

Abstract
Studying the evolution of catalytically promiscuous enzymes like those from the N-succinylamino acid racemase/o-succinylbenzoate synthase (NSAR/OSBS) subfamily can reveal mechanisms by which new functions evolve. Some enzymes in this subfamily only have OSBS activity, while others catalyze OSBS and NSAR reactions. We characterized several NSAR/OSBS subfamily enzymes as a step toward determining the structural basis for evolving NSAR activity. Three enzymes were promiscuous, like most other characterized NSAR/OSBS subfamily enzymes. However, Alicyclobacillus acidocaldarius OSBS (AaOSBS) efficiently catalyzes OSBS activity but lacks detectable NSAR activity. Competitive inhibition and molecular modeling show that AaOSBS binds N-succinylphenylglycine with moderate affinity in a site that overlaps its normal substrate. Based on possible steric conflicts identified by molecular modeling and sequence conservation within the NSAR/OSBS subfamily, we identified one mutation, Y299I, which increased NSAR activity from undetectable to 1.2 x 102 M−1s−1 without affecting OSBS activity. This mutation does not appear to affect binding affinity, but instead affects kcat, by reorienting the substrate or modifying conformation changes to allow both catalytic lysines to access the proton that is moved during the reaction. This is the first site known to affect reaction specificity in the NSAR/OSBS subfamily. However, this gain of activity was obliterated by a second mutation, M18F. Epistatic interference by M18F was unexpected because a phenylalanine at this position is important in another NSAR/OSBS enzyme. Together, modest NSAR activity of Y299I AaOSBS and epistasis between sites 18 and 299 indicate that additional sites influenced the evolution of NSAR reaction specificity in the NSAR/OSBS subfamily.
Graphical Abstract
Introduction
Predicting enzyme specificity on the basis of sequence and structure is currently challenging. Studying enzyme evolution can address this challenge by associating reaction specificity with amino acid changes. Mounting evidence indicates that enzymes evolve new specificity by transitioning through promiscuous intermediates, which can catalyze more than one reaction in the same active site.1-4 For example, many members of the N-succinylamino acid racemase/o-succinylbenzoate synthase (NSAR/OSBS) subfamily are catalytically promiscuous. This subfamily originated in a larger family of OSBS enzymes, which catalyze a step in menaquinone synthesis. The OSBS family belongs to the functionally diverse enolase superfamily, whose members share a common fold, a set of conserved catalytic residues, and a partial chemical reaction leading to formation of a metal-stabilized enolate anion intermediate (Figure 1).5 Phylogenetic analysis of the OSBS family showed that it is comprised of several large, divergent subfamilies, which primarily correspond to the phylum from which the OSBS enzymes originated.6
Figure 1.
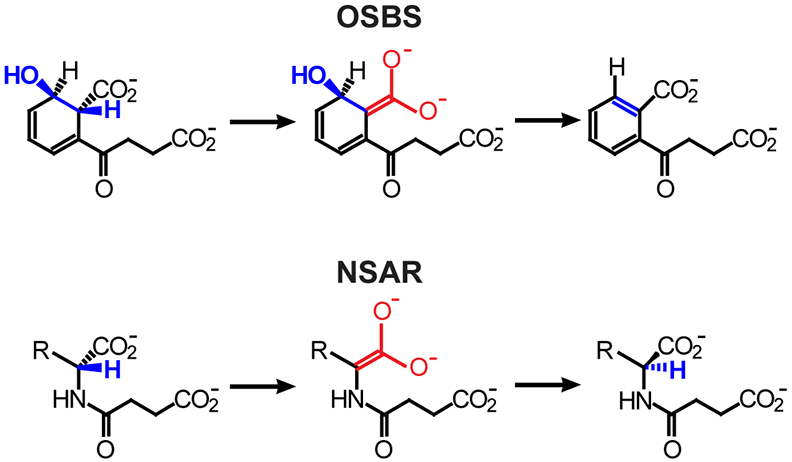
The o-succinylbenzoate synthase (OSBS) and N-succinylamino acid racemase (NSAR) reactions. Structural similarities of the intermediates are red; blue atoms are lost or rearranged in the reactions. R, hydrophobic amino acid side chain.
Previous studies to determine the structural basis for NSAR activity compared several promiscuous NSAR/OSBS subfamily enzymes to OSBS enzymes from other subfamilies, including Escherichia coli OSBS and Thermobifida fusca OSBS from the γ-Proteobacteria and Actinobacteria OSBS subfamilies, respectively.6-8 Both genome context and biochemical assays indicate that members of these other OSBS subfamilies lack NSAR activity.8, 9 While the sequence identities within the NSAR/OSBS subfamily are typically >40%, the sequence identities between the NSAR/OSBS subfamily and other OSBS subfamilies is usually <25%, which is similar to the degree of divergence between the NSAR/OSBS subfamily and other, functionally diverse families in the enolase superfamily. We previously showed that the divergence of the other OSBS subfamilies was due to their loss of quaternary structure and accumulation of insertions and deletions that were associated with an increased rate of amino acid substitution.8 Compared to the monomeric OSBS subfamilies, structurally characterized members of the NSAR/OSBS subfamily are dimers or octamers whose symmetry and overall structure is more similar to other, functionally diverse families in the enolase superfamily. In addition, NSAR/OSBS subfamily enzymes bind o-succinylbenzoate in a different conformation than OSBS enzymes from E. coli and T. fusca, which requires some different active site residues.10 Because of the high level of sequence and structural divergence between the NSAR/OSBS subfamily and other OSBS subfamilies, distinguishing between sequence differences that were relevant for gain of NSAR activity and those reflecting overall sequence and structural divergence has been difficult.
Consequently, this study has turned to comparison of enzymes within the NSAR/OSBS subfamily to understand how NSAR activity evolved. Only nine proteins out of >1100 in the NSAR/OSBS subfamily have been experimentally characterized. Of these, only the OSBS enzymes from Bacillus subtilis and Staphylococcus aureus lack detectable NSAR activity.11 Another subfamily member from Exiguobacterium sp. AT1b catalyzes the OSBS reaction as its biological function, but also exhibits inefficient, promiscuous NSAR activity.12 A fourth subfamily member from Geobacillus kaustophilus is bifunctional, requiring OSBS activity for menaquinone synthesis and NSAR activity for a pathway that converts D-amino acids to L-amino acids.13 The other five enzymes also catalyze both OSBS and NSAR reactions efficiently.8, 14 In several cases, OSBS activity is a promiscuous, non-biological side reaction, while NSAR or another activity that is yet to be determined is the primary biological function.9 Thus, the NSAR/OSBS subfamily is an excellent model system for investigating the evolution of enzyme specificity.
Although differences in NSAR reaction specificity among NSAR/OSBS subfamily enzymes have been identified, the sequence and structural basis for differences in reaction specificity are unknown. In this study, we approached this question by characterizing four additional NSAR/OSBS subfamily enzymes from Amycolatopsis mediterranei S699 (AmedNSAR), Lysinibacillus varians GY32 (LvNSAR/OSBS), Roseiflexus castenholzii HLO8 (RcNSAR/OSBS) and Alicyclobacillus acidocaldarius LAA1 (AaOSBS). Genome context analysis and activity assays showed that one enzyme, AaOSBS, is an efficient OSBS enzyme that lacks NSAR activity. Inhibition experiments, structural analysis, and mutagenesis enabled us to identify the first known residue, Y299, that strongly influences NSAR reaction specificity. Significantly, the effect of this residue on reaction specificity of AaOSBS and other NSAR/OSBS subfamily enzymes is influenced by epistasis, which occurs when mutations have different effects in different sequence contexts. Evidence that epistasis plays an important role in determining the route of protein evolution is accumulating quickly, so understanding how the role of residues like Y299 vary in different sequence contexts is essential for improving the ability to predict enzyme specificity.15-20
Materials and Methods
Protein Production and Crystallization.
The genes encoding AmedNSAR (Uniprot ID: G0FPT7), LvNSAR/OSBS (Uniprot ID: X2GR01) and RcNSAR/OSBS (Uniprot ID: A7NLX0) were obtained via PCR from genomic DNA and cloned into the modified pET-21a expression vectors pMCSG7 or pMCSG8, which encode an N-terminal His6-tag, via ligation independent cloning.21 Lysinibacillus varians GY32 and Roseiflexus castenholzii HLO8 genomic DNA were purchased from the Leibniz Institute DSMZ-German collection of Microorganisms and Cell Cultures (Braunschweig, Germany), while genomic DNA for Amycolatopsis mediterranei S699 was kindly provided by Dr. Rup Lal from the University of New Delhi (New Delhi, India). Sequences of primers used for cloning are included in supporting information (Table S1).
The gene encoding AaOSBS (Uniprot ID: B7DSY7) from A. acidocaldarius (encoding amino acids 3-379) was obtained via synthesis (GeneScript), amplified via PCR, cloned into a C-terminal His6-tag expression vector, tested for expression and solubility, fermented at large scale, purified and crystallized using the published methods employed by the NYSGXRC.22 Detailed protocols for this target are publicly available from the NIGMS PSI archival repository of protocols (https://zenodo.org/record/821654#.WmC5YK6nHyM), under the Target ID NYSGXRC-9710a; the expression clone is available from the PSI Materials Repository (http://dnasu.org/DNASU/GetCloneDetail.do?cloneid=537073).
Se-Met labeled material was generated by a 1 L fermentation of HY medium (Medicilon, Inc.) yielding 16.7 mg of purified protein. The purity and Se incorporation were confirmed by SDS-PAGE analysis of chromatographic fractions and mass spectrometry (ESI and MALDI) of the final pool. This sample was used for structure determination of this protein.
Diffraction quality crystals were obtained by mixing 1 μL of protein at 10.9 mg/mL with 1 μL of 30% PEG 8K, and 0.2 M ammonium sulfate, pH 7.0, and equilibrating by vapor diffusion against 100 μL of the same precipitant at room temperature. The resulting crystals were cryoprotected and flash-cooled in liquid nitrogen.
Data Collection and Structure Determination.
Single wavelength anomalous diffraction data, consistent with space group P21 and extending to 1.85 Å resolution, were collected in the vicinity of the selenium anomalous peak wavelength (λ=0.9793 Å) at Brookhaven National Laboratory National Synchrotron Light Source X29A beamline. Matthews coefficient calculations were consistent with the presence of two molecules in the asymmetric unit (ASU).
Sixteen of sixteen ordered selenium sites were located using the SHELXD program, and density modified SAD phases were calculated with SHELXE.23 Following several rounds of automated model building using ARP/wARP in CCP424, 25 and manual adjustment using Coot26, refinement using Refmac527 converged at Rwork=18.3% and Rfree=22.5%.
The final model consists of 5,859 protein atoms including two chains of AaOSBS (chain A, Gln3 to L379; chain B, Gln3 to Thr378), 235 waters, N-terminal “SL” cloning artifacts (SerLeu; both chains), C-terminal cloning artifact/tag (EGHH: chain A), and 5 sulfate ions. Observed methionine residues were modeled as selenomethionines. Several segments, including Ser83 to Gln86 of chain B, Leu379 of chain B, and C-terminal cloning artifacts (His4 of chain A and the entire EGHHHHHH of chain B), were not observed in the electron density presumably due to disorder. Crystallographic data collection and refinement statistics are shown in Table 1. The coordinates and structure factors have been deposited in the Protein Data Bank as entry 3QLD.
