

ABSTRACT
The decision to pursue a monoclonal antibody (mAb) as a therapeutic for disease intervention requires the assessment of many factors, such as target-biology, including the total target burden and its accessibility at the intended site of action, as well as mAb-specific properties like binding affinity and the pharmacokinetics in serum and tissue. Interleukin-36 receptor (IL-36 R) is a member of the IL-1 family cytokine receptors and an attractive target to treat numerous epithelial-mediated inflammatory conditions, including psoriatic and rheumatoid arthritis, asthma, and chronic obstructive pulmonary disease. However, information concerning the expression profile of IL-36 R at the protein level is minimal, so the feasibility of developing a therapeutic mAb against this target is uncertain. Here, we present a characterization of the properties associated with absorption, distribution, metabolism, and excretion of a high-affinity IL-36 R-targeted surrogate rat (IgG2a) mAb antagonist in preclinical mouse models. The presence of IL-36 R in the periphery was confirmed unequivocally as the driver of non-linear pharmacokinetics in blood/serum, although a predominant site of tissue accumulation was not observed based upon the kinetics of radiotracer. Additionally, the contribution of IL-36 R-mediated catabolism of mAb in kidney was tested in a 5/6 nephrectomized mouse model where minimal effects on serum pharmacokinetics were observed, although analysis of functional mAb in urine suggests that target can influence the amount of mAb excreted. Our data highlight an interesting case of target-mediated drug disposition (TMDD) where low, yet broadly expressed levels of membrane-bound target result in a cumulative effect to drive TMDD behavior typical of a large, saturable target sink. The potential differences between our mouse model and IL-36 R target profile in humans are also presented.
KEYWORDS: Cytokine, IL-36R, IL-1Rrp2, TMDD, monoclonal antibody, biodistribution, pharmacokinetics, radiolabel
Introduction
Development of therapeutic antagonists of cytokine receptor signaling is an attractive strategy to treat myriad immune-mediated disorders, including chronic respiratory illness,1–3 hyperproliferative epidermal conditions such as psoriasis,4,5 and other diseases linked to chronic inflammation.6 For instance, interleukin-1 (IL-1) family cytokine signaling is implicated in a wide variety of disease etiologies, and, consequently, receptors and associated ligands of the IL-1 family have long been considered drug targets.7 IL-1 receptor-related protein 2 (IL-1 Rrp2), also known as IL-36 R, is a functionally distinct IL-1 receptor family member:8–10 the gene encoding IL-36 R was identified from a homology-based PCR screen, where it was found that the encoded protein maintains 42% homology with IL-1 R, although there is no evidence that canonical IL-1 R ligands IL-1α, IL-1β, IL-1 Ra, and IL-18 bind IL-36 R.8,11 Consistent with IL-1R biology, however, signaling via IL-36 R requires formation of a heterodimer to a common accessory protein, IL-1RAcP, which binds subsequent to cognate agonist ligands IL-36α/β/ɣ; in turn, ligand binding and signaling can be blocked by the cognate antagonist IL-36 Ra.9 IL-36 R gene expression has been characterized on lymphocytes,10,12–14 and multiple epithelial and fibroblastic cell types,10,15 but quantitative data informing IL-36 R protein levels in tissues are limited or non-existent.16 Interest in IL-36 R-targeted therapeutics stems from its implication in chronic inflammatory conditions that affect psoriatic disorders,17 and a potential role in pulmonary pathology.10 Importantly, low sequence homology between the cognate IL-36 ligands (α, β, and ɣ) renders the more attractive strategy of developing a single antagonist to broadly target the three soluble cytokines effectively with a single reagent difficult.18
Monoclonal antibodies (mAbs) are an established therapeutic modality to treat multiple disease indications.19 From a developmental standpoint, the attraction of mAbs is due to their potential for high affinity and specificity for a target, as well as their long serum half-life (up to 25 d for IgG1) that allows infrequent clinical dosing. However, mAb development is often limited by aspects of the target biology, and in certain cases, target-mediated drug disposition (TMDD) imparts non-linearity to the serum pharmacokinetics (PK) that can hinder accurate translation of dose requirements in humans. TMDD is due to mAb binding highly expressed membrane-bound receptors, where cellular retention and uptake become significant, and this process manifests as non-linear clearance (CL) as measured from the blood compartment.20,21 One strategy to assess the “druggability” of a given target is to use mAb surrogate molecules specific for the homologous receptor in a lower order species to characterize the degree that target influences overall biodisposition, and by extension, efficacy.22–24 The overarching goals of an early stage mAb development campaign are thus: 1) to characterize of the effects of both target expression and mAb-target affinity on mAb circulating half-life; 2) to understand how the specific target expression profile in preclinical species compares to humans; and 3) to identify the dominant sites of target-driven clearance that might render simplified physiology-based pharmacodynamic models amenable to the prediction of clinical dosing requirements.25
The goal of this study was preclinical characterization of the absorption, distribution, metabolism, and excretion of an anti-IL-36 R mAb as a potential human therapeutic by using a surrogate mAb in mouse models. We first characterized the single-dose serum PK of a high-affinity rat IgG2a mAb (M616) specific for muIL-36 R in C57BL/6 mice over multiple dose levels by intravenous (IV) and subcutaneous (SC) routes of administration. A dose-dependent PK profile suggested TMDD, which was rationalized via modeling. Next, we used a second surrogate mAb, murine/rat chimeric anti-IL-36 R surrogate IgG1 (chM616) dosed in combination with M616. The serum exposure of M616 was selectively analyzed to reveal a linear PK profile consistent with saturation of IL-36 R by chM616, confirming the non-linear PK of M616 was IL-36 R-mediated. To quantitate exposure and specific catabolism in tissue, we conducted a multi-organ biodistribution analysis of M616 harboring either non-residualizing 125I, or residualizing 111In tracer after low (0.3 mg/kg) IV dose alone or co-administered as a tracer dose with excess (10 mg/kg) unlabeled M616. The results of the biodistribution analysis prompted a subsequent investigation in 5/6 nephrectomized mouse model to determine the precise role of kidney in M616 clearance. These results present an interesting case of TMDD potentially overlooked during preclinical to human translation in which apparent low, yet broad target expression can manifest as non-linear mAb PK in blood.
Results
Characterization of surrogate anti-mouse IL-36 R mAbs
To characterize the intrinsic antigen binding affinity of the two antibodies used in this study, equilibrium binding in solution was measured by a Kinetic Exclusion Assay (KinExA).26 Briefly, for each binding interaction, analyte solutions were prepared as pairwise 12-point rIL-36 R (extracellular domain, ECD) dilution series titrated against 50 pM or 1000 pM fixed concentration of IL-36 R mAb to properly characterize equilibrium binding in both ‘KD-controlled’ and ‘receptor-controlled’ binding regimes, respectively.26,27 Equilibrium binding isotherms given in Figure 1reveal that both antibodies bind muIL-36 R with very high affinity, yet the fully rat IgG2a (M616) binds the receptor with >3-fold higher affinity (KD = 6.3 ± 0.1 pM) than the rat/mu chimeric mAb (chM616) (KD = 21.0 ± 0.8 pM) It is worth mention that the KD values reported represent the intrinsic binding affinity between a single mAb variable domain and IL-36 R, that is, these values are not confounded by avidity, which is typical when deriving KD values from kinetic measurements binding surface-immobilized antigen during standard surface plasmon resonance (SPR)-based experiments. Thus, given the high-affinity nature of these antibodies, plus the potential for avidity in the context of cell surface binding, we anticipated the potential for non-linear PK behavior, but this would depend on the overall expression level and turnover properties of IL-36 R (vide infra).
Figure 1.
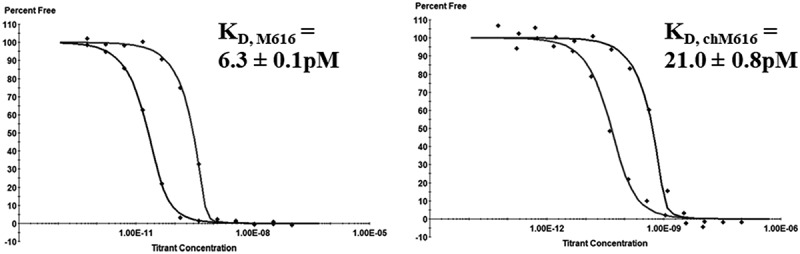
muIL-36 R targeted mAbs bind with high affinity. Solution phase equilibrium binding measurements of rat anti-muIL-36 R and chimeric mouse/rat anti-muIL-36 R surrogate mAbs to recombinant muIL-36 R (titrant) show that M616 binds mu rIL-36 R 3.3-fold tighter than the chimeric chM616. Note: these values represent the intrinsic binding affinity to IL-36 R target in solution in the absence of avidity.
MAbs that target mouse IL-36 R receptor exhibit non-linear plasma PK
Scant data exist in the literature regarding IL-36 R protein quantitation in tissue, and most studies aimed toward the characterization of IL-36 R levels are limited to mRNA expression (Supplemental S1).9,28 To assess the potential impact of IL-36 R target on mAb PK, a rat IgG2a (wild type (WT)) mAb with high affinity for murine IL-36 R (clone M616; hereafter, M616) was administered as a single IV dose to female C57BL/6 mice at multiple dose levels to cover a 100-fold range (0.04–4 mg/kg). Additionally, due to the putative IL-36 R target burden in skin, extravascular (SC and intraperitoneal (IP)) dosing routes were investigated at 4 mg/kg to assess the absorption kinetics and overall bioavailability. Serum concentration versus time profiles is given in Figure 2, with the PK parameter estimates obtained through noncompartmental analysis (NCA) given in Table 1. It is obvious from the comparison of the serum concentration profiles, as well as the dose-dependent NCA parameter estimates that M616 PK is inherently non-linear. Specifically, the examination of the serum PK after 4 mg/kg IV dose reveals elements of classic TMDD behavior (see Discussion).
Figure 2.
