

Abstract
In the present study, a practical method to prepare piperazinyl amides of 18β-glycyrrhetinic acid was developed. Two main procedures for the construction of important intermediate 8 are discussed. One procedure involves the amidation of 1-Boc-piperazine with 3-acetyl-18β-glycyrrhetinic acid, prepared by the reaction of 18β-glycyrrhetinic acid with acetic anhydride without any solvent at 130 °C. The other procedure to prepare compound 8 involves the amidation of 18β-glycyrrhetinic acid followed by the esterification with acetic anhydride. Finally, compound 8 underwent N-Boc deprotection to prepare product 4. To ascertain the scope of the reaction, another C-3 ester derivative 17 was tested under the optimized reaction conditions. Furthermore, the reasons for the appearance of byproducts were elucidated. Crystallographic data of a selected piperazinyl amide is reported.
Keywords: 18β-glycyrrhetinic acid, piperazinyl amides, synthesis
Introduction
Glycyrrhizin was the major bioactive component in Glycyrrhiza uralensis fisch root. 18β-glycyrrhetinic acid (1, Figure 1) was then obtained by hydrolysis of glycyrrhizin. 18β-glycyrrhetinic acid and its derivatives have been extensively investigated in medicinal chemistry for their various biological activities, including anti-inflammatory [1], antiulcer [2], antioxidative [3], antitumor [4], and antibacterial [5] activity and their proberties as hepatic protective agents [6].
Figure 1.

Chemical structure of 18β-glycyrrhetinic acid and known derivatives.
Over the past few decades, the emphasis was placed primarily on explaining structure–activity relationships of 18β-glycyrrhetinic acid derivatives to enhance their biological activity. The structure of 18β-glycyrrhetinic acid offered numerous possibilities to chemical modifications, thereby leading to the formation of novel derivatives [7–9]. There are three functional groups at C-3, C-11 and C-30 in the structure of 18β-glycyrrhetinic acid amenable to chemical modifications.
The modifications at the C3-OH group of 18β-glycyrrhetinic acid are identified to be relatively common and effective. The modification of the C3-OH group, altering the molecular polarity of 18β-glycyrrhetinic acid, may be an advantage in achieving better cytotoxicity or antiproliferative activity [8–10]. For instance, the hydroxy group can be converted into an oxime, acyloxyimino, alkoxyimino, alkoxy and 3-oxo group [9]. As a proteasome inhibitor, compound a suppresses the chymotrypsin-like activity of the proteasome in MT4 cells with an IC50 of 0.22 μM, nearly 100-fold more potent than 18β-glycyrrhetinic acid.
Besides, the C-30 carboxyl group is often esterified or amidated in order to enhance the antitumor or other efficacy of 18β-glycyrrhetinic acid derivatives [11]. A novel piperazinyl amide b exhibits the optimal inhibitory activity against MCF-7 and can be further developed as a potent VEGFR2 and antitumor agent [12].
Piperazinyl amide fragments have the ability to form several hydrogen bonds, modulate the acid–base equilibrium constant and change the octanol–water partition coefficient [13]. They are considered as the basic motif for designing many biologically active molecules [14–15]. Some piperazinyl amides of 18β-glycyrrhetinic acid (c) are synthesized using various methods. A method involves the C30-position of the acyl chloride with symmetric piperazine [16–17]. In this case, the acyl chloride can be prepared without purification, and the total yield over two steps can reach 81% [16]. Such structures (c) can also be formed by treating the C30 carboxyl group with piperazine in the presence of activators (e.g., 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (EDCl), 1-hydroxybenzotriazole (HOBt) and trimethylamine [18]), the yield of piperazinyl amide derivatives was 80.6% [19]. These two methods are complicated through the side formation of bisamide, obviously, due to the competitive attack on the N atoms of the symmetric diamine. An alternative method [12] to prepare such piperazinyl amides (c) involves the amidation of 18β-glycyrrhetinic acid with 4-substituted phenylpiperazines in the presence of N,N-dicyclohexylcarbodiimide (DDC) and HOBt. Under such reaction conditions, the bisamide as byproduct can be evitable, whereas the substituted piperazine compounds will not be readily available.