Table 1. Crystallographic data collection and refinement statistics.
Data collection and refinement statistics for 3QLD. Values in parentheses are for the highest resolution shell.
| Data Collection | |
|---|---|
| PDB Code | 3QLD |
| Space group | P21 |
| Cell dimension (Å) | a = 54.70 |
| b = 82.05 | |
| c = 77.95 | |
| β = 104.60 | |
| Molecules/ASU | 2 |
| Wavelength (Å) | 0.9793 |
| Resolution (Å) | 50 – 1.85 (1.88 – 1.85) |
| Unique reflections | 56,819 |
| Completeness (%) | 100 (99.9) |
| Rsym (%) | 0.090 (0.764) |
| <I/σI> | 12.8 (2.0) |
| Redundancy | 7.5 (7.4) |
| Refinement | |
| R / Rfree | 18.3 / 22.5 |
| No. of Atoms | |
| Protein/Solvent/Salt | 5,859 / 235 / 25 |
| Average B Factor (Å2) | |
| Protein/Solvent/Salt | 31.9 / 33.9 / 62.5 |
| Ramachandran Statistics (%) | |
| Favored/Allowed/Outlier | 97.0 / 3.0 / 0 |
| Rms deviation from ideal | |
| Bonds (Å) | 0.023 |
| Angles (°) | 1.921 |
Mutagenesis.
Site directed mutagenesis was performed using the Q5 Site-Directed Mutagenesis protocol (New England Biolabs). Primers were designed using NEBaseChanger, NEB’s online design software (NEBasechanger.com). Mutations were confirmed by sequencing in each direction (Eurofins Genomics). Sequences of primers used for mutagenesis are included in supporting information (Table S2).
Protein Expression and Purification for Size Exclusion and Activity Assays.
The AaOSBS protein and variants were expressed with a C-terminal His6-tag in the vector pSGX3. Wild type AaOSBS was transformed into BL21 (DE3), while the mutants were transformed into E. coli BW25113 strain (ΔmenC, DE3) to ensure that the purified protein would not be contaminated with the native E. coli OSBS, which is encoded by the menC gene.6 This strain was derived from E. coli BW25113 strain (menC::kan, DE3) by deleting the kanamycin resistance gene using the FLP helper plasmid pCP20.28 Bacterial cultures were grown for 20 hr at 37 °C (AaOSBS wild type) or 48 hr at 30 °C (AaOSBS mutants) without induction in Luria-Bertani broth supplemented with kanamycin at a final concentration of 50 μg/ml.
The expression plasmids encoding AmedNSAR, LvNSAR/OSBS and RcNSAR/OSBS were transformed into E. coli BW25113 strain (menC::kan, DE3), and bacterial cultures were grown for 20 hr at 37 °C without induction in Luria-Bertani broth supplemented with both kanamycin and carbenicillin at a final concentration of 50 μg/ml.
Cells were pelleted by centrifugation, resuspended in 10 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole, 0.02 mg/mL DNase (GoldBio) and 2 μM phenylmethanesulfonyl fluoride (PMSF; Thermo Scientific), and lysed by sonication. The lysate was centrifuged to remove cell debris, filtered using a 0.22 μm Steriflip filter (Millipore) and loaded into a 5 mL HisTrap FF column charged with Ni2+ (GE Healthcare). The protein was eluted using a buffer containing 10 mM Tris pH 8.0, 500 mM NaCl, and 500 mM imidazole with a step to 15% elution buffer to elute loosely bound proteins, followed by a linear gradient to 100% elution buffer over 20 column volumes. The protein was homogeneous as determined by SDS-PAGE, and fractions containing the protein were pooled and concentrated using an Amicon Ultra-15 centrifugal filter (10 kD MW cutoff; Millipore), supplemented with glycerol to a final concentration of 25% and stored at −20 °C.
Size Exclusion Chromatography.
Size exclusion chromatography of AaOSBS was performed using a Superdex 200 PG 16/60 gel filtration column (GE Healthcare) connected to an AKTA Explorer 10 FPLC (GE Healthcare). Column pre-equilibration was performed with the mobile phase buffer containing 150 mM NaCl, 10 mM Tris pH 8.0, and 5 mM MgCl2. 1.5 mg of protein was separated at a flow rate of 1 mL/min. The size exclusion column was standardized by plotting log10 molecular weights of thyroglobulin (669 kDa), apoferritin (443 kDa), IgG (160 kDa), BSA (65 kDa), ovalbumin (43 kDa), myoglobulin (17.5 kDa), and lysozyme (14.3 kDa) against their elution volumes. The equation derived from the linear plots was then used to estimate the molecular weights of the observed peaks. Peak fractions observed at 280 nm were assayed for OSBS activity (see below) to verify that AaOSBS was present in the fractions.
Substrate synthesis.
2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC), N-succinyl-L-phenylglycine (L-NSPG), and N-succinyl-D-phenylglycine (D-NSPG) were prepared as previously described.6, 29 Synthesis of other succinylamino acids is described in Supporting Information.
Enzymatic Assays.
OSBS activity was measured in 50 mM Tris pH 8.0 and 0.1 mM MnCl2 at 25 °C with final enzyme concentrations ranging from 0.001 to 0.02 μM and various substrate concentrations. OSBS activity was determined by measuring the disappearance of SHCHC at 310 nm (Δε = −2400 M−1cm−1) using a SpectraMax Plus384 UV-VIS microplate spectrophotometer (Molecular Devices).11, 14 NSAR activity of AaOSBS was measured using 10 μM enzyme and 7.5 mM N-succinylamino acid substrates in 200 mM Tris pH 8.0 and 0.1 mM MnCl2. NSAR activities of AmedNSAR, LvNSAR/OSBS and RcNSAR/OSBS were measured using 0.05 μM enzyme and various concentrations of D-succinylphenylglycine in 200 mM Tris pH 8.0 and 0.1 mM MnCl2. The substrate’s change in optical rotation was measured at 405 nm in a sample cell with path length of 5 cm using a P-2000 Polarimeter (Jasco). The measurements were integrated for 10 seconds with readings every 30 seconds for a total of 30 minutes for AaOSBS and 5 minutes for AmedNSAR, LvNSAR/OSBS and RcNSAR/OSBS.29 The initial rates were determined by fitting a line to the portion of the curve that was linear, and the initial rates at varying substrate concentrations were fit to the Michaelis-Menten equation using Kaleidagraph (Synergy Software).
Inhibition of AaOSBS activity was measured by supplementing the reaction with 0-8.0 mM L-NSPG or D-NSPG under the conditions above. After determining the initial rates, KI was determined by fitting the observed rates to the equation below, in which [I] is the concentration of the inhibitor and KM is a constant determined in the absence of inhibitor:
The reported KI is the average of KI determined at 3 inhibitor concentrations (3 mM, 5 mM and 8 mM).
Isotopic Exchange Experiments Using 1H NMR Spectroscopy.
AaOSBS variants were exchanged into D2O using a Vivaspin Turbo 15 centrifugal filter (Sartorius). A 10 mL aliquot of protein was concentrated to 1 mL; 4 mL of 50 mM Tris (pD 8.0) was added, and the protein solution was again concentrated to 1 mL. This process was repeated three times to maximize the exchange. Samples for 1H NMR (600 μL) contained 20 mM L- or D-N-succinylphenylglycine, 50 mM Tris (pD 8.0), 0.1 mM MnCl2, and 0.5 mg/mL (11.7 μM) AaOSBS WT or AaOSBS Y299I in D2O. The exchange was monitored by 1H NMR (500 MHz Bruker NMR), following the change in intensity of the α-proton as it is exchanged with deuterium over time. The peak for the α-proton (δ = 5.15 ppm) was integrated relative to that of the five aromatic protons (δ = 7.40 ppm). The relative peak area was converted to concentration based on the initial substrate concentration. The slopes of the plots of the NSPG substrate concentration as a function of time were used to obtain the isotopic exchange rates (kex).
Bioinformatics and Molecular Modeling.
The genomes of A. acidocaldarius, A. mediterranei, L. varians, and R. castenholzii were examined in the PubSEED database to determine whether the menaquinone synthesis pathway was present and to determine which genes flank the NSAR/OSBS subfamily gene.30 The genomes were also examined in the ProOpDB and DOOR2.0 databases to determine if the NSAR/OSBS subfamily gene and the flanking genes were predicted to be co-transcribed as an operon.31-34 Structural analysis was performed using UCSF Chimera.35 PISA (“Protein interfaces, surfaces and assemblies” service at the European Bioinformatics Institute, http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) was used to analyze the dimeric interface.36 Sequence logos were generated by the Weblogo 3.0 server (http://weblogo.threeplusone.com) using an alignment of 571 NSAR/OSBS subfamily enzymes that share <80% identity.37 The phylogeny was constructed from a subset of this sequence alignment using RaxML.38, 39
Two types of structural models of AaOSBS were constructed to examine potential conformation changes and ligand binding. The first model was based on the AaOSBS experimental structure (3QLD). To estimate how ligands would bind to the structure, OSB was placed into the active site in the conformation observed in Amycolatopsis sp. T-1-60 NSAR (AmyNSAR) bound to OSB (1SJB:B) by aligning the catalytic residues of the AaOSBS experimental structure (3QLD:A) with AmyNSAR (1SJB:B).7 The aligned coordinates of AaOSBS and OSB were used as a template in MODELLER.40, 41 The backbone of a loop from residues 17-25 (the 20s loop) was modeled with the DOPE loop algorithm, while all other residues were modeled with a fixed backbone.42 100 loop conformations were produced, and the lowest energy structure was selected for docking. Compared to 3QLD:A, the 20s loop extends farther over the active site, reminiscent of the closed 20s loop observed in related, ligand-bound enzymes.7, 43, 44 Other side chains show little deviation from the experimental structure. This model is designated “3QLD with remodeled 20s loop” in Figure 7.
Second, we constructed homology models based on ligand-bound NSAR/OSBS enzyme structures from Deinococcus radiodurans (1XPY:C) and Amycolatopsis sp. T-1-60 (1SJB:B), to examine potential conformation changes and ligand binding in a closed active site.7, 44 Four homology models were constructed using these two proteins as templates in the presence of each substrate and product of the NSAR and OSBS reactions. The coordinates of OSB were from the experimental AmyNSAR structure (1SJB:B), as described above. The coordinates of SHCHC, D-NSPG, and L-NSPG were from previous computational ligand docking experiments with AmyNSAR.12 The N-acetyl-Gln ligand from 1XPY:C was not included in the model. Constructing separate models in the presence of each ligand reduced the number of clashes and improved the likelihood of obtaining reasonable ligand conformations in subsequent docking experiments (below). The homology models were built using MODELLER, as described above, using the DOPE loop algorithm to model the 20s loop and modeling other residues with a fixed backbone. These models are designated “AaOSBS model” in Figure 7.
