencing suppressor HC-Pro that interferes with the RNA silencing machinery and blocks its action. The ancestral TuMV genotypes simply take advantage of this situation by coinfecting cells along with mutant viruses. Third, the ancestral TuMV sequence is being constantly created by back mutation from the other more abundant mutant genotypes. We find this third possibility less plausible than the others. Using a recent estimate of TuMV mutation rate in A. thaliana (De la Iglesia et al. 2012) of ∼6 × 10−5 per replication event and the fact that TuMV replication mostly proceeds by a stamping machine (Martínez et al. 2011), the expected population frequency of reversion mutants produced by backward mutation during replication of the numerically dominant resistant haplotype should be ∼0.006%, a value about 115-fold smaller than the observed frequency. Therefore, we can conclude that the observed frequency of TuMV genomes carrying the ancestral amiR159-HCPro target sequence is much higher than expected by backward mutation and, therefore, likely to be a consequence of inefficient RNA silencing machinery or of complementation with escape mutants during cell coinfection.
Finally, figure 2A shows that the temporal dynamics of the 50 most abundant haplotypes is complex, with frequent stochastic fluctuations and haplotypes that appear, disappear (i.e., went beyond experimental detection limit), and, in some cases, bounce back later in time.
Next, we investigated the evolution of variability at the different positions of the amiR target. Figure 2B shows the evolution of diversity in each of the 21 nt in the amiR159-HCPro target. During the 19 passages in WT plants, all positions of the amiR159-HCPro target showed variability. Under the hypothesis of neutral accumulation of mutations, we should expect all sites to show approximately the same variability. Furthermore, under the same assumption, we should expect this variability to fluctuate around a value resulting from a balance between mutation and the stochastic genetic drift associated with the passage events. To explore whether the first expectation would hold up, we fitted the frequency data to an ANCOVA model in which variability at each nucleotide site was treated as a random variable and passage as a covariable. This analysis detected significant differences among nucleotide sites (F21,357 = 38.343, P < 0.001), thus rejecting the null hypothesis of all sites accumulating mutations at equal frequency. Further, a two-step cluster analysis found that the optimal number of groups into which sites could be classified was 6, with site 1 being the least variable one (average frequency across passages: 0.02%) and site 11 accumulating almost twice as many variants (average frequency across passages: 0.05%). The ANCOVA analysis also revealed that the frequency of variants per site remained constant along the experimental evolution process (F1,357 = 0.458, P = 0.499), which supports our second expectation. However, this equilibrium composition changed radically on successful infection of the transgenic 12-4 plant (passage 20; fig. 2B). In this new situation, 52.52% of the sequences, regardless their haplotype, had a change at position 11 of the amiR159-HCPro target. This change is a synonymous replacement. The second most variable site was position 12, with 22.24% of the sequences (again, regardless their haplotype) containing a mutation. This mutation results in the conservative amino acid replacement V to M. Positions 3, 9, 16, 17, 18, and 19 had frequencies of variation ∼1%. This distribution of variation along the amiR159-HCPro target within a single evolutionary lineage matches the distribution of mutations found by Lafforgue et al. (2011) for 25 independent lineages. In that case, positions 11 and 12 were overwhelmingly associated with resistance breaking, and positions 17, 18, and 19 were also frequently associated with escapes. Using an heterologous system of transgenic A. thaliana plants expressing amiR159-P69 targeting an engineered TuMV genome that contained 21 nucleotides from the TYMV P69 cistron, Lin et al. (2009) showed that positions 3, 9, and 12 were critical for resistance breaking, although other sites, qualified as crucial (4, 5, and 6), did not show particularly high frequencies of variants in our study. In contrast, position 11 was considered as of moderate importance for resistance breaking (Lin et al. 2009), but turned out to be the most important one in our study. A critical difference between Lin et al. (2009) study and ours is that in their case the amiR159-P69 target sequence was neutral for the virus, whereas here the target is a coding region of TuMV HC-Pro cistron, and, consequently, mutations in successful escape variants must result from a balance between avoiding recognition by amiR159-HCPro and retaining biological function. Indeed, this neutrality effect may explain why Lin et al. (2009) observed an excess of critical positions at the 5′-end of the amiR159-P69. The importance of the two central positions may be explained by the fact that imperfect pairing with central mismatches in small-RNA-target hybrids promotes translational repression as it excludes slicing (Brodersen et al. 2008). This observation suggests the possibility that imperfect pairing between the amiR159-HCPro and mutated targets might lead to translational repression rather than to viral RNA cleavage. In contrast to the catalytic effect of amiR-mediated viral RNA cleavage, translational repression requires stoichiometric amounts of amiRs and, therefore, is not as efficient as antiviral mechanism that may allow for residual viral replication.