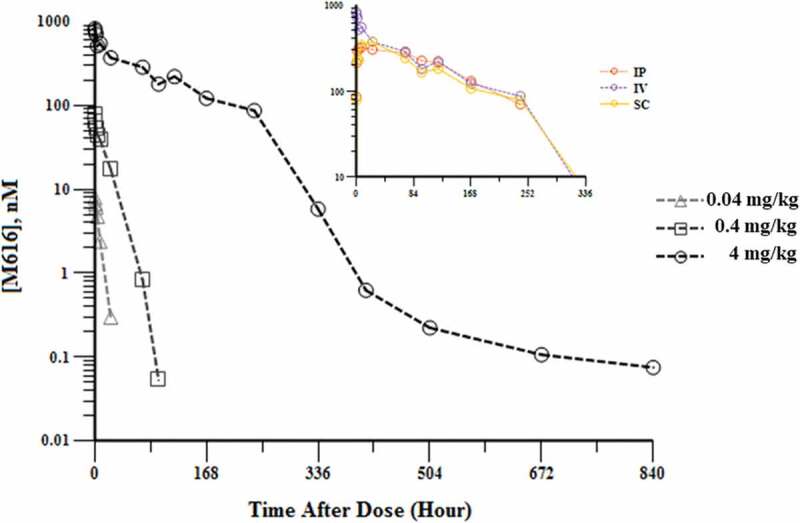
Characterization of dose-dependent single-dose serum PK of rat anti-muIL-36 R (M616) in female C57BL/6 mice. Main: Log-linear mean (n = 3/time point) serum concentration profile observed after 0.04, 0.4, and 4 mg/kg IV doses. Note the several distinct phases of non-linear behavior captured at 4 mg/kg by the extended duration of blood sampling. Inset: Three separate routes of administration (IP, IV, and SC) at 4 mg/kg dose are shown for comparison. Bioavailability >80% was observed by both IP and SC dosing routes, suggesting minimal catabolism at the site of injection.
Table 1.
Serum PK parameter estimates for single-dose M616 (multiple routes) in C57BL/6 mice obtained by NCA. Serum PK of M616 is not dose proportional. High bioavailability was observed for M616 based on comparison of AUC for IV vs. extravascular dose.
| Dose (mg/kg) | Route | AUCinf(nM*hr)/Dose (mg/kg)* |
MRTlast (hr) | CL* (mL/hr/kg) | Vss (mL/kg) | %F |
|---|---|---|---|---|---|---|
| 0.04 | IV | 1570 | 5.5 | 4.2 | 27.0 | |
| 0.4 | IV | 3230 | 15.0 | 2.1 | 31.1 | |
| 4 | IV | 14700 | 95.9 | 0.45 | 43.8 | |
| 4 | IP | 12800 | 103 | 0.52 | - | 87 |
| 4 | SC | 12200 | 102 | 0.55 | - | 83 |
*Note: There is a dose-dependent decrease in clearance indicating non-linearity. However, this violates the assumptions of the moment analysis method used to determine these parameters; hence, these estimates should be interpreted with caution.
We next turned to mathematical modeling, where the objective was to examine the non-linearity observed in the anti-muIL-36 R (M616) mAb concentration-time data in more detail as it relates to interaction parameters of the biological system. Simultaneous fitting of the PK data (IV dosing) using a TMDD model (Figure 3) captured the data effectively (Figure 4). The model estimate for the IL-36 R internalization rate, kint, was found to be ~4-fold higher than an estimate of its degradation rate, kdeg. Using the literature reported estimate for Ro, which represents the overall IL-36 R concentration in mouse irrespective of localization of expression, combined with physiological plasma volume29 V, and our experimentally determined KD, the model is able to capture the dose dependence of the PK profile. The systematic deviation in the model fitting of the terminal phase from the observed PK data could be due to lack of experimental information on the koff value of the mAb,30 which could not be estimated within the model due to lack of parameter identifiability between kon and koff. Overall, the fitted parameters k12, kel, kint and kdeg have a coefficient of variation % of less than 15, suggesting the good degree of precision (Table 2).
Figure 3.
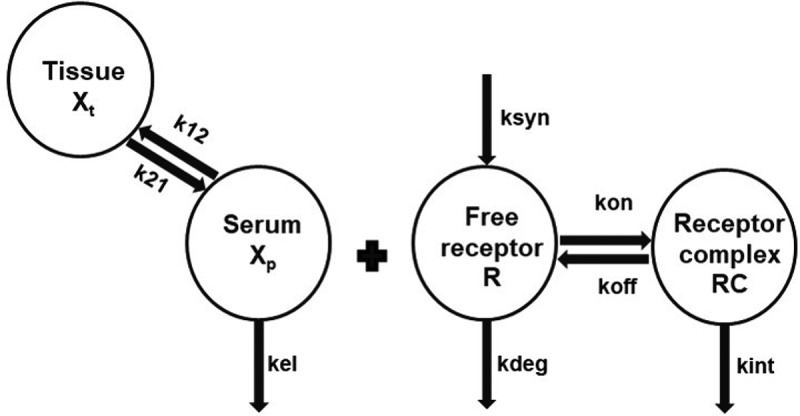
Non-linear serum PK for M616 is captured reasonably well by a TMDD model, consisting of target binding parameters limited to the central compartment (see Methods). k12 and k21 first-order distribution rate constants, k21 = k12, kel first-order elimination rate constant, kint mAb-target internalization rate constant, kdeg first-order degradation rate constant for the target, V is the volume of distribution of the drug in the central compartment, Ro baseline target concentration. For the model, the second-order association rate (kon) was derived in the model using relation kon = First-order dissociation rate (koff)/Dissociation rate constant (KD).
Figure 4.
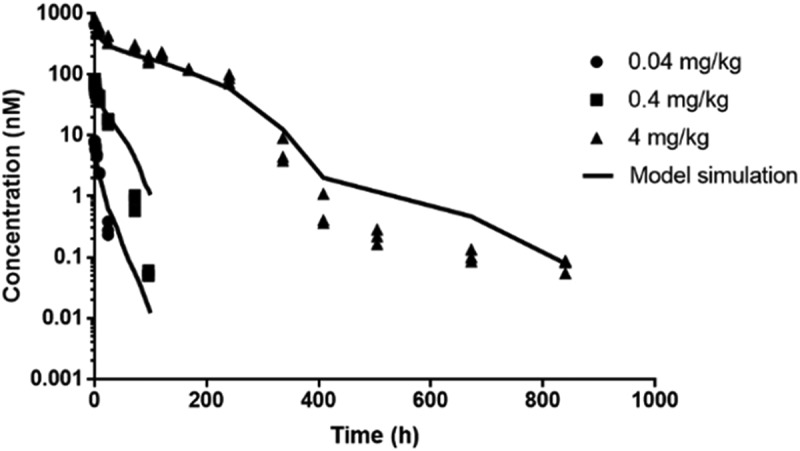
Serum concentration (nM) vs. time (h) for anti-muIL-36 R (M616) in female C57BL/6 mice after intravenous injection of 0.04, 0.4 and 4 mg/kg fitted to a TMDD model. Solid lines represent model fitted profile. Data points represent raw data (n = 3 mice/time point).
Table 2.
Summary of the parameter estimates generated by fitting anti-muIL-36 R (M616) serum concentration-time data (0.04, 0.4 and 4 mg/kg, IV) in TMDD model.
| Parameter name (unit) | Parameter estimate | CV% | Source |
|---|---|---|---|
| k12 (h−1) | 0.0880 | 11.1 | Estimated |
| kel (h−1) | 0.01 | 9.02 | Estimated |
| kint (h−1) | 5.15 | 10.1 | Estimated |
| kdeg (h−1) | 1.27 | 7.86 | Estimated |
| V (L) | 0.000944 | - | Fixeda |
| Ro (M) | 7.94e-10 | - | Fixedb |
| koff(h−1) | 0.00127 | - | Fixedc |
| KD (M) | 6.3e-12 | - | Fixedd |
aPhysiological mouse plasma volume was used (see Shah & Betts, J Pharmacokinet Pharmacodyn, 2012).
bRo was taken from Ahlberg et al., mAbs, 2019.
ckoff was fixed to average mAb value.
dKD was experimentally determined.
Although the data fit the TMDD model reasonably well, dose normalization of our data (Figure 5) revealed a conserved distribution, or α-phase across dose groups, suggesting dose-independent PK during this interval. This type of PK behavior is typical of high-affinity mAb-target systems where engagement in the periphery is rate-limited by extravasation into tissue (see Discussion).25
Figure 5.
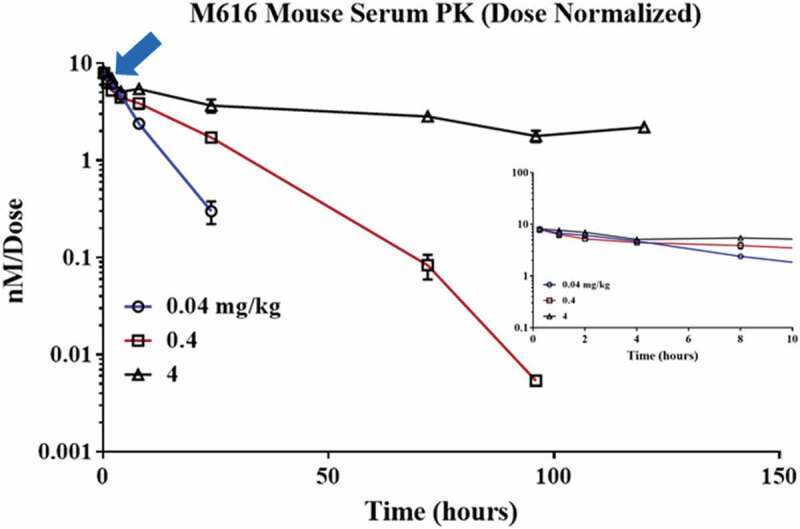
The initial distribution (α) phase in serum is conserved across all dose groups. Dose normalized M616 single-dose PK (IV) across a 100-fold concentration range reveals that serum profiles overlap until ~8 h.
Competitive saturation of IL-36 receptor in vivo confirms M616 cleared by a target-mediated pathway
The serum PK of M616 surrogate mAb is suggestive of TMDD; however, the degree of non-linearity was unexpected based on the putative low expression of IL-36 R in mouse. Additionally, M616 is WT IgG2a, so functional binding with additional Fc receptors could, theoretically, play a role in M616 serum disposition. To confirm the mechanism driving the observed non-linear CL in mouse serum, we designed a subsequent PK study to assess the effect of competitive saturation of IL-36 R target in vivo on the serum PK profile of M616 at a low IV dose (0.5 mg/kg). To this end, we took advantage of the distinct mouse/rat chimeric IgG framework of a second surrogate, chM616, harboring M616 complementarity-determining regions grafted to mouse IgG1 constant regions, to achieve analytical selectivity by way of species-selective immunoassay (see Methods) for M616 despite including excess chM616 as a cassette dose. Importantly, chM616 was engineered to lack effector function by way of N297G mutation of the Fc CH2 domain, and therefore competition binding will occur at the level of IL-36 R only.31,32 When we administered a cassette dose containing 0.5 mg/kg of M616 plus 10 mg/kg chM616, a striking effect on the M616 serum profile was observed (Figure 6) as an extension of the β-elimination half-life from <1 d to ~6 d. The resultant PK parameter estimates obtained by NCA are provided in Supplemental Table 1.