Although AaOSBS was modeled in the presence of ligand, we discovered that removing the ligand from the model and re-docking was necessary to achieve ligand conformations that had no steric clashes. Substrates and products were docked into each model using the Autodock Vina tool of Chimera using the Opal web server.45, 46 Coordinates of ligands (OSB, SHCHC, L-NSPG and D-NSPG) were the same as used in our previous models of Exiguobacterium sp. AT1b OSBS (ExiOSBS).12 Some torsion angles of active site residues were manually adjusted following docking as described in the results below. Previously published models of ligand-bound Amycolatopsis sp. T-1-60 NSAR/OSBS (AmyNSAR) were used for comparison.12
Results
Biological Functions of AmedNSAR, LvNSAR/OSBS, RcNSAR/OSBS, and AaOSBS.
To improve understanding about the range of specificity in the NSAR/OSBS subfamily, we selected four proteins to experimentally characterize based on their phylogenetic distribution and genome context (Figure 2). AmedNSAR, LvNSAR/OSBS, RcNSAR/OSBS, and AaOSBS share 83%, 48%, 49%, and 34% identity with the extensively characterized AmyNSAR (known as AmyNSAR/OSBS in previous publications; note that all of these enzymes are named according to their predicted biological functions, as described below).7, 11, 14, 29 Compared to the outgroup species in Figure 2, all four enzymes share about 32% sequence identity with Exiguobacterium AT1b OSBS (ExiOSBS) and 25% sequence identity with Staphylococcus aureus OSBS. While this demonstrates the range of sequence identities within the NSAR/OSBS subfamily, members of the NSAR/OSBS subfamily are much more similar to each other than to members of other OSBS subfamilies, which frequently have sequence identities <20% and large unalignable regions due to insertions and deletions.8 RcNSAR/OSBS (the first NSAR/OSBS subfamily member to be characterized from the Chloroflexi phylum, from species R. castenholzii) and AmedNSAR (found in the Actinobacteria phylum species A. mediterranei) originated from horizontal gene transfer from a Firmicutes phylum ancestor, which was a common occurrence in the NSAR/OSBS subfamily.9 It is difficult to determine whether LvNSAR/OSBS, from the Firmicutes species L. varians, originated from vertical or horizontal gene transmission, because a widely accepted Firmicutes species tree is not available to compare with the NSAR/OSBS subfamily tree. AaOSBS is also found in a Firmicutes species, but the fact that it clusters with NSAR/OSBS subfamily enzymes found in non-Firmicutes species and its gene’s position far from the menaquinone operon suggest that it could also have originated by horizontal gene transfer, perhaps replacing the OSBS gene that was originally in the menaquinone synthesis operon.
Figure 2.
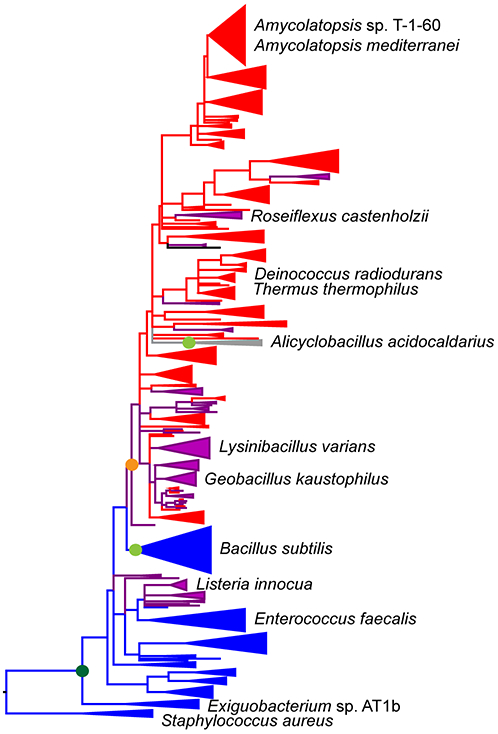
Phylogenetic distribution of characterized members of the NSAR/OSBS subfamily. Enzymes characterized in this study are marked with asterisks. The phylogeny (solid lines) was constructed using a representative set of sequences from the NSAR/OSBS subfamily. Groups of sequences were collapsed into wedges if they had the same predicted function and the branch support values (SH scores) were >0.85. The dashed lines illustrate how the NSAR/OSBS subfamily is nested within the OSBS family and enolase superfamily, based on separate phylogenetic analyses of representative sequences from the family and superfamily.6, 9 Blue branches consist of proteins that are encoded in menaquinone operons, indicating that OSBS is their biological function. Red branches consist of proteins whose biological function is expected to be NSAR activity because their species do not require OSBS activity to make menaquinone or there is a separate OSBS gene encoded in the menaquinone operon. Purple branches consist of proteins that are expected to be bifunctional, because OSBS activity is required for menaquinone synthesis but the NSAR/OSBS subfamily gene is not in the menaquinone operon. Many of these proteins are encoded in operons with genes from the D-amino acid conversion pathway, but some, like the aaOSBS gene (gray branch), are in novel genome contexts and require further analysis to determine their biological functions. Based on phylogenetic relationships among characterized sequences, the dark green circle indicates initial appearance of NSAR activity as a promiscuous reaction, and light green circles indicate loss of NSAR activity within the indicated group.12, 32-34, 47 Based on genome context of contemporary NSAR/OSBS subfamily enzymes, the orange symbol indicates the earliest point when promiscuous NSAR activity was recruited into a biological pathway. NSAR activity might have also been recruited into a different pathway on the L. innocua branch.
Genome context suggests that the NSAR/OSBS subfamily enzymes from L. varians and R. castenholzii are bifunctional, as observed in G. kaustophilus (Figure 3). The GkNSAR/OSBS gene is in an operon encoding a pathway for converting D-amino acids to L-amino acids, and it is also required to complete the menaquinone synthesis pathway (encoded by the menA, menB, menC, menD, menE, menF, and menH genes).13 Both L. varians and R. castenholzii have the menaquinone synthesis pathway, and the only enzyme they encode that could supply OSBS activity for this pathway is the NSAR/OSBS subfamily enzyme. Consistent with its position in a phylogenetic clade with GkNSAR/OSBS, LvNSAR/OSBS is encoded in a similar operon, along with homologs of the G. kaustophilus succinyltransferase (GNAT superfamily) and L-desuccinylase (M20 family). RcNSAR/OSBS is in an operon with a member of the GNAT superfamily and an α/β hydrolase superfamily member. It is possible that this α/β hydrolase serves the same function as the M20-family L-desuccinylase of G. kaustophilus.
Figure 3.
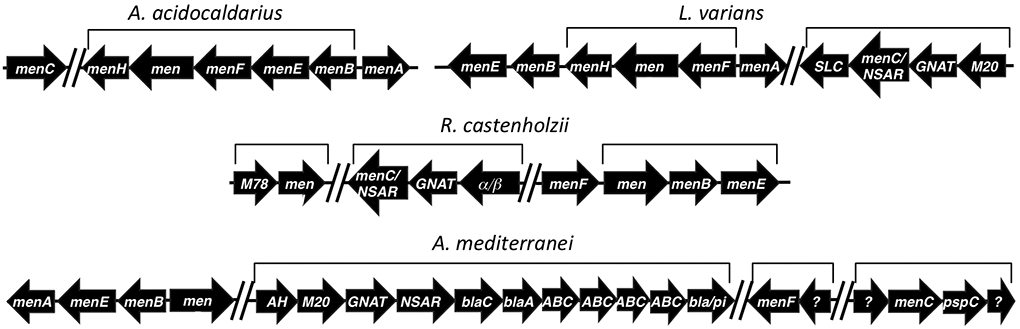
Genome context of NSAR/OSBS genes. Genes that are predicted to be co-transcribed as an operon are designated with brackets (for L. varians, operon predictions were taken from the closely related species L. sphaericus).32-34 Gaps indicate regions of >150 kilobases between gene clusters. Menaquinone synthesis genes are designated menA-menH. menH and menA were not annotated and could not be unambiguously identified in some species. Other genes are annotated by their family or superfamily.48 M20 = M20 peptidase family; GNAT = GCN5-related N-acyltransferase; SLC = solute carrier families 5 and 6-like solute binding domain; M78 = M78 peptidase family; α/β = α/β hydrolase superfamily; AH = amidohydrolase superfamily; blaC = beta-lactamase class C; blaA = beta-lactamase class A; ABC = subunits of dipeptide/oligopeptide ABC transporter; bla/pi = beta lactamase/prolyl oligopeptidase; pspC = phage shock protein C transcriptional regulator; ? = hypothetical protein.
Although the sequence similarity between AmedNSAR and the well-characterized AmyNSAR is high, we chose to characterize AmedNSAR because, unlike AmyNSAR, its species’ genome has been sequenced, and its gene has a different genome context than other NSAR/OSBS genes. The genome context of AmedNSAR suggests that NSAR activity is its only biological function. Although A. mediterranei has the menaquinone synthesis pathway and requires OSBS activity, it encodes an OSBS enzyme from the Actinobacteria OSBS subfamily (labeled by its gene name, menC, in Figure 3), which shares <30% sequence identity with enzymes in the NSAR/OSBS subfamily.6 The amedNSAR gene is in an operon that appears to be an elaboration of the G. kaustophilus D-amino acid conversion pathway operon. In addition to the GNAT and M20 family enzymes, this operon encodes an amidohydrolase superfamily enzyme, members of three different beta-lactamase families, and four subunits of an ABC transporter. The annotations of the amidohydrolase superfamily enzyme (D-glutamate deacylase), the M20 family enzyme (glutamate carboxypeptidase), the transporter subunits (dipeptide/oligopeptide ABC transporter), and the third beta-lactamase protein (prolyl oligopeptidase), suggest that these proteins are involved in peptidoglycan degradation. Because several Amycolatopsis species have an NSAR/OSBS subfamily enzyme in this genome context and because of their high sequence similarity, the characterized NSAR/OSBS subfamily enzyme from Amycolatopsis sp. T-1-60 was rechristened AmyNSAR, instead of AmyNSAR/OSBS, which was used in previous studies about this enzyme.
A. acidocaldarius also has a menaquinone synthesis operon, but the gene that encodes AaOSBS, (which is shown as menC in Figure 3) is over 1300 kilobases away from the rest of the menaquinone synthesis genes. Also, the AaOSBS enzyme clusters in the phylogeny with other proteins whose biological functions are predicted to be NSAR activity, based on genome context (Figure 2). This raised the possibility that AaOSBS could be bifunctional, like GkNSAR/OSBS. However, the aaOSBS gene in A. acidocaldarius is between some ribosomal RNA genes and an operon comprised of putative cell division (ftsE, ftsX and minJ) and flagellar motor genes (motA and motB). The aaOSBS gene is not predicted to be co-transcribed with any of these genes.32-34 Furthermore, no homolog of the G. kaustophilus succinyltransferase could be identified, and the two M20 family proteins encoded by A. acidocaldarius share only 28% and 33% identity with the G. kaustophilus L-desuccinylase. Thus, unless AaOSBS has an undiscovered activity, OSBS activity is likely to be its only biological function.
