

We identified inhibitors targeting COUP-TFII for the treatment of diseases including prostate cancer.
Abstract
The orphan nuclear receptor COUP-TFII is expressed at a low level in adult tissues, but its expression is increased and shown to promote progression of multiple diseases, including prostate cancer, heart failure, and muscular dystrophy. Suppression of COUP-TFII slows disease progression, making it an intriguing therapeutic target. Here, we identified a potent and specific COUP-TFII inhibitor through high-throughput screening. The inhibitor specifically suppressed COUP-TFII activity to regulate its target genes. Mechanistically, the inhibitor directly bound to the COUP-TFII ligand-binding domain and disrupted COUP-TFII interaction with transcription regulators, including FOXA1, thus repressing COUP-TFII activity on target gene regulation. Through blocking COUP-TFII’s oncogenic activity in prostate cancer, the inhibitor efficiently exerted a potent antitumor effect in xenograft mouse models and patient-derived xenograft models. Our study identified a potent and specific COUP-TFII inhibitor that may be useful for the treatment of prostate cancer and possibly other diseases.
INTRODUCTION
Nuclear receptors are important pharmaceutical targets because they are key regulators of many diseases and have druggable ligand-binding sites. Approximately 13% of the U.S. Food and Drug Administration–approved drugs target nuclear receptors (1). Recently, chicken ovalbumin upstream promoter–transcription factor II (COUP-TFII; NR2F2), an orphan nuclear receptor, has been identified as an important molecular target for disease treatment. COUP-TFII protein is expressed in the mesenchymal compartment during embryonic development and regulates essential genes in organogenesis (2, 3) through binding to direct repeats of DNA elements or interacting with other transcription factors and cofactors (4). As development proceeds, the expression of COUP-TFII is greatly reduced after the completion of organogenesis in adulthood (3, 4). However, increasing evidence has shown that COUP-TFII expression is elevated under pathological conditions, resulting in the development of multiple diseases (5, 6). COUP-TFII has been shown to promote cancer growth and metastasis in several cancer types, including prostate (7–9), lung (10), and colon (11) cancer. In addition, COUP-TFII promotes angiogenesis to enhance cancer progression (12, 13). Increased COUP-TFII expression also plays important promoting roles in heart failure (14) and muscular dystrophy (15). Reduced dosage of COUP-TFII expression in prostate cancer, heart failure, and muscular dystrophy disease models slows down disease progression, suggesting that COUP-TFII can serve as an intriguing therapeutic target for treatment of these diseases. Thus, identification of small molecular inhibitors targeting COUP-TFII will greatly benefit these patients. Although some progress has been made toward identification of compounds that can modulate COUP-TFII activity (16, 17), a potent COUP-TFII inhibitor has yet to be identified.
Prostate cancer is the most common cancer and the second leading cause of cancer death in American men. Androgen deprivation therapy or androgen receptor (AR) antagonist, which targets androgen signaling, has been widely used for prostate cancer treatment (18). However, patients under this treatment strategy eventually develop castration-resistant prostate cancer (CRPC) and succumb to death (19, 20). Therefore, it is important to identify previously unidentified molecular therapeutic targets for the future treatment of prostate cancer. COUP-TFII has been found to be highly up-regulated in prostate cancer tissues and positively correlated with tumor progression and recurrence (7). Moreover, COUP-TFII plays important oncogenic roles in promoting prostate cancer development and metastasis through regulating transforming growth factor–β (TGFβ) signaling, forkhead box protein M1 (FOXM1)/Centromere Protein F (CENPF) pathway, and cancer metabolism (7–9). Suppression of COUP-TFII expression substantially hinders prostate cancer progression and inhibits metastasis, implicating its potential as an important therapeutic target for prostate cancer.
Here, through high-throughput screening and medicinal chemistry optimization, we have identified a potent COUP-TFII inhibitor that represses COUP-TFII activity on target gene expression in a COUP-TFII–dependent manner. We showed that the inhibitor directly bound to COUP-TFII protein and disturbed COUP-TFII binding to its associated transcription regulators, thus suppressing COUP-TFII in target gene regulation. Moreover, we showed that the COUP-TFII inhibitor successfully exerted a potent effect on reducing prostate cancer tumor growth using both xenograft tumor and patient-derived xenograft (PDX) models. Our study identifies a potent COUP-TFII inhibitor and provides a novel drug for treatment of diseases caused by increased activity of COUP-TFII.