Dynamics of Molecular Evolution in the TuMV Target of amiR159-HCPro: Passages in Partially Resistant 10-4 Plants
Figure 3 A–C shows the change in frequency of the 50 most common haplotypes for each of three independent lineages evolved in the partially resistant 10-4 plants. The patterns described in these figures are qualitatively homogeneous and similar to that previously discussed for lineage L20.Col-0, although the event of resistance breaking took place much earlier. In all cases, haplotypes containing mutations in the amiR159-HCPro target were present in the populations since the very first experimental passage. Another remarkable difference is that although in the L20.Col-0 lineage the frequency of haplotypes containing putative escape mutations was in the range 0.0001–0.001, in the three lineages evolved in 10-4 plants, this frequency fluctuated between 0.001 and 0.01, ∼10-fold higher, an observation that is consistent with the hypothesis that in these partially resistant plants escape mutations confer a fitness advantage to the viral genotypes bearing them (Lafforgue et al. 2011).
In the case of lineage L1.10-4 (fig. 3A), the resistance of 12-4 plants was broken after only two serial passages in 10-4 plants. The most abundant escape mutant haplotype contained an A19C mutation in the amiR159-HCPro target, and it completely dominated the population infecting the 12-4 plant (99.59% frequency). The other 17 haplotypes identified in the end-point population (among the 50 more abundant during the evolution process) had frequencies that were in the range 0.01–0.03%, including the ancestral TuMV genotype, which was retained at a frequency of ∼0.02%. All-but-one nonancestral haplotypes present in this population contained the A19C mutation plus an additional one, an observation that is compatible with the notion that they were produced by mutation during replication of the numerically dominant haplotype. The remaining haplotype carried mutation A19U, and it was present at a frequency slightly lower than 0.02%.
As illustrated in figure 3B, lineage L10.10-4 was able to successfully infect 12-4 plants after three serial passages. In this case, the most abundant haplotype in the successful population had mutation G12A (99.48%), but the population still retained 17 additional haplotypes at frequencies between 0.04% and 0.02%. The ancestral TuMV genotype was also present in the population at a frequency of ∼0.03%. In this case, all the low-frequency haplotypes (aside from the ancestral one) carried two mutations in the target, with G12A always being present, thus suggesting that they are being generated from replication of the dominant haplotype.
Lineage L11.10-4 showed a similar evolutionary pattern (fig. 3C). In this case, it took four consecutive passages in partially susceptible 10-4 plants to break the resistance imposed by plants 12-4. As in previous cases, a single haplotype containing a point mutation in the amiR159-HCPro target dominated (98.06%) the population found in the 12-4 plant. In this case, the escape mutation was C11U, described in detail earlier. Twenty other haplotypes were identified, with frequencies in the range 0.01–0.76%, including the ancestral-sensitive TuMV genotype (0.04% frequency). As in the two previous lineages, all minority haplotypes also contained the C11U mutation plus an additional one.