Assaying the four enzymes for OSBS and NSAR activities largely confirmed their predicted activities (Table 2). LvNSAR/OSBS has very efficient OSBS activity, but has lower NSAR activity with N-succinylphenylglycine than most other promiscuous or bifunctional NSAR/OSBS enzymes. Likewise, RcNSAR/OSBS efficiently catalyzes both reactions, although its NSAR activity is slightly lower than most other promiscuous or bifunctional NSAR/OSBS enzymes. While N-succinylphenylglycine was chosen for this study because it has the highest structural similarity to SHCHC (the substrate in the OSBS reaction), it is possible that LvNSAR/OSBS and RcNSAR/OSBS would exhibit higher activities with succinylamino acids that have other side chains. OSBS activity is not expected to be a necessary biological function of AmedNSAR, but it promiscuously catalyzes OSBS activity along with the expected, biologically-relevant NSAR activity. Efficient promiscuity for the ancestral OSBS activity is typical of other characterized NSAR/OSBS subfamily enzymes, such as Deinococcus radiodurans NSAR/OSBS (DrNSAR), whose species do not need OSBS activity for menaquinone synthesis.8, 9
Table 2.
Enzymatic activities of AaOSBS and related enzymes in the NSAR/OSBS subfamily.
| Enzyme | OSBS activity | NSAR activity a | ||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) |
kcat/KM (M−1s−1) |
kcat (s−1) | KM (μM) |
kcat/KM (M−1s−1) |
|
| AaOSBS | 64 ± 3 | 118 ± 18 | 5.4 × 105 | <0.0025 b | - | - |
| LvNSAR/OSBS | 179 ± 8 | 96 ± 13 | 1.9 × 106 | 2.2 ± 0.2 b | 2700 ± 540 | 8.1 × 102 |
| RcNSAR/OSBS | 38 ± 3 | 155 ± 22 | 2.5 × 105 | 15 ± 4 b | 1800 ± 230 | 8.3 × 103 |
| AmedNSAR | 242 ± 11 | 415 ± 39 | 5.8 × 105 | 74 ± 7 b | 2800 ± 550 | 2.6 × 104 |
| ExiOSBS 12 | 51 ± 5 | 20 ± 7 | 2.6 × 106 | 0.07 ± 0.006 c | 1700 ± 500 | 41 |
| GkNSAR/OSBS 13 | 180 | 95 | 1.9 × 106 | 19 ±1 d | 800 ± 200 | 2.3 × 104 |
| AmyNSAR 29 | 46 ± 5 | 550 ± 120 | 8.3 × 104 | 42 ± 2 c | 1000 ± 10 | 4.2 × 104 |
| DrNSAR 8 | 8.1 ± 0.6 | 26 ± 7 | 3.1 × 105 | 520 ± 30 c | 1400 ± 200 | 3.7 × 105 |
Enzymes were assayed with the designated substrates. Note that previously characterized NSAR/OSBS enzymes exhibited comparable rates with D- and L-succinylamino acids, making it reasonable to compare NSAR activities of these enzymes.13, 14
N-succinyl-D-phenylglycine was the substrate.
N-succinyl-L-phenylglycine was the substrate.
N-succinyl-L-phenylalanine was the substrate.
In contrast, AaOSBS is an efficient OSBS enzyme, with NSAR activity below the limit of detection, using the substrates L-NSPG, D-NSPG, N-succinyl-L-phenylalanine, N-succinyl-D-phenylalanine, N-succinyl-L-valine, or N-succinyl-L-tryptophan. Thus, given the position of AaOSBS in the phylogeny, we hypothesize that it lost its ancestral NSAR activity. To test this hypothesis, we characterized the mechanism and structure of AaOSBS to determine if the loss of activity could be understood and reversed.
To determine if AaOSBS can bind D- or L-NSPG, we measured the inhibition of the OSBS reaction by these compounds. Lineweaver-Burk and Dixon plots are consistent with competitive inhibition (Figure 4). The KI of L-NSPG and D-NSPG are 2600 ± 400 μM and 3600 ± 400 μM, respectively. The KI values are ~20-fold higher than the KM of SHCHC, indicating that the affinity for succinylamino acids is lower than that for SHCHC. However, we would have expected to detect activity if only binding affinity were affected. In fact, the inhibition constants are only slightly higher than the KM of NSAR reactions catalyzed by several NSAR/OSBS subfamily enzymes (Table 2).8, 14, 29
Figure 4.
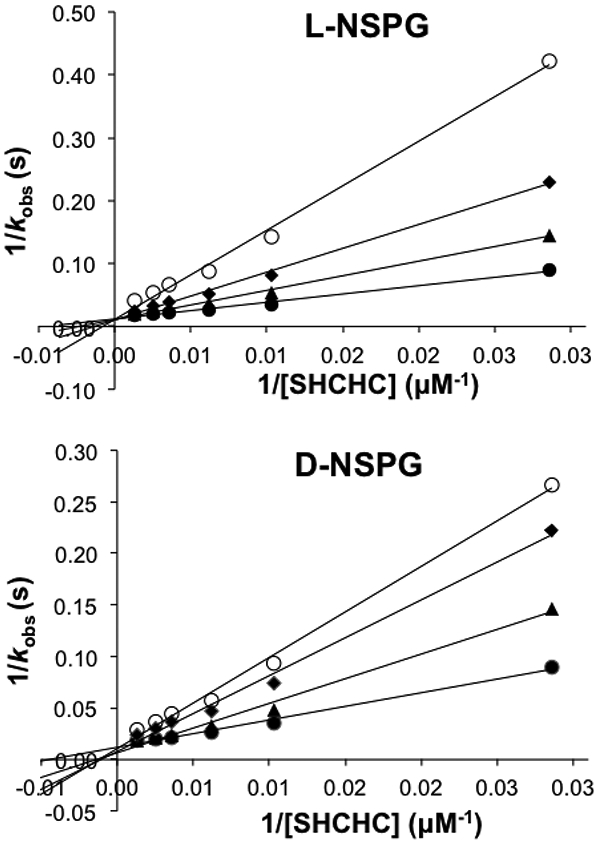
Lineweaver-Burk plot of OSBS activity in the presence of 0 mM (●), 3 mM (▲), 5 mM (♦), or 8 mM (O) L- or D-NSPG.
Structure of AaOSBS.
Because AaOSBS is one of only two known members of the NSAR/OSBS subfamily that lack NSAR activity, we determined its structure to discover why its specificity differs from its homologs. AaOSBS adopts the canonical enolase superfamily fold, consisting of an α/β-barrel domain that includes the catalytic amino acids and an α + β capping domain consisting of the N-terminal third of the enzyme and a short section of the C-terminus. The crystal structure of AaOSBS suggests that it is a dimer, with an interface that is very similar to that of other members of the NSAR/OSBS subfamily (Figure 5A). The interface consists of the 50s loop, which is one of two active site loops from the capping domain, part of the α-helix after the 50s loop (Cap-α1), another loop from the capping domain, and the 4th and 5th α-helices of the barrel domain. We verified the quaternary structure of AaOSBS by size exclusion chromatography. It eluted with an apparent molecular weight of 60 kD, halfway between the calculated molecular weight of the monomer (43 kD) and dimer (86 kD). In previous experiments, we found that this size exclusion column underestimated the molecular weight of a 43 kD L. innocua NSAR/OSBS which eluted in two peaks with estimated molecular weights of 71 kD and 39 kD, corresponding to dimer and monomer, respectively 8. Thus, the molecular weight estimation of AaOSBS by size exclusion chromatography is more consistent with it being a dimer.
Figure 5.
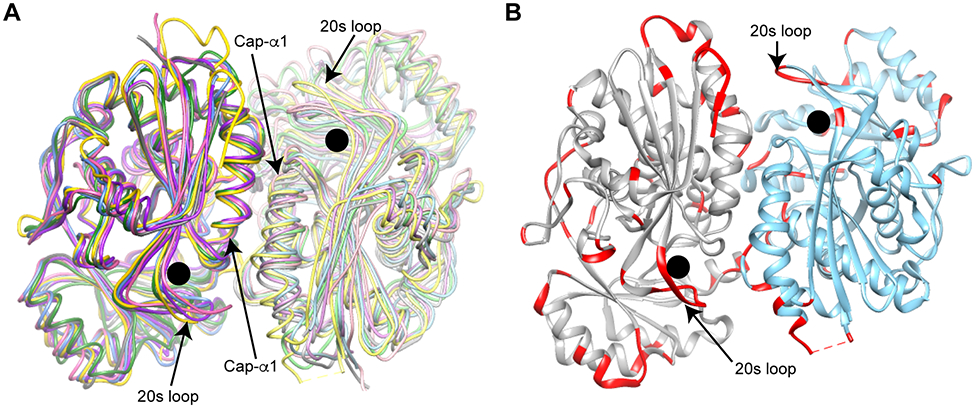
Structure of AaOSBS. The active site is marked with a black circle. A) Conserved dimeric interface in the NSAR/OSBS subfamily. The first helix of the capping domain and a flexible loop around position 20 are labeled. AaOSBS (3QLD chains A and B) is gold/yellow; AmyNSAR (1SJB chains A and B) is green/light green; Listeria innocua NSAR/OSBS (1WUF chains A and B) is blue/light blue; EfNSAR/OSBS (1WUE chains A and B) is dark pink/light pink; Thermus thermophilus NSAR/OSBS (2ZC8 chains A and B) is purple/lavender; and DrNSAR (1XPY chains C and A) is dark gray/light gray.7, 8, 44, 49 B) Predicted flexibility of AaOSBS. B-factors of atoms in residues shown in red is > 50. Chain A is gray and chain B is cornflower blue.
AaOSBS is structurally very similar to other NSAR/OSBS subfamily enzymes. Throughout this manuscript, we compared root mean square deviations (RMSD) between structures by matching 330 Cα-atoms (~90% of the sequence length). This eliminated the structurally divergent N- and C-termini, as well as some surface loops, that skew the RMSD toward outlying values. RMSDs between chain A of AaOSBS (3QLD:A) and NSAR/OSBS subfamily enzymes from Listeria innocua (1WUF), Enterococcus faecalis (EfNSAR/OSBS; 1WUE), Thermus thermophilus (2ZC8), and D. radiodurans (1XS2) are 0.9-1.1 Å (Figure 5A). 3QLD:B is less similar, with RMSDs of 1.4-1.6 Å. None of these structures are bound to a substrate or product analog. In contrast, chains in the structure of D. radiodurans NSAR/OSBS (DrNSAR; 1XPY:C) that are bound to N-acetylglutamine are less similar to AaOSBS (3QLD:A), with an RMSD of 1.4 Å. Likewise, the RMSD between AaOSBS (3QLD:A) and AmyNSAR (1SJB), which is bound to OSB, is also higher, at 1.6 Å. Thus, the higher RMSD between AaOSBS and the ligand bound AmyNSAR and DrNSAR probably reflects conformation changes upon ligand binding.