RESULTS
Identification of COUP-TFII inhibitors through high-throughput screening
To identify a small molecular inhibitor that targets COUP-TFII, we performed a luminescence-based cell-based high-throughput screening assay. We evaluated candidate compounds on their ability to inhibit COUP-TFII activity in inducing its target promoter nerve growth factor–induced clone A (NGFIA)-based luciferase reporter expression in 293T cells (Fig. 1A). In the meantime, a counter screen was set up in 293T cells transfected with a Gal4-vp16 fusion protein expression vector and a upstream activating sequence (UAS)-Human herpesvirus 1 thymidylate kinase (TK) luciferase reporter vector. The Molecular Libraries Probe Production Centers Network (MLPCN) chemical library containing 370,276 compounds was screened (PubChem AID: 686940). The fully automated screening was complete after three batches of screening and validation, and we obtained several COUP-TFII inhibitor candidates that showed a potent and specific inhibition of COUP-TFII activity (Fig. 1B and fig. S1A). We found that two candidates shared a similar structure (Fig. 1C) and named them CIA1 and CIA2, which were classified as CIA (COUP-TFII inhibitor A) inhibitors. The purity and characterization of CIA1 and CIA2 were confirmed by liquid chromatography (LC), high-resolution mass spectrometry (MS), as well as 1H and 13C nuclear magnetic resonance assays (fig. S1, B to E). Both CIA1 and CIA2 substantially inhibited COUP-TFII–driven NGFIA reporter expression but showed limited effects on Gal4-vp16–driven UAS-TK reporter expression in accordance with high-throughput screening results (Fig. 1D and fig. S2A). The median inhibitory concentration (IC50) values of CIA1 and CIA2 in NGFIA reporter luciferase assay were 3.2 and 2.8 μM, respectively. Moreover, CIA1 or CIA2 repressed NGFIA reporter expression in a COUP-TFII–dependent manner, as COUP-TFII knockdown abrogated CIA1- or CIA2-induced inhibition (fig. S2, B and C). Furthermore, CIA1 or CIA2 reduced prostate cancer cell growth while barely showing an effect when COUP-TFII was silenced (Fig. 1E), supporting COUP-TFII as the target of CIA1 and CIA2. To confirm the dependency of CIA inhibitors on COUP-TFII, we tested endogenous COUP-TFII target genes in prostate cancer cells, including COUP-TFII–activated genes, FOXM1 and CDK1, and COUP-TFII–repressed gene, CDKN1A. CIA1 or CIA2 treatment substantially inhibited the expression of COUP-TFII–activated target genes but promoted expression of COUP-TFII–repressed genes. As expected, the expression of these genes was hardly affected by CIA1 or CIA2 when COUP-TFII was silenced (Fig. 1F), indicating that CIA1 and CIA2 target COUP-TFII. To evaluate further the specificity of inhibitors on COUP-TFII, we compared genome-wide expression profiling of COUP-TFII target genes using RNA sequencing analysis upon CIA1 treatment and upon silencing of COUP-TFII expression (fig. S2D). Notably, gene expression profiles in which COUP-TFII activity was inhibited by CIA1 highly resembled the signature of silencing of COUP-TFII, where the expression of COUP-TFII–repressed genes was enhanced and the expression of COUP-TFII–induced genes was suppressed (Fig. 1G and fig. S2E). Gene set enrichment analysis (GSEA) consistently showed that CIA1 treatment and silencing of COUP-TFII regulated similar pathways (fig. S2, F and G). Together, these results support the notion that CIA1 and CIA2 act as specific inhibitors of COUP-TFII.
Fig. 1. Identification of COUP-TFII small-molecule inhibitor through high-throughput screening.