Similar to what has been described earlier for the L20.Col-0 lineage, also for these lineages Tajima’s D was significantly negative in all cases (supplementary fig. S1, Supplementary Material online), thus suggesting that bottlenecks played a major role in configuring the genetic composition of these lineages. Putting together this observation and the previous one of a higher frequency of escape mutants in the partially resistant plants provide support to the hypothesis of Lafforgue et al. (2011) that TuMV populations evolving in this host were at mutation–selection–drift balance: mutation and selection being relevant within a plant (and specially after the resistance-breaking event) and drift being relevant during the serial transmission events.
Figure 3 D–F shows the evolution in diversity for each of the 21 nt in the amiR target for the three 10-4-evolved lineages. In all cases, variability per site fluctuated both among sites and along time, although an ANCOVA test using lineage and nucleotide site as random factors and time as covariate showed that variation among sites did not have a net significant effect. This can be interpreted as indicating that no site is particularly variable or conserved, but only in the context of interaction with passage (F20,126 = 3.067, P < 0.001) and in the three-way interaction (F40,126 = 3.414, P < 0.001), meaning that in some particular passages and lineages, certain sites are more polymorphic than others. This is quite obvious for instance in lineage L1.10-4 (fig. 3D), where sites 11, 12, and 16 are more variable (∼1%) at the second passage than at any other, whereas site 19 is the most variable in the population replicating in the 12-4 plant. A similar situation can be described for passage 3 of lineage L10.10-4 (fig. 3E), in which sites 3, 11, 12, 16, 17, and 19 had frequencies of variants in the range 0.1–1%.
Evolutionary Molecular Dynamics Outside the amiR159HC-Pro Target
The Illumina approach used in this study generates reads that are longer than the amiR159-HCPro 21-nt target analyzed in the two previous sections. Indeed, the analysis of the flanking sequences (50 nt in total) may still provide useful and interesting information. For example, we can hypothesize that if the amiR159-HCPro target is what drives the evolutionary transition from sensitivity to the resistance observed in the last passage of figures 2A and 3A–C, then variation outside the target would not be the object of selection and thus would simply reflect the mutation–selection balance. In the case of lineage L20.Col-0, the ancestral sequence was preserved along the entire evolution experiment, with its frequency oscillating in the narrow range 99.07–99.62% (median value 99.50%). Only three other haplotypes remained in all the passages, with frequencies in the range 0.02–0.04%. Looking at the per site variability, all sites showed variation, but it was lower than 0.4% for all sites and passages (median 0.02%). A similar situation was found in the three lineages evolved in the partially resistant 10-4 plants. For lineage L1.10-4, the ancestral TuMV haplotype had a median frequency across the three passages of 99.36%, with an average frequency of variants per site of ∼0.02%. In the case of lineage L10.10-4, the median frequency of the ancestral haplotype was 99.38% across the four passages, and the average frequency of variants per site in the amiR159-HCPro target was ∼0.02%. Finally, the ancestral sequence also dominated all passages of lineage L11.10-4 (99.42% on median), and the average variation per site was equivalent to that observed in the other lineages (∼0.02%).
Interestingly, all the haplotypes characterized outside the target contained a single-nucleotide substitution, even those found in the population infecting the 12-4-resistant plants, thus confirming our interpretation that the additional mutations found in the target were produced within the 12-4 plant during replication of a genome that already contained a point mutation that facilitated escape from the RISC machinery and providing further support for the strong effects of drift.
Patterns of Nucleotide Substitution
Figure 4 shows the pattern of nucleotide substitutions estimated by maximum likelihood from the 50 most abundant haplotypes identified in the four evolutionary lineages. A first remarkable observation from this figure is the significant heterogeneity among the substitution patterns estimated for each lineage (χ2 = 97.136, 15 df, P < 0.001). However, this heterogeneity is completely driven by the peculiar pattern observed for lineage L1.10-4, characterized by an excess of AC/UG and GU/CA transversions and a defect of transitions. After removing this lineage from the analysis, no heterogeneity among the remaining three lineages was observed (χ2 = 17.225, 10 df, P = 0.070).