The most notable difference between the AaOSBS structure and ligand-bound NSAR/OSBS enzyme structures is the openness of the active site. The active site of OSB-bound AmyNSAR is a closed tunnel of about 580 Å3, with two small openings only large enough for water, while the active site of N-acetlyglutamine-bound DrNSAR adopts a similar shape but is about 750 Å3 due to an additional, water-filled channel to the protein surface. In contrast, the active sites of AaOSBS and EfNSAR/OSBS are open clefts of about 800-900 Å3, and the active site of apo-DrNSAR (1XPY:B) is ~1300 Å3, because an active site loop around position 20 (the 20s loop) is disordered. The active site volume estimates for DrNSAR, EfNSAR/OSBS, and AaOSBS are less precise because defining the boundaries of surface-accessible channels and open clefts are less precise. Nevertheless, the active site volumes of the two ligand bound structures are smaller, and both enclose their ligands in constraining tunnels. Because EfNSAR/OSBS and DrNSAR efficiently catalyze both NSAR and OSBS reactions, active site size or openness of apo-AaOSBS does not explain its inability to catalyze the NSAR reaction. Structural comparisons do support the idea that ligand-binding induces a conformation change that decreases the active site volume. Complete active site closure might not be necessary, because distantly related OSBS enzymes from other subfamilies only partially close their active sites.10, 43 Ligand-bound structures of AaOSBS and EfNSAR/OSBS are necessary to determine if the extent of active site closure affects the ability to catalyze the NSAR reaction.
Several types of motions could contribute to this conformation change, including repositioning the 20s loop, a flexible active site loop that is typically disordered or has high B-factors in structures of enolase superfamily enzymes (Figure 5B).8, 43, 44, 49-51 Another type of motion would narrow the active site between the two catalytic lysines in the barrel domain. In the OSBS reaction, K170 abstracts a proton from the substrate, while K269 presumably stabilizes the transition state through a cation-π interaction.14 In AaOSBS, the distance between the two catalytic lysines (K170 and K269) is rather wide at 8.8 or 10.4 Å in chains A and B, respectively. This is similar to the distance between the catalytic lysines in the apo-subunits of DrNSAR (8.8 and 12.8 Å in 1XPY:A and 1XPY:B, respectively). In contrast, the distance is only 6.1 Å in both OSB-bound AmyNSAR (1SJB) and N-acetylglutamine-bound subunits of DrNSAR (1XPY:C and 1XPY:D). Narrowing the AaOSBS active site would be expected to promote OSBS activity, and it could be required for NSAR activity, since both lysines must act as general acid or base catalysts. Attempts to determine the magnitude of these conformation changes in AaOSBS by determining its structure bound to Mg2+ and o-succinylbenzoate or N-succinylphenylglycine have not been successful yet.
Active Site Comparison and Ligand Docking.
Most active site residues of AaOSBS are conserved with those in other NSAR/OSBS subfamily enzymes, with a few exceptions (Figure 6). In the 20s loop, M18 in AaOSBS aligns with F19 of AmyNSAR, which is important for both NSAR and OSBS activities.29 Notably, the vast majority of enzymes in the NSAR/OSBS subfamily have phenylalanine at this position. Two exceptions are AaOSBS, which has no NSAR activity, and ExiOSBS, which has very little NSAR activity.
Figure 6.
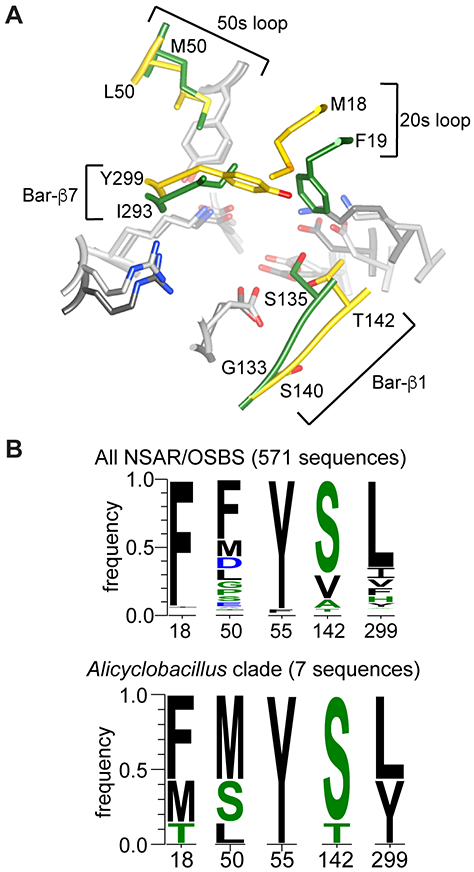
Comparison of AaOSBS to other NSAR/OSBS enzymes. A) Superposition of the active sites of AaOSBS and AmyNSAR (1SJB:B).7 Conserved amino acids are light gray (AaOSBS) or dark gray (AmyNSAR). Nonconserved residues (labeled) are gold (AaOSBS) or green (AmyNSAR). Bar-β1 and Bar-β7 are strands in the barrel domain. Alternate conformations of M18, M298, and Y299 in AaOSBS and some conserved active site residues have been omitted for clarity. B) Conservation of active site residues that differ between AaOSBS and most other NSAR/OSBS subfamily enzymes or which were predicted to affect specificity based on potential steric conflicts observed by in silico ligand docking. Height of the letters is proportional to their frequency in the data set. Positions are numbered according to AaOSBS.
Across the active site from M18 at the end of the seventh β-strand of the barrel domain (Bar-β7) is Y299, which corresponds to I293 in AmyNSAR and L299 in DrNSAR. ExiOSBS, like AaOSBS, has a tyrosine at this position.12 Nearly all other experimentally characterized members of the NSAR/OSBS subfamily have leucine or isoleucine at this position; almost all of these enzymes efficiently catalyze both NSAR and OSBS activities. Overall, however, this position is more variable than most other active site residues, with about 80% of the NSAR/OSBS subfamily having branched chain hydrophobic amino acids and the remainder having aromatic, polar or charged residues at this site.
Two other positions (50 and 142) that differ between AaOSBS and AmyNSAR also vary within the NSAR/OSBS subfamily. Position 50 contacts the cyclohexyl ring of SHCHC and the side chain of succinylamino acids. It is thus expected to be hydrophobic in enzymes with OSBS activity or which racemize hydrophobic succinylamino acids, as observed in GkNSAR/OSBS.13 Position 142 is usually polar and forms a hydrogen bond with the carbonyl of the succinyl group. This does not appear to be strictly necessary, however, since alanine and valine are common at this site.
To investigate how active site differences among these proteins could affect specificity, we computationally docked the substrates and products of the NSAR and OSBS reactions into the structure of AaOSBS. A minimal conformational change was modeled by using the AaOSBS crystal structure as a template and remodeling the 20s loop in a more extended and closed conformation, making the active site a partially closed cleft. While this enabled OSB to be docked in a similar conformation to that observed in AmyNSAR, the distances to active site residues are larger. In particular, the catalytic lysine K170 is more than one ångstrom farther from the alpha carbon from which the proton was abstracted. The succinyl carboxylate is also farther from R305 (Figure 7A,C). This supports the idea mentioned above that an additional conformation change contracts the active site when the substrate binds.
Figure 7.
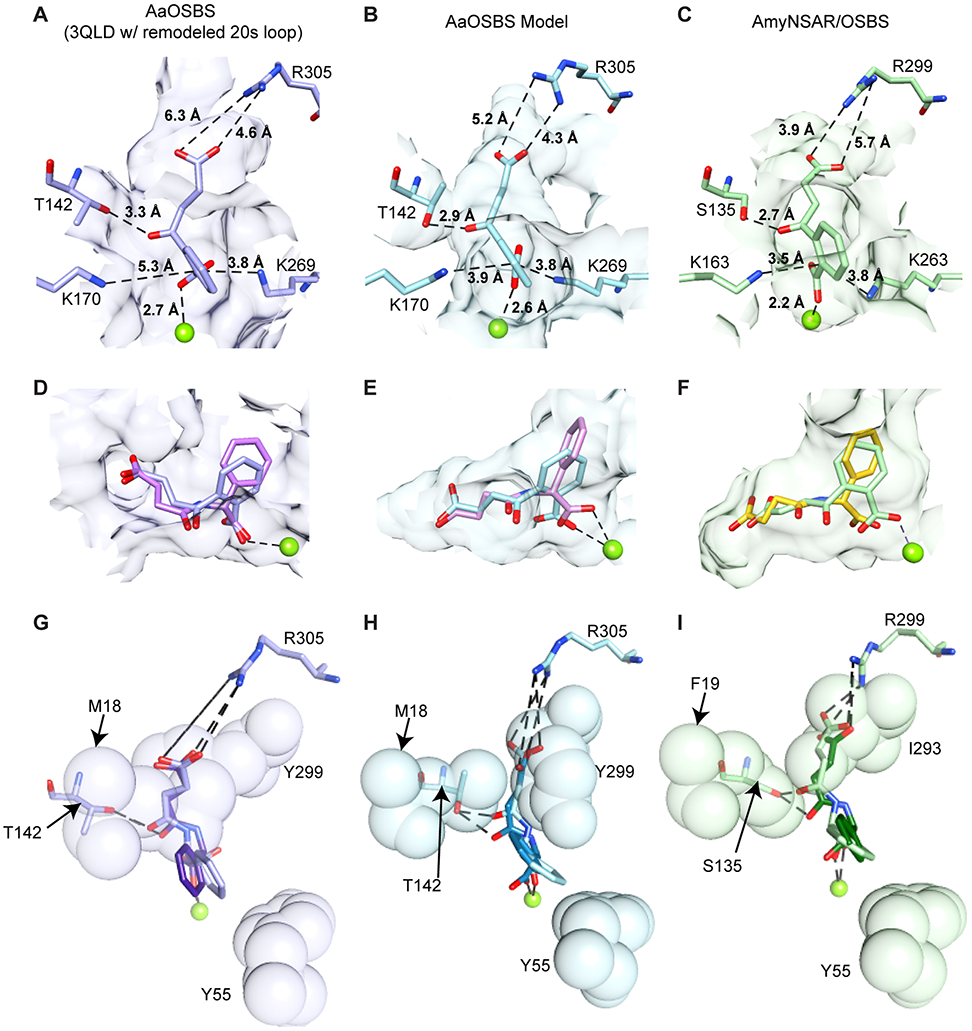
Comparison of ligand binding to experimental structures or docked models of AaOSBS and AmyNSAR. Mg2+ is shown as a lime sphere. Surfaces of active site residues that are < 5.0 Å from OSB in A-F are shown in transparent purple, blue, or green. In the first column (A, D, and G), ligands were docked into the AaOSBS experimental structure (3QLD:A) after remodeling the 20s loop. In the second column (B, E, and H), ligands were docked into a homology model of AaOSBS generated using ligand-bound crystal structures of AmyNSAR and DrNSAR as templates. In the third column, panel C shows the experimental structure of AmyNSAR bound to OSB (1SJB:B), and panels F and I show the other ligands docked into AmyNSAR (1SJB:B). (A-C) OSB. D) OSB (lavender) and D-NSPG (pink). E) OSB (light blue) and D-NSPG (pink). F) OSB (light green) and D-NSPG (gold). In G-I, residues shown as transparent spheres are in the foreground and residues and ligands shown as sticks are in the background. G) D-NSPG (lavender) and L-NSPG (purple). M18 is above Y299. H) D-NSPG (light blue) and L-NSPG (turquoise). I) D-NSPG (light green) and L-NSPG (dark green).