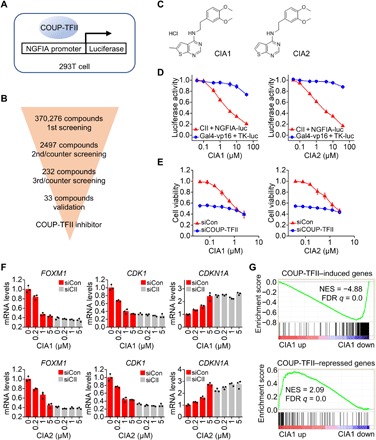
(A) Screening strategy. 293T cells were transfected with COUP-TFII and NGFIA-Luc (or Gal4-VP16 and TK-Luc as negative control). Luciferase activity was detected 18 hours after compound treatment. (B) Schematic of three rounds of screening. (C) Structure of COUP-TFII inhibitor CIA1 and CIA2. (D) Dose-dependent inhibition of NGFIA reporter expression by the inhibitor. 293T cells that transfected with vectors were treated with CIA1 or CIA2 for 18 hours. n = 3 per group. (E) LNCaP cells were transfected with siCOUP-TFII for 24 hours and then treated with CIA1 or CIA2 for 72 hours. Cell viability was measured. n = 3 per group. (F) CIA1 and CAI2 function in a COUP-TFII–dependent manner. LNCaP cells were transfected with siCOUP-TFII (siCII) or control small interfering RNA (siRNA) (siCon) for 48 hours and then treated with CIA1 or CIA2 for 18 hours. Target gene expression was measured by quantitative polymerase chain reaction (qPCR). n = 3 per group. (G) GSEA showed that CIA1 reduced COUP-TFII–induced genes and increased COUP-TFII–repressed genes. NES, normalized enrichment score; FDR, false discovery rate.
Direct interaction between the inhibitor and COUP-TFII protein
Next, we investigated whether CIA inhibitors directly interact with COUP-TFII protein. Through the cellular thermal shift assay (CETSA), we found that CIA1 treatment resulted in a thermal stabilization of COUP-TFII (Fig. 2A), suggesting that CIA inhibitors may bind to COUP-TFII protein. To determine the interaction between inhibitor and COUP-TFII protein, we performed pulldown assay using biotinylated inhibitor (fig. S3A). Our results showed that the biotinylated CIA inhibitor could pull down both overexpressed COUP-TFII protein in 293T cells and endogenous COUP-TFII protein in prostate cancer cells (Fig. 2, B and C). Moreover, free CIA1 could dose-dependently compete in the interaction between biotinylated inhibitor and COUP-TFII protein, leading to impaired pulldown (fig. S3B). In addition, other tested active CIA analogs all could work as a competitor (fig. S3C).
Fig. 2. Direct interaction between the inhibitor and COUP-TFII protein.
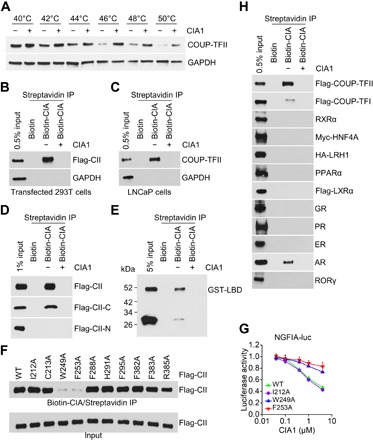
(A) CETSA was performed using LNCaP cells. COUP-TFII overexpressed 293T cells (B) or LNCaP cells (C) were used for biotinylated inhibitor pulldown assay. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IP, immunoprecipitation. Twenty micromolar CIA1 was used as competitor. (D) Biotinylated inhibitor pulldown assay using COUP-TFII fragments overexpressed 293T cells. Flag-CII-C (C-terminal 147 to 414 amino acids) and Flag-CII-N (N-terminal 1 to 182 amino acids). (E) Biotinylated inhibitor pulldown assay using purified glutathione S-transferase–tagged COUP-TFII ligand-binding domain (GST-LBD; 172 to 414 amino acids). GST antibody was used for Western blot assay. (F) Biotinylated inhibitor pulldown assay using overexpressed COUP-TFII with amino acid mutation in 293T cells. WT, wild type. (G) 293T cells that transfected with NGFIA-Luc and mutated COUP-TFII were treated with CIA1 for 18 hours. Luciferase assay was performed. n = 3 per group. (H) Biotinylated inhibitor pulldown assay using overexpressed nuclear receptors in 293T cells. Twenty micromolar CIA1 was used as competitor. HA, hemagglutinin.