Figure 6.
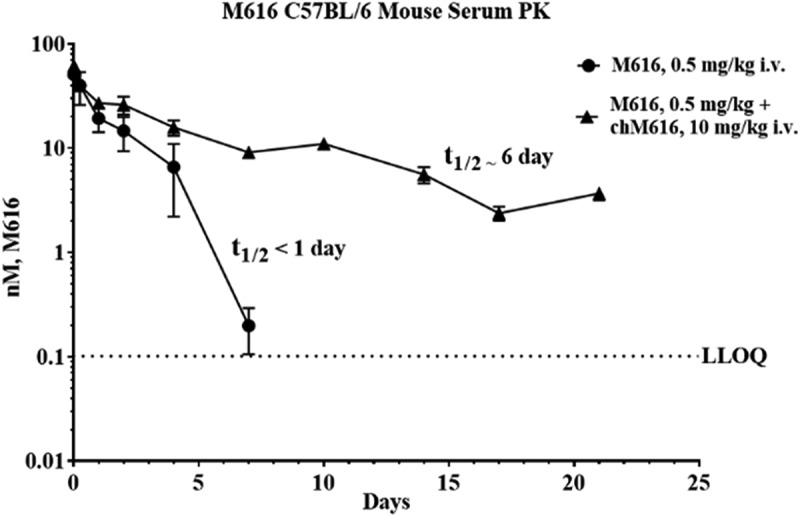
Saturation of IL-36 R target in vivo normalizes M616 serum pharmacokinetics in C57BL/6 mouse. M616 was administered IV at 0.5 mg/kg alone, or in combination with 10 mg/kg competitive mAb chM616 dosed as a cassette. A species-selective immunoassay was used to monitor the pharmacokinetics of M616 in the presence of chM616.
111In/125I-labeled anti-IL-36 R mAb biodistribution study used to characterize tissue PK and identify primary tissues sites of M616 catabolism
Next, we characterized the whole-organ biodistribution of M616 in C57BL/6 mice to facilitate the identification of the primary tissues of IL-36 R target engagement and sites of catabolism. To this end, we measured M616 PK in whole blood and select tissues via administration of 125I or 111In-labeled M616 conjugate at low dose (0.3 mg/kg, IV). The biodistribution properties of 125I/111In-M616 conjugates are anticipated to deviate only in tissue harboring significant levels of IL-36 R because 111In shows a strong propensity to residualize in lysosomes upon catabolic processing of the mAb conjugate.33,34 The biodistribution study design is shown in Table 3 and included two IV dosing arms with 125I/111In-M616 given at 0.3 mg/kg alone, or in combination with a large excess (9.7 mg/kg) of unlabeled, or “cold” M616 material to assess the influence of target saturation on the tissue exposure.
Table 3.
Biodistribution study design incorporating both non-residualizing (125I) and residualizing (111In) radio-tracers for M616 for quantitation.
| Group (n = 24) | Total M616 Dose (mg/kg) | Tracer (% of M616 Material Labeled) | Route# | Tissues Sampled |
|---|---|---|---|---|
| 1 | 0.3 | 125I (100) | IV | Whole-blood, heart, liver, lung, kidney, spleen, thymus, brain, skin (middle between shoulders), muscle, femur, lymph nodes |
| 2 | 10 | 125I (3) | IV | |
| 3 | 0.3 | 111In (100) | IV | |
| 4 | 10 | 111In (3) | IV |
# Time Points: 0.5, 2, 4, 12, 24, 48, 96, 120 h.
Whole blood PK profiles for both 125I-M616 and 111In-M616 are plotted as %ID/mL vs. time in Figure 7. Several key observations can be made from the blood data by contrasting the behavior of M616 under different dosing conditions. First, a simple visual comparison of the two 125I-M616 study arms (Figure 7, top left) illustrates the large effect that excess unlabeled (“cold”) M616 material (9.7 mg/kg cold) has on the PK of 125I-M616 (0.3 mg/kg), consistent with saturation of IL-36 R target, and this result is similar to that previously observed after administration of low dose (0.5 mg/kg) M616 plus competitive chM616 mAb (10 mg/kg) as a cassette. Additionally, the inclusion of excess cold M616 does not alter the 125I/111In-M616 PK profiles relative to 0.3 mg/kg tracer dose alone until >12 h, and this PK behavior is consistent with our original dose-ranging study results where dose-proportional PK was observed during the α-phase, indicating rate-limiting extravasation into tissue (see Discussion). Finally, there is a striking similarity between the 111In-M616 (Figure 7. Bottom left) and 125I-M616 blood profiles despite the distinct chemical nature of 125I and 111In-probes that suggests insignificant IL-36 R mediated uptake and catabolic processing of M616 in the blood compartment. Also shown in Figure 7 are the results of trichloroacetic acid (TCA)-precipitation analysis performed throughout the study to verify the integrity of M616 in circulation, where the data indicate both 125I and 111In-M616 remain largely intact.
Figure 7.
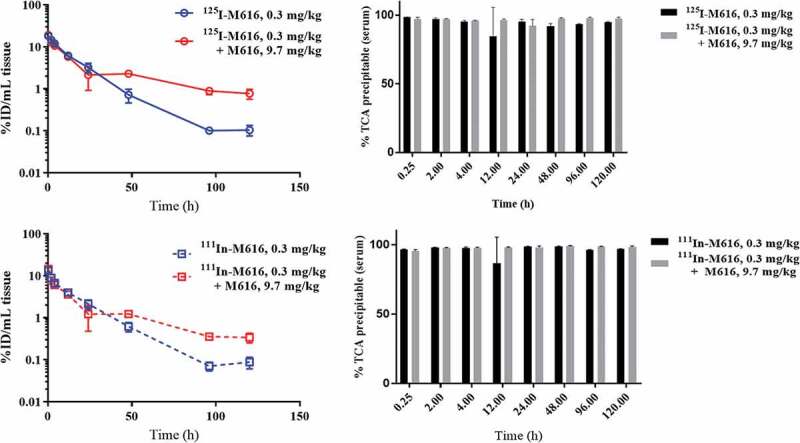
PK in whole blood for 125I-M616 (top left), 111In-M616 (bottom left), in C57BL/6 mice dosed 0.3 mg/kg IV alone, or as a cassette plus 9.7 mg/kg (10 mg/kg total M616) unlabeled M616. Note the lack of effect of a large excess of unlabeled M616 on the PK of the 0.3 mg/kg tracer dose until >12 h, consistent with a lack of significant IL-36 R target in the central (blood) compartment. Additionally, consistent PK profiles across distinct labeling strategies suggest insignificant uptake and catabolism within the blood compartment. (Top/bottom right) TCA precipitation analysis confirms that both conjugates remain fully intact in blood during the course of the study in all dose groups.
The combined serum/blood PK results suggest that the primary driver of TMDD resides in the periphery. So, we turned to a comparison of tissue PK between 125I and 111In-labeled M616 conjugates across several organs to identify the primary sites of IL-36 R-mediated uptake and catabolism. Tissue distribution time courses over 120 h (0.3 mg/kg IV dose) expressed as %ID/g tissue for both 125I-M616 and 111In-M616 arms of the study is given in Figure 8. Interestingly, the biodistribution profile of 125I-M616 (Figure 8, left) did not illustrate a preponderant tissue site of uptake that would implicate target because the highest level of exposure was maintained in blood over the duration of the study. However, when the time course was plotted as a tissue:blood (T:B) ratio (Figure 9, top left) the role of kidney as a site of 125I-M616 accumulation is emphasized, as is some retention in skin. Specificity at the level of the kidney was confirmed by the inclusion of excess (9.7 mg/kg) cold M616 material in the dose (Figure 9, top right), which resulted in the 125I-M616 T:B for kidney to be disproportionally mitigated over the entire time course relative to all other organs, presumably due to saturation at the level of IL-36 R target. The results from the 111In-M616 study arm were largely complementary, as the highest degree of 111In signal was observed in kidney (Figure 8, right), and this suggests specific M616-mediated uptake and turnover. Additional target-specific distribution of 111In-M616 to liver, spleen, and skin is evidenced as well, where both the 111In-M616 tissue time course and its representative T:B profile suggests that 111In levels in these organs increase over time (kidney) or remain relatively constant (liver, spleen, skin) through 120 h due to the residualizing nature of the label.
Figure 8.
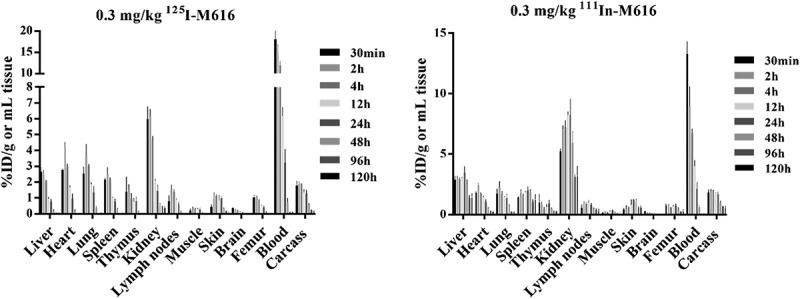
Tissue pharmacokinetics of 125I-M616 (left) and 111In-M616 (right) after administration of 0.3 mg/kg IV dose in male C57BL/6 mice (n = 3/time point). Quantitation of 125I-M616 PK in tissues suggests kidney, lung, and heart are the primary sites of tissue exposure in C57BL/6 mouse. A complementary analysis using 111In-M616 confirms the role of the kidney as a site of elevated M616 catabolism based on probe residualization in this organ. Note: both absolute levels (%ID/g) and a shift in tmax for 111In-M616 in kidney relative to other tissues indicates specific catabolism.
Figure 9.
