

Abstract
Autosomal recessive spastic paraplegia 52 is caused by biallelic mutations in AP4S1 which encodes a subunit of the adaptor protein complex 4 (AP‐4). Using next‐generation sequencing, we identified three novel unrelated SPG52 patients from a cohort of patients with cerebral palsy. The discovered variants in AP4S1 lead to reduced AP‐4 complex formation in patient‐derived fibroblasts. To further understand the role of AP4S1 in neuronal development and homeostasis, we engineered the first zebrafish model of AP‐4 deficiency using morpholino‐mediated knockdown of ap4s1. In this model, we discovered several phenotypes mimicking SPG52, including altered CNS development, locomotor deficits, and abnormal neuronal excitability.
Introduction
The hereditary spastic paraplegias (HSP) are a group of rare and genetically heterogeneous neurodegenerative disorders characterized by progressive spasticity. 1 , 2 Autosomal recessive spastic paraplegia‐52 (SPG52) is a form of childhood‐onset complex hereditary spastic paraplegia characterized by early developmental delay, microcephaly, epilepsy, and progressive spasticity. Given the slowly progressive nature of this disease, patients are often diagnosed with cerebral palsy (CP) before genetic testing is pursued. SPG52 is caused by bi‐allelic loss‐of‐function variants in AP4S1, which encodes the sigma subunit of the adaptor protein complex 4 (AP‐4), and has been described in less than 10 individuals, thus far. 3 AP‐4 role in endosome membrane trafficking was evaluated in a previous study. 4
Here we screened a large cohort of patients with a diagnosis of CP for mutations in genes related to HSP and identified three novel unrelated individuals with SPG52. To gain further insights into the role of mutant AP4S1 in neuronal development and function, we developed the first model of AP‐4 deficiency in zebrafish (Danio rerio) using morpholino‐mediated knockdown of ap4s1, which recapitulate the main aspects of the patients’ phenotype.
Materials and Methods
All participants, including relatives involved in the segregation studies, provided written informed consent in accordance with the Italian National Health Service guidelines and the Declaration of Helsinki. Ethics committee approval was available. Animal experiments were performed in accordance with the IACUC at the University of Pisa. Inclusion criteria were a clinical diagnosis of CP 5 and accurate recording of at least one neurologic examination and brain MRI. Variants in AP4S1 (ENST00000216366.8) were detected using a targeted next‐generation sequencing (NGS) panel approach designed to cover reported HSP and spastic ataxia genes 2 or, in 5 cases, using whole exome sequencing. Variants were confirmed by Sanger sequencing. Pathogenicity was scored in silico using CADD. 6 Fibroblasts were derived via diagnostic punch biopsies. However, qPCR, RT‐PCR and Western blotting were carried out using published protocols. Adult male and female zebrafish AB and Tg (Neurod1‐GcAMP6f) were maintained according to standard procedures. 7 Morpholino antisense oligonucleotides (MO) targeting transcription at the exon 3 acceptor splice site or ATG start codon and a standard scramble MO 8 were designed as reported in Data S1. Touch‐evoked escape response was measured at 48 hpf on a semi‐quantitative scale ranging from severe (=no movement), to mild (=flicker of movement but no swimming), or normal (=normal swimming). 9 Coiling frequency was evaluated at 30 hpf using the Leica M205FA microscope (Leica, Wetzlar, Germany) and Danio Scope software (Noldus, Wageningen, The Netherlands). Behavioral and movement data were acquired using Danio Vision at 120 hpf and analyzed using EthoVision software (Noldus). The transgenic NeuroD1 line was used to assess brain/head morphology after MO injection at 72 hpf. Electrophysiological forebrain recordings were performed at 120 hpf. Statistical analyses used GraphPad Prism v.7.1 with significance set at P < 0.05.