Fig. 4.

Observed rates for the different types of nucleotide substitutions. Each column groups mutations rendering complementary pairs and, thus, can occur during the synthesis of TuMV genomic or antigenomic strains. Each lineage is represented by a different color, as indicated in the legend. For any given lineage, rates have been made relative so they add up to one.
Consistent with the principle that transitions are biochemically more likely than transversions and that they are more often silent at the codon level, the maximum likelihood estimates of the overall rate of transitions to transversions κ were >4 for lineages L20.Col-0 (κ = 4.26), L10.10-4 (κ = 4.50), and L11.10-4 (κ = 4.42). This excess also occurred when purines or pyrimidines were considered separately. Indeed, the observed frequencies of transitions among purines and among pyrimidines were similar (fig. 4) and by far the most frequent type of mutation in these three lineages. In sharp contrast, but consistent with the aforementioned excess of transversions over transitions, κ = 0.97 for lineage L1.10-4. Overall, we can conclude that TuMV replicase produces, on average, ∼4/5 transitions and ∼1/5 transversions. This result is in qualitative agreement with the mutational spectrum described for another potyvirus, tobacco etch virus (TEV), for which 2/3 of the observed point mutations were transitions and 1/3 transversions (Tromas and Elena 2010).
Under the observed mutational spectrum, the equilibrium base composition was homogenous among the four lineages (χ2 = 0.799, 9 df, P = 1) and equal to 24.25% A, 20.25% U, 18.75% C, and 36.75% G. This distribution significantly deviated from the expectation by sheer chance (χ2 = 32.720, 15 df, P = 0.005), and the deviation was mainly driven by the 47% excess in G that compensated the defect in both pyrimidines.
Next, we sought to evaluate the impact of nucleotide substitutions in the TuMV HC-Pro protein amino acid sequence. In all four lineages, the number of nonsynonymous mutations was larger than that of synonymous mutations; the largest observed difference corresponded to lineage L20.Col-0 (23 nonsynonymous and 6 synonymous) and the smallest to L1.10-4 (18 nonsynonymous and 9 synonymous), although no significant difference in the distribution of both types of substitutions was found among lineages (χ2 = 1.584, 3 df, P = 0.663). To evaluate whether this pattern of synonymous and nonsynonymous changes was compatible with a model of neutral evolution, we estimated the difference between substitutions rates per nonsynonymous and synonymous sites, dN − dS. For lineages L20.Col-0 (–0.182 ± 0.128), L1.10-4 (–0.026 ± 0.055), and L10.10-4 (–0.077 ± 0.055), the differences between rates were negative but not large enough as to reject the null hypothesis of neutral evolution (in all cases z-score, P ≥ 0.078). In the case of lineage L11.10-4, however, the difference was positive (0.249 ± 0.131) and significant (P = 0.028), although the test was not significant when the sequential Bonferroni correction for multiple tests of the same null hypothesis was applied. Therefore, we may conclude that selection at the amino acid level has played a minor role, if any, in shaping the observed patterns of nucleotide substitutions.
Conclusion
We had previously shown that the durability of plant resistance to RNA viruses generated by transgenic expression of amiRs targeting viral genomes can be jeopardized by mutations in the viral genome (Lafforgue et al. 2011). In this previous study, we showed the emergence of escape alleles containing mutations in the target that are likely to affect the binding of the amiR and consequently slicing by RISC. Now, we have characterized the molecular evolutionary dynamics of TuMV populations evolving either in a fully susceptible or in a partially resistant host. Using Illumina ultradeep sequencing, we have reached an unprecedented detection limit, allowing us to detect variants in the viral populations at a frequency as low as 2 × 10−6. We have shown that variation in the amiR159-HCPro target was generated and maintained along evolutionary time and that every nucleotide in the target sequence underwent mutation. In fully susceptible plants, new variants emerged to frequencies below 0.001. Some variants persisted for several passages, whereas others had a transient existence. The escape variant that finally succeeded in infecting fully resistant 12-4 plants was present in the evolving population from the very beginning of the evolution experiment. These results are compatible with a mutation–drift model (substantiated by significant, negative Tajima’s D). TuMV populations evolving in partially resistant plants showed a qualitatively similar behavior although with a quantitatively important difference: the frequency of escape variants in the evolving populations was more than one order of magnitude higher in partially resistant than in fully susceptible plants. These results are compatible with a mutation–selection–drift model of evolution and explain why resistance was broken much more easily in the former case. In both cases, the selective fixation of variants was due to changes in the nucleotide sequences and not by changes in the amino acid composition of the HC-Pro, as the rates of synonymous and nonsynonymous substitutions were equivalent.