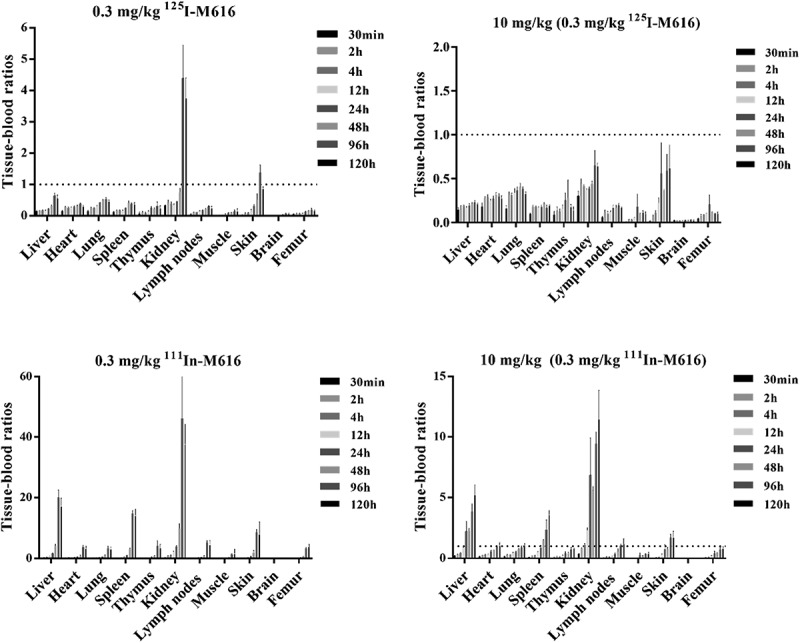
Anti-muIL-36 R mAb (M616) biodistribution in C57BL/6 mouse plotted as tissue:blood ratios over 120 h. (Top left) 125I-M616 (0.3 mg/kg) PK in tissue highlights kidney and skin as sites of tissue retention/accumulation, while 125I-M616 dosed in combination with 9.7 mg/kg unlabeled M616 (top right) confirms specific uptake in these organs. (Bottom left) 111In-M616 was utilized as a more sensitive tracer to identify sites of M616 catabolism based on residualization properties of the 111In probe in tissue. Here, the kidney, liver, spleen, and skin are implicated as possible sites of IL-36 R-mediated uptake clearance. (bottom right) Excess cold M616 significantly mitigates tissue catabolism of 111In-M616, again indicating specific uptake/catabolism in these organs.
Detection of IL-36 R in primary mouse proximal tubule epithelial cells by Western Blot
The kidney exhibited the highest level of 125I-M616 exposure based on %ID/g tissue consistent with the observed accumulation of 111In-M616 in the biodistribution studies. To add further confidence in our assessment that IL-36 R target engagement and cellular catabolism were possible in kidney, we examined C57BL/6 mouse kidney cell lysates for IL-36 R protein. Western Blot (WB) analysis detected IL-36 R in both mouse primary glomerular endothelial and mixed kidney cell lysates using a human/mouse cross-reactive anti-IL-36 R antibody as detection reagent (Supplemental Figure S2). As controls for IL-36 R protein expression, we also analyzed normal human dermal fibroblast (NHDF) and MCF-7 human breast cancer cell line lysates. The human and mouse proteins share approximately 67% sequence homology and are similar in molecular weight. IL-36 R protein expression in primary C57BL/6 mouse glomerular endothelial cells appeared independent of 24 hr pre-stimulation with 10 µg/mL M616 (lanes 2 and 3) based on luminescence intensity. The specific cell types contained within the “mixed” primary mouse kidney cell lysates were unknown, and we did not confirm the presence of an analogous protein band at ~65 kDa, although there was a large amount of a smaller (~48 kDa) protein common (faint bands) between the two kidney cell isolates that may represent truncated IL-36 R forms.
Single Dose PK study in 5/6 Nephrectomized Mouse to assess the contribution of IL-36 R in kidney to TMDD in serum
A final in vivo study was designed to interrogate the kidney’s contribution to TMDD of M616. We used a 5/6 nephrectomy model35 (see Materials and Methods) to test whether near-complete kidney ablation could affect M616 clearance in male C57BL/6 mice. Figure 10 (left) shows the serum PK profiles of WT mice dosed 4 mg/kg IV and mice that underwent 5/6 nephrectomy and were dosed 5 mg/kg IV. For comparison, the figure also includes the exposure time course from the original dose-ranging PK study at 4 mg/kg IV in female animals for reference at a consistent dose level to WT. We observed no significant effect of sex on IL-36 R-mediated TMDD of M616. Qualitatively, the effect of 5/6 nephrectomy manifests as only a slight alteration of the PK behavior at the curve inflection, as acceleration of CL over this timeframe is more gradual than in WT animals. However, due to a slight mismatch in dose within this study, and because of the sparse serial sampling used across studies, we are unable to reliably quantitate this effect. It is clear, however, that a strong TMDD effect is still observed even after near-total kidney ablation.
Figure 10.
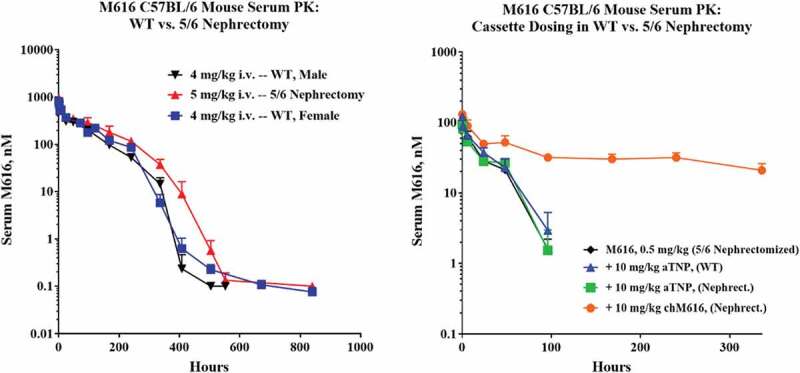
(Left) 5/6 nephrectomy in C57BL/6 mice does not dramatically impact the TMDD serum PK profile of M616. Shown are overlaid results from two separate PK studies where either male or female mice were given 4 mg/kg IV dose of M616 highlighting a lack of gender influence on IL-36 R-mediated TMDD in serum. Also shown (red) is a 5 mg/kg IV arm to illustrate that partial kidney ablation has only a minor impact on TMDD. (Right) 5/6 nephrectomy has no impact on M616 TMDD at 0.5 mg/kg IV dose and does not alter the impact of IL-36 R saturation as observed by a cassette dosing strategy. Additionally, cassette dosing of anti-TNP, rat IgG2a does not change the pharmacokinetics of M616, suggesting fast CL at low dose (0.5 mg/kg) is predominantly driven by target binding.
Our study also tested the effect that 5/6 nephrectomy had on our ability to “normalize” low dose (0.5 mg/kg) M616 plasma PK with cassette dosing of chimeric chM616, as well as an assessment of the effect of including a 10 mg/kg IV dose of non-targeted rat IgG2a mAb (anti-trinitrophenol (TNP)) as an off-target control. Figure 10 (right) illustrates that 5/6 nephrectomy has no perceptible effect on M616 PK at low dose, and that a significant reservoir of systemic IL-36 R target is still accessible to mAb after kidney ablation where again we see a dramatic “normalization” of M616 half-life with the administration of chM616 as a cassette, consistent with previous results (Figures 6 and 7; Supplemental Table 1 for NCA parameter estimates). Urine was collected over the first 24 h (α-phase) post-dose to assess functional M616 and associated degradation products. Urine was assayed for total M616-derived products using a species-specific anti-Fc sandwich assay (Supplemental Figure S3, left). M616 was detected at concentrations up to 7.3 ng/mL (5 mg/kg dose group) from the nephrectomized animals and increased with dose. Consistent with a minor role of kidney in M616 elimination, we see a large increase in average M616-Fc concentration up to 22.5 ng/mL in urine collected between 0 and 24 h for WT animals at 4 mg/kg IV dose. Strictly adjusted for dose and not accounting for potential differences in urine volume collected, this suggests that 5/6 nephrectomized animals clear roughly ~25% as much M616 through kidney in WT animals. An attempt was made to standardize the measured M616-Fc/M616 in terms of total excreted protein using a bicinchoninic acid (BCA) assay. Interestingly, when we compare levels (0–24 h) after normalization (Supplemental Figure S3, bottom), the nephrectomized mice appear to excrete a higher proportion of total protein as M616-Fc than WT, even after accounting for the slight mismatch in dose between groups. We also assayed urine for functional M616 material using recombinant muIL-36 R target as a capture reagent and expressed this as a percentage of the total Fc concentration (Supplemental Figure S3, right). Surprisingly, we observed up to 87% of the detectable M616-derived material to be functionally intact (5 mg/kg, 5/6 nephrectomized group). There was also some indication of IL-36 R-mediated M616 catabolism at the level of the kidney, as the percentage of a 0.5 mg/kg dose of M616 material that was found intact doubled (36→73%) when 10 mg/kg chM616 was given as a cassette.
Discussion
IL-36 R signals via three known cognate cytokines (α, β, γ) in addition to binding the IL-1RAcP co-receptor specific to the IL-1 family.9 Therefore, an IL-36 R-targeted therapeutic antagonist should be specific and exhibit high affinity to maintain receptor blockade over a range of circulating drug concentrations. MAbs are ideally suited as broadly neutralizing antagonists, especially given their long half-life, although mAb PK can be dominated by TMDD depending on the biological setting, which can hinder a practical clinical dosing regimen. Myriad factors play a role in the manifestation of TMDD PK behavior, including, but not limited to, overall target expression, accessibility to mAb in tissue, and rate of turnover of the resultant mAb-target complex, so an assumption of TMDD based on expression data alone is not tenable.30 We explored these properties of IL-36 R using an intrinsically high affinity (KD = 6.3 pM) murine surrogate mAb, M616, to test whether modest target levels in tissue might drive quantitative binding and affect non-linear plasma CL, as well as to capture the relationship between systemic exposure and tissue PK.