To determine which domain of COUP-TFII is important for binding to the inhibitor, we generated flag-tagged COUP-TFII constructs and found that COUP-TFII C-terminal region (147 to 414 amino acids), including ligand-binding domain (LBD), interacted well with the inhibitor, while the N-terminal region (1 to 182 amino acids), including the DNA binding domain, barely showed interaction (Fig. 2D). Furthermore, the purified glutathione S-transferase (GST)–tagged COUP-TFII LBD (172 to 414 amino acids) protein could be pulled down by the CIA inhibitor (Fig. 2E), indicating that the CIA inhibitor directly interacts with COUP-TFII LBD. X-ray structure of COUP-TFII LBD illustrates a potential ligand-binding pocket and some surface cavities that might directly bind to the inhibitor (16, 17). To test potential binding sites for the inhibitor, we mutated some key amino acids on the corresponding surface (16, 17). Mutation of amino acids W249 and F253, key residues proposed to be important for ligand binding, notably disrupted interactions between COUP-TFII protein and inhibitor (Fig. 2F) and rendered COUP-TFII becoming more resistant to inhibitor treatment (Fig. 2G), suggesting that the inhibitor interacts with COUP-TFII through the ligand-binding pocket. Together, all these results indicate that the inhibitor directly interacts with the COUP-TFII LBD.
To further determine the binding specificity, we measured inhibitor interaction with other nuclear receptors that have a similar structure as COUP-TFII. Most receptors such as retinoid X receptor α (RXRα), hepatocyte nuclear factor 4α (HNF4A), liver receptor homolog–1 (LRH-1), peroxisome proliferator–activated receptor α (PPARα), liver X receptor α (LXRα), glucocorticoid receptor (GR), progesterone receptor (PR), estrogen receptor (ER), and retinoic acid receptor (RAR)–related orphan receptor γ (RORγ) did not show detectable interaction with the inhibitor, except COUP-TFI and AR, which showed a much weaker interaction in comparison to COUP-TFII (Fig. 2H). We found that COUP-TFII contributed to the inhibitor pulldown of AR. Since it was reported that AR can interact with COUP-TFII (21), we asked whether AR pulldown by inhibitor is due to its association with COUP-TFII. We found that silencing of COUP-TFII notably impaired the inhibitor-induced pulldown of AR (fig. S3D). We did not detect any pulldown of purified AR protein by the inhibitor (fig. S3E). Hence, these results suggest that the CIA inhibitor does not directly interact with AR and demonstrate the specificity of CIA inhibitor binding to COUP-TFII protein. In addition, we found that silencing of AR or COUP-TFI did not affect the expression of COUP-TFII target genes (fig. S3, F and G), further supporting that the effects of inhibitors are through COUP-TFII.
Screening of CIA1 and CIA2 analogs
To investigate which group of the CIA inhibitor is important for repressing COUP-TFII activity, we measured various CIA analogs. CIA inhibitor structure has one thienopyrimidine ring, one dimethoxybenzene ring, and the linker chain (fig. S4A). We measured the efficacy of CIA analogs on inhibiting COUP-TFII–driven NGFIA reporter luciferase activity and compared the IC50 values of different analogs to original CIA1 and CIA2 (fig. S4B). We found that most of the modifications on the thienopyrimidine ring did not affect the inhibitor activity (fig. S4, B and C), only adding certain larger groups that slightly impaired the inhibitor activity. However, modifications on the dimethoxybenzene ring with replacing or closing the dimethoxy groups substantially reduced the inhibitor activity as indicated by the IC50 values of inhibitor analogs in luciferase assay (fig. S4, B and D). In addition, modifications of the linker chain with shortening, extending, or fixing also slightly impaired the inhibitor activity (fig. S4, B and E). Together, the data suggest that the dimethoxybenzene group is important for the CIA inhibitor activity.