Results
Through NGS studies of 112 individuals with a clinical diagnosis of CP, we identified three children harboring biallelic variants in AP4S1. These variants are predicted to cause an early truncation of the protein (Fig. S1A) and segregate in the families (Fig. S2). Clinical, molecular and neuroimaging findings are in Table 1. All three patients presented with developmental delay and later intellectual disability as well as slowly progressive spasticity. Patient #3 required a wheelchair at the age of 9 years, whereas patients #1 and #2 are still ambulant (with aid) at age 18 and 4 years, respectively. Epilepsy was found in all three cases: patient #1 developed focal seizures at age 10 months; patient #2 had epileptic spasms at 14 months, and patient #3 experienced generalized seizures starting at 12 months. In all three cases, seizures were well controlled with standard antiepileptic drugs. Fibroblasts from patients #1 and #2 (Fig. 1A and B) showed reduced levels of AP4E1, consistent with the notion that a loss of the sigma subunit of AP‐4 destabilizes the protein complex leading to reduced levels of other AP‐4 subunits. Moreover, levels of ATG9A, the major cargo of AP‐4, were significantly increased consistent with prior reports. 10 Zebrafish ap4s1 shares 78% identity with the human gene and protein (Fig. S1C). In situ hybridization analysis (WISH) confirmed that ap4s1 mRNA is ubiquitously expressed in 48 hpf embryos and 72 hpf larvae, with a high expression in the CNS (Fig. S1D). However, at earlier stages, WISH and qPCR demonstrated ap4s1 mRNA expression mainly after 24 hpf (Fig. S1E and F). To knock‐down gene expression, we designed two different MOs, ATG‐ and splice‐MO; after testing both, we noticed a stronger effect using the splice‐MO, whereas the ATG‐MO induced a milder yet significant phenotype as compared to uninjected fish (Fig. S1G). MO‐mediated knockdown of ap4s1 (Fig. S1H and I) lead to several morphological defects including decreased head size, ventriculomegaly, altered eye development, cardiac edema, and a curved tail (Fig. 1C and D). Whilst Ap4e1 protein levels were reduced (Fig. 1E), we could not study ATG9A because commercial antibodies failed to recognize the fish protein (not shown).
Table 1.
Clinical features of three patients with SPG52.
| Patient | Patient #1 | Patient #2 | Patient #3 |
|---|---|---|---|
| Sex | Female | Female | Male |
| AP4S1 Variant 1 | c.47insT/c.234insG p.Ser17*/p.Ala79Glyfs*4 | c.234insG/c.234insG p.Ala79Glyfs*4/p.Ala79Glyfs*4 | c.138 + 2T>G/c.138 + 2T>G |
| CADD Score | 28.9/28.1 | 28.1 | 25.9 |
| Age at last evaluation | 18 years | 4 years, 10 months | 14 years |
| Consanguinity | No | Yes | Unknown |
| Ethnicity | Caucasian | Caucasian | Caucasian |
| Spasticity | Spastic tetraplegia | Spastic tetraplegia | Spastic tetraplegia |
| Level of ambulation | Walks without support | Walks with support | Wheelchair‐dependent |
| Developmental Delay/Intellectual Disability | Severe | Severe | Moderate |
| Speech | Simple sentences | Nonverbal | Nonverbal |
| Short Statue | Yes | Yes | No |
| Microcephaly | Yes (postnatal) | Yes (postnatal) | No |
| Thin Corpus Callosum | Yes | Yes | Yes |
| Ventriculomegaly | No | No | Yes |
| Cerebral Atrophy | Yes | No | No |
| Cerebellar Atrophy | Yes | No | No |
| Seizures | Focal (onset at 10 years) | Epileptic spasms (onset at 14 months) | Focal and generalized (onset at 12 months) |
| EEG | Sharp waves on bilateral anterior regions | Generalized sharp waves | Diffuse epileptiform abnormalities |
AP4S1 Reference Sequence ENST00000313566.
Figure 1.