Consequently, we docked the substrates and products of the NSAR and OSBS reactions into a homology model of AaOSBS which was constructed based on ligand-bound crystal structures of AmyNSAR and DrNSAR (Figure 7B). The active site in this model is a closed tunnel of about 550 Å, as observed in the crystal structure of AmyNSAR. Although the conformation change predicted by the AaOSBS homology model could be overestimated, distances between OSB and active site residues are similar to those observed in AmyNSAR.
OSB, SHCHC (not shown), and D-NSPG (Figure 7D-F) dock into the AaOSBS crystal structure (with remodeled 20s loop) and AaOSBS homology model with very similar conformations to those seen in the AmyNSAR OSB-bound crystal structure (1SJB) and the docked model of AmyNSAR bound to SHCHC or D-NSPG. In the AaOSBS models, automated docking procedures had more difficulty placing L-NSPG into a conformation comparable to OSB. In the highest-scoring docked conformations, the carbonyl of the succinyl group was rotated away from T142, its putative hydrogen-bonding partner. However, we manually adjusted the bond torsion angles and position of L-NSPG in both models, so that the succinyl carbonyl could contact the T142 hydroxyl (Figure 7G-I). Neither the automatically docked nor manually adjusted ligands exhibited steric clashes with the enzyme.
The lack of steric conflicts in the ligand-docked models are consistent with competitive inhibition of OSBS activity by L-NSPG and D-NSPG. Explaining the lack of NSAR activity is more difficult, but modeling suggests a few possibilities. If the apo-crystal structure undergoes only a small conformation change when binding the substrate, there do not appear to be any steric reasons to prevent the transition from D- to L-NSPG during racemization. However, the distance between the catalytic lysines and the α-carbon of the substrate might be too large for both of them to efficiently participate in acid-base catalysis. The distance might be less problematic for the OSBS reaction, in which only one lysine acts as a general base. If a larger conformation change occurs, as suggested by the AaOSBS homology model, the active site could impose some steric restrictions on the transition between D- and L-NSPG. Manually adjusting the position and torsion angles of L-NSPG to match D-NSPG suggested that Y55 and Y299 could sterically constrain the movement of the phenyl ring during catalysis. Y299 also restricts the conformation of the succinyl group, which could limit the ability to properly position succinylphenylglycine for racemization. Alternatively, the electrostatic environment around K269, which plays different roles in the OSBS and NSAR reactions, could differ between AaOSBS and promiscuous NSAR/OSBS subfamily enzymes, so that it cannot effectively act as a general acid/base catalyst in the NSAR reaction.
Y299 helps determine specificity.
Based on sequence analysis and results from ligand docking, we selected several residues for mutagenesis (Table 3). M18 was chosen because conservation of phenylalanine at this position in most other NSAR/OSBS subfamily members suggested it could be responsible for loss of NSAR activity. Y299 was chosen because molecular modeling suggested potential steric conflicts with D- and L-NSPG. Also, ExiOSBS, which has tyrosine at this position, has little NSAR activity, while 8 out of 9 known promiscuous NSAR/OSBS subfamily enzymes have leucine or isoleucine at this position. Finally, Y55 was also chosen because molecular modeling suggested potential steric conflicts with D- and L-NSPG. The other two residues mentioned above, L50 and T142, were not mutated because sequence variation at these sites does not appear to correlate with specificity differences between AaOSBS and promiscuous NSAR/OSBS subfamily enzymes.
Table 3.
Enzymatic activities of AaOSBS variants. Activities of wild type AmyNSAR, LvNSAR/OSBS, and AaOSBS are shown for comparison.
| OSBS activity | NSAR activity a | |||||
|---|---|---|---|---|---|---|
| Variant | kcat (s−1) |
KM (μM) |
kcat/KM (M−1s−1) |
kcat (s−1) | KM (μM) |
kcat/KM (M−1s−1) |
| AmyNSAR 29 | 46 ± 5 | 550 ± 120 | 8.3 × 104 | 42 ± 2 c | 1000 ± 10 | 4.2 × 104 |
| LvNSAR/OSBS WT | 179 ± 8 | 96 ± 13 | 1.9 × 106 | 2.2 ± 0.2 | 2700 ± 540 | 8.1 × 102 |
| AaOSBS WT | 64 ± 3 | 118 ± 18 | 5.4 × 105 | <0.0025 | - | - |
| AaOSBS M18F | 1.3 ± 0.04 | 34 ± 6 | 3.8 × 103 | < 0.0025 | - | - |
| AaOSBS Y55A | n.d. b | n.d. | 6.9 × 103 | < 0.0025 | - | - |
| AaOSBS Y299I | 91 ± 3 | 220 ± 18 | 4.2 × 105 | 0.22 ± 0.02 | 1800 ± 330 | 1.2 × 102 |
| AaOSBS M18F/Y299I | n.d. | n.d. | 7.0 × 102 | < 0.00002 | - | - |
N-succinyl-D-phenylglycine was the substrate.
Not determined because substrate saturation could not be achieved.
M18F reduces OSBS reaction efficiency more than 100-fold without conferring detectable NSAR activity. In AmyNSAR, mutating the phenylalanine at the same site to alanine is equally deleterious.29 This suggests that the important contacts between the 20s loop and the rest of the enzyme are different in AaOSBS and that replacing methionine with the larger phenylalanine in AaOSBS disrupts the 20s loop so that it cannot close properly to orient the substrate for catalysis.
The second mutation, Y55A, was chosen because it could potentially interfere with the reorientation of the phenylglycine side chain in the NSAR reaction. However, this mutation also reduced OSBS reaction efficiency by ~100-fold without conferring detectable NSAR activity. In addition, Y55A significantly reduced protein expression. This residue is in the 50s loop, which is at the dimer interface, where mutations could compromise the quaternary structure, leading to a protein folding or stability defect.
Remarkably, the third mutation, Y299I, increased NSAR activity with D-succinylphenylglycine from undetectable to 1.2 × 102 M−1s−1 without reducing OSBS activity. The increase in NSAR activity appears to be primarily from increasing kcat. Because the KMD-NSPG of Y299I is only marginally lower than the KID-NSPG, replacing a bulky tyrosine with an isoleucine appears to allow the substrate to adopt a more appropriate orientation for catalysis, rather than merely increasing its binding affinity. This mutation increases the active site volume by about 50 Å3, but it is difficult to model how changing the shape of the active site alters the orientation of D- or L-NSPG because Y299 occurs in several alternative orientations in the crystal structure. The NSAR activity of AaOSBS Y299I is still one to two orders of magnitude lower than that of other NSAR/OSBS subfamily enzymes, in which NSAR is a biological function. While the activity threshold for biologically relevant activity is uncertain, the relatively low NSAR activity of AaOSBS Y299I suggests that additional mutations are required to achieve efficient NSAR activity.
Inspection of the NSAR/OSBS subfamily sequence alignment shows that most subfamily members, including the majority of characterized enzymes that have NSAR activity, have leucine or isoleucine at the position equivalent to Y299. In fact, of the seven NSAR/OSBS subfamily members from Alicyclobacillus species, only three have a tyrosine at this position. Leucine occurs at this position in the other Alicyclobacillus NSAR/OSBS subfamily enzymes, as well as enzymes that share a common ancestor with them, suggesting that the leucine to tyrosine mutation was relatively recent. To determine whether this position is a general specificity determinant in the NSAR/OSBS subfamily, we made the reverse mutation, I293Y, in AmyNSAR. No soluble protein of this mutant could be isolated, suggesting that I293Y causes a severe folding defect. The role of the amino acid at this site depends on sequence context in other members of the NSAR/OSBS subfamily, too. Although Bacillus subtilis OSBS has a leucine at this position, it lacks NSAR activity, suggesting that there are other important specificity determinants in some members of the NSAR/OSBS subfamily. Also, Enterococcus faecalis NSAR/OSBS, which catalyzes both OSBS and NSAR reactions efficiently, has a phenylalanine at this position, as do nearly all NSAR/OSBS subfamily enzymes from Enterococcus species.8 This demonstrates that Y299 is under epistatic constraints, helping determine reaction specificity in some sequence contexts but not others.
We also observed epistasis between M18 and Y299 in AaOSBS. Although Y299I had no effect on OSBS activity by itself, the double mutant M18F/Y299I exhibited positive epistasis for OSBS activity, in which the M18F mutation became even more deleterious in combination with Y299I. M18F/Y299I also eliminated the NSAR activity conferred by Y299I. This was surprising, because modeling the double mutation in the crystal structure, crystal structure with remodeled 20s loop, and homology model suggested feasible rotamers of both mutant residues that could avoid steric clashes. As a result, we expected that mimicking the active site of AmyNSAR by swapping the positions of the aromatic and branched-chain amino acids would be compensatory (Fig. 7H,I). However, a possible explanation for the actual result is that introducing a bulky phenylalanine at position 18 reduces activity by limiting the ability of the 20s loop to close. If transient loop closure is mediated by interaction of the phenylalanine at position 18 with Y299, replacing the tyrosine with a smaller isoleucine at position 299 could exacerbate the activity defect.
Effect of Y299 on proton abstraction.
To gain insight into the effect of Y299I, we measured the exchange (kex) of the proton on the α-carbon of D- or L-NSPG with deuterated solvent, which corresponds to the first step in the reaction mechanism (Table 4). In wild type AaOSBS, we observed slow proton-deuterium exchange with L-NSPG only. In contrast, a previous study reported that kex of AmyNSAR was >380 s−1 for both isomers.14 AaOSBS Y299I catalyzes slow exchange with both D- and L-NSPG. These results indicate that AaOSBS binds L-NSPG in a position proximal to K269, which abstracts a the α-proton from L-NSPG. The Y299I mutation does not alter kex when L-NSPG is the substrate, suggesting that the substrate is in similar positions relative to K269 in wild type and Y299I AaOSBS.
Table 4.
Rate of proton-deuterium exchange (kex) for AaOSBS.
| D-NSPG | L-NSPG | |
|---|---|---|
| AaOSBS WT | - | 0.29 s−1 |
| AaOSBS Y299I | 0.59 s−1 | 0.28 s−1 |
At first glance, it was surprising that K170, which is the base in the OSBS reaction, did not catalyze proton exchange with the substrate in wild type AaOSBS. However, K269 is surrounded by L50, Y55, M298, and Y299, which form a neutral surface on the back wall of the active site that interacts with the succinyl methylenes and the phenylglycine side chain, while the two carboxylates of the substrate are pinned against R305 and the metal ion. These interactions are likely to pull the substrate closer to K269. Thus, the question is what prevents K170 from getting into the right position for the NSAR reaction? K170 is near the active site opening, and the B-factors of its the three terminal atoms are well above average, suggesting some flexibility in the absence of bound substrate. Furthermore, the distance between K170 and K269 is larger than observed in ligand-bound AmyNSAR (1SJB) and DrNSAR (1XPY:C). One possibility is that the bulky tyrosine, in combination with L- or D-NSPG, which are slightly larger than the substrate of the OSBS reaction (SHCHC), could limit a conformation change that would bring K170 closer to the substrate by narrowing the active site. Replacement of tyrosine with a smaller isoleucine could permit this conformation change and thus allow catalysis. Alternatively, Y299I could have little effect on conformation changes, but instead relieve steric constraints to adjust the position of L- or D-NSPG, so that both catalytic lysines are accessible.