Quantitative IL-36 R expression data have been mostly limited to the mRNA level (Supplemental, S3), where the receptor is reportedly expressed significantly in only a few human cell types, including keratinocytes and dermal fibroblasts of skin, while lung and kidney epithelium are known to produce, as well as respond to, IL-36 cytokines.10,36 Additional reports show IL-36 R is expressed on hematopoietic bone marrow-derived dendritic cells and CD4 + T-cells as well.10,13,37 In mouse there are data to suggest that IL-36 R is more widely expressed (Supplemental, S3);28 however, the overall target burden is still in question even with expression data such as RNA-seq.38 Interestingly, although IL-36 R expression and signaling have been demonstrated in mouse skin, including several mouse models of psoriasis described in the literature,10,17,39 we observed fast absorption kinetics typical of rat IgG2a dosed to mouse. Furthermore, M616 bioavailability was >80% suggesting the lack of a significant IL-36 R sink due to binding and/or receptor-mediated tissue uptake and catabolism at the injection site. Mouse FcRn is known to bind rat IgG2a with similar efficiency relative to mouse IgG, so the possibility that a competitive binding pathway could protect M616 from local IL-36 R-mediated catabolism is minimal.40,41
At doses from 0.04 to 4 mg/kg, we observed unambiguous non-linear PK behavior of M616 in serum. As a precaution, we also assessed the integrity of our stock dosing solution (less than 5% aggregate), and verified the thermostability of our construct (data not shown). Importantly, because we collected blood past 4 weeks at the highest IV dose (4 mg/kg), several distinct PK phases that are consistent with TMDD were fully captured.30 Notably, an initial rapid change in plasma concentrations at early times (<72 h) that is typical of a distribution, or α phase, was followed by a period best described by a first-order β-elimination dominated by RES-mediated CL, and this led to yet a third phase of rapid acceleration of drug clearance where the inflection of the curve is governed by both KD and target concentration under a condition of partial target saturation. Terminally, we see the classic rebound phase where the apparent CL rate is between that of β-elimination and the maximum CL observed at inflection (phase 3), and this process is governed both by koff and kelim of the RL complex.30
A TMDD model defined with target binding and elimination of mAb-target complex from the central compartment, including target turnover kinetics, was able to capture the observed data and the parameters were estimated with sufficient precision. A previously published report that characterized the serum PK of a surrogate anti-muIL-36 R mAb (vide infra) was of lower affinity and monitored over a narrower dose range (0.3–10 mg/kg) in C57BL/6 mice.42 In that study, the authors observed dose-dependent behavior as well, although all major phases of TMDD were not captured by the data. Recent work from Ahlberg et al. used a similar modeling approach to characterize the PK profile of this same chimeric rat/mouse anti-muIL-36 R (MAB04) antibody, while extending the results to a retrospective analysis of a clinical candidate human anti-IL-36 R mAb.28 However, there are some inherent differences in the underlying assumptions of their model and that used in our study. Ahlberg et al. reported an in vitro KD = 240 pM for MAB0428 that was significantly greater than the KD reported for their human IL-36 R mAb (MAB92) of 24 pM,42 as well as mAb M616 investigated in this study (KD 6.3 pM), and they concluded that KD was not a sensitive parameter by their method of analysis. Ahlberg et al. also assumed kint = kdeg within their semi-mechanistic two-compartment model, while in this study, the TMDD model was able to distinguish between kint and kdeg parameters. This distinction revealed an increase in kint rate relative to kdeg suggesting that M616 binding may accelerate internalization of the membrane-bound IL-36 R. The authors also estimated the volume of central compartment to be 0.403 mL, whereas in this study the volume was fixed to physiological mouse plasma volume (0.944 mL).28,29 Overall, both models take a mechanistic approach to characterize non-linearity in the data when limited information on target concentration and turnover is available, yet the empirical nature of the models does not allow an examination of the effects of target organ expression on mAb biodistribution. Hence, a radiolabeled biodistribution study was undertaken to identify the sites of M616 distribution and elimination.
It should be noted that additional sources of non-linear CL for antibodies unrelated to target are documented, including scavenger receptor-mediated uptake CL in liver.43 Even normally advantageous mAb-FcRn interactions can be a source of poor PK properties, as was observed with briakinumab where endothelial recycling efficiency is compromised due to charge-driven Fab interactions with the receptor.44,45 By co-dosing M616 with a mouse chimeric version of M616 lacking glycans at N297, and therefore unable to interact with FcγR as a cassette, we were able to modulate the PK behavior of M616 ruling out these additional CL pathways. Our ability to discriminate M616 from chM616 bioanalytically with a species-specific immunoassay for quantitation was key to establishing a common saturable CL mechanism. We observed a dramatic extension of the elimination half-life of M616 from less than 1 d to over 6 d by this approach. This result is strong evidence that the IL-36 R target drives TMDD of M616, where competitive saturation of target with chM616 essentially “normalizes” M616 serum PK in mice so that only general catabolic CL mechanisms typical of circulating rat IgG2a in mouse are dominant. An important observation during modeling of the M616 serum data was that the distribution, or α-phase, of the PK time course across dose levels was dose-independent. Cao et al. have demonstrated by way of minimal physiologically based PK modeling with TMDD incorporated that a dose-independent α-phase indicates rate-limiting perivascular extravasation to the interstitial space prior to target binding.25 Thus, we inferred that the primary IL-36 R target sink driving non-linear PK was limited to the periphery based on serum data alone.
When used in tandem, 125I/111In mAb-labeling strategies are meant to provide complementary information: 125I is rapidly cleared by the cell upon tissue uptake and degradation within lysosomes,46 so 125I as conjugation tracer is used to track tissue PK of the intact mAb. Conversely, the residualizing nature of 111In renders this probe useful for identification of the primary sites of antibody catabolism marked by its tendency to accumulate in tissue.33,34 Comparison of the 125I/111In-M616 profiles in blood supports our hypothesis that IL-36 R in the periphery drive non-linear PK in two ways. First, there is near perfect agreement of the PK profiles at low dose (0.3 mg/kg) for both 125I and 111In-M616 dose arms (Figure 7), indicating a lack of uptake/catabolism in the blood compartment. Second, when including excess (9.7 mg/kg) “cold” M616 material, we do not see an effect on the PK of a tracer dose of M616 conjugate until after ~12 h, signifying that the interaction with IL-36 R target is limited by the kinetics of tissue extravasation.
Given the degree of non-linearity of the serum/blood PK observed, it was surprising that biodistribution analysis failed to reveal a major site of M616 accumulation. Skin has been implicated as a primary site of IL-36 R expression, consistent with the pharmacology of anti-IL-36 R agents.39,47 However, we found only moderate accumulation in skin with some residualization when presented on a %ID/g tissue scale that was most evident when plotted as a tissue-to-blood concentration ratio (T:B), although delayed mAb extravasation within skin is often due to tight junctions of the blood vessels in this organ. However, based on the levels observed, skin is not an obvious sink driving non-linear elimination in blood, but it may be possible that, on a per gram tissue basis, low to moderate levels of IL-36 R are capable of inducing non-linear serum PK behavior because the total amount of skin present in an animal is significant relative to all other tissues.
The trafficking of IL-36 R receptor is also unusual in that it has been shown to escape degradation in lysosomes during cellular recycling to and from the plasma membrane,48 so this pathway may be perceived as being the high capacity for uptake of bound cargo. Several tissues did appear to stand out, however, based on three criteria: 1) a high T:B concentration ratio >1 due to shifts in the PK profiles relative to blood, 2) evidence of competitive displacement of 125I/111In tracer when including a high dose of unlabeled M616, and 3) evidence for residualization of 111In due to catabolism. Of all the tissues analyzed, kidney was the most prominent. This result was unexpected given the presumed limited accessibility of the IL-36 R target in kidney parenchyma due to the mAb’s high molecular weight (~150 kDa), which should limit kidney exposure to intact mAbs by glomerular filtration.23 Kidney exposure to circulating molecules larger than the molecular weight limit (~60 kDa) is not unprecedented, as there are known transport mechanisms by which large proteins gain access to kidney parenchyma.49,50 Consistent with this finding, Chi et al. describe inducible IL-36α expression in renal tubules in mouse models of renal disease, and demonstrated an IL-36 R dependence by the observation of reduced fibrosis, collagen IV accumulation, as well as immune cell (dendritic cells, CD4+/CD8+ tumor-infiltrating lymphocytes) infiltration in draining renal lymph node of knockout animals.51
We confirmed IL-36 R expression in mouse primary kidney glomerular endothelial cells, as well as mixed cell isolates likely to contain proximal tubule cells by Western Blot (Supplemental, S2). Therefore, we proceeded to directly test this hypothesis by performing an additional analysis in a 5/6 nephrectomized mouse model, where the animals, although viable, maintain less than 20% normal kidney function. However, we were unable to confirm a dominant effect of partial kidney ablation on the overall serum PK of M616, although there were some notable effects that potentially implicate IL-36 R. Analysis of collected urine during the first 24 hr interval after dosing revealed functionally intact M616 at levels normalized to total protein content indicate that 5/6 nephrectomized mice excrete a greater proportion of functional M616 relative to WT animals. Additionally, the inclusion of competitive mAb chM616 seemed to increase the amount of functional M616 in urine, possibly due to saturation of the IL-36 R receptor. Overall, the combination of studies focused on mouse kidney suggests that M616 can reach kidney parenchyma where it can interact with IL-36 R target, and that a significant fraction of M616 can access the collecting duct and tubules intact before elimination in the urine.
The results presented in this work, combined with evidence from the literature,28,42 suggest that anti-IL-36 R mAbs are characterized by dose-dependent PK with elimination at non-saturating doses mediated predominantly by target expressed in peripheral tissues. Typically, semi-mechanistic mathematical TMDD models are applied to understand the dose required to saturate target, to design dosing regimens for further characterization in non-human primates (NHP), and to guide preliminary first-in-human (FIH) dose predictions, as TMDD models are generally amenable to scaling across species.52 Here, our extensive characterization applies to murine-specific surrogate, M616, so accurate clinical dose projection assuming a suitable human antibody was obtained would include multiple considerations. Drug-specific parameters V, kel, k12 and k21 (Table 2) can be scaled by standard rules of allometry, with PK data obtained in NHP for NHP/human cross-reactive mAbs considered the gold standard for clinical translation. Ideally, experimental measurements inform system-specific parameters. Differences in total target burden (Ro) across species expressed as molar concentration may be obtained through a combination of IHC and cytometry methods, with a correction for differences between healthy vs. disease states.52 MAb-target binding affinity, KD, can be measured in vitro assuming the availability of recombinant target and/or applicable cell expression model. Additionally, target turnover properties defined by kdeg and kint are often obtained from a suitable in vitro cell assay.