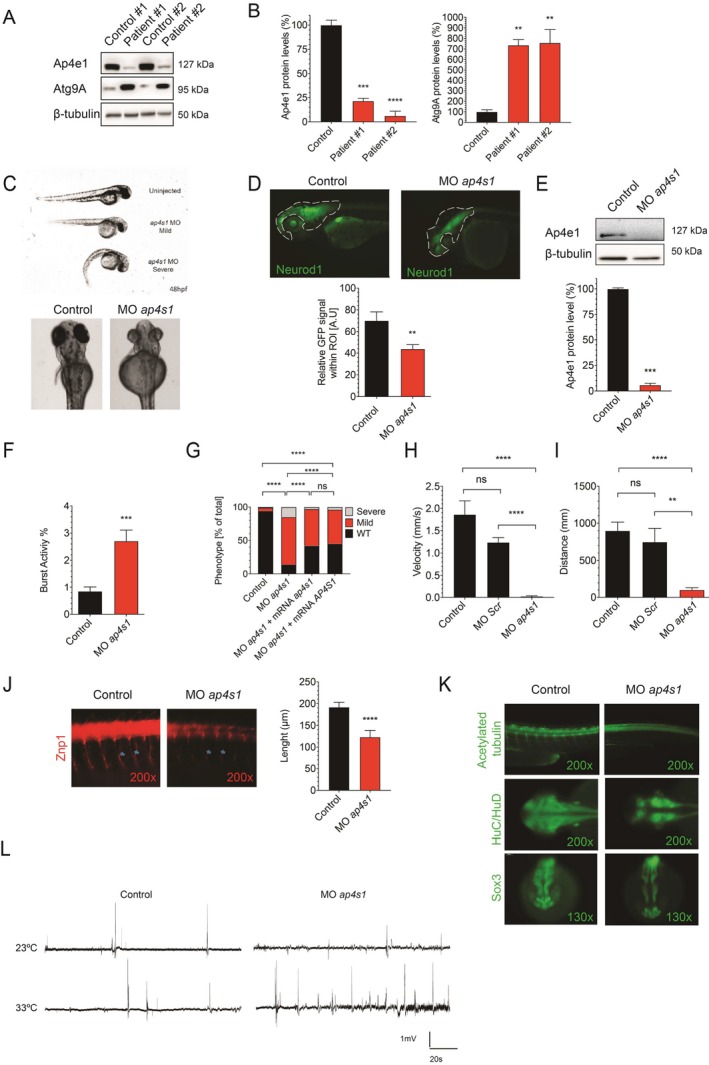
(A and B) Western blot analysis from whole cell lysates of patient #1 and patient #2 as well as two healthy, unrelated controls. Levels of AP4E1 are reduced, consistent with the notion that loss of AP4S1 destabilizes the AP‐4 complex and lowers levels of the other subunits. ATG9A, the main cargo of AP‐4, is increased in fibroblasts from patient #1 and patient #2, consistent with prior reports. This indicates a loss of AP‐4 function (one‐way ANOVA with multiple comparison, n = 2–6 samples per condition per experiment, P‐value: ****<0.0001; ***0.0002; **0.0008). (C) Morphology of zebrafish embryos injected with MOap4s1 at 48 hpf. Mild and severe phenotypes are found. Several dysmorphic features are present, including an abnormal eye shape, smaller head size, cardiac edema and curved tail. (D) CNS morphology as assessed by GFP fluorescence in NeuroD1‐GFP zebrafish at 72 hpf. Dotted line outlines head area (ROI). GFP‐fluorescence within the ROI is significantly reduced (Mann–Whitney test, n = 10; P‐value: **0.0052). (E) Western blot analysis from lysates of ap4s1‐depleted zebrafish at 72 hpf shows reduced Ap4e1 expression (unpaired t‐test, n = 2; P‐value: ***0.0004). (F) Coiling frequency in zebrafish embryos at 30 hpf is increased in ap4s1‐depleted zebrafish (Mann–Whitney test, n = 80 in 4 independent experiments; P‐value: ***0.0002). (G) Touch‐evoked escape response was measured at 48 hpf on a semi‐quantitative scale ranging from severe (=no movement), to mild (=flicker of movement but no swimming), or normal (=normal swimming). About 80% of ap4s1‐depleted zebrafish show a mild or severe impairment and about 40% of morphants showed partial rescued phenotypes, after co‐injection with either human or zebrafish mRNA (Chi Square Test, n = >100 in 2 independent experiments; P value: ****<0.0001). (H and I) Automated analysis of spontaneous motor activity revealed a reduction in swim distance and velocity in ap4s1‐depleted zebrafish at 120 hpf (Mann–Whitney test, n = 37 per condition; P‐value: ****=<0.0001; **=0.0011). (J) Immunocytochemistry with motor neuron marker znp1 demonstrates a reduction in axon length of spinal motor neurons in ap4s1‐depleted zebrafish at 48 hpf (Mann‐Whitney test, n = 20; P‐value: ****=<0.0001). (K) Immunocytochemistry using the pan‐neural marker sox3 and the postmitotic neuronal marker Huc/Hud in 24 hpf embryos. A reduction in sox3 staining, suggesting an impairment in the maintenance of the neuronal progenitor pool, is found in ap4s1‐depleted zebrafish. Huc/Hud staining was globally reduced in ap4s1‐depleted zebrafish indicating loss of postmitotic neurons, in particular at the level of diencephalon, telencephalon and in the optic tectum. (L) Local field potentials recorded from 120 hpf zebrafish. At an ambient temperature of 23°C there was no difference observed. When the temperature was raised to 33°C frequent bursts were found in ap4s1‐depleted zebrafish, indicating abnormal excitability and a lower threshold for epileptiform activity in the setting of stressors.