As discussed above, the relatively low NSAR activity of AaOSBS Y299I suggests that this mutation is necessary, but not sufficient to achieve the more efficient NSAR activity observed in most other NSAR/OSBS subfamily enzymes. The insufficiency of Y299I is even more evident from the low proton exchange rates observed with both L- and D-NSPG. While kex for both NSPG isomers using AaOSBS wild type and Y299I was < 1 s−1, the lower limit of kex by K170 using SHCHC must be equal to kcatOSBS (64 s−1). Given the structural similarities between the substrates and the observation that kcatNSAR and kcatOSBS of many promiscuous NSAR/OSBS enzymes differ by less than 10-fold, a difference in the reactivity of SHCHC and L- or D-NSPG is unlikely to explain the low kexNSAR of AaOSBS wild type and Y299I variants. Instead, it seems likely that introduction of Y299I rotates or laterally shifts L- or D-NSPG so that K170 can access the substrate, albeit at a suboptimal distance or angle, relative to SHCHC in the OSBS reaction. Ongoing experiments are attempting to identify other mutations that account for the additional ~100-fold lower kcatNSAR and kcat/KMNSAR of AaOSBS Y299I relative to NSAR/OSBS subfamily members that efficiently catalyze the NSAR reaction.
Discussion
Decades of research into protein structure-function relationships has improved prediction methods.52 However, predicting precise molecular functions like enzyme specificity is still difficult, contributing to high functional annotation error rates.53 Our research aims to address this problem by delving into evolutionary and biochemical mechanisms for changing enzyme specificity. The NSAR/OSBS subfamily is a particularly challenging and interesting target for this research because of the relatively high sequence similarity, catalytic promiscuity, and divergent biological functions of its members.9 In this paper, we offer the first description of the structure and activity of a non-promiscuous member of the NSAR/OSBS subfamily. Moreover, we identified one residue that determines NSAR activity in this enzyme. Strikingly, Y299I only affected NSAR activity and had no detectable effect on OSBS activity, demonstrating that a tradeoff between catalytic efficiency and reaction specificity is not inevitable. In contrast, some other enzymes exhibit a strong tradeoff between efficiency and reaction specificity. For example, almost any mutation in the sulfatase SpAS1 substantially increases its phosphatase activity at the expense of its sulfatase activity.54
At first glance, the Y299I mutation might be expected to enable NSAR activity by enlarging the active site to improve binding affinity of L- or D-NSPG, which are slightly larger than SHCHC. However, competitive inhibition and positions of the ligands predicted by in silico docking suggest that L- and D-NSPG can bind both wild-type and Y299I AaOSBS in overlapping positions with SHCHC and with similar affinities to those observed for enzymes which have efficient NSAR activity. This indicates that loss of binding affinity did not significantly contribute to loss of NSAR activity by AaOSBS. Instead, gain of NSAR activity in Y299I AaOSBS appears to be due to subtle repositioning of the substrate in the active site, leading to a large increase in kcat. Reorienting the substrate might be sufficient on its own, or it could affect conformation changes that are necessary for catalysis. Discovering how a new activity can evolve by improving kcat is significant, because, despite many successful attempts to design proteins with high ligand-binding affinity, enzyme design has been limited by insufficient strategies to improve kcat.55
Because the NSAR activity and kex of AaOSBS Y299I is still two orders of magnitude lower than most other characterized NSAR/OSBS subfamily enzymes, other active site features are expected to affect relative specificity of NSAR and OSBS activities. In particular, we hypothesize that other residues within or distant from the active site affect the alignment of the substrate between the two catalytic lysines or the ability of K269, whose function differs between the OSBS and NSAR reactions, to efficiently act as a catalytic base, due to its electrostatic environment or position. This lysine is thought to stabilize the transition state of the OSBS reaction via a cation-π interaction, but would need to function as general acid or base in the NSAR reaction.14 Identification of other sites that affect specificity and the structural basis for their effects are currently under investigation.
The mechanism of evolving new specificity has also been investigated in other enzymes. For example, lactate dehydrogenase evolved from malate dehydrogenase by changing the substrate specificity to prefer lactate, which lacks a carboxylate found in malate. Instead of merely losing the arginine that interacts with malate’s extra carboxylate, lactate dehydrogenase evolved once by insertion of a loop that displaces the arginine.18 Lactate dehydrogenase evolved a second time by a different mechanism involving loss of the arginine plus additional second-shell mutations.19 A separate study about the catalytic specificity of alkaline phosphatase predicted that electrostatic interactions with the more negatively charged phosphoryl group helped discriminate between phosphate and sulfate monoesters. However, mutagenesis demonstrated very little discrimination on the basis of electrostatics, leaving the nature of phosphatase specificity unknown.56 These studies, along with our results, show that changes in specificity often occur by non-intuitive mechanisms, even when only the substrate specificity changes.
Along with identifying the first site that significantly affects reaction specificity in a member of the NSAR/OSBS subfamily, we discovered that epistatic interactions affect its role. Epistasis occurs when mutations have different effects in different sequence contexts. The frequency, impact and biophysical basis of epistasis are at the forefront of protein evolution research.57 The number of studies demonstrating that epistasis is an important constraint in protein evolution is growing.15-20 However, the biophysical basis of epistasis has only been determined in a few proteins.15, 58-61 Within AaOSBS, the identity of position 18 determines how position 299 affects OSBS activity and NSAR reaction specificity. Among other NSAR/OSBS subfamily enzymes, an aromatic residue at position 299 does not always preclude NSAR activity, nor does a branched-chain hydrophobic amino acid guarantee NSAR activity. Together, these results are a stepping stone for comparing how specificity determinants vary within the NSAR/OSBS subfamily and determining the structural basis for epistasis. Understanding epistasis is a fundamental issue in deciphering protein structure-function relationships, which could lead to development of experimental strategies and predictive models for determining and designing enzyme specificity.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the efforts of all NYSGXRC and NYSGRC personnel who contributed to the structure determination and manuscript preparation. We would also like to thank Dr. Rup Lal from the University of New Delhi for providing us the Amycolatopsis mediterranei S699 genomic DNA and Dr. Jamison P. Huddleston for assistance with deuterium exchange experiments.
Funding Sources
This work was funded by NSF CAREER Award 1253975 to M.E.G. The NYSGXRC was supported by NIH Grant U54 GM074945 (Principal Investigator: S.K. Burley). The NYSGRC is supported by NIH Grant U54 GM094662 (Principal Investigator: S.C. Almo). The Center for Synchrotron Biosciences, where diffraction data were collected, was supported by grant P30-EB-009998 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. Synthetic chemistry performed at the Natural Products LINCHPIN Laboratory at Texas A&M was supported by the Dept. of Chemistry, College of Science, and Office of the Vice President of Research and was continued at the CPRIT Synthesis and Drug Lead Discovery Laboratory at Baylor University supported by the College of Arts and Sciences and Baylor University.
ABBREVIATIONS
- OSBS
o-succinylbenzoate synthase
- NSAR
N-succinylamino acid racemase
- SHCHC
2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate
- OSB
o-succinylbenzoate
- D-NSPG
N-succinyl-D-phenylglycine
- L-NSPG
N-succinyl-L-phenylglycine
- PMSF
phenylmethylsulfonyl fluoride
- AaOSBS
Alicyclobacillus acidocaldarius OSBS
- AmyNSAR
Amycolatopsis sp. T-1-60 NSAR
- DrNSAR
Deinococcus radiodurans NSAR
- EfNSAR/OSBS
Enterococcus faecalis NSAR/OSBS
- GkNSAR/OSBS
Geobacillus kaustophilus NSAR/OSBS
- AmedNSAR
Amycolatopsis mediterranei S699 NSAR
- LvNSAR/OSBS
Lysinibacillus varians NSAR/OSBS
- RcNSAR/OSBS
Roseiflexus castenholzii NSAR/OSBS
- ExiOSBS
Exiguobacterium sp. AT1b OSBS
Footnotes
Supporting Information. Supporting information consisting of supplemental materials and methods is available free of charge in the file Supplemental.pdf.
Accession Codes. The structural coordinates of AaOSBS have been deposited in the RCSB PDB as 3QLD. The UniProt accession numbers of the enzymes characterized in this study are AaOSBS (B7DSY7), AmedNSAR (G0FPT7), LvNSAR/OSBS (X2GR01) and RcNSAR/OSBS (A7NLX0).