One important caveat of TMDD models is the assumption that the target is located at a site in rapid equilibrium with plasma. Our biodistribution data implicate the source of TMDD to be driven solely by target binding in the periphery over a wide range of tissues. Hence, a TMDD model is not suitable to understand the specific tissue elimination kinetics or receptor occupancy (RO) across tissue sites, factors of critical importance for accurate interpretation of preclinical toxicities, or when leveraging pharmacology-driven FIH dose selection, e.g., minimal anticipated biological effect level (MABEL)-based approaches.53 As such, future investigations should focus on defining both IL-36 R protein expression and turnover kinetics to inform PBPK models, as this approach is expected to provide more realistic depiction of mAb disposition within peripheral tissues leading to more reliable a priori predictions.23,54 Finally, our robust tissue biodistribution data in mouse may be informative for the anticipation of specific tissue sites at risk for elevated exposure. Although no single tissue sink was observed in our study, the data implicate kidney, liver, spleen, and skin, as potential sites warranting increased scrutiny during preclinical toxicity studies and early clinical trials alike.
In summary, we have defined the non-linear PK of surrogate anti-muIL-36 R antibody, M616, to be the result of TMDD. Importantly, our results suggest a lack of a preponderant IL-36 R target sink based on radio-label biodistribution analysis. Thus, we conclude that TMDD of IL-36 R mAbs in mouse is driven by low-level, high CL capacity expression of the receptor across a wide range of tissues/organs. It is unclear whether this biodistribution profile for a mAb exhibiting pronounced TMDD such as M616 is more common, or an exception due to the unique turnover properties of IL-36 R. Additional characterization of IL-36 R biology as it pertains to the disposition of target therapeutic antibodies is warranted given the interest of IL-36 R in the etiology of numerous disease states.
Materials and methods
Reagents
Rabbit anti-rat Fc mAb was purchased from abcam (clone R18-2; # ab125900). Recombinant mouse IL-1Rrp2/IL-1R6 (mu IL-36 R) protein was purchased from R&D systems (# 2354-RP). Both rabbit anti-mouse (H + L) polyclonal antibody (pAb) (# 315-605-045), and donkey anti-rat (H + L) pAb Alexa Fluor-647 conjugates (cat# 712-605-150) used as detection antibodies for the affinity measurements were purchased from Jackson ImmunoResearch. For Meso™ Scale Discovery (MSD) detection, goat anti-rat IgG (H + L chain) pAb maintaining minimal cross-reactivity with mouse IgG was obtained from Jackson ImmunoResearch (cat# 112-005-167). For WB analysis of cell lysates, rabbit anti-IL-36 R pAb (human/mouse cross-reactive) was purchased from ABclonal (#A10090). Criterion™ TGX Stain-Free Precast gels (4–15%) were purchased from BioRad (#5678083). Goat anti-rabbit (H + L)-horseradish peroxidase (HRP) conjugate used for visualization of the WB was obtained from Jackson ImmunoResearch (#111-035-144). Total protein assays were conducted using a Pierce™ BCA assay kit (Thermo Scientific, #23225). Radioimmunoprecipitation assay (RIPA) Buffer (Thermo Scientific, #89901) supplemented with Halt™ Protease Inhibitor Cocktail (Thermo Scientific, #78410) were used for cell lysis and IL-36 R protein extraction. NHS-Activated Sepharose 4 Fast Flow was obtained from GE LifeSciences (#17090601). Primary mouse (C57BL/6) kidney glomerular endothelial (Cat. # C57-6014 G) and mixed population kidney cells (Cat. # C57-6227) were purchased from Cell Biologics. MCF7 cell line was obtained from American Type Culture Collection (Manassas, VA). NHDF cells were obtained from Lonza (Williamsport, PA). 4x Laemmli Sample Buffer (BioRad, #161-0747) and SeeBlue™ Plus2 protein standard (Thermo Scientific, #LC5925) were used in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). 125I was obtained as sodium iodide in 10−5 N sodium hydroxide from Perkin Elmer (Boston, MA). 111InCl3 obtained from Nordion, Inc. (Ontario, Canada) (IPG-In-111, No. IN-17-025). Bio-gel® P6 spin columns were purchased from BioRad. All common buffer additives media of the highest purity were purchased through VWR.
Anti-muIL-36 R mAbs
Rat IgG2a (M616) and mouse/rat chimeric IgG1 (rat variable regions, murine Fc; chM616) anti-IL-36 R mAbs specific and with high affinity for murine IL-36 R were constructed using an in-house protocol (data not shown).
Measurement of muIL-36 R binding affinity M616 (rat IgG2a, WT), and chM616 (mu/rat IgG1, N297 G)
Equilibrium binding affinity of M616 and chM616 to mu-IL-36 R was characterized in solution by Kinetic Exclusion assay (KinExA; Sapidyne Instruments Inc.). To assess free IL-36 R mAb in solution at equilibrium, recombinant mu-IL-36 R (Asp22-Arg338) protein (R&D Systems) was coupled to activated NHS-Sepharose (GE Life Sciences) at a density of 30 µg protein to 1 mL of packed resin per the manufacturer’s recommended coupling protocol and used as the assay capture substrate. All equilibrium titrations were conducted at 25°C in running buffer (phosphate-buffered saline (PBS), 1 mg/mL bovine serum albumin, 0.01% sodium azide, pH = 7.4). For detection of captured IL-36 R mAb, the appropriate anti-species pAb (Jackson ImmunoResearch) coupled to Alexa Fluor® 647 was used at a concentration of 0.5 µg/mL in running buffer. Two fixed concentrations of anti-IL-36 R mAb (50 pM and 1000 pM) were titrated with IL-36 R protein spanning a 3-fold dilution range between 500 and 0.9 pM. The samples were left to equilibrate at room temperature for 48 h prior to analysis. The percent free antibody binding sites as a function of IL-36 R concentration were used to quantify the KD and confidence intervals for M616 and chM616 using a global fitting routine (n-curve analysis) contained within the KinExA Pro Software environment (Sapdiyne Instruments Inc.).
Radiochemistry
For the generation of 125I-M616, 75 µg mAb was labeled at tyrosine residues with 1 mCi iodine-125 (125I) using a modification of the Iodogen method (Pierce Chemical Co., Rockford, IL) as previously described.55 125I was obtained as sodium iodide in 10−5 N sodium hydroxide solution. The material was purified with Bio-gel® P6 spin columns with Dulbecco’s PBS (DPBS) using the manufacturer’s provided protocol. After purification, the product contained 2.48 mCi with a concentration of 1.27 mg/mL protein. The radiochemical purity was 99% as assessed by thin-layer chromatography.
Radiosynthesis of the 111In-M616 was performed by random conjugation of SCN-DOTA to free lysine residues using established protocols (~1.8 DOTA molecules per antibody). After conjugation, 111InCl3 (IPG-In-111, Nordion Inc., Canada, No. IN-17-025) was added and incubated for 45 min to effect chelation of the metal. Conjugation efficiency and radiochemical purity (90%, 1.40 mCi at 0.67 mg/mL protein, specific activity = 540 Ci/mmol) were assessed by size exclusion chromatography. To assess label effects on protein binding, recombinant mouse IL-36 R binding kinetics were assessed by SPR (Biacore, T-200 instrument) and found to be negligible for 125I-M616, and minimal for 111In-M616 (less than 10% reduction in protein activity; data not shown). 125I and DOTA-conjugated IL-36 R showed ≤80% binding activity relative to the unconjugated antibody.
Single-dose mouse PK analysis
M616 (anti-muIL-36 R) serum concentrations were determined by ELISA. Wells of 96-well microtiter plates (“Maxisorp,” Nunc) were coated with muIL-36 R-huFc and incubated overnight at 2–8°C. The coated wells were then blocked with a solution of 10% nonfat dried milk in PBS pH 7.2 with 0.05% Tween-20 (PBST/NFDM). The blocked wells were washed with PBS pH 7.2 with 0.05% Tween-20 (PBST) prior to use. M616 concentration standards and quality control samples (QCs) prepared in 100% mouse serum were diluted 20-fold in PBST with 0.5 M NaCl and dispensed into wells. Mouse serum concentration was normalized at 5% (1:20 dilution) in sample and standard titrations. Samples were incubated for 1 h with agitation and the wells were washed before the addition of peroxidase-labeled goat anti-rat IgG:HRP (Jackson ImmunoResearch) diluted in PBST. Following another 1-h incubation without agitation, the wells were again washed and developed with tetramethylbenzidine (TMB) peroxidase substrate (Kirkegaard & Perry Laboratories). The reaction was quenched with acid and optical densities were determined at a wavelength of 450 nm. The dose–response curve, constructed of concentration standards in the range of 0.313–20 ng/mL, was fitted to a four-parameter logistic model (GraphPad Prism). Sample concentrations were interpolated from the fitted curve and corrected for dilution factor. The assay limit of quantitation was 6.26 ng/mL.
Species-specific immunoassay for quantitation of M616 dosed as a cassette with chM616
To quantitate M616 in serum in the presence of chM616 dosed as a cassette, an immunoassay based on the Gyrolab® xP platform (Gyros Protein Tech., Uppsala, Sweden) was developed. Selectivity for M616 was achieved by the use of rabbit anti-rat Fc mAb (abcam, clone R18-2), which was verified prior to analysis to exhibit no cross-reactivity with chimeric mouse/rat IgG1, chM616 (data not shown) over the working range of the PK assay. Briefly, biotinylated R18-2 mAb was loaded as capture antibody on a BioAffy 200 CD at a concentration of 1 µg/mL using a standard Gyrolab® automated method prior to the capture of M616 in serum samples at the appropriate dilution. Alexa Fluor 647-conjugated R18-2 (0.5 µg/mL) was used to detect bound M616 by fluorescence. The fluorescence signal was converted to concentration via regression to a standard curve analyzed by a four-parameter (4PL) Logistic regression model.