Moreover, behavioral analysis revealed that at 30 hpf ap4s1 morphant embryos showed an increased coiling frequency compared to uninjected ones (Fig. 1F; Videos S1 and S2). Touch evoked response was impaired in over 80% of injected morphants at 48 hpf (Fig. 1G). Rescue experiments were performed through co‐injection of 200 pg of either human WT or zebrafish mRNA together with spliceMO at the same concentration used for the knockdown experiments (Fig. 1G). At 120 hpf ap4s1‐deficient zebrafish larvae showed locomotor impairment (Fig. 1H and I). To further explore the nature of this motor deficit, we investigated the morphology of spinal motor neurons using the motor axon marker Znp1. Immunolabeling of spinal motor neuron axons in 48 hpf morphants and control embryos showed abnormal axon outgrowth and a reduction in axon length (Fig. 1J). Staining of acetylated α‐tubulin confirmed truncated axons that appeared thinner than in controls and were hardly visible in the most severely affected embryos (Fig. 1K). Knockdown of ap4s1 resulted in not only an alteration of ventrally projecting motor neurons at 48 hpf but also a strong reduction in axon distribution in the anterior region of the CNS. To further characterize the neurodevelopmental consequences of ap4s1 depletion, we studied the pan‐neural marker Sox3 and the postmitotic neuronal marker Huc/Hud (Fig. 1K). Morphants (24 hpf) showed a reduction in Sox3 staining suggesting an impairment in the maintenance of the neuronal progenitor pool (Fig. 1K). Huc/Hud staining was globally reduced indicating loss of postmitotic neurons, in particular at the level of diencephalon, telencephalon and in the optic tectum (Fig. 1K). Overall, these results suggested an impairment of primary neurogenesis in ap4s1 zebrafish morphants. This corroborates the altered CNS morphology observed in 72 hpf ap4s1‐depleted Neurod1‐GFP embryos (Fig. 1D). To further explore the functional implications of loss of ap4s1, we measured local field potentials in the forebrain of control and ap4s1‐depleted embryos at 120 hpf. We detected no difference at room temperature (23°C) (Fig. 1L). When the temperature was raised to 33°C, frequent bursts were observed in ap4s1 morphant larvae (Fig. 1L), indicating a lower threshold for abnormal excitability in the setting of stressors, mimicking in part what is experienced by SPG52 children. 5
Discussion
We here describe three additional cases of SPG52 discovered by genetic screening of a cohort of patients with a diagnosis of CP. Clinical and imaging features of these patients resemble those reported previously. 3 , 5 , 11 , 12 , 13 , 14 Interestingly, patient #1 presented with a relatively mild phenotype and preserved independent walking at the age of 18 years. On a cellular level, fibroblasts derived from two patients showed reduced levels of the AP4E1 subunit consistent with reduced stability of the AP‐4 complex. In line with prior reports, 10 we also observed an accumulation of ATG9A, further corroborating the pathogenicity of the discovered SPG52 variants. In humans, AP4S1 encodes a protein whose function is, only in part, understood, also because of the absence of in vivo models of SPG52, though initial iPSC studies 15 appear helpful to study axon outgrowth. To explore the consequence of loss of AP4S1 in vivo, we generated the first zebrafish model of AP‐4‐deficiency. Antisense morpholino‐mediated knockdown of ap4s1 resulted in a robust reduction in expression. Loss of formation of a functional AP‐4 complex was confirmed by a reduction in protein levels of the Ap4e1 subunit, a surrogate for complex formation, 10 a finding already seen in SPG52 disease. Various neurodevelopmental brain malformations are found in patients with AP‐4‐associated hereditary spastic paraplegia (SPG47, SPG50, SPG51, and SPG52). 