REFERENCES
- (1).O’Brien PJ, and Herschlag D (1999) Catalytic promiscuity and the evolution of new enzymatic activities, Chem Biol 6, R91–R105. [DOI] [PubMed] [Google Scholar]
- (2).Khersonsky O, and Tawfik DS (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective, Annu Rev Biochem 79, 471–505. [DOI] [PubMed] [Google Scholar]
- (3).Patrick WM, Quandt EM, Swartzlander DB, and Matsumura I (2007) Multicopy suppression underpins metabolic evolvability, Mol Biol Evol 24, 2716–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim J, Kershner JP, Novikov Y, Shoemaker RK, and Copley SD (2010) Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5’-phosphate synthesis, Mol Syst Biol 6, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gerlt JA, Babbitt PC, and Rayment I (2005) Divergent evolution in the enolase superfamily: the interplay of mechanism and specificity, Arch Biochem Biophys 433, 59–70. [DOI] [PubMed] [Google Scholar]
- (6).Zhu WW, Wang C, Jipp J, Ferguson L, Lucas SN, Hicks MA, and Glasner ME (2012) Residues required for activity in Escherichia coli o-succinylbenzoate synthase (OSBS) are not conserved in all OSBS enzymes, Biochemistry 51, 6171–6181. [DOI] [PubMed] [Google Scholar]
- (7).Thoden JB, Taylor Ringia EA, Garrett JB, Gerlt JA, Holden HM, and Rayment I (2004) Evolution of enzymatic activity in the enolase superfamily: structural studies of the promiscuous o-succinylbenzoate synthase from Amycolatopsis, Biochemistry 43, 5716–5727. [DOI] [PubMed] [Google Scholar]
- (8).Odokonyero D, Sakai A, Patskovsky Y, Malashkevich VN, Fedorov AA, Bonanno JB, Fedorov EV, Toro R, Agarwal R, Wang C, Ozerova ND, Yew WS, Sauder JM, Swaminathan S, Burley SK, Almo SC, and Glasner ME (2014) Loss of quaternary structure is associated with rapid sequence divergence in the OSBS family, Proc Natl Acad Sci U S A 111, 8535–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Glasner ME, Fayazmanesh N, Chiang RA, Sakai A, Jacobson MP, Gerlt JA, and Babbitt PC (2006) Evolution of structure and function in the o-succinylbenzoate synthase/N-acylamino acid racemase family of the enolase superfamily, J Mol Biol 360, 228–250. [DOI] [PubMed] [Google Scholar]
- (10).Odokonyero D, Ragumani S, Swaminathan S, Lopez MS, Bonanno JB, Ozerova ND, Woodard DR, Machala BW, Burley SK, Almo SC, and Glasner ME (2013) Divergent Evolution of Ligand Binding in the o-Succinylbenzoate Synthase Family, Biochemistry 52, 7512–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Palmer DR, Garrett JB, Sharma V, Meganathan R, Babbitt PC, and Gerlt JA (1999) Unexpected divergence of enzyme function and sequence: “N-acylamino acid racemase” is o-succinylbenzoate synthase, Biochemistry 38, 4252–4258. [DOI] [PubMed] [Google Scholar]
- (12).Brizendine AM, Odokonyero D, McMillan AW, Zhu M, Hull K, Romo D, and Glasner ME (2014) Promiscuity of Exiguobacterium sp. AT1b o-succinylbenzoate synthase illustrates evolutionary transitions in the OSBS family, Biochem Biophys Res Commun 450, 679–684. [DOI] [PubMed] [Google Scholar]
- (13).Sakai A, Xiang DF, Xu C, Song L, Yew WS, Raushel FM, and Gerlt JA (2006) Evolution of enzymatic activities in the enolase superfamily: N-succinylamino acid racemase and a new pathway for the irreversible conversion of D- to L-Amino Acids, Biochemistry 45, 4455–4462. [DOI] [PubMed] [Google Scholar]
- (14).Taylor Ringia EA, Garrett JB, Thoden JB, Holden HM, Rayment I, and Gerlt JA (2004) Evolution of enzymatic activity in the enolase superfamily: functional studies of the promiscuous o-succinylbenzoate synthase from Amycolatopsis, Biochemistry 43, 224–229. [DOI] [PubMed] [Google Scholar]
- (15).Bridgham JT, Ortlund EA, and Thornton JW (2009) An epistatic ratchet constrains the direction of glucocorticoid receptor evolution, Nature 461, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Field SF, and Matz MV (2010) Retracing Evolution of Red Fluorescence in GFP-Like Proteins from Faviina Corals, Molecular Biology and Evolution 27, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gong LI, Suchard MA, and Bloom JD (2013) Stability-mediated epistasis constrains the evolution of an influenza protein, eLife 2, e00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Boucher JI, Jacobowitz JR, Beckett BC, Classen S, and Theobald DL (2014) An atomic-resolution view of neofunctionalization in the evolution of apicomplexan lactate dehydrogenases, eLife 3, e02304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Steindel PA, Chen EH, Wirth JD, and Theobald DL (2016) Gradual neofunctionalization in the convergent evolution of trichomonad lactate and malate dehydrogenases, Protein Sci 25, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Noor S, Taylor MC, Russell RJ, Jermiin LS, Jackson CJ, Oakeshott JG, and Scott C (2012) Intramolecular epistasis and the evolution of a new enzymatic function, PLoS One 7, e39822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, and Mark ID (2009) A family of LIC vectors for high-throughput cloning and purification of proteins, Methods in molecular biology 498, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sauder MJ, Rutter ME, Bain K, Rooney I, Gheyi T, Atwell S, Thompson DA, Emtage S, and Burley SK (2008) High throughput protein production and crystallization at NYSGXRC, Methods in molecular biology 426, 561–575. [DOI] [PubMed] [Google Scholar]
- (23).Sheldrick GM (2008) A short history of SHELX, Acta crystallographica. Section A, Foundations of crystallography 64, 112–122. [DOI] [PubMed] [Google Scholar]
- (24).Perrakis A, Morris R, and Lamzin VS (1999) Automated protein model building combined with iterative structure refinement, Nat Struct Biol 6, 458–463. [DOI] [PubMed] [Google Scholar]
- (25).Collaborative Computational Project Number 4. (1994) The CCP4 suite: programs for protein crystallography, Acta crystallographica. Section D, Biological crystallography 50, 760–763. [DOI] [PubMed] [Google Scholar]
- (26).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta crystallographica. Section D, Biological crystallography 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (27).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta crystallographica. Section D, Biological crystallography 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (28).Datsenko KA, and Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products, Proc Natl Acad Sci U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McMillan AW, Lopez MS, Zhu M, Morse BC, Yeo IC, Amos J, Hull K, Romo D, and Glasner ME (2014) Role of an active site loop in the promiscuous activities of Amycolatopsis sp. T-1–60 NSAR/OSBS, Biochemistry 53, 4434–4444. [DOI] [PubMed] [Google Scholar]
- (30).Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, and Vonstein V (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes, Nucleic Acids Res 33, 5691–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Taboada B, Ciria R, Martinez-Guerrero CE, and Merino E (2012) ProOpDB: Prokaryotic Operon DataBase, Nucleic Acids Res 40, D627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dam P, Olman V, Harris K, Su Z, and Xu Y (2007) Operon prediction using both genome-specific and general genomic information, Nucleic Acids Res 35, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mao F, Dam P, Chou J, Olman V, and Xu Y (2009) DOOR: a database for prokaryotic operons, Nucleic Acids Res 37, D459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, Mao F, Lai W, and Xu Y (2014) DOOR 2.0: presenting operons and their functions through dynamic and integrated views, Nucleic Acids Res 42, D654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis, J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (36).Krissinel E, and Henrick K (2007) Inference of macromolecular assemblies from crystalline state, J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (37).Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004) WebLogo: a sequence logo generator, Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models, Bioinformatics 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- (39).Stamatakis A, Hoover P, and Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers, Syst Biol 57, 758–771. [DOI] [PubMed] [Google Scholar]
- (40).Sali A, and Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints, J Mol Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- (41).Webb B, and Sali A (2014) Comparative Protein Structure Modeling Using MODELLER, Current protocols in bioinformatics 47, 5.6.1–5.6.32. [DOI] [PubMed] [Google Scholar]
- (42).Fiser A, Do RK, and Sali A (2000) Modeling of loops in protein structures, Protein Sci 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Thompson TB, Garrett JB, Taylor EA, Meganathan R, Gerlt JA, and Rayment I (2000) Evolution of enzymatic activity in the enolase superfamily: structure of o-succinylbenzoate synthase from Escherichia coli in complex with Mg2+ and o-succinylbenzoate, Biochemistry 39, 10662–10676. [DOI] [PubMed] [Google Scholar]
- (44).Wang WC, Chiu WC, Hsu SK, Wu CL, Chen CY, Liu JS, and Hsu WH (2004) Structural basis for catalytic racemization and substrate specificity of an N-acylamino acid racemase homologue from Deinococcus radiodurans, J Mol Biol 342, 155–169. [DOI] [PubMed] [Google Scholar]
- (45).Trott O, and Olson AJ (2010) AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading, J Comput Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ren JY, Williams N, Clementi L, Krishnan S, and Li WW (2010) Opal web services for biomedical applications, Nucleic Acids Res 38, W724–W731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Marchler-Bauer A, and Bryant SH (2004) CD-Search: protein domain annotations on the fly, Nucleic Acids Res 32, W327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Akiva E, Brown S, Almonacid DE, Barber AE 2nd, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, Mischel D, Morris JH, Ojha S, Schnoes AM, Stryke D, Yunes JM, Ferrin TE, Holliday GL, and Babbitt PC (2014) The Structure-Function Linkage Database, Nucleic Acids Res 42, D521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hayashida M, Kim SH, Takeda K, Hisano T, and Miki K (2008) Crystal structure of N-acylamino acid racemase from Thermus thermophilus HB8, Proteins 71, 519–523. [DOI] [PubMed] [Google Scholar]
- (50).Gulick AM, Hubbard BK, Gerlt JA, and Rayment I (2000) Evolution of enzymatic activities in the enolase superfamily: crystallographic and mutagenesis studies of the reaction catalyzed by D-glucarate dehydratase from Escherichia coli, Biochemistry 39, 4590–4602. [DOI] [PubMed] [Google Scholar]
- (51).Landro JA, Gerlt JA, Kozarich JW, Koo CW, Shah VJ, Kenyon GL, Neidhart DJ, Fujita S, and Petsko GA (1994) The role of lysine 166 in the mechanism of mandelate racemase from Pseudomonas putida: mechanistic and crystallographic evidence for stereospecific alkylation by (R)-alpha-phenylglycidate, Biochemistry 33, 635–643. [DOI] [PubMed] [Google Scholar]
- (52).Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A, Pandey G, Yunes JM, Talwalkar AS, Repo S, Souza ML, Piovesan D, Casadio R, Wang Z, Cheng J, Fang H, Gough J, Koskinen P, Toronen P, Nokso-Koivisto J, Holm L, Cozzetto D, Buchan DW, Bryson K, Jones DT, Limaye B, Inamdar H, Datta A, Manjari SK, Joshi R, Chitale M, Kihara D, Lisewski AM, Erdin S, Venner E, Lichtarge O, Rentzsch R, Yang H, Romero AE, Bhat P, Paccanaro A, Hamp T, Kassner R, Seemayer S, Vicedo E, Schaefer C, Achten D, Auer F, Boehm A, Braun T, Hecht M, Heron M, Honigschmid P, Hopf TA, Kaufmann S, Kiening M, Krompass D, Landerer C, Mahlich Y, Roos M, Bjorne J, Salakoski T, Wong A, Shatkay H, Gatzmann F, Sommer I, Wass MN, Sternberg MJ, Skunca N, Supek F, Bosnjak M, Panov P, Dzeroski S, Smuc T, Kourmpetis YA, van Dijk AD, ter Braak CJ, Zhou Y, Gong Q, Dong X, Tian W, Falda M, Fontana P, Lavezzo E, Di Camillo B, Toppo S, Lan L, Djuric N, Guo Y, Vucetic S, Bairoch A, Linial M, Babbitt PC, Brenner SE, Orengo C, Rost B, Mooney SD, and Friedberg I (2013) A large-scale evaluation of computational protein function prediction, Nat Methods 10, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Schnoes AM, Brown SD, Dodevski I, and Babbitt PC (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies, PLoS Comput Biol 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bayer CD, van Loo B, and Hollfelder F (2017) Specificity Effects of Amino Acid Substitutions in Promiscuous Hydrolases: Context-Dependence of Catalytic Residue Contributions to Local Fitness Landscapes in Nearby Sequence Space, Chembiochem 18, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Korendovych IV, and DeGrado WF (2014) Catalytic efficiency of designed catalytic proteins, Curr Opin Struct Biol 27, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Andrews LD, Zalatan JG, and Herschlag D (2014) Probing the origins of catalytic discrimination between phosphate and sulfate monoester hydrolysis: comparative analysis of alkaline phosphatase and protein tyrosine phosphatases, Biochemistry 53, 6811–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Starr TN, and Thornton JW (2016) Epistasis in protein evolution, Protein Sci 25, 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ortlund EA, Bridgham JT, Redinbo MR, and Thornton JW (2007) Crystal structure of an ancient protein: evolution by conformational epistasis, Science 317, 1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Yokoyama S, Xing J, Liu Y, Faggionato D, Altun A, and Starmer WT (2014) Epistatic adaptive evolution of human color vision, PLoS genetics 10, e1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Olson CA, Wu NC, and Sun R (2014) A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain, Curr Biol 24, 2643–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dellus-Gur E, Elias M, Caselli E, Prati F, Salverda ML, de Visser JA, Fraser JS, and Tawfik DS (2015) Negative Epistasis and Evolvability in TEM-1 beta-Lactamase--The Thin Line between an Enzyme’s Conformational Freedom and Disorder, J Mol Biol 427, 2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.