Supplementary Material
Acknowledgments
The authors thank Francisca de la Iglesia for technical assistance and the useful comments from two anonymous reviewers. This research was supported by the Human Frontier Science Program Organization grant RGP0012/2008, Generalitat Valenciana grant PROMETEO/2010/019, Ministerio de Ciencia e Innovación grants BFU2009-06993 (to S.F.E.) and BFU2011-24112 (to F.G.-C.) and by CSIC grant 2010TW0015 and Ministerio de Ciencia e Innovación grants BIO2008-01986 and BIO2011-26741 (to J.-A.D.). F.M. was supported by a fellowship from the Universidad Politécnica de Valencia. M.J.M. was supported by a SYSBIO postdoctoral grant (BB/F005733/1) from the UK BBSRC.
References
- Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–1190. doi: 10.1126/science.1159151. [DOI] [PubMed] [Google Scholar]
- Bunnik EM, Swenson LC, Edo-Matas D, et al. (11 co-authors) Detection of inferred CCR5- and CXCR4-using HIV-1 variants and evolutionary intermediates using ultra-deep pyrosequencing. PLoS Pathog. 2011;7:e1002106. doi: 10.1371/journal.ppat.1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn GA, Cullen BR. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J Virol. 2002;76:9225–9231. doi: 10.1128/JVI.76.18.9225-9231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordey S, Junier T, Gerlach D, Gobbini F, Farinelli L, Zdobnov EM, Winther B, Tapparel C, Kaiser L. Rhinovirus genome evolution during experimental human infection. PLoS One. 2010;5:e10588. doi: 10.1371/journal.pone.0010588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, Berkhout B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Iglesia F, Martínez F, Hillung J, Cuevas JM, Gerrish PJ, Daròs JA, Elena SF. Luria-Delbrück estimation of turnip mosaic virus mutation rate in vivo. J Virol. 2012;86:3386–3388. doi: 10.1128/JVI.06909-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle LD, Becker MM, Halpin RA, et al. (12 co-authors) Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Martínez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson N, Pachter L, Mitsuya Y, Rhee SY, Wang C, Bharizadeh B, Ronaghi M, Shafer RW, Beerenwinkel N. Viral population estimation using pyrosequencing. PLoS Comp Biol. 2008;4:e1000074. doi: 10.1371/journal.pcbi.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. ARLEQUIN ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinf Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN, Chen J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci U S A. 2003;100:2718–2723. doi: 10.1073/pnas.0437841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Stone JK, Andino R. Poliovirus escape from RNA interference: short interfering RNA-target recognition and implications for therapeutic approaches. J Virol. 2005;79:1027–1035. doi: 10.1128/JVI.79.2.1027-1035.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Vorwald AC, Alt DP, Lager KM, Bayles DO, Faaberg KS. Large scale parallel pyrosequencing technology: PRRSV strain VR-2332 nsp2 deletion mutant stability in swine. Virus Res. 2011;161:162–169. doi: 10.1016/j.virusres.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Guo HS, Xie Q, Fei JF, Chua NH. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell. 2005;17:1376–1386. doi: 10.1105/tpc.105.030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Krönke J, Kittler R, Buchholz F, Windisch MP, Pietschmann T, Bartenschlager R, Frese M. Alternative approaches for efficient inhibition of hepatitis C virus RNA replication by small interfering RNAs. J Virol. 2004;78:3436–3446. doi: 10.1128/JVI.78.7.3436-3446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafforgue G, Martínez F, Sardanyés J, de la Iglesia F, Niu QW, Lin SS, Solé RV, Chua NH, Daròs JA, Elena SF. Tempo and mode of plant RNA virus escape from RNA interference-mediated resistance. J Virol. 