3 These include a thinning of the corpus callosum, loss of periventricular white matter with resulting ventriculomegaly, as well as polymicrogyria in some. In ap4s1‐depleted zebrafish, we observed a reduction in head size and a complex CNS malformation. In combination with the finding of reduced staining for neuronal progenitors and postmitotic neurons, these results demonstrate the crucial role of AP‐4 for neuronal development. Mimicking the human phenotype, we found locomotor deficits in ap4s1‐depleted zebrafish. Behavioral assays in morphant larvae revealed significant defects inverall mobility, suggesting that ap4s1‐related motor neuron development and function is required for normal motility in zebrafish. We next examined spinal motor axons and found that morphants exhibited shorter axonal length, indicating that SPG52 may be important for the outgrowth of motor axons. This is consistent with impaired neuron outgrowth found in cultured neurons from Ap4e1 knockout mice 16 , 17 and similar to results seen in several zebrafish models of HSP including spastin, 18 atl1, 19 and spatacsin. 20 Seizures are found in about two‐thirds of AP‐4‐HSP patients and febrile seizures are common early in life. 3 Local field potentials recordings of forebrain in ap4s1‐depleted zebrafish revealed no spontaneous seizures at basal temperature. With hyperthermia, however, burst of abnormal activity were induced, suggesting the possibility for a lower threshold for epileptiform activity in the setting of stressors.
Summarizing, ap4s1 knockdown in zebrafish leads to several phenotypes that resemble AP4S1‐associated hereditary spastic paraplegia. Thus, this first zebrafish model of AP‐4 deficiency represents a new tool for dissecting the role of AP‐4 in neurodevelopment and neurodegeneration and could be the starter point for future drug screening, along with a KO model.
Conflict of Interest
Ebrahimi‐Fakhari reports grants from Thrasher Research Fund, grants from Spastic Paraplegia Foundation, grants from CureSPG47 Foundation, during the conduct of the study. All other authors declare no conflict of interest related to this study.
Author Contribution
Conception and design of the study: A.D., F.G., F.M.S. Acquisition and analysis of data: A.D., A.T., V.N., G.F., D.G., M.M., S.M., G.B., A.C., M.T.B., F.S., J.B., R.R., R.B., I.R., A.R., and D.E.F. Drafting a significant portion of the manuscript or figures: A.D., J.H., M.M., D.E.F, M.S., and F.M.S.
Supporting information
Figure S1. (A) Three‐dimensional model of human AP4S1 (qmean4 global score: 0.710).
Figure S2. Family tree and segregation of mutations in the kindred. Circles and squares are women and men, respectively. Filled symbols are affected individuals.
Data S1. Additional methods.
Video S1. Coiling frequency in uninjected zebrafish embryos at 30 hpf.
Video S2. Coiling frequency in zebrafish embryos injected withMOap4s1 at 30 hpf.
Acknowledgments
The authors sincerely thank the patients and their families for their help and willingness to participate in this study. We also thank Professor Margaret Robinson (Cambridge, UK) for donating AP4E1 antibody. This work was supported by the University of Siena “Pegaso Scholarship” (to AD), the Italian Ministry of Health, Ricerca 5x1000 (to FMS, RB, and FS), the Italian Ministry of Health, grant #RC2017‐2018‐2019 (to MTB), and funds from CureSPG47 Inc., the Spastic Paraplegia Foundation (SPF) Inc., the Thrasher Foundation and the Lovejoy Award (all to DEF).
Funding Information
This work was supported by the University of Siena “Pegaso Scholarship” (to AD), the Italian Ministry of Health, Ricerca 5x1000 (to FMS, RB, and FS), the Italian Ministry of Health, grant #RC2017‐2018‐2019 (to MTB), and funds from CureSPG47 Inc., the Spastic Paraplegia Foundation (SPF) Inc., the Thrasher Foundation and the Lovejoy Award (all to DEF).
Funding Statement
This work was funded by Thrasher Foundation grant ; Spastic Paraplegia Foundation (SPF) Inc. grant ; CureSPG47 Inc grant ; Lovejoy Award grant ; University of Siena “Pegaso Scholarship” grant ; the Italian Ministry of Health grant #RC2017‐2018‐2019.