Assay of urine in 5/6 nephrectomized mouse PK study
For quantitation of M616 and derived IgG Fc-fragments in mouse urine, two formats of electrochemiluminescent (ECL) immunoassay were developed on the MSD platform (Rockville, Maryland). Briefly, MSD Gold streptavidin 96-well plates were functionalized with differing biotinylated capture reagent immobilized at 1 µg/mL in Blocker™ blotto (Thermo Scientific) for >2 h: rabbit anti-rat Fc (abcam clone R18-2) to capture both intact and Fc-containing M616 catabolites (total M616 assay), or mouse rIL-36 R ECD (R&D Systems) to capture intact M616 (functional M616 assay). Next, a standard curve was prepared with M616 spiked into control C57BL/6 urine, with the concentration ranging from 500 ng/mL to 25 pg/mL (LLOQ = 76, and 25 pg/mL, for total vs. intact assays, respectively). After thorough washing (KPL buffer) of the coated MSD plates, the plates were prepared similarly: 30 µL of pooled urine sample (n = 3/group, assayed in triplicate), as well as full standard curves in triplicate, plus blanks and QCs, were plated prior to the addition of 30 µL goat anti-rat IgG (H + L chain) detection antibody (0.25 µg/mL) in blotto that had been previously labeled with Sulfo-NHS ECL detection label following the manufacturer’s labeling protocol. The assays were left to incubate on an orbital shaker overnight at 4°C. The next day, the plates were brought to room temperature, and washed three times (KPL buffer) prior to the addition of 200 µL MSD read buffer before data acquisition on a QuickPlex Meso™ QuickPlex SQ 120 instrument. MSD counts for all samples were interpolated from the resultant standard curves analyzed using non-linear regression to a 4-parameter logistic function. Finally, to normalize the M616 content in urine, matched urine samples were assayed for total protein content using a standard Pierce BCA assay (Thermo Scientific).
Noncompartmental analysis
Nominal doses and sampling times were used for PK analysis. Noncompartmental analysis was performed for sparsely sampled serum concentration-time data using Phoenix® WinNonlin® (v6.4; Certara, NJ). Total exposure was assessed as AUCinf (Area under the curve from time zero to infinity), and primary PK parameters CL (total body clearance) and Vss (volume of distribution at steady state) were used for the assessment of non-linear PK.
Mathematical PK model
The TMDD originally proposed by Mager and Jusko20 was used to characterize the non-linearity in the data (Figure 2). The model assumes that the mAb has the ability to interact with the target receptor (R) in the central compartment to form mAb-receptor complex (RC). The experimentally determined dissociation rate constant KD of 6.3 pM suggests the antibody had a high affinity to the target. The first-order dissociation constant koff of 0.00127 hr−1 was assumed and the second-order rate association constant kon was derived by the relation kon = koff/kD. The target kinetics was determined by a zero-order production rate ksyn and first-order degradation rate kdeg. In the model, ksyn was defined as RO (the baseline receptor concentration) x kdeg, with RO fixed to the value reported by Ahlberg et al.28 The baseline receptor concentration RO was fixed in the based from Ahlberg et al.28 The mAb-target complex (RC) is assumed to undergo internalization as described by a first-order rate constant kint. The free mAb can also distribute to the peripheral tissue compartment via first-order rate processes (k12 and k21; assuming k12 = k21) and it may also undergo linear elimination (kel). The volume of the central compartment was fixed to the reported mouse plasma physiological volume of 0.944 mL from Shah and Betts et al.29
Differential equations describing the model are:
Xp is mass of antibody in the central compartment.
Xt is mass of antibody in the tissue compartment.
V is the volume of distribution of the drug in the central compartment.
PK model fitting
The serum PK data from dose group 0.04, 0.4 and 4 mg/kg (given IV) were fitted simultaneously using the above described TMDD model. The modeling fittings were conducted using the Ubiquity package in R software.56
In vivo experimental protocols
Male or female C57BL/6 mice were purchased from Charles River (Hollister, CA, USA) or the Jackson Laboratory (Sacramento, CA, USA). Animals were allowed at least 1-week acclimation to the facility prior to any procedures. All research protocols were approved by the Amgen, Inc. Institutional Animal Care and Use Committee. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition. Animals were individually or group-housed at an Association for Assessment and Accreditation of Laboratory Animal Care, an internationally accredited facility, in ventilated caging on corn cob bedding or in metabolism caging. Animals had ad libitum access to feed (Harlan 2020X, IN, USA) and reverse osmosis-purified water via water bottles or automatic watering system. Animals were maintained on a 12:12 hlight:dark cycle in rooms at 18°C to 26°C and a 30% to 70% humidity range and had access to enrichment opportunities.
M616 dose-ranging PK study
A PK dose-ranging study with muIL-36 R (M616) was carried out in female C57BL/6 mice (6–8 weeks old; Charles River Laboratories). M616 was formulated in 10 mM sodium acetate with 9% sucrose, pH 5.2 (A5Su) and dosed via IV bolus administration at 0.04, 0.4, and 4 mg/kg via the lateral tail vein. Extravascular doses of M616 were administered via IP injection at 4 mg/kg in the lower right-hand quadrant and via SC injection in the mid-scapular region. Blood samples were collected at pre-determined time points up to 840-h post-dose by submandibular venipuncture using a sparse serial sampling scheme (n = 3 mice/group/time point). Whole blood was collected, placed into Sarstedt Microvette® 500 serum separator tubes, allowed to clot at room temperature for 20 min prior to centrifugation at 11,500 x g for 15 min. The resulting serum was stored at −70°C until analysis.
M616/chM616 cassette dosing study
A PK study with M616 and chM616 was performed in male C57BL/6 mice (6–8 weeks old, Charles River Laboratories). M616 and chM616 were formulated in A5Su and administered via IV injection into the lateral tail vein. M616 was administered individually at 0.5 mg/kg or as a combined solution (cassette) containing chM616 (5 mg/kg of M616 and 10 mg/kg of chM616). Blood samples were collected at pre-determined time points up to 504-h post-dose and processed for serum as outlined above.
5/6 nephrectomy study
Surgical manipulation was similar to the 5/6 nephrectomy model described by Wang et al. with a few modifications.35 Aseptic surgical technique was used in all surgical procedures. Briefly, mice were anesthetized with 4–5% isoflurane (Abbott Laboratories, North Chicago, IL, USA). Two-thirds of renal mass was ablated by resection of 1/3 of the left kidney at the upper and lower poles. Gelfoam® (Pharmacia and Upjohn Co. Kalamazoo, MI, USA) was applied to renal tissue before the kidney was placed back into the abdomen. One week later the entire right kidney was removed, and the mice underwent an additional 1-week recovery period prior to being placed on study. An SC dose of buprenorphine (0.05 mg/kg) was given preoperatively and post-operatively twice a day for 48 h during each surgical session.
A PK study with M616, chM616, and rat IgG2a mAb (anti-TNP) was carried out in male 5/6 nephrectomized C57BL/6 or WT C57BL/6 mice (10–12 weeks old, Jackson Laboratory). M616 was administered at 0.5 and 5 mg/kg via IV injection into the lateral tail vein to 5/6 nephrectomized mice and at 4 mg/kg to intact C57BL/6 mice. A cassette of M616 and chM616 was administered via IV injection (5 mg/kg of M616 and 10 mg/kg of chM616) to 5/6 nephrectomized mice. Additionally, a cassette of M616 and anti-TNP was administered via IV injection (5 mg/kg of M616 and 10 mg/kg of anti-TNP). Blood samples were collected at pre-determined time point up to 840 h and processed for serum as outlined above. Urine samples were collected in 24-h intervals at pre-determined time points, weighed, and stored at −70°C until analysis.
Tissue distribution analysis
Twenty-four C57BL/6 male mice (6–8 weeks old, Charles River Laboratories) weighing between 22 and 26 g received a single bolus IV injection via the lateral tail vein containing either 125I-anti-IL-36 R and 111In-anti-IL-36 R at tracer-only dose or mixed with unlabeled anti-IL-36 R to complete a total antibody dose of 10 mg/kg. To prevent thyroid sequestration of 125I, 100 L of 30 mg/mL of sodium iodide was administered via IP injection 1 and 24 h before dosing.
Tissue distribution was assessed by tissue dissection at terminal time points (0.083, 24, 48, 96, and 168 h post-dose, n = 3 mice/time point). Blood was collected via cardiac puncture under general anesthesia for plasma preparation. Tissues collected were heart, kidneys, liver, lungs, lymph nodes, spleen, thymus, bone marrow, brain, and skin from the mid-scapular region (hairless). Upon harvest, all tissues were rinsed with 1X PBS and blotted dry, weighed, and frozen at −70°C until radioactivity counting. Radioactivity quantification was obtained as count per minute in dissected tissues using a gamma counter (Wallac 1470; PerkinElmer Life and Analytical Sciences) with different channels to separate each radioisotope. The resulting values were used to calculate the percentage of injected dose normalized to a gram of tissue (%ID/g) or milliliter of fluid. All tissue/blood ratios were calculated by taking the %ID/g in tissues divided by the %ID/g in blood over each respective time point.
Plasma TCA was performed as a surrogate of protein-bound radioactivity stability in circulation. Radioactivity before and after TCA precipitation (x100) was used to determine the percentage of antibody-bound 125I or 111In for each plasma sample at each time point.
Western blot analysis
IL-36 R protein was isolated from confluent T-25 flask cultures of mouse primary or control cell lines using a standard RIPA buffer lysis and extraction protocol. Total protein isolates were assayed by BCA assay prior analysis and ranged between 0.6 and 0.9 mg/mL total protein in 300 µL final volume. For SDS-PAGE and blotting, 20–30 µg total protein was reconstituted in Laemmli buffer supplemented with dithiothreitol. Protein was resolved by SDS-PAGE on a Criterion TGX Stain-Free Precast (4–15%) gel with lanes reserved for protein ladder control. Total protein was detected via activation and colorimetric imaging on a ChemDoc SRS Imaging system (BioRad, Hercules, CA). Protein bands were next transferred to nitrocellulose membranes using a Trans-Blot® Turbo™ (BioRad, Hercules, CA) transfer system per the manufacturer’s suggested protocol. IL-36 R protein was developed by application of primary antibody (anti-IL-36 R, ABclonal) at 1:5000 overnight, washed 3x with Tris buffer, prior to addition of 1:1000 goat anti-rabbit HRP for 2 h. Subsequent to additional wash steps, Pierce TMB Substrate (Thermo Scientific) was added for 10 min prior to imaging on the ChemDoc SRS Imaging system.
IL-36 R expression analysis
For comparison of mouse and human IL-36 R expression profiles at the RNA level, an internal Amgen, Inc. RNA-Seq database was queried. All RNA-seq data were generated using Illumina sequencer using an internal library of tissue specimens. Data processing was done by the Amgen Genome Analysis Unit (GAU) using an internal analysis pipeline. Expression levels are measured in FPKQ for each gene/sample. FPKQ is upper-quartile-normalized fragments per kilobase of transcript per million mapped reads (FPKM), developed by GAU, to counter the skewing effect on FPKM values by highly expressed transcripts. As a general guideline, FPKQ of 1 roughly translates to one transcript per cell, genes with values below that should be considered not expressed.