Suppression of prostate tumor growth by COUP-TFII inhibitor
Next, we asked whether CIA inhibitor suppresses COUP-TFII activity to function in cells. On the basis of our previous findings that COUP-TFII promotes prostate cancer development and progression, we investigated whether CIA inhibitor would interfere with prostate cancer cell growth. Our result showed that CIA1 or CIA2 treatment inhibited growth of multiple prostate cancer cell lines including androgen-sensitive LNCaP cells, AR-negative PC3 cells, as well as CRPC C4-2, abl, and AR variant 22Rv1 cells (Fig. 3A). The IC50 values ranged from 1.2 to 7.6 μM for CIA1 and from 2.2 to 10.2 μM for CIA2 in inhibiting growth of tested prostate cancer cells. In contrast, CIA1 or CIA2 treatment had little effect on the growth of “normal” prostate cells RWPE-1 and PrEC, consistent with the elevated COUP-TFII expression in prostate cancer cells (Fig. 3A). Likewise, CIA1 or CIA2 also notably inhibited the colony formation ability of prostate cancer cells in a dose-dependent manner (Fig. 3B). In addition, analogous to previous observation that COUP-TFII promotes prostate cancer metastasis, CIA1 or CIA2 treatment greatly reduced prostate cancer cell invasion (Fig. 3C) but had little effect when COUP-TFII was silenced (fig. S5A). In addition, COUP-TFII has been reported to promote angiogenesis to enhance tumor growth (12, 13). Accordingly, our result showed that CIA1 or CIA2 reduced the angiogenic ability of endothelial cells as observed in sprouting assays (Fig. 3D). Collectively, these results indicate that COUP-TFII inhibitor CIA1 and CIA2 can phenocopy COUP-TFII silencing and exhibit potent inhibitory effects on prostate cancer cell growth. Furthermore, we found that CIA1 or CIA2 did not show an apparent effect on COUP-TFII protein levels in prostate cancer cells when treated with varying doses or different time periods (fig. S5, B to E).
Fig. 3. Reduced prostate cancer cell growth, invasion, and angiogenesis by COUP-TFII inhibitor.
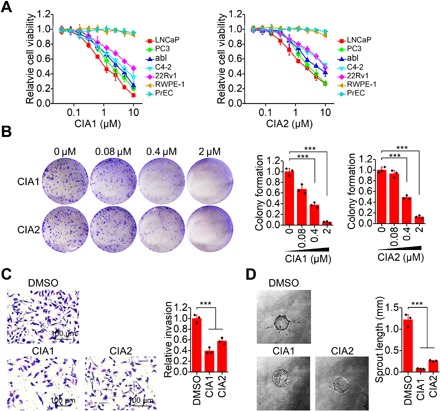
(A) CIA1 and CIA2 reduced cell growth of prostate cancer cells. Cell viability was measured 96 hours after CIA1 or CIA2 treatment. n = 3 per group. (B) CIA1 and CIA2 reduced colony formation ability of prostate cancer cells. PC3 cells were treated with inhibitor for 12 days. n = 3 per group. Two-way analysis of variance (ANOVA). (C) CIA1 and CIA2 reduced prostate cancer cell invasion. PC3 cells were treated with 1 μM CIA1 or CIA2 for 48 hours. Invasion was measured by transwell assay. n = 3 per group. One-way ANOVA. DMSO, dimethyl sulfoxide. (D) Angiogenesis was measured by human umbilical cord endothelial cell sprouting assay. n = 3 per group. One-way ANOVA. ***P < 0.001.
Subsequently, we tested the effect of CIA inhibitors in vivo to evaluate the clinical relevance of COUP-TFII inhibitors in the context of prostate cancer. First, we measured the antitumor activity of CIA1 in prostate cancer xenograft mouse models (fig. S6A). In the LNCaP xenograft model, CIA1 treatment induced a marked and robust inhibition of prostate cancer tumor growth (Fig