References
- 1. Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol 2018;148:633–652. [DOI] [PubMed] [Google Scholar]
- 2. D'Amore A, Tessa A, Casali C, et al. Next generation molecular diagnosis of hereditary spastic paraplegias: an Italian cross‐sectional study. Front Neurol 2018;9:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ebrahimi‐Fakhari D, Behne R, Davies AK, Hirst J.AP‐4‐associated hereditary spastic paraplegia In: Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., et al. GeneReviews® [Internet]. Seattle: University of Washington, 1993–2020. [PubMed] [Google Scholar]
- 4. Behne R, Teinert J, Wimmer M, et al. Adaptor protein complex 4 deficiency: a paradigm of childhood‐onset hereditary spastic paraplegia caused by defective protein trafficking. Hum Mol Genet 2020;29:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tessa A, Battini R, Rubegni A, et al. Identification of mutations in AP4S1/SPG52 through next generation sequencing in three families. Eur J Neurol 2016;23:1580–1587. [DOI] [PubMed] [Google Scholar]
- 6. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Westerfield M. Zebrafish book. A guide for the laboratory use of Zebrafish (Danio rerio), 4th ed Eugene: University of Oregon Press, 2000. [Google Scholar]
- 8. Peng SX, Yao L, Cui C, et al. Semaphorin4D promotes axon regrowth and swimming ability during recovery following zebrafish spinal cord injury. Neuroscience 2017;351:36–46. [DOI] [PubMed] [Google Scholar]
- 9. Marchese M, Pappalardo A, Baldacci J, et al. Dolichol‐phosphate mannose synthase depletion in zebrafish leads to dystrophic muscle with hypoglycosylated alpha‐dystroglycan. Biochem Biophys Res Commun 2016;477:137–143. [DOI] [PubMed] [Google Scholar]
- 10. Davies AK, Itzhak DN, Edgar JR, et al. AP‐4 vesicles contribute to spatial control of autophagy via RUSC‐dependent peripheral delivery of ATG9A. Nat Commun 2018;9:3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abou Jamra R, Philippe O, Raas‐Rothschild A, et al. Adaptor protein complex 4 deficiency causes severe autosomal‐recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 2011;88:788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmona S, Marecos C, Amorim M, et al. AP4S1 splice‐site mutation in a case of spastic paraplegia type 52 with polymicrogyria. Neurol Genet 2018;4:e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hardies K, May P, Djemie T, et al. Recessive loss‐of‐function mutations in AP4S1 cause mild fever‐sensitive seizures, developmental delay and spastic paraplegia through loss of AP‐4 complex assembly. Hum Mol Genet 2015;24:2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vill K, Muller‐Felber W, Alhaddad B, et al. A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov Disord 2017;32:797–799. [DOI] [PubMed] [Google Scholar]
- 15. Teinert J, Behne R, D'Amore A, et al. Generation and characterization of six human induced pluripotent stem cell lines (iPSC) from three families with AP4B1‐associated hereditary spastic paraplegia (SPG47). Stem Cell Res 2019;40:101575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Pace R, Skirzewski M, Damme M, et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP‐4 deficiency syndrome. PLOS Genet 2018;14:e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivankovic D, Drew J, Lesept F, et al. Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP‐4 deficiency syndrome. Autophagy 2020;16(3):391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Butler R, Wood JD, Landers JA, Cunliffe VT. Genetic and chemical modulation of spastin‐dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech 2010;3(11–12):743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fassier C, Hutt JA, Scholpp S, et al. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat Neurosci 2010;13:1380–1387. [DOI] [PubMed] [Google Scholar]
- 20. Southgate L, Dafou D, Hoyle J, et al. Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 2010;11:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Three‐dimensional model of human AP4S1 (qmean4 global score: 0.710).
Figure S2. Family tree and segregation of mutations in the kindred. Circles and squares are women and men, respectively. Filled symbols are affected individuals.
Data S1. Additional methods.
Video S1. Coiling frequency in uninjected zebrafish embryos at 30 hpf.
Video S2. Coiling frequency in zebrafish embryos injected withMOap4s1 at 30 hpf.