In principle, an efficient approach to the synthesis of structural analogues of (c) is based on the reaction of carboxylic acid with N-Boc-protected aliphatic diamine in the presence of activators and then deprotection of the tert‐butoxycarbonyl (Boc) group using trifluoroacetic acid (TFA) [20]. Compared with the mentioned methods, the latter method achieves the higher yield (up to 90%) without the formation of byproducts [21].
Given the flexible 18β-glycyrrhetinic acid scaffold in the design of biologically active compounds, a novel and efficient method was reported to prepare C3 ester derivatives or/and C30 piperazinyl amides (c).
18β-Glycyrrhetinic acid belongs to the class of ursane-type pentacyclic triterpenoids, and the synthetic methods studied here can also apply to the modification of structurally similar other triterpenoic acids, but further experimental verification is needed.
Results and Discussion
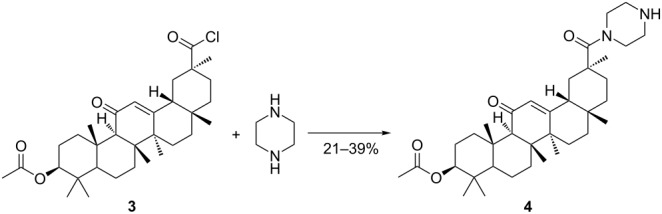
The synthetic route to 18β-glycyrrhetinic acid piperazinyl amide 4 was originally reported by Sommerwerk [16] (Scheme 1). In this case, 18β-glycyrrhetinic acid reacted with acetic anhydride in the presence of triethylamine to give 3-acetyl-18β-glycyrrhetinic acid (2), which by successive chlorination with oxalyl chloride yielded acyl chloride 3. Without isolation, the intermediate 3 reacted with piperazine to give 18β-glycyrrhetinic acid piperazinyl amide 4. The total yield of chlorination and amidation reactions was 67%.
Scheme 1.

Synthesis of compound 4. Reagents and conditions: (a) Ac2O, NEt3, DMF (cat.), DCM, 25 °C, 1 day; (b) oxalyl chloride, NEt3, DMF (cat.), DCM, 25 °C, 5 h, then piperazine, DCM, NEt3, DMAP, 0/25 °C, 30 min.
In the beginning, the reaction of acyl chloride 3 with piperazine was taken as a prototypical case to ascertain and screen the experimental conditions. First, the solution with intermediate 3 was cooled to 0 °C, and triethylamine (1.1 equiv) as well as piperazine (3.0 equiv) were added. After 30 min of stirring, the solvent was removed in vacuo and the residue was purified by column chromatography to afford amide 4 and bisamide 5. When increasing the amount of piperazine, relatively high yields of amide 4 would be achieved (Table 1, entries 1–3). Given the high reactivity of the acyl chloride with piperazine, we decided to verify the feasibility of the feeding sequence in a model system. The solution with intermediate 3 was added dropwise to a solution of piperazine in 30 mL of dichloromethane (DCM). After the solutions were mixed, intermediate 3 was surrounded by piperazine molecules. The excess of piperazine could sufficiently avoid the formation of bisamide 5 (Table 1, entry 4). When intermediate 3 was dissolved in more DCM (100 mL), less side reactions occurred and the product was obtained in higher yield (Table 1, entries 4 and 5). By altering the amidation reaction temperature, the yield of amide 4 showed an obvious variation. A reaction temperature led to excessive bisamide 5 (Table 1, entry 6). The results of the experiments demonstrated that the side reaction was inevitable, and the amount of bisamide 5 produced was dependent on the reaction temperature and material ratio. The isolation and purification of product 4 were significantly more difficult, causing an unavoidable loss of target compound as well.
Table 1.
Formation of piperazinyl amide 4: selected optimization reactions.
 | ||
| entry | conditionsa | yield of 4b |
| 1 | piperazine (3 equiv), 0 °C | 21% |
| 2 | piperazine (6 equiv), 0 °C | 28% |
| 3 | piperazine (10 equiv), 0 °C | 33% |
| 4 | compound 3 (in 30 mL DCM) was added dropwise to the solution of piperazine (10 equiv), 0 °C | 36% |
| 5 | compound 3 (in 100 mL DCM) was added dropwise to the solution of piperazine (10 equiv), 0 °C | 39% |
| 6 | compound 3 (in 100 mL DCM) was added dropwise to the solution of piperazine (10 equiv), 25 °C | 32% |
aReaction performed on a 0.90 mmol scale of acyl chloride 3. bIsolated yield.
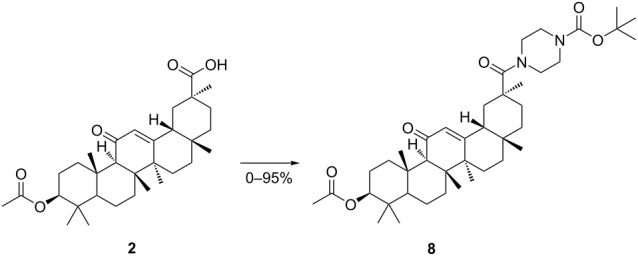
The observation described in Scheme 1 and Table 1 led us to consider a new approach for preparing the piperazinyl amide 4 (Scheme 2). Two general procedures for the synthesis of compound 8 are documented. One method involves acetylation of 18β-glycyrrhetinic acid, followed by amidation of the resulting ester 2 to give compound 8. Finally, the piperazinyl amide 4 was synthesized by of N-Boc deprotection.
Scheme 2.

Synthesis of compound 4. Reagents and conditions: (a) Ac2O, 130 °C, 1 h; (b) 1-Boc-piperazine, CH3CN, NEt3, EDCl, HOBt, reflux, 24 h; (c) TFA, DCM, 0/25 °C.
Another procedure for the synthesis of compound 8 involves acylation of compound 9, which was prepared by the reactions of 18β-glycyrrhetinic acid with 1-Boc-piperazine under similar reaction conditions. Compound 8 was prepared in virtually quantitative yield from compound 9 with acetic anhydride without solvent at 130 °C. The acetic acid formed in the reaction need not be neutralized and can escape from the reaction system. The reaction gave the optimal yield at 125–135 °C. When the temperature was below 125 °C, compound 9 did not reacted completely after 48 h (TLC monitoring).
The reaction from 2 to 8 proceeded via the intermediate 6, which can be isolated. The intermediate 6 did not react with 1-Boc-piperazine in low boiling point solvents even after an extended reaction time (up to 72 h). Heating the reaction using conventional or microwave methods had no effect on the conversion of intermediate 6 to compound 8.
To explain the stable structure of the intermediate 6 (Figure 2a), energy minimization by MM2 was performed using the ChemBio3D Ultra 14.0 (CambridgeSoft Corporation, 2014) software force field [22]. The estimated structure was supported by crystal structures of similar structural compounds [23]. Figure 2b suggests that the steric hindrance around the C30 ester group of intermediate 6 has a remarkable influence on the aminolysis reaction. The mechanism of the aminolysis reaction revealed that a higher reaction temperature of the solvent mixture contributed to the synthesis of compound 8.
Figure 2.

a) Estimated structure of the intermediate 6; b) Possible aminolysis process.
To promote the yield of compound 8 and improve the overall yield of the reaction, an optimization of the reaction conditions was performed (Table 2). First, by using low boiling point solvents, only low or no yields (<19%) of the desired compound 8 were obtained after a longer reaction time (Table 2, entries 1–4). A further investigation of the reaction using microwave irradiation to heat at reflux temperature also failed to produce the target product (Table 2, entry 2). The intermediate 6 exhibited very low solubility in ethyl acetate solution, thereby directly causing the incomplete conversion of the substrate over several days (Table 2, entry 5). The reaction performed in acetonitrile as solvent achieved an excellent yield within 10 h but still with a small amount of intermediate 6 (Table 2, entry 6). Besides, N,N-dimethylformamide (DMF), a solvent with a higher boiling point than acetonitrile, was also tested, yet the reaction led to the formation of unknown compounds (TLC monitoring, Table 2, entry 7).
Table 2.
Optimization of the reaction conditions for the coupling of compound 2 with 1-Boc-piperazine.
 | ||||
| entry | solvent | conditionsa | yield of 6b | yield of 8b |
| 1 | CH2Cl2 | reflux, 48 h | 93% | trace |
| 2 | CH2Cl2 | reflux, 48 h, microwave | 93% | trace |
| 3 | acetone | reflux, 48 h | 93% | trace |
| 4 | THF | reflux, 48 h | 81% | 19% |
| 5 | EtOAc | reflux, 48 h | 37% | 61% |
| 6 | acetonitrile | reflux, 10 h | 12% | 88% |
| 7 | DMF | reflux, 5 h, 130 oC | 0 | 0 |
| 8 | acetonitrile | reflux, 10 h, 1-Boc-piperazine (2.5 mmol) |
0 | 95% |
a2 (1.0 mmol), EDCl (1.2 mmol), NEt3 (1.2 mmol), HOBt (1.2 mmol) and 1-Boc-piperazine (1.2 mmol) were dissolved in 10 mL of solvent for the indicated time. bIsolated yield.
After screening several solvents, the optimal yield was obtained in acetonitrile, though a small amount of unconverted intermediate 6 was observed in the reaction mixture (Table 2, entry 6). Next, the reactivity of compound 2 with 1-Boc-piperazine in a 1:1.5 ratio was investigated. The reaction did not proceed completely in acetonitrile at reflux temperature, even after the period was prolonged to 48 h. In contrast, 1-Boc-piperazine (2.5 equiv) exhibited high performance, thereby leading to the preparation of compound 8 in 95% yield (Table 2, entry 8), which essentially completed conversion of the intermediate 6 within 10 h.
Alternatively, the aminolysis reaction of the isolated intermediate 6 with 1-Boc-piperazine (2.5 equiv) without any other additives can readily proceed in acetonitrile at reflux temperature in 95% yield.
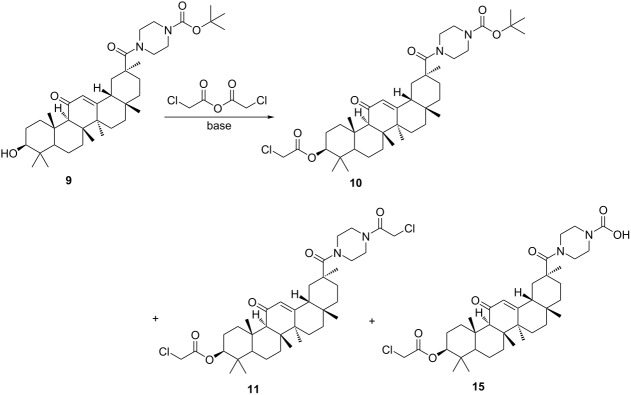
Having demonstrated the effectiveness of the new procedure, we decided to apply this method towards the synthesis of compound 10. But our investigations proved that the above experimental conditions seem not to be suitable for the synthesis of compound 10. Interestingly, treatment of compound 9 with chloroacetic anhydride by heating did not afforded compound 10, but obtained another compound, 11 (Scheme 3).
Scheme 3.

Synthesis of byproduct 11. Reagents and conditions: (a) chloroacetic anhydride, 130 °C, 1 h.
In our analysis, compound 9 reacted with acetic anhydride to give compound 8 and acetic acid. At the reaction temperature, the acetic acid (boiling point 117–118 °C) boiled and vapors escaped from the reaction mixture during the reaction process. However, in the reaction of compound 9 with chloroacetic anhydride (Scheme 3), the chloroacetic acid (boiling point 189 °C) formed during the reaction process cannot escape from the reaction system in the form of a gas, therefore, the initially formed compound 10 undergo N-Boc deprotection in an acidic medium to give byproduct 11. The structure of the byproduct 11 was confirmed by HRMS analysis.
Another noteworthy factor is that the acidity of chloroacetic acid (pKa = 2.87) was stronger than that of acetic acid (pKa = 4.76). The procedure for the generation of byproduct 11 was similar to N-Boc-deprotection using trifluoroacetic acid.
The effective synthesis of compound 10 was then explored (Table 3). In our initial attempt, compound 9 was reacted with chloroacetic anhydride (Table 3, entry 1) at 130 °C, whereas the reaction was complicated by the side formation of byproduct 11, which was obviously due to the presence of chloroacetic acid. Next, the reactions were performed with organic or inorganic bases to tie up or neutralize the chloroacetic acid released in the reaction. The base was provided in an amount or in a slight excess corresponding to the quantity required to tie up or neutralize the theoretical amount of acid released. The excess base did not affect the reaction and was washed off with water after the reaction. Triethylamine is a weak organic base that cannot completely neutralize the acid generated in the reaction. It may also volatilize at this reaction temperature (Table 3, entry 2). When the inorganic base was used as an acid-binding agent, the H2O, produced during the neutralization reaction, consumed considerable quantities of chloroacetic anhydride (Table 3, entries 3 and 4). Thus, when the amount of chloroacetic anhydride was elevated to 20 equiv, compound 9 were not completely converted within 48 h under reflux conditions. Next, compound 9 was reacted with chloroacetic anhydride in refluxing toluene in the presence of K2CO3 in a Dean–Stark apparatus to remove the water formed during the reaction (Table 3, entry 5). Under such conditions, the reaction was completed in 1 h to give product 15, though a small amount of byproduct 11 was observed in the reaction mixture. Examples of suitable inorganic bases include Na2CO3, NaHCO3, and especially K2CO3. The synthesis of compound 9 was described in a similar manner in [11].
Table 3.
Formation of amide 10: selected optimization reactions.
 | ||||
| entry | conditionsa | yield of 10b | yield of 11b | yield of 15b |
| 1 | chloroacetic anhydride (10 equiv) | 0 | 99 | 0 |
| 2 | chloroacetic anhydride (10 equiv), NEt3, acetonitrile | 0 | 90 | trace |
| 3 | chloroacetic anhydride (10 equiv), K2CO3, acetonitrile | 0 | 34 | trace |
| 4 | chloroacetic anhydride (20 equiv), K2CO3, acetonitrile | 0 | 36 | trace |
| 5 | chloroacetic anhydride (8 equiv), K2CO3, toluene, water separator | 0 | trace | 96 |
| 6 | chloroacetic anhydride (8 equiv), Na2CO3, toluene, water separator | 0 | trace | 92 |
| 7 | chloroacetic anhydride (8 equiv), NaHCO3, toluene, water separator | 0 | trace | 91 |
aReaction performed on a 10 mmol scale. bIsolated yield.
In another reaction, 18β-glycyrrhetinic acid was smoothly esterified with chloroacetic anhydride at the C3-OH group to form compound 12 in a quantitative yield. The reaction was completed in 30 min. The principal advantages of this method are that there is no solvent required, and that the reaction time is shorter than in previous cases [16], since the acid formed during the reaction may catalyze the esterification reaction of 18β-glycyrrhetinic acid with chloroacetic anhydride.
Having accessed compound 12, many other new compounds can also be prepared by replacing an amine with the halogen atom. With compounds 12 and 15 in hand, we then tried to synthesize target compound 17 (Scheme 4), which is a novel 18β-glycyrrhetinic acid derivative to biologically active substances.
Scheme 4.

Synthesis of compound 17. Reagents and conditions: (a) chloroacetic anhydride, 130 °C, 1 h; (b) morpholine, K2CO3/ I2, C2H5OH; (c)1-Boc-piperazine, CH3CN, NEt3, EDCl, HOBt, reflux, 24 h; (d) TFA, DCM, 0/25 °C; (e) chloroacetic anhydride, K2CO3, toluene, 130 °C, 1 h.
According to the first method, compound 12 reacted with morpholine in the presence of K2CO3/I2 to give compound 13, followed by successive amidation and N-Boc deprotection gave compound 17. It is noteworthy that the carboxyl group of compound 12 did not react with inorganic bases, such as K2CO3, Na2CO3, NaHCO3, etc.
Another approach for the synthesis of compound 17 is the amination of compound 15 to give compound 16. Subsequently, compound 17 was obtained by hydrolysis of compound 16 using TFA in DCM. The synthesis of compounds 15–17 was described in a similar manner in [11].
After the C3 position has been converted to a stable ester structure, the piperazinyl amides of 18β-glycyrrhetinic acid can be also converted to novel unsymmetrical amides. For example, treatment of compound 4 with acyl chloride in dry DCM under alkaline conditions directly afforded 18β-glycyrrhetinic acid derivative 18. The molecular structure of compound 18 in the crystalline state is shown in Figure 3. In this crystal structure, there is an orientational disorder of the m-fluorophenyl moiety due to the rotation of a single bond. Details for the crystal structure determinations are given in Table 4.
Figure 3.

Crystal structure of conpound 18.
Table 4.
Crystallographic data of compound 18.
| CCDC number | 1904891 |
| formula | C43H59FN2O5 |
| formula weight | 702.92 |
| crystal colour, shape | colourless |
| Solution | CH3OH |
| crystal system | monoclinic |
| space group | P212121 |
| a / Å | 11.581 (3) |
| b / Å | 12.216 (3) |
| c / Å | 27.749 (7) |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| V / Å3 | 3925.7 (17) |
| T / K | 273 |
| Z | 4 |
| density (calculated) / g·cm−3 | 1.189 |
| F(000) | 1520 |
| R (reflections) | 0.0439 (5028) |
| wR2 (reflections) | 0.1130 (7118) |
Conclusion
To sum up, piperazinyl amides of 18β-glycyrrhetinic acid were prepared from the 18β-glycyrrhetinic acid or its corresponding acyl chloride and piperazine. In this case, a considerable number of byproducts were inevitably produced. To reduce and/or avoid the formation of byproducts, efficient procedures were developed to prepare 18β-glycyrrhetinic acid derivatives that contain a terminal piperazinyl amide fragment. Due to the steric hindrance around the C30 ester group of intermediates, the intermediates 6 and 8 do not react with 1-Boc-piperazine at lower reaction temperature. The results showed the temperature dependence of the reaction between 18β-glycyrrhetinic acid and 1-Boc-piperazine. Besides, the Boc-piperazinyl amides of 18β-glycyrrhetinic acid reacted with chloroacetic anhydride in the presence of a base to give product 15. If no base is present, the Boc-piperazinyl amide of 18β-glycyrrhetinic acid 9 will undergo N-Boc deprotection and amidation to give compound 11. These results can be used to produce N-substituted piperazines of 18β-glycyrrhetinic acid in high yield, and further to develop more asymmetric piperazinamide derivatives. The structure of the asymmetric piperazinyl amide was confirmed by X-ray analysis.
Experimental
Detailed synthetic procedures for all new compounds including copies of their NMR spectra can be found in Supporting Information File 1.
Supporting Information
Preparation procedures and analytical data of compounds 1, 4–9, 11, 13, 15–18.
Funding Statement
This research was funded by the Natural Science Foundation of the Liaoning Province (No. 20170540396 and No. 2019-ZD-0693) and the Undergraduate innovation and entrepreneurship training program (No. 2019052).
Contributor Information
DianShen Yang, Email: 86384370@qq.com.
YiXia Gong, Email: gongyixia_2006@163.com.
References
- 1.Su L, Wang Z, Huang F, Lan R, Chen X, Han D, Zhang L, Zhang W, Hong J. Environ Toxicol Pharmacol. 2018;60:82–90. doi: 10.1016/j.etap.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Yano S, Harada M, Watanabe K, Nakamaru K, Hatakeyama Y, Shibata S, Takahashi K, Mori T, Hirabayashi K, Takeda M, et al. Chem Pharm Bull. 1989;37:2500–2504. doi: 10.1248/cpb.37.2500. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal M K, Iqbal M, Athar M. Redox Rep. 2005;10:151–157. doi: 10.1179/135100005x57346. [DOI] [PubMed] [Google Scholar]
- 4.Jin L, Huang R, Huang X, Zhang B, Ji M, Wang H. Bioorg Med Chem. 2018;26:1759–1775. doi: 10.1016/j.bmc.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 5.Kim H K, Park Y, Kim H N, Choi B H, Jeong H G, Lee D G, Hahm K-S. Biotechnol Lett. 2002;24(22):1899–1902. doi: 10.1023/a:1020900124997. [DOI] [Google Scholar]
- 6.Jeong H G, You H J, Park S J, Moon A R, Chung Y C, Kang S K, Chun H K. Pharmacol Res. 2002;46:221–227. doi: 10.1016/s1043-6618(02)00121-4. [DOI] [PubMed] [Google Scholar]
- 7.Tolstikov G A, Baltina L A, Shul'Ts E E, Pokrovskił A G. Bioorg Khim. 1997;23:691–709. [PubMed] [Google Scholar]
- 8.Lallemand B, Gelbcke M, Dubois J, Prévost M, Jabin I, Kiss R. Mini-Rev Med Chem. 2011;11:881–887. doi: 10.2174/138955711796575443. [DOI] [PubMed] [Google Scholar]
- 9.Xu B, Wu G-R, Zhang X-Y, Yan M-M, Zhao R, Xue N-N, Fang K, Wang H, Chen M, Guo W-B, et al. Molecules. 2017;22:924. doi: 10.3390/molecules22060924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma R, Guru S K, Jain S K, Pathania A S, Vishwakarma R A, Bhushan S, Bharate S B. Med Chem Commun. 2015;6:564–575. doi: 10.1039/c4md00344f. [DOI] [Google Scholar]
- 11.Dong C, YuQi S, DianShen Y, YiXia G, YunHong J, inventors. The piperazine amide derivatives and the preparation method and application thereof of 18 β-enoxolone. CN201811582206.4A. Chin. Patent. 2018 Dec 24;
- 12.Yan T-L, Bai L-F, Zhu H-L, Zhang W-M, Lv P-C. ChemMedChem. 2017;12(13):1087–1096. doi: 10.1002/cmdc.201700271. [DOI] [PubMed] [Google Scholar]
- 13.Szakács Z, Béni S, Varga Z, Örfi L, Kéri G, Noszál B. J Med Chem. 2005;48:249–255. doi: 10.1021/jm049546c. [DOI] [PubMed] [Google Scholar]
- 14.Gang-Yue L, Sheng-Gang Y, Shan J, Xu-Hong Q, Qing-Chun H, Rong Z. Chin J Org Chem. 2008;28:2001–2006. [Google Scholar]
- 15.Kowalski P, Kowalska T, Mokrosz M, Bojarski A, Charakchieva-Minol S. Molecules. 2001;6:784–795. doi: 10.3390/60900784. [DOI] [Google Scholar]
- 16.Sommerwerk S, Heller L, Kerzig C, Kramell A E, Csuk R. Eur J Med Chem. 2017;127:1–9. doi: 10.1016/j.ejmech.2016.12.040. [DOI] [PubMed] [Google Scholar]
- 17.Kurono M, Unno R, Kimura H, Oka M, Hasegawa K, Ikeda S, Kuboyama N, Ito T, Sawai K, Ito S, inventors. Glycyrrhetic acid derivatives and use thereof. US4871740. U.S. Patent. 1989 Oct 3;
- 18.Chen C, Li X, Li T, Zhou X. Acta Chim Sin (Chin Ed) 2012;70:852–858. doi: 10.6023/a1111051. [DOI] [Google Scholar]
- 19.Couxi C, Mengxue W, Xueqiang L, Tiancai L, Xuezheng Z. Chin J Org Chem. 2015;35:835–842. [Google Scholar]
- 20.Lallemand B, Chaix F, Bury M, Bruyère C, Ghostin J, Becker J-P, Delporte C, Gelbcke M, Mathieu V, Dubois J, et al. J Med Chem. 2011;54:6501–6513. doi: 10.1021/jm200285z. [DOI] [PubMed] [Google Scholar]
- 21.Asakawa C, Ogawa M, Fujinaga M, Kumata K, Xie L, Yamasaki T, Yui J, Fukumura T, Zhang M-R. Bioorg Med Chem Lett. 2012;22:3594–3597. doi: 10.1016/j.bmcl.2012.04.049. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda H, Ikuta N, Nakata D, Ishida Y, Terao K. Bull Chem Soc Jpn. 2015;88:1123–1127. doi: 10.1246/bcsj.20140335. [DOI] [Google Scholar]
- 23.Beseda I, Czollner L, Shah P S, Khunt R, Gaware R, Kosma P, Stanetty C, del Ruiz-Ruiz M C, Amer H, Mereiter K, et al. Bioorg Med Chem. 2010;18(1):433–454. doi: 10.1016/j.bmc.2009.10.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Preparation procedures and analytical data of compounds 1, 4–9, 11, 13, 15–18.