Acknowledgments
The authors would like to thank Dr. John Harrold for helpful discussion on target-mediated disposition modeling. We would also like to thank Dr. Josh Pearson for his helpful suggestions of experimental design.
Declaration of Interest
The authors declare no financial or other interests related to the submitted work that has affected our objectivity in the interpretation of the results.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Slager RE, Hawkins GA, Ampleford EJ, Bowden A, Stevens LE, Morton MT, Tomkinson A, Wenzel SE, Longphre M, Bleecker ER, et al. IL-4 receptor alpha polymorphisms are predictors of a pharmacogenetic response to a novel IL-4/IL-13 antagonist. J Allergy Clin Immunol. 2010;126:875–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M.. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–31. [DOI] [PubMed] [Google Scholar]
- 3.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, et al. Lebrikizumab Treatment in Adults with Asthma. N Engl J Med. 2011;365:1088–98. [DOI] [PubMed] [Google Scholar]
- 4.Hänel KH, Cornelissen C, Lüscher B, Baron JM. Cytokines and the Skin Barrier. Int J Mol Sci. 2013;14:6720–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol. 2012;12:486–90. [DOI] [PubMed] [Google Scholar]
- 6.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. [DOI] [PubMed] [Google Scholar]
- 7.Kovach MA, Singer B, Martinez-Colon G, Newstead MW, Zeng X, Mancuso P, Moore TA, Kunkel SL, Peters-Golden M, Moore BB, et al. IL-36gamma is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal Immunol. 2017;10:1320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovenberg TW, Crowe PD, Liu C, Chalmers DT, Liu X-J, Liaw C, Clevenger W, Oltersdorf T, De Souza EB, Maki RA. Cloning of a cDNA encoding a novel interleukin-1 receptor related protein (IL1R-rp2). J Neuroimmunol. 1996;70:113–22. [DOI] [PubMed] [Google Scholar]
- 9.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 Signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-κB and MAPKs. J Biol Chem. 2004;279:13677–88. [DOI] [PubMed] [Google Scholar]
- 10.Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol. 2015;97:645–52. [DOI] [PubMed] [Google Scholar]
- 11.Yi G, Ybe JA, Saha SS, Caviness G, Raymond E, Ganesan R, Mbow ML, Kao CC. Structural and functional attributes of the Interleukin-36 receptor. J Biol Chem. 2016. Aug 5;291(32):16597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813–23. [DOI] [PubMed] [Google Scholar]
- 13.Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E, Olleros ML, Vesin D, Garcia I, Ronchi F, et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood. 2012;120:3478–87. [DOI] [PubMed] [Google Scholar]
- 14.Penha R, Higgins J, Mutamba S, Barrow P, Mahida Y, Foster N. IL-36 receptor is expressed by human blood and intestinal T lymphocytes and is dose-dependently activated via IL-36beta and induces CD4+ lymphocyte proliferation. Cytokine. 2016;85:18–25. [DOI] [PubMed] [Google Scholar]
- 15.Green H, Easley K, Iuchi S. Marker succession during the development of keratinocytes from cultured human embryonic stem cells. Proc Natl Acad Sci U S A. 2003;100:15625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carle B, Knieling F, Claussen J, Klimowicz AC, Zheng J, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. 2019;156:1082–1097.e1011. [DOI] [PubMed] [Google Scholar]
- 17.Blumberg H, Dinh H, Dean C Jr., Trueblood ES, Bailey K, Shows D, Bhagavathula N, Aslam MN, Varani J, Towne JE, et al. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J Immunol. 2010;185:4354–62. [DOI] [PubMed] [Google Scholar]
- 18.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, Sims JE. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsumoto K, Isozaki Y, Yagami H, Tomita M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy. 2019;11:119–27. [DOI] [PubMed] [Google Scholar]
- 20.Mager D, Jusko W. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–32. [DOI] [PubMed] [Google Scholar]
- 21.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72:1–10. [DOI] [PubMed] [Google Scholar]
- 22.Tabrizi M, Bornstein G, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. Aaps J. 2010;12:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glassman PM, Abuqayyas L, Balthasar JP. Assessments of antibody biodistribution. J Clin Pharmacol. 2015;55:S29–S38. [DOI] [PubMed] [Google Scholar]
- 24.Luu KT, Bergqvist S, Chen E, Hu-Lowe D, Kraynov E. A model-based approach to predicting the human pharmacokinetics of a monoclonal antibody exhibiting target-mediated drug disposition. J Pharmacol Exp Ther. 2012;341:702–08. [DOI] [PubMed] [Google Scholar]
- 25.Cao Y, Jusko W. Incorporating target-mediated drug disposition in a minimal physiologically-based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn. 2014;41:375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darling RJ, Brault PA. Kinetic exclusion assay technology: characterization of molecular interactions. Assay Drug Dev Technol. 2004;2:647–57. [DOI] [PubMed] [Google Scholar]
- 27.Glass TR, Winzor DJ. Confirmation of the validity of the current characterization of immunochemical reactions by kinetic exclusion assay. Anal Biochem. 2014;456:38–42. [DOI] [PubMed] [Google Scholar]
- 28.Ahlberg J, Giragossian C, Li H, Myzithras M, Raymond E, Caviness G, Grimaldi C, Brown S-E, Perez R, Yang D, et al. Retrospective analysis of model-based predictivity of human pharmacokinetics for anti-IL-36R monoclonal antibody MAB92 using a rat anti-mouse IL-36R monoclonal antibody and RNA expression data (FANTOM5). mAbs. 2019 Jul;11(5):956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39:67–86. [DOI] [PubMed] [Google Scholar]
- 30.Peletier LA, Gabrielsson J. Dynamics of target-mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn. 2012;39:429–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radaev S, Sun PD. Recognition of IgG by Fcγ Receptor: the role of fc glycosylation and the binding of peptide inhibitors. J Biol Chem. 2001;276:16478–83. [DOI] [PubMed] [Google Scholar]
- 32.Jacobsen FW, Stevenson R, Li C, Salimi-Moosavi H, Liu L, Wen J, Luo Q, Daris K, Buck L, Miller S, et al. Engineering an IgG scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292:1865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorpe SR, Baynes JW, Chroneos ZC. The design and application of residualizing labels for studies of protein catabolism. Faseb J. 1993;7:399–405. [DOI] [PubMed] [Google Scholar]
- 34.Wall DA, Maack T. Endocytic uptake, transport, and catabolism of proteins by epithelial cells. Am J Physiol Cell Physiol. 1985;248:C12–C20. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Chaudhry MA, Nie Y, Xie Z, Shapiro JI, Liu J. A mouse 5/6th nephrectomy model that induces experimental uremic cardiomyopathy. J Visualized Exp. 2017 Nov 7;(129). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sørensen KK, McCourt P, Berg T, Crossley C, Couteur DL, Wake K, Smedsrød B. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1217–R1230. [DOI] [PubMed] [Google Scholar]
- 37.Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, Young DA, Fouser LA, Nickerson-Nutter C, Collins M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–37. [DOI] [PubMed] [Google Scholar]
- 38.Edfors F, Danielsson F, Hallström BM, Käll L, Lundberg E, Pontén F, Forsström B, Uhlén M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12:883–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, Ward NL, Johnston A. IL-36 promotes myeloid cell infiltration, activation and inflammatory activity in skin. J Immunol. 2014;192:6053–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs. 2015;7:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–59. [DOI] [PubMed] [Google Scholar]
- 42.Ganesan R, Raymond EL, Mennerich D, Woska JR Jr., Caviness G, Grimaldi C, Ahlberg J, Perez R, Roberts S, Yang D, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. mAbs. 2017;9:1143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datta-Mannan A, Croy JE, Schirtzinger L, Torgerson S, Breyer M, Wroblewski VJ. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. mAbs. 2016;8:969–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A. 2015;112:5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD. A two-pronged binding mechanism of IgG to the neonatal Fc receptor controls complex stability and IgG serum half-life. Mol Cell Proteomics. 2017;16:451–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boswell CA, Bumbaca D, Fielder PJ, Khawli LA. Compartmental tissue distribution of antibody therapeutics: experimental approaches and interpretations. Aaps J. 2012;14:612–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The Significance of IL-36 Hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20:3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saha SS, Singh D, Raymond EL, Ganesan R, Caviness G, Grimaldi C, Woska JR, Mennerich D, Brown S-E, Mbow ML, et al. Signal transduction and intracellular trafficking by the interleukin 36 receptor. J Biol Chem. 2015. Sep 25;290(39):23997–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagai J, Sato K, Yumoto R, Takano M. Megalin/Cubilin-mediated Uptake of FITC-labeled IgG by OK kidney epithelial cells. Drug Metab Pharmacokinet. 2011;26:474–85. [DOI] [PubMed] [Google Scholar]
- 50.Akilesh S, Huber TB, Wu H, Wang G, Hartleben B, Kopp JB, Miner JH, Roopenian DC, Unanue ER, Shaw AS. Podocytes use FcRn to clear IgG from the glomerular basement membrane. Proc Natl Acad Sci. 2008;105:967–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY, Hsu YJ, Ka SM, Tsai YL, Liu FC, Chen A. IL-36 signaling facilitates activation of the NLRP3 inflammasome and IL-23/IL-17 axis in renal inflammation and fibrosis. J Am Soc Nephrol. 2017;28:2022–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh AP, Krzyzanski W, Martin SW, Weber G, Betts A, Ahmad A, Abraham A, Zutshi A, Lin J, Singh P. Quantitative prediction of human pharmacokinetics for mAbs exhibiting target-mediated disposition. Aaps J. 2015;17:389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotechnol. 2009;20:722–29. [DOI] [PubMed] [Google Scholar]
- 54.Glassman PM, Balthasar JP. Physiologically-based modeling of monoclonal antibody pharmacokinetics in drug discovery and development. Drug Metab Pharmacokinet. 2019;34:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chizzonite R, Truitt T, Podlaski FJ, Wolitzky AG, Quinn PM, Nunes P, Stern AS, Gately MK. IL-12: monoclonal antibodies specific for the 40-kDa subunit block receptor binding and biologic activity on activated human lymphoblasts. J Immunol. 1991;147:1548. [PubMed] [Google Scholar]
- 56.Harrold JM (2019) Ubiquity: PKPD model development and deployment tools. R package version 1.0.0. https://ubiquity.tools.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.