2011;85:9686–9695. doi: 10.1128/JVI.05326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SS, Wu HW, Elena SF, Chen KC, Niu QW, Yeh SD, Chen CC, Chua NH. Molecular evolution of a viral non-coding sequence under the selective pressure of amiRNA-mediated silencing. PLoS Pathog. 2009;5:e1000312. doi: 10.1371/journal.ppat.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez F, Sardanyés J, Elena SF, Daròs JA. Dynamics of a plant RNA virus intracellular accumulation: stamping machine vs. geometric replication. Genetics. 2011;188:637–646. doi: 10.1534/genetics.111.129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia PR, Baillie GJ, Daly J, et al. (20 co-authors) Intra- and interhost evolutionary dynamics of equine influenza virus. J Virol. 2010;84:6943–6954. doi: 10.1128/JVI.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu QW, Lin SS, Reyes JL, Chen KC, Wu HW, Yeh SD, Chua NH. Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat Biotech. 2006;24:1420–1428. doi: 10.1038/nbt1255. [DOI] [PubMed] [Google Scholar]
- Qu J, Ye J, Fang R. Artificial microRNA-mediated virus resistance in plants. J Virol. 2007;81:6690–6699. doi: 10.1128/JVI.02457-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabariegos R, Giménez-Barcons M, Tàpia N, Clotet B, Martínez MA. Sequence homology required by human immunodeficiency virus type 1 to escape from short interfering RNAs. J Virol. 2006;80:571–577. doi: 10.1128/JVI.80.2.571-577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schwab R, Ossowski S, Riester M, Warthmann N, Weigel D. Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell. 2006;18:1121–1133. doi: 10.1105/tpc.105.039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmone M, Vincenti D, Prosperi MCF, Bruselles A, Ippolito G, Capobianchi MR. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naïve patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J Virol. 2009;83:1718–1726. doi: 10.1128/JVI.02011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Tamura S. MEGA5: molecular evolutionary genetics using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N, Elena SF. The rate and spectrum of spontaneous mutations in a plant RNA virus. Genetics. 2010;185:983–989. doi: 10.1534/genetics.110.115915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucheret H, Vázquez F, Crete P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Eije KJ, ter Brake O, Berkhout B. human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J Virol. 2008;82:2895–2903. doi: 10.1128/JVI.02035-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warthmann N, Chen H, Ossowski S, Weigel D, Hervé P. Highly specific gene silencing by artificial microRNAs in rice. PLoS One. 2008;3:e1829. doi: 10.1371/journal.pone.0001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Berkhout B. A systematic analysis of the effect of target RNA structure on RNA interference. Nucleic Acids Res. 2007;35:4322–4330. doi: 10.1093/nar/gkm437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willerth SM, Pedro HAM, Pachter L, Humeau LM, Arkin AP, Schaffer DV. Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS One. 2010;5:e13564. doi: 10.1371/journal.pone.0013564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrigth CF, Morelli MJ, Thébaud G, Knowles NJ, Herzyk P, Paton DJ, Haydon DT, King DP. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol. 2011;85:2266–2275. doi: 10.1128/JVI.01396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O, Bhattacharya A, Eriksson N, Beerenwinkel N. ShoRAH: estimating the genetic diversity of a mixed sample of next-generation sequencing data. BMC Bioinformatics. 2011;12:119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O, Klein R, Däumer M, Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.