

Abstract
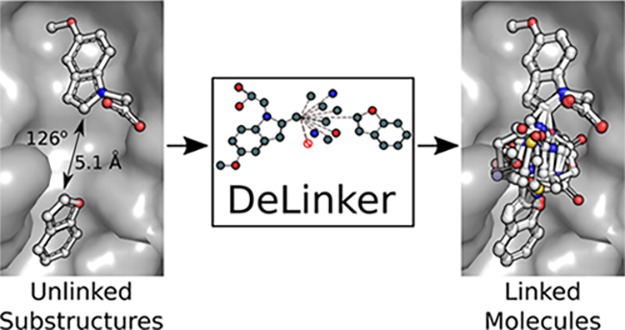
Rational compound design remains a challenging problem for both computational methods and medicinal chemists. Computational generative methods have begun to show promising results for the design problem. However, they have not yet used the power of three-dimensional (3D) structural information. We have developed a novel graph-based deep generative model that combines state-of-the-art machine learning techniques with structural knowledge. Our method (“DeLinker”) takes two fragments or partial structures and designs a molecule incorporating both. The generation process is protein-context-dependent, utilizing the relative distance and orientation between the partial structures. This 3D information is vital to successful compound design, and we demonstrate its impact on the generation process and the limitations of omitting such information. In a large-scale evaluation, DeLinker designed 60% more molecules with high 3D similarity to the original molecule than a database baseline. When considering the more relevant problem of longer linkers with at least five atoms, the outperformance increased to 200%. We demonstrate the effectiveness and applicability of this approach on a diverse range of design problems: fragment linking, scaffold hopping, and proteolysis targeting chimera (PROTAC) design. As far as we are aware, this is the first molecular generative model to incorporate 3D structural information directly in the design process. The code is available at https://github.com/oxpig/DeLinker.
Introduction
Drug design is an iterative process that requires potential compounds to be optimized for specific properties, ranging from binding affinity to pharmacokinetics. This process is challenging, in part, due to the size of the search space1 and discontinuous nature of the optimization landscape.2 Typically, molecule design is undertaken by human experts and therefore is a subjective process.
Machine learning models for molecule generation have been proposed as an alternative to human-led design and rule-based transformations.3−5 Generative models have adopted either the SMILES string representation of molecules6−10 or, more recently, graph representations.11−16 Existing generative models have primarily been used in two ways. First, methods have been developed to generate molecules that follow the same distribution as the training set, whether a general set of molecules10 such as ZINC17 or ChEMBL,18 or a more focused one such as inhibitors for a particular protein target.7,19 Second, generative models have been proposed to perform molecular optimization, taking an input molecule and attempting to modify one or several chemical properties, typically subject to a similarity constraint.16,20,21
While substantial progress has been made for these two problems, current methods have inherent limitations, in particular, for structure-based design. Only one approach to date has attempted to include any three-dimensional (3D) information in the generative process,22 despite its importance for designing potent and selective compounds. In this work, Skalic et al.22 proposed a SMILES-based model for generating molecules from 3D representations.22 A shape variational autoencoder using convolutional neural networks (CNNs) was coupled with a shape captioning network consisting of a separate CNN used to condition a recurrent neural network (RNN). In this formulation, 3D information was only provided implicitly to seed the RNN, and the method did not allow further control over generated compounds. As a result, their generative model frequently changed the entire molecule and recovered fewer than 2% of the seed molecules. This is undesirable in many practical settings, such as the design problems described below.
Fragment-based drug discovery (FBDD) has become an increasingly important tool for finding hit compounds, in particular, for challenging targets and novel protein families. FBDD utilizes smaller than drug-like compounds (typically <300 Da) to identify low potency, high-quality leads, which are then matured into more potent, drug-like compounds. One common way of maturing fragment hits is through a linking strategy, joining fragments together that bind to distinct sites via a linker. It is crucial for successful fragment linking that a linker does not disturb the original binding poses of each fragment.23,24 Thus, compound suggestions have strong 3D constraints, determined by the binding mode of the fragments.
Scaffold hopping, though a distinct problem, shares some characteristics with fragment linking. The aim of scaffold hopping is to discover structurally novel compounds starting from a known active compound by modifying the central core structure of the molecule.25 Such a change can result in much improved molecular properties, such as solubility, toxicity, synthetic accessibility, affinity, and selectivity.25,26
Numerous computational methods have been proposed for fragment linking or scaffold hopping.27−32 However, almost all methods published to date rely exclusively on a database of candidate fragments from which to select a linker, with the differences between approaches arising solely from how the database is searched, how the linked compounds are scored, or the contents of the database itself. As a result, these methods are inherently constrained to a set of predetermined rules or examples, limiting exploration of chemical space. In addition, they can only incorporate additional structural knowledge (e.g., the fragment’s binding mode) via filtering or search mechanisms.
Current machine-learning-based molecule generation methods have not been designed to effectively handle the structure-based design tasks of fragment linking and scaffold hopping. These scenarios require proposed molecules to contain specific substructures, with the goal to design a molecule that maintains the binding mode of the original compound or fragments.
Numerous challenges using SMILES-based methods have been previously noted, largely arising from using language to represent inherently graph-based objects.12,33 In particular, the grammar makes working with existing structures especially difficult and has even led to alternative language-based representations being proposed.34,35 We therefore do not believe that design tasks requiring proposed molecules to contain specific substructures naturally suit a SMILES-based representation, although this remains an active area of research.36
Graph-based methods have become more widely used, in particular, for molecular optimization.16,20,21 Due to the nature of these tasks, the shapes of the suggested molecules often differ greatly from the starting points, and many of the proposed changes are R-group modifications. In addition, most methods do not afford a high level of control over the generated molecule. Li et al.37 recently proposed a scaffold-based molecular generator that designs molecules retaining particular scaffolds as their core structures.37 This allows greater control over generated molecules than most existing methods. However, none of the graph-based methods to date incorporate 3D structural information, with only one SMILES-based approach, including any 3D information in the generative process.22
Finally, Ståhl et al.38 introduced a fragment-based reinforcement learning approach for multiparameter optimization.38 At each step, their model selects a fragment contained in the molecule and replaces it with a similar one. This typically leads to minor changes in the compounds and could be utilized for scaffold hopping. However, their model is not able to perform fragment linking and is not designed for tasks requiring 3D shape optimization.
In this work, we introduce the first graph-based deep generative method that incorporates 3D structural information directly into the design process. Our method takes as input two molecular fragments and designs a molecule incorporating both substructures, either generating or replacing the linker between them. This allows our method to handle structure-based design tasks such as fragment linking and scaffold hopping effectively. The generation process integrates 3D structural information, specifically the distance between the fragments and their relative orientations. This 3D information is vital to successful compound design, and we demonstrate the limitations of omitting such information, both quantifying its impact in large-scale assessments and empirically showing how our model uses the structural information.
We first demonstrate the effectiveness of our proposed deep generative approach over a database method through large-scale computational assessments. We show that our method, DeLinker, designs 60% more compounds with high 3D similarity to the original molecule compared to a database-based approach on an independent test set. DeLinker outperforms the database approach by 200% when the evaluation is restricted to linkers with at least five atoms. We then apply our method to several case studies encompassing fragment linking, scaffold hopping, and PROTAC design. DeLinker frequently recovers the experimental end point, even in cases where the linker was not present in the training set, and produces many novel designs with high 3D similarity to the original molecules.
Methods
The method takes two fragments and their relative position and orientation and generates or replaces the linker between them. This is achieved by building new molecules in an iterative manner “bond by bond” from a pool of atoms that can be initialized with partial structures (Figure 1). In this framework, the user is able to control the generation process by specifying both the substructures that should be linked and the maximum length of the linker between them. The starting substructures are always retained in the generated molecule, with changes only from the specified exit vectors. In addition, 3D structural information in the form of the distance and angle between the starting substructures is provided to the model to inform the design process. Molecules are encoded by a set of 14 permitted atom types, and our model enforces simple atomic valency rules via a masking procedure to ensure chemical validity. This is the only chemical knowledge incorporated directly into our model; all other decisions required to generate molecules are learnt through a supervised training procedure.
Figure 1.

Overview of the generation process. The initial fragments (a) are iteratively expanded bond by bond (c–e) to produce a molecule, including both fragments (f). Atoms are represented by nodes in a graph, with the color of the nodes representing different atom types, while bonds are represented by edges, with different edge types for single, double, and triple bonds.
Generative Process
The generative process is illustrated in Figure 1 and is similar to Liu et al.,14 in that our method builds molecules bond by bond in a breadth-first manner. Generation is initialized with two fragments or substructures that are to be linked together with structural information providing the distance and angle between the substructures. The fragments are converted to a graph representation, where atoms and bonds are represented by nodes and edges, respectively. Each node is associated with a hidden state, zv, and label, lv, representing the atom type of the node. A list of the 14 permitted atom types can be found in the Supporting Information. The graph is passed through an encoder network, a standard gated graph neural network (GGNN),39 and the hidden states of the nodes are updated to incorporate their local environment (Figure 1a).
Next, a set of expansion nodes
are initialized at random, with hidden states zv drawn from the h-dimensional
standard normal distribution, 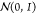 , where h is the
length
of the hidden state (Figure 1b). The nodes are then labeled with an atom type according
to their hidden state, zv, and the structural information by sampling from the softmax output
of a learned mapping f. Here, f is
implemented as a linear classifier but could be any function mapping
a node’s hidden state to an atom type. The number of expansion
nodes determines the maximum length of the linker and is a parameter
chosen by the user.
, where h is the
length
of the hidden state (Figure 1b). The nodes are then labeled with an atom type according
to their hidden state, zv, and the structural information by sampling from the softmax output
of a learned mapping f. Here, f is
implemented as a linear classifier but could be any function mapping
a node’s hidden state to an atom type. The number of expansion
nodes determines the maximum length of the linker and is a parameter
chosen by the user.
The new molecule is constructed from this set of nodes via an iterative process consisting of edge selection, edge labeling, and node update (Figure 1c–e). At each step, we consider whether to add an edge between one of the nodes, v, and another node in the graph. v is chosen according to a deterministic first-in-first-out queue that is initialized with the exit vectors of each fragment. When a node is connected to the graph for the first time, it is added to the queue. New edges are added to node v until an edge to the stop node is selected. The node then becomes “closed” with no additional edges with that node permitted.
All possible edges between the node v and other nodes in the graph are considered (Figure 1c), subject to basic valency constraints. A single-layer neural network assesses the candidate edges using a feature vector. The feature vector for the edge between node v and candidate node u is given by
where svt = [zv, lv] is the concatenation of the hidden state of node v after t steps and its atomic label, dv,u is the graph distance between v and u, H0 is the average initial representation of all nodes, Ht is the average representation of nodes at generation step t, and D represents the 3D structural information.
As such, when choosing which edge to add to the graph, the model utilizes (1) local information about the nodes, (2) global information regarding the unlinked fragments and the current graph state, and (3) 3D structural information.
Once a node u has been selected, the edge between v and u is labeled as either a single, double, or triple bond (subject to valency constraints) by another single-layer neural network taking as input the same feature vector ϕv,ut (Figure 1d).
Finally, the hidden states of all
nodes are updated according to
a GGNN (Figure 1e).
At each step, we discard the current hidden states  and compute new representations
and compute new representations  , taking their (possibly changed) neighborhood
into account. Note that
, taking their (possibly changed) neighborhood
into account. Note that 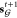 is computed from
is computed from 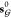 rather than
rather than 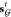 . This means that the state
of each node
is independent of the generation history of the graph and depends
only on the current state of the graph.
. This means that the state
of each node
is independent of the generation history of the graph and depends
only on the current state of the graph.
Steps c–e in Figure 1 are repeated for each node in the queue, until the queue is empty, at which point the generation process terminates. At termination (Figure 1f), all unconnected nodes are removed and the largest connected component is returned as the generated molecule. We note that the stereochemistry of generated molecules is not assigned during the generative process.
Multimodal Encoder–Decoder Setup
Our goal is to learn a multimodal mapping from unconnected fragments to connected molecules. During training, we utilized a data set of paired fragments and molecules and trained our model in a supervised manner to reconstruct known linkers. While in this data set there may be a unique molecule associated with two fragments, in practice, there are many ways to link two fragments. As such, given a new pair of starting points, a model should be able to generate a diverse set of output compounds.
To this end, we took inspiration from Jin et al.16 and augmented the basic encoder–decoder model with
a low-dimensional latent vector z to explicitly encode
the multimodal aspect of the distribution of suitable linkers. The
generative mapping is converted from F: X → Y to F: (X, z) → Y, where X represents the starting substructures and Y the
connected molecule, with latent code z drawn from a prior
distribution, chosen to be the standard normal distribution, 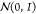 .
.
There are two challenges in learning this mapping. First, as shown in the image domain,40 the latent codes are often ignored by the model unless they are forced to encode meaningful variations. Second, the latent codes should be suitably regularized so that the model does not produce invalid outputs. That is, the generated molecule F(X, z) should belong to the domain of the target molecule Y (i.e., connected and able to satisfy the structural constraints provided) given a latent code drawn from the prior distribution. We overcame both of these challenges through our training procedure, where we derived z during training from the embedding of the linked molecule, but regularized the latent vector to follow a standard normal distribution so that we can sample z during generation.
Training
We trained our generative model under a variational autoencoder (VAE) framework on a collection of fragment–molecule pairs (Figure 2). For a given pair of fragments X and linked molecule Y, the model is trained to reconstruct Y from (X, z), while enforcing the standard regularization constraint on both z and the encoding of X, zX ≔ {zv: v ∈ X}.
Figure 2.

Illustration of training and generation procedures. (a) Pairs of fragments and linked molecules are provided as input. The model is trained to reproduce the linked molecule from a combination of the encodings of the fragments and linked molecule. (b) At generation time, the model is given only the unlinked fragments and structural information and is able to sample a diverse range of linked molecules by combining the encoding of the fragments with random noise.
To encode meaningful variations, the latent code z is derived via a learnt mapping from the average of the node embeddings of the ground truth molecule Y, the linked molecule. Crucially, z is constrained to be a low-dimensional vector to prevent the model from ignoring input X and degenerating to an autoencoder for Y. The decoder is trained to reconstruct Y when taking as input a combination of the low-dimensional vector z and the node embeddings zX of the unlinked fragments X (Figure 2).
The training objective is similar to the standard VAE loss, including a reconstruction loss and a Kullback–Leibler (KL) regularization term
The reconstruction loss, 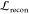 , is composed of two cross-entropy
loss
terms, resulting from the error in predicting the atom types and in
reconstructing the sequence of steps required to produce the target
molecule.
, is composed of two cross-entropy
loss
terms, resulting from the error in predicting the atom types and in
reconstructing the sequence of steps required to produce the target
molecule.
The KL regularization loss, 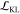 , contains
two terms, one for the encoding
of the unlinked fragments, X, and the other for the
low-dimensional vector z derived from the linked molecule Y. These terms are the standard VAE terms capturing the
KL divergence between the encoder distributions and the standard Gaussian
prior.
, contains
two terms, one for the encoding
of the unlinked fragments, X, and the other for the
low-dimensional vector z derived from the linked molecule Y. These terms are the standard VAE terms capturing the
KL divergence between the encoder distributions and the standard Gaussian
prior.
We performed limited hyperparameter tuning, measuring performance via the validation loss and not generative performance directly. We found that overall the model was fairly robust to the choice of hyperparameters. Full details of the model architecture and hyperparameters can be found in the Supporting Information.
Data Sets
There have only been a limited number of examples of successful fragment linking or scaffold hopping reported. As such, for training and large-scale evaluation, we constructed sets of fragment–molecule pairs using standard transformations from matched-molecular pair analysis.5
ZINC
To construct our training set, we used the subset of ZINC17 selected at random by Gómez-Bombarelli et al.10 that contains 250 000 molecules. We constructed possible fragmentations of each molecule by enumerating all double cuts of acyclic single bonds that were not within functional groups, the same procedure adopted by Hussain and Rea.5 Fragmentations satisfying basic criteria regarding the number of atoms in the linker and fragments were retained, removing trivial and unrealistic scenarios (see the Supporting Information for further details).
The remaining fragment–molecule pairs were filtered for several two-dimensional (2D) properties, namely, synthetic accessibility,41 ring aromaticity, and pan-assay interference compounds (PAINS),42 to remove unwanted examples. Full details of the property filters can be found in the Supporting Information.
By filtering the training set for specific 2D properties, we are also able to assess whether the model is able to learn to generate linkers with certain properties implicitly from the data alone. Since these properties are not input explicitly into the model, these could easily be tailored to a specific project or other requirements.
To provide structural information, we generated 3D conformers for the ZINC set using RDKit,43 adopting the filtering and sampling procedure proposed by Ebejer et al.44 We took the lowest-energy conformation as the reference 3D structure for each molecule.
These preprocessing and filtering steps resulted in a data set of 418 797 example fragment elaborations, with linkers of between 3 and 12 atoms. We selected 800 fragment–molecule pairs at random for model validation (400) and testing (400) and used the remainder to train our model, ensuring no overlap between the molecules in the training and held-out sets.
CASF
A major limitation of the ZINC data set is the use of generated conformers, as opposed to experimentally verified active ones. To address this, we used the CASF-2016 data set,45 also known as the PDBbind core set, which consists of 285 protein–ligand complexes from PDBbind with high-quality crystal structures from a diverse set of proteins, as an independent test set. We followed the same preprocessing procedure as for the ZINC data set (except for conformer generation), resulting in a set of 309 examples.
We performed large-scale evaluations of our method on both the held-out ZINC test data and the CASF data set. For each example, we generated 250 molecules from each pair of unlinked fragments, assuming that the linker length was equal to the linker length of the original molecule.
Assessment Metrics
We assessed the generated molecules with a range of 2D and 3D metrics. As is standard in the assessment of models for molecule generation,46 we first checked the generated molecules for validity, uniqueness, and novelty. We then determined if the generated linkers were consistent with the 2D property filters used to produce the training set. In addition, we recorded in how many cases the original molecule used to produce the fragments was recovered by the generation process.
Molecules that passed the 2D property filters were assessed on the basis of their 3D shape. Conformers of the generated molecules and the original molecule were compared using two distinct methods: (i) a shape and color similarity score (SCRDKit) and (ii) root-mean-square deviation (RMSD).
The shape and color similarity score (SCRDKit) uses two RDKit functions, based on the methods described in Putta et al.47 and Landrum et al.48 The color similarity function scores two 3D conformers against each other based on the overlap of their pharmacophoric features, while the shape similarity measure is a simple volumetric comparison between the two conformers. Each produces a score between 0 (no match) and 1 (perfect match), which are averaged to produce a final score between 0 and 1. Scores above 0.7 indicate a good match, while scores above 0.9 suggest an almost perfect match. An illustration of several conformers and their similarity scores can be seen in Figure 3.
Figure 3.
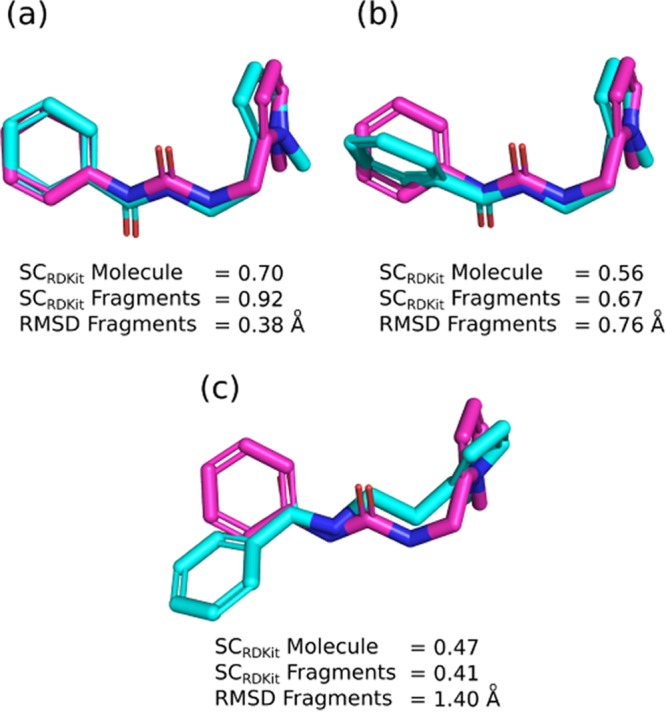
Examples of the 3D metrics used to assess the similarity of conformers. The reference conformer is shown in magenta, while conformers of the generated molecule are shown in cyan. (a) Represents very strong alignment by both fragment-based metrics but lower similarity by SCRDKit Molecule due to the different linker. (b) Shows modest similarity by all three metrics, while (c) represents poor similarity by all three measures.
SCRDKit can either be calculated by comparing only the atoms of the starting fragments (SCRDKit Fragments) or by comparing the entire generated molecule to the original molecule (SCRDKit Molecule). The first measure assesses how closely the conformations of the fragments match, whereas the second also incorporates whether the generated linker matches the original (Figure 3). Our method is trained to output a diverse range of linkers and not to map exactly to a previously observed linker. However, in the case of scaffold hopping, this metric is important as typically the new linker should match the shape and pharmacophoric features of the original core.26
RMSD between the coordinates of atoms in the starting fragments in the original and generated molecules can be calculated to give a different measure of 3D similarity (RMSD Fragments). A perfect match has an RMSD of 0 Å, with a higher figure indicating greater deviation. An RMSD of below 0.5 Å suggests an almost perfect match, while an RMSD of above 1.0 Å corresponds to a poor match given the alignment procedure and number of heavy atoms. An illustration of several conformations and their RMSDs can be seen in Figure 3. Due to the need to match specific atoms, RMSD can only be (reliably) calculated between the atoms of the fragments that are linked and not the entire molecule.
For each proposed molecule, we generated 3D conformers using RDKit,43 adopting the filtering and sampling procedure proposed by Ebejer et al.,44 and scored all conformers. The score for each similarity measure was the best score among all generated conformers for a particular molecule.
Comparison to Other Methods
Several traditional methods exist for linking fragments or replacing the core of a molecule.27−32 Almost all methods rely on a database from which to select linkers. As a baseline with which to compare our method, we created a set of all linkers from the training data and sampled from this set, joining the linker in one of the two possible orientations at random. This setup ensures that both methods are constructed using the same data and allows us to assess whether the generated molecules have better shape complementarity than using linkers from the database, while still obeying 2D chemical constraints.
Liu et al.14 proposed constrained graph variational autoencoders to generate molecular graphs. They sought to generate arbitrary molecules that conformed to the distribution observed in the training data. Similar to our method, they assumed a sequential ordering of graph extension steps, constructing molecules in a bond-by-bond manner. To compare their work to our method, it was necessary to modify their implementation to perform the molecular design tasks described.
Our work differs from the model of Liu et al.14 in three key ways. First, our model takes as input structural information in the form of the distance and angle between the starting substructures and uses this to augment the feature vector used in the generation process. Second, our model is designed to be trained for molecular translation, in particular, taking as input unlinked substructures and generating linked molecules. While others have developed methods for molecular translation,16,21 none have taken two unlinked substructures as input. Third, we have modified the training procedure from a standard encoder–decoder framework to explicitly encode the set of possible linkers. The training process combines encodings of both the unlinked substructures and the linked molecule, as opposed to solely the starting point (Figure 2). In addition, there are several minor architectural differences between DeLinker and their model (further details can be found in the Supporting Information). The impact of these changes is detailed in Tables S2 and S3.
Experimental Setup
In all of our experiments, we used the same training set derived from the ZINC data set to train DeLinker and construct the database for the Database baseline. In both of the large-scale evaluations on the held-out ZINC test data and the CASF data set, we generated 250 linkers for each pair of starting fragments to be assessed for both DeLinker and the Database. The number of atoms in the linker was set equal to the linker length of the original molecule for both DeLinker and the Database baseline in each of the large-scale evaluations. We also demonstrated the applicability of DeLinker using case studies from fragment linking, scaffold hopping, and PROTAC design. There are minor differences in the evaluation of our generative method for the case studies. These deviations are detailed in the Results and Discussion section with the appropriate case study.
Generated molecules were first assessed by the 2D metrics described above. Molecules that passed the 2D property filters were assessed on the basis of their 3D shape, as described above. We reported the proportion of molecules that pass the 2D metrics that meet 3D similarity thresholds.
Results and Discussion
We demonstrate DeLinker, a deep generative method that designs a molecule incorporating two starting substructures using 3D structural information. We first checked the impact of the structural information and then assessed our generative method in three experiments: (i) large-scale validation on ZINC (generated conformers), (ii) large-scale validation on CASF (experimentally determined active conformations), and (iii) three case studies covering fragment linking,49 scaffold hopping,50 and PROTAC design.51
Importance of Structural Information
To assess the importance of including structural information, we empirically examined its impact on the generation process (Table 1). We considered three almost identical fragment–molecule pairs based on ZINC7670105 from the held-out ZINC test set (see Methods). In all three cases, the starting substructures remained constant, but the substitution pattern of the benzene linker differed. This resulted in the distance and angle between the fragments changing but no other differences between the input data to our model. We generated 1000 linkers with a maximum of six atoms for each set of structural information and assessed the substitution pattern of generated molecules that contained a six-membered ring as the linker (Table 1).
Table 1. Impact of Structural Information on Generated Ring Substitution Patternsa.

The generated compounds closely followed the true substitution pattern, with only the structural information provided differing between examples.
DeLinker generated a high number of six-membered rings in all three cases (33–54%). Most rings were generated with the para structural information. This is consistent with the chemical knowledge since there are fewer possibilities given these structural constraints. The generated molecules closely followed the substitution pattern of the molecule used to calculate the structural information, with between 83 and 98% of the rings produced following the same pattern (Table 1). The effect of the structural information on the performance of DeLinker in a large-scale evaluation is discussed below and can be found in Tables S2 and S3.
Validation on ZINC
We next evaluated our method on the held-out test set from the ZINC data set, consisting of 400 pairs of fragments. We primarily compared DeLinker to a method based on database lookup (“Database”). The Database samples linkers from the same set of data used to train our method, joining the fragments in one of the two possible orientations at random. This setup ensures that both methods are constructed using the same data and allows a direct comparison to be made between database lookup and our deep-learning-based generative approach.
We generated 250 linkers for each pair of fragments, resulting in 100 000 generated molecules to be assessed for both DeLinker and the Database (see Methods for details). For the evaluation of the ZINC test set, the number of atoms in the linker was set equal to the linker length of the original molecule. This is an easier test for both methods than if the linker length was assumed to be unknown but allows us to assess whether the two methods presented are able to generate molecules that possess desired 2D chemical properties and high 3D structural similarity.
DeLinker generated a high proportion of valid molecules that passed the 2D chemical property filters (Table 2) and substantially outperformed the Database method by all 3D similarity measures (Table 3). DeLinker displayed a similar improvement over the graph-based molecular generative model of Liu et al.,14 which performed broadly comparably to the Database method (Tables S2 and S3). Further metrics and an ablation study showing the effects of including different structural information can be found in Tables S2 and S3. Without any structural information, the deep generative model performed similarly to the Database method by the 3D similarity measures (Table S3). Including only the distance between the fragments substantially improved performance, with further benefit from including the angle between fragments.
Table 2. 2D Performance Metrics for Molecules Generated by DeLinker, Our Deep Generative Model, Compared to a Database Baseline on the Held-Out ZINC Test Set and the Independent CASF Data Set.
| ZINC |
CASF |
CASF
≥5 atoms |
||||
|---|---|---|---|---|---|---|
| metric | Database (%) | DeLinker (%) | Database (%) | DeLinker (%) | Database (%) | DeLinker (%) |
| valid | 100.0 | 98.4 | 99.0 | 95.5 | 98.4 | 94.7 |
| unique | 38.8 | 44.2 | 43.0 | 51.9 | 58.3 | 72.9 |
| novel | 0.0 | 39.5 | 0.0 | 51.0 | 0.0 | 68.7 |
| recovered | 78.0 | 79.0 | 42.8 | 53.7 | 14.9 | 29.8 |
| pass 2D filters | 97.0 | 89.8 | 95.0 | 81.4 | 93.6 | 71.7 |
Table 3. 3D Performance Metrics for Molecules Generated by DeLinker, Our Deep Generative Model, Compared to a Database Baseline on the Held-Out ZINC Test Set and the Independent CASF Data Seta.
| ZINC |
CASF |
CASF
≥5 atoms |
||||
|---|---|---|---|---|---|---|
| metric | Database (%) | DeLinker (%) | Database (%) | DeLinker (%) | Database (%) | DeLinker (%) |
| SCRDKit Molecule | ||||||
| >0.7 | 33.5 | 47.1 | 14.9 | 22.3 | 7.8 | 16.3 |
| >0.8 | 8.5 | 14.2 | 3.3 | 5.2 | 1.1 | 3.6 |
| >0.9 | 1.3 | 1.8 | 0.5 | 0.8 | 0.3 | 0.8 |
| SCRDKit Fragments | ||||||
| >0.7 | 60.2 | 71.3 | 28.4 | 39.1 | 24.2 | 38.7 |
| >0.8 | 24.7 | 35.8 | 8.7 | 12.7 | 6.1 | 12.3 |
| >0.9 | 4.5 | 8.2 | 1.4 | 2.3 | 0.5 | 1.6 |
| RMSD Fragments | ||||||
| <1.00 Å | 46.9 | 58.6 | 19.4 | 28.1 | 14.6 | 26.6 |
| <0.75 Å | 20.5 | 30.0 | 7.1 | 10.2 | 4.3 | 9.3 |
| <0.50 Å | 5.7 | 9.3 | 2.0 | 3.1 | 0.9 | 2.4 |
See Methods, assessment metrics for a description of the metrics.
A molecule is deemed “valid” if it contains both starting fragments (i.e., the fragments have been linked), and its SMILES representation can be parsed by RDKit43 (i.e., satisfies atomic valency rules). The small proportion of invalid molecules produced by DeLinker (Table 2) was due to the fragments remaining unlinked, rather than failing atomic valency. This is a design choice by the deep learning system and is beneficial in reducing the number of unsuitable linkers suggested.
A fundamental benefit of our deep generative method over any database is evident in the proportion of novel linkers. The Database method is unable to suggest linkers not in the database, and thus 0% of the proposals were novel. In contrast, DeLinker proposed a linker not in the training set in around 40% of suggestions, despite the training set of linkers containing over 5000 unique linkers. Examples of novel linkers proposed by DeLinker are shown in Figure S1.
Both methods recovered over 75% of the original molecules (Table 2), demonstrating that they are able to sample from the distribution of linkers effectively. However, this is in part due to the chemical redundancy of molecules in ZINC. Indeed, all of the original linkers in the held-out ZINC test set were present in the training set.
DeLinker was able to learn the 2D filters implicitly, although it produced slightly fewer molecules passing these filters than the Database method (Table 2). Both methods had high success rates of 95% or above for all of the individual filters (Table S2).
For all of the 3D measures at all thresholds assessed, DeLinker produced a substantially higher proportion of linkers with the required 3D similarity than the Database (Table 3). In particular, at the highest levels of similarity, DeLinker generated over 80% more molecules, scoring >0.9 for SCRDKit Fragments and over 60% more molecules with an RMSD < 0.5Å.
Performance of both methods is impacted by the length of the generated linkers and in particular the number of short (three/four atoms) linkers in the test set (Table S1), where there are a limited number of possibilities. The degree of outperformance of DeLinker over the Database increased substantially when only considering linkers with at least five atoms (Table S4). In this setting, DeLinker generated around 190% more molecules, scoring >0.9 for SCRDKit Fragments and 130% more molecules with RMSD Fragments <0.5 Å or SCRDKit Molecule >0.9.
Validation on CASF
We saw similar performance when we evaluated the methods on the CASF data set (Tables 2 and 3). Both methods found producing 3D similar molecules more challenging for the CASF set than for the held-out ZINC set. Two possible explanations are the lower molecular similarity and use of experimentally determined structures in the evaluation. The average Tanimoto similarity of the Morgan fingerprints52 (radius 2, 1024 bits) of the 250 most similar starting fragments in the training set to each starting point in the test set was 0.36 for the held-out ZINC set but only 0.26 for the CASF set. However, our method was still frequently able to generate compounds with high similarity to the original molecule (Table 3).
In particular, DeLinker generated around 60% more molecules than the Database at the highest 3D similarity threshold (>0.9 SCRDKit Fragments and SCRDKit Molecule, <0.5 Å RMSD Fragments). When restricting the evaluation to linkers with at least five atoms, the degree of outperformance substantially increased, with DeLinker producing 200% more molecules that scored >0.9 by SCRDKit Fragments than the Database (Table 3).
DeLinker recovered 54% of the original linkers, compared to only 43% for the Database method, while around 50% of molecules generated by DeLinker were novel. The proportion recovered was lower than in the evaluation on ZINC; however, this set is more challenging with an average length of the true linker 5.9 atoms, compared to 4.9 for the held-out ZINC test set and 4.7 for the ZINC training set. In addition, only around 70% of the true linkers were present in the training set, providing an upper bound for the Database method. Similar to the ZINC set, DeLinker substantially outperformed the Database method for longer linkers; DeLinker recovered around 30% of molecules with a linker of at least five atoms, twice as many as the Database method that only recovered 15% (Table 2).
As previously noted, a fundamental limitation of a database method is an inability to generate linkers that are not present in the database. Despite being trained on the same database of linkers, DeLinker has learnt to extrapolate from this set to novel linkers. The following is an example of when this is crucial for successful compound design.
Dequalinium is a nanomolar binder (Ki: 70 nM) of chitinase A (PDB ID: 3ARP, Figure 4b).53 One possible fragmentation of the dequalinium–chitinase complex is shown in Figure 4a. To recover dequalinium from these fragments requires joining them with a decane linker, which is not present in the training set of linkers and thus the Database is unable to recover the original molecule. We generated 250 molecules with DeLinker, which included several highly similar novel linkers. The five most similar measured by SCRDKit Fragments are shown in Figure 4c. While DeLinker did not recover the decane linker within 250 generated compounds, simple chain linkers that closely resemble the true decane linker are prevalent. We compared this to an exhaustive search of linkers in the Database of the same length as the true decane linker (790 unique molecules). None of the Database-generated molecules are highly similar to dequalinium (Figure 4d), with only one molecule with SCRDKit Fragments >0.7. In contrast, DeLinker generated 34 unique molecules with SCRDKit Fragments >0.7. This illustrates the importance of de novo design and the limitations of any database-based solution.
Figure 4.

Comparison of DeLinker with an exhaustive Database search. A fragmention of dequalinium (PDB ID: 3ARP, (b)) is shown in (a). The most 3D similar molecules by SCRDKit Fragments proposed by DeLinker and the Database method are shown in (c) and (d), respectively, together with the 3D similarity score. (c) DeLinker was able to produce several very similar molecules, despite limited sampling (250 samples). (d) Exhaustive search of the database was not able to recover the original molecule or produce any highly similar molecules.
Finally, we showed the applicability of our method in three diverse examples from the literature, covering fragment linking,49 scaffold hopping,50 and PROTAC design.51 Due to the availability of independent experimental structural data for both the initial and optimized complexes, this represents the most realistic evaluation, albeit with limited sample size.
Fragment Linking Case Study
Trapero et al.49 considered both growing and linking strategies to create potent inhibitors of inosine 5-monophosphate dehydrogenase (IMPDH, UniProt: G7CNL4), a tuberculosis drug target. Linking proved most successful, with the authors identifying several promising compounds, the most potent with more than 1000-fold improvement in affinity over the initial fragment hits. Three direct elaborations of the initial fragments were reported (compounds 29–31 in Table 4 of Trapero et al.49), with structures of both the initial fragments (PDB ID: 5OU2) and most potent linked compound (PDB ID: 5OU3) available (Figure 5a).
Figure 5.

Fragment linking case study. (a) Left: the initial fragment hits (5OU2). Right: the most potent experimentally verified linked molecule of Trapero et al.49 (PDB ID: 5OU3). DeLinker recovered this and two other experimentally verified active molecules. (b) 3D similarity metrics and AutoDock Vina Minimized Affinities. Unique ligands with SCRDKit Fragments >0.8 were docked with AutoDock Vina using a local minimization. DeLinker produced more than twice as many molecules than the Database method with better Vina scores than the most potent reported binder (Lead).
In previous experiments, we chose the linker length based on the number of atoms in the linker of the original molecule. To reflect prospective use more accurately, we assumed that the linker length was unknown and generated 1000 linkers for each length between 3 and 11 atoms, inclusively. We assessed the generated linkers using the same criteria as before.
Both DeLinker and the Database method recovered all three experimentally validated compounds. However, DeLinker identified more than twice as many unique compounds as the Database with high 3D similarity (>0.8 SCRDKit Fragments) to the initial fragments (301 vs 129, Figure 5b and Table S5). The compounds meeting the above 3D similarity threshold were docked with AutoDock Vina54,55 via a local minimization after alignment with the starting fragments. This allows us to understand whether the proposed molecules are complementary to the active site and are able to maintain the binding mode of the original fragments. Around 40% of molecules generated by both DeLinker and the Database were scored better than the most potent experimentally validated compound. As a result, DeLinker suggested more than twice as many unique compounds as the Database with better docking scores than the active compound (123 vs 51, Figure 5b).
Scaffold Hopping Case Study
Kamenecka et al.50 designed JNK3-selective (UniProt: P53779) inhibitors that had >1000-fold selectivity over p38 (UniProt: Q16539), another closely related mitogen-activated protein kinase family member. Starting with an indazole class of compounds, they were not able to establish significant selectivity for JNK3 over p38. However, changing scaffolds led to an aminopyrazole linker that afforded compounds with >2800-fold selectivity. The two inhibitors displayed nearly identical binding mode (RMSD 0.33 Å, Figure 6a) and affinity for JNK3 (indazole: IC50 12 nM, aminopyrazole: IC50 25 nM) but significantly different binding affinity to p38 (indazole: IC50 3.2 nM, aminopyrazole IC50 3.6 μM).
Figure 6.

Scaffold hopping case study. (a) Overlay of the indazole (PDB ID 3FI3, magenta carbons) and aminopyrazole (PDB ID 3FI2, cyan carbons) structures, with JNK3 shown in green. DeLinker recovered both active molecules, despite neither linker being in the training set. (b) Structures of the indazole (left) and aminopyrazole (right) linkers and their Murcko scaffolds. (c) Overlay of the indazole compound (PDB ID 3FI3, magenta carbons) and example linkers (yellow carbons) from several highly 3D similar scaffolds.
Starting with the indazole-based inhibitor (PDB ID: 3FI3), we explored the ability of our method to change molecular scaffold, in particular, toward the aminopyrazole-based inhibitor (PDB ID: 3FI2). We generated 5000 linkers with both eight and nine atoms and assessed the generated linkers using the same criteria as before. In particular, we focused on the diversity of molecular scaffolds proposed by DeLinker that satisfied the 3D structural information and could adopt a highly similar conformation to the original indazole-based inhibitor.
Of the 10 000 compounds generated by DeLinker, there were 2688 unique compounds that satisfied the 2D chemical filters (Table S6). Six hundred and ninety-nine of these had an SCRDKit fragment score of above 0.75, of which 627 were not in the training set (89.7% novel). These compounds covered 182 unique generic Murko scaffolds.56 Five of the most common are shown in Figure 6b, together with an example linker and the number of unique linkers generated with the same generic Murko scaffold that also met the 3D similarity threshold. The examples from all five scaffolds show an almost perfect overlap with the indazole linker while maintaining the conformation of the remainder of the molecule.
In addition, DeLinker recovered both the indazole- and aminopyrazole-based linkers, despite neither being present in the training set.
PROTAC Case Study
Farnaby et al.51 developed PROTAC degraders of the BAF ATPase subunits SMARCA2 (UniProt: P51531) and SMARCA4 (UniProt: P51532) using a bromodomain ligand and recruitment of the E3 ubiquitin ligase VHL (UniProt: P40337). They first designed a PROTAC by combining known binders of SMARCA2/4 and E3 ubiquitin ligase VHL using poly(ethylene glycol)-based linkers (PDB ID: 6HAY, Figure 7a). The linker was then optimized to improve interactions with the lipophilic face created in part by Y98 of the VHL protein. In particular, they designed the linker to mimic the conformation observed in the ternary complex structure, resulting in improved molecular recognition (PDB ID: 6HAX). This was confirmed by the two crystal structures displaying near-identical ternary complexes (Figure 7b).
Figure 7.

PROTAC design case study. (a) Two-dimensional chemical structures of PROTAC 1 and PROTAC 2. (b) Overlays of ternary crystal structures of PROTAC 1 (PDB ID 6HAY, magenta carbons) and PROTAC 2 (PDB ID 6HAX, yellow carbons), with SMARCA2 shown in orange and VHL in blue. (c) Overlays of three linkers with different scaffolds produced by DeLinker (green carbons); all three accurately recapitulate the linker geometry observed in PROTAC 2 (yellow carbons). None of these linkers were present in the training set.
We investigated the ability of our model to design alternative linkers to the known poly(ethylene glycol)-based linker (PDB ID: 6HAY) that could maintain the same conformation observed in the ternary complex. We generated 5000 linkers with a maximum of either 9 or 10 atoms. There were almost 3000 unique linkers that passed the 2D chemical filters (Table S7).
Due to the size and complexity of the PROTAC, we generated conformers constraining the two starting substructures (Figure 7a) to adopt poses close to their known binding conformation, removing any high-energy poses. DeLinker produced 2150 unique compounds with SCRDKit Fragments >0.85, of which three novel linkers that accurately recapitulate the linker geometry observed in PROTAC 2 are shown in Figure 7c. In all three cases, the aromatic systems perfectly align with that of PROTAC 2 and are likely to fulfill the goal of improving interactions with the lipophilic face compared to PROTAC 1. In particular, the pyrrole-based linker (Figure 7c, left) appears to be making an NH−π interaction with the Y98 residue, possibly improving the CH−π interaction being made by the benzene in PROTAC 2. When the structures were minimized using AutoDock Vina,54,55 the compound with the pyrrole-based linker scored comparably to PROTAC 2 (−14.17 vs −14.32), with both scoring substantially better than PROTAC 1 (−12.66). However, we note that AutoDock Vina does not model electrostatics explicitly and thus does not capture NH−π or CH−π interactions. The other two compounds shown (Figure 7c, center, right) scored better than PROTAC 2 (−14.81 and −14.33 respectively), as did almost 20% of all of the minimized compounds (536).
For each of the three case studies, the top 20 molecules generated by DeLinker that met the 3D similarity threshold (SCRDKit Fragments >0.80) ranked by AutoDock Vina54,55 score are shown in Figures S2–S4.
Conclusions
We have developed a graph-based deep generative method for fragment linking or scaffold hopping that integrates 3D structural information, utilizing the relative distance and orientation between the starting substructures in the design process. Unlike previous attempts at computational fragment linking or scaffold hopping, our method does not rely on a database of fragments from which to select a linker but instead designs one given the fragments provided and 3D information.
Through two large-scale assessments, we have demonstrated that our generative method is able to learn to produce a distribution of linkers that matches the constraints present in the training set, while being able to generalize to novel linkers that satisfy both 2D and 3D constraints. In addition, the generated molecules consistently have high 3D similarity to both the initial fragments and the original molecules, outperforming a Database baseline by 60% in the evaluation on CASF, increasing to 200% when restricting the evaluation to linkers with at least five atoms.
Finally, through three case studies, we have shown that our method can be applied to fragment linking, scaffold hopping, and PROTAC design. In the fragment linking example, our method reproduced all of the reported potent molecules using only the crystal data of the initial fragment hits. In addition, in docking-based evaluation, many of the generated molecules were scored more highly than the original hits while maintaining similar binding modes. In the scaffold hopping case study, our method reproduced both the starting and final molecules, while suggesting many other scaffolds with high 3D similarity to the initial crystal data. Finally, in the PROTAC design case study, our method suggested a range of novel linkers that met the design goal of maintaining the linker geometry of PROTAC 1, while improving interactions with the lipophilic face created in part by residue Y98 of the VHL protein.
Our generative model can be readily combined with previous methods for fragment linking or scaffold hopping.27−32 This can be achieved by using the compounds generated by DeLinker to augment the database that is used as input for the search and filtering methods adopted by existing database-based methods.
As is frequently the case with machine-learning-based generative models, some of the molecules generated by DeLinker might have unstable functional groups or be hard to synthesize. We have demonstrated that our method was able to learn to produce molecules that frequently passed the 2D filters used to construct the training set through implicit supervision alone, thus, altering the training set composition would likely reduce the number of undesirable molecules generated. The properties of generated molecules could also potentially be improved through direct supervision with reinforcement learning57 or filtered via a postprocessing approach.58
As far as we are aware, this is the first molecular generative model to incorporate 3D structural information directly in the design process. Currently, the only 3D information utilized by the model is the distance between the fragments or starting substructures and their relative orientations. This provides explicit constraints for a given compound but only implicit information about the shape of the binding site. Despite this minimal parametrization, there is a substantial impact on the generated molecules.
Extending our method to use additional structural information that incorporates further constraints from the protein is a direction for future research. This could be achieved directly by providing our method with a richer representation of the protein–ligand binding site or indirectly through the combination of reinforcement learning8,59,60 and existing methods from structure-based drug discovery.54,55,61,62 Both directions promise substantial benefits for structure-based molecular generative methods.
While we have shown that our method can be applied to fragment linking, scaffold hopping, and PROTAC design, we believe that our framework is general and readily extendable to other design tasks. Applications of DeLinker to other design tasks such as fragment growing, R-group optimization, and macrocycle design would be interesting avenues for further studies.
The code is available at https://github.com/oxpig/DeLinker.
Acknowledgments
The authors thank Veerabahu Shanmugasundaram for helpful discussions on PROTACs. F.I. is supported by the EPSRC (Reference: EP/N509711/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.9b01120.
Details on the implementation of DeLinker and data curation; distribution of number of atoms contained in the original linkers in the data sets utilized (Table S1); ablation study for DeLinker on the held-out ZINC test set (Tables S2 and S3); 2D and 3D metrics for molecules generated by DeLinker compared to a Database baseline on the held-out ZINC test set (Table S4); fragment linking case study (Table S5); scaffold hopping case study (Table S6); PROTAC design case study (Table S7); a random sample of 50 novel linkers generated by DeLinker during testing on the held-out ZINC data set (Figure S1); and 20 example molecules generated by DeLinker for each of the case studies (Figures S2–S4) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Polishchuk P. G.; Madzhidov T. I.; Varnek A. Estimation of the Size of Drug-Like Chemical Space Based on GDB-17 Data. J. Comput.-Aided Mol. Des. 2013, 27, 675–679. 10.1007/s10822-013-9672-4. [DOI] [PubMed] [Google Scholar]
- Stumpfe D.; Bajorath J. Exploring Activity Cliffs in Medicinal Chemistry. J. Med. Chem. 2012, 55, 2932–2942. 10.1021/jm201706b. [DOI] [PubMed] [Google Scholar]
- Venkatasubramanian V.; Chan K.; Caruthers J. Computer-Aided Molecular Design Using Genetic Algorithms. Comput. Chem. Eng. 1994, 18, 833–844. 10.1016/0098-1354(93)E0023-3. [DOI] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain J.; Rea C. Computationally Efficient Algorithm to Identify Matched Molecular Pairs (MMPs) in Large Data Sets. J. Chem. Inf. Model. 2010, 50, 339–348. 10.1021/ci900450m. [DOI] [PubMed] [Google Scholar]
- Guimaraes G. L.; Sanchez-Lengeling B.; Outeiral C.; Farias P. L. C.; Aspuru-Guzik A.. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models, arXiv preprint:1705.10843. arXiv.org e-Print archive. https://arxiv.org/abs/1705.10843 (submitted May 30, 2017).
- Segler M. H. S.; Kogej T.; Tyrchan C.; Waller M. P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M.; Isayev O.; Tropsha A. Deep Reinforcement Learning for De Novo Drug Design. Sci. Adv. 2018, 4, eaap7885 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arús-Pous J.; Blaschke T.; Ulander S.; Reymond J.-L.; Chen H.; Engkvist O. Exploring the GDB-13 Chemical Space Using Deep Generative Models. J. Cheminf. 2019, 11, 20 10.1186/s13321-019-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Vinyals O.; Dyer C.; Pascanu R.; Battaglia P.. Learning Deep Generative Models of Graphs, arXiv preprint:1803.03324. arXiv.org e-Print archive. https://arxiv.org/abs/1803.03324 (submitted Mar 8, 2018).
- Jin W.; Barzilay R.; Jaakkola T. S.. Junction Tree Variational Autoencoder for Molecular Graph Generation, International Conference on Machine Learning (ICML), 2018; pp 2323–2332.
- You J.; Ying R.; Ren X.; Hamilton W.; Leskovec J.. GraphRNN: Generating Realistic Graphs with Deep Auto-regressive Models, International Conference on Machine Learning (ICML), 2018; pp 5708–5717.
- Liu Q.; Allamanis M.; Brockschmidt M.; Gaunt A.. Constrained Graph Variational Autoencoders for Molecule Design, Advances in Neural Information Processing Systems 31 (NeurIPS), 2018; pp 7795–7804.
- Li Y.; Zhang L.; Liu Z. Multi-objective De Novo Drug Design with Conditional Graph Generative Model. J. Cheminf. 2018, 10, 33 10.1186/s13321-018-0287-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Yang K.; Barzilay R.; Jaakkola T.. Learning Multimodal Graph-to-Graph Translation for Molecule Optimization, International Conference on Learning Representations (ICLR), 2019.
- Sterling T.; Irwin J. J. ZINC 15 - Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Hersey A.; Nowotka M.; Bento A. P.; Chambers J.; Mendez D.; Mutowo P.; Atkinson F.; Bellis L. J.; Cibrián-Uhalte E.; Davies M.; Dedman N.; Karlsson A.; Magariños M. P.; Overington J. P.; Papadatos G.; Smit I.; Leach A. R. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Müller A. T.; Huisman B. J. H.; Fuchs J. A.; Schneider P.; Schneider G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inf. 2018, 37, 1700111 10.1002/minf.201700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Barzilay R.; Jaakkola T.. Hierarchical Graph-to-Graph Translation for Molecules, arXiv preprint:1907.11223. arXiv.org e-Print archive. https://arxiv.org/abs/1907.11223 (submitted June 11, 2019).
- Zhou Z.; Kearnes S.; Li L.; Zare R. N.; Riley P. Optimization of Molecules via Deep Reinforcement Learning. Sci. Rep. 2019, 9, 10752 10.1038/s41598-019-47148-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalic M.; Jiménez J.; Sabbadin D.; De Fabritiis G. Shape-Based Generative Modeling for De Novo Drug Design. J. Chem. Inf. Model. 2019, 59, 1205–1214. 10.1021/acs.jcim.8b00706. [DOI] [PubMed] [Google Scholar]
- Ichihara O.; Barker J.; Law R. J.; Whittaker M. Compound Design by Fragment-Linking. Mol. Inf. 2011, 30, 298–306. 10.1002/minf.201000174. [DOI] [PubMed] [Google Scholar]
- Bienstock R. J. In Fragment-Based Methods in Drug Discovery; Klon A. E., Ed.; Springer: New York, 2015; pp 119–135. [Google Scholar]
- Böhm H.-J.; Flohr A.; Stahl M. Scaffold Hopping. Drug Discovery Today: Technol. 2004, 1, 217–224. 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Langdon S. R.; Ertl P.; Brown N. Bioisosteric Replacement and Scaffold Hopping in Lead Generation and Optimization. Mol. Inf. 2010, 29, 366–385. 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- Böhm H.-J. The Computer Program LUDI: A New Method for the De Novo Design of Enzyme Inhibitors. J. Comput.-Aided Mol. Des. 1992, 6, 61–78. 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- Böhm H.-J. LUDI: Rule-Based Automatic Design of New Substituents for Enzyme Inhibitor Leads. J. Comput.-Aided Mol. Des. 1992, 6, 593–606. 10.1007/BF00126217. [DOI] [PubMed] [Google Scholar]
- Maass P.; Schulz-Gasch T.; Stahl M.; Rarey M. Recore: AFast and Versatile Method for Scaffold Hopping Based on Small Molecule Crystal Structure Conformations. J. Chem. Inf. Model. 2007, 47, 390–399. 10.1021/ci060094h. [DOI] [PubMed] [Google Scholar]
- Thompson D. C.; Aldrin Denny R.; Nilakantan R.; Humblet C.; Joseph-McCarthy D.; Feyfant E. CONFIRM: Connecting Fragments Found in Receptor Molecules. J. Comput.-Aided Mol. Des. 2008, 22, 761 10.1007/s10822-008-9221-8. [DOI] [PubMed] [Google Scholar]
- Dey F.; Caflisch A. Fragment-Based De Novo Ligand Design by Multiobjective Evolutionary Optimization. J. Chem. Inf. Model. 2008, 48, 679–690. 10.1021/ci700424b. [DOI] [PubMed] [Google Scholar]
- Vainio M. J.; Kogej T.; Raubacher F.; Sadowski J. Scaffold Hopping by Fragment Replacement. J. Chem. Inf. Model. 2013, 53, 1825–1835. 10.1021/ci4001019. [DOI] [PubMed] [Google Scholar]
- Elton D. C.; Boukouvalas Z.; Fuge M. D.; Chung P. W. Deep Learning for Molecular Design-A Review of the State of the Art. Mol. Syst. Des. Eng. 2019, 4, 828–849. 10.1039/C9ME00039A. [DOI] [Google Scholar]
- O’Boyle N.; Dalke A.. DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures, 2018.
- Krenn M.; Häse F.; Nigam A.; Friederich P.; Aspuru-Guzik A.. SELFIES: A Robust Representation of Semantically Constrained Graphs with an Example Application in Chemistry, abs/1905.13741. arXiv.org e-Print archive. https://arxiv.org/abs/1905.13741 (submitted on May 31, 2019).
- Arús-Pous J.; Patronov A.; Bjerrum E. J.; Tyrchan C.; Reymond J.-L.; Chen H.; Engkvist O.. SMILES-Based Deep Generative Scaffold Decorator for De-Novo Drug Design, ChemRxiv preprint:chemrxiv.11638383.v1. arXiv.org e-Print archive. https://doi.org/10.26434/chemrxiv.11638383.v1 (submitted Jan 17, 2020). [DOI] [PMC free article] [PubMed]
- Li Y.; Hu J.; Wang Y.; Zhou J.; Zhang L.; Liu Z. DeepScaffold: A Comprehensive Tool for Scaffold-Based De Novo Drug Discovery Using Deep Learning. J. Chem. Inf. Model. 2020, 60, 77–91. 10.1021/acs.jcim.9b00727. [DOI] [PubMed] [Google Scholar]
- Ståhl N.; Falkman G.; Karlsson A.; Mathiason G.; Boström J. Deep Reinforcement Learning for Multiparameter Optimization in De novo Drug Design. J. Chem. Inf. Model. 2019, 59, 3166–3176. 10.1021/acs.jcim.9b00325. [DOI] [PubMed] [Google Scholar]
- Li Y.; Tarlow D.; Brockschmidt M.; Zemel R.. Gated Graph Sequence Neural Networks, International Conference on Learning Representations (ICLR), 2016.
- Zhu J.-Y.; Zhang R.; Pathak D.; Darrell T.; Efros A. A.; Wang O.; Shechtman E., Toward Multimodal Image-to-Image Translation, Advances in Neural Information Processing Systems 30 (NeurIPS), 2017; pp 465–476.
- Ertl P.; Schuffenhauer A. Estimation of Synthetic Accessibility Score of Drug-Like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminf. 2009, 1, 8 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Landrum G.RDKit: Open-Source Cheminformatics. http://www.rdkit.org/ (accessed November 4, 2019).
- Ebejer J.-P.; Morris G. M.; Deane C. M. Freely Available Conformer Generation Methods: How Good Are They. J. Chem. Inf. Model. 2012, 52, 1146–1158. 10.1021/ci2004658. [DOI] [PubMed] [Google Scholar]
- Su M.; Yang Q.; Du Y.; Feng G.; Liu Z.; Li Y.; Wang R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. 10.1021/acs.jcim.8b00545. [DOI] [PubMed] [Google Scholar]
- Brown N.; Fiscato M.; Segler M. H. S.; Vaucher A. C. GuacaMol: Benchmarking Models for De Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. 10.1021/acs.jcim.8b00839. [DOI] [PubMed] [Google Scholar]
- Putta S.; Landrum G. A.; Penzotti J. E. Conformation Mining: An Algorithm for Finding Biologically Relevant Conformations. J. Med. Chem 2005, 48, 3313–3318. 10.1021/jm049066l. [DOI] [PubMed] [Google Scholar]
- Landrum G. A.; Penzotti J. E.; Putta S. Feature-map Vectors: A New Class of Informative Descriptors for Computational Drug Discovery. J. Comput.-Aided Mol. Des. 2007, 20, 751–762. 10.1007/s10822-006-9085-8. [DOI] [PubMed] [Google Scholar]
- Trapero A.; Pacitto A.; Singh V.; Sabbah M.; Coyne A. G.; Mizrahi V.; Blundell T. L.; Ascher D. B.; Abell C. Fragment-Based Approach to Targeting Inosine-5-monophosphate Dehydrogenase (IMPDH) from Mycobacterium tuberculosis. J. Med. Chem 2018, 61, 2806–2822. 10.1021/acs.jmedchem.7b01622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T.; Habel J.; Duckett D.; Chen W.; Ling Y. Y.; Frackowiak B.; Jiang R.; Shin Y.; Song X.; LoGrasso P. Structure-Activity Relationships and X-ray Structures Describing the Selectivity of Aminopyrazole Inhibitors for c-Jun N-terminal Kinase 3 (JNK3) over p38. J. Biol. Chem. 2009, 284, 12853–12861. 10.1074/jbc.M809430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnaby W.; Koegl M.; Roy M. J.; Whitworth C.; Diers E.; Trainor N.; Zollman D.; Steurer S.; Karolyi- Oezguer J.; Riedmueller C.; Gmaschitz T.; Wachter J.; Dank C.; Galant M.; Sharps B.; Rumpel K.; Traxler E.; Gerstberger T.; Schnitzer R.; Petermann O.; Greb P.; Weinstabl H.; Bader G.; Zoephel A.; Weiss- Puxbaum A.; Ehrenhöfer-Wölfer K.; Wöhrle S.; Boehmelt G.; Rinnenthal J.; Arnhof H.; Wiechens N.; Wu M.-Y.; Owen-Hughes T.; Ettmayer P.; Pearson M.; McConnell D. B.; Ciulli A. BAF Complex Vulnerabilities in Cancer Demonstrated via Structure-Based PROTAC Design. Nat. Chem. Biol. 2019, 15, 672–680. 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Pantoom S.; Vetter I. R.; Prinz H.; Suginta W. Potent Family-18 Chitinase Inhibitors: X-ray Structures, Affinities, and Binding Mechanisms. J. Biol. Chem. 2011, 286, 24312–24323. 10.1074/jbc.M110.183376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D. R.; Baumgartner M. P.; Camacho C. J. Lessons Learned in Empirical Scoring with smina From the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem 1996, 39, 2887–2893. 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lengeling B.; Outeiral C.; Guimaraes G. L.; Aspuru-Guzik A.. Optimizing Distributions over Molecular Space. An Objective-Reinforced Generative Adversarial Network for Inverse-Design Chemistry (ORGANIC), ChemRxiv preprint:chemrxiv.5309668.v3. arXiv.org e-Print archive. https://doi.org/10.26434/chemrxiv.5309668.v3 (submitted Jan 1, 2017).
- Sumita M.; Yang X.; Ishihara S.; Tamura R.; Tsuda K. Hunting for Organic Molecules with Artificial Intelligence: Molecules Optimized for Desired Excitation Energies. ACS Cent. Sci. 2018, 4, 1126–1133. 10.1021/acscentsci.8b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. J. Simple Statistical Gradient-Following Algorithms for Connectionist Reinforcement Learning. Mach. Learn. 1992, 8, 229–256. 10.1007/BF00992696. [DOI] [Google Scholar]
- Olivecrona M.; Blaschke T.; Engkvist O.; Chen H. Molecular De-Novo Design Through Deep Reinforcement Learning. J. Cheminf. 2017, 9, 48 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragoza M.; Hochuli J.; Idrobo E.; Sunseri J.; Koes D. R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrie F.; Bradley A. R.; van der Schaar M.; Deane C. M. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model. 2018, 58, 2319–2330. 10.1021/acs.jcim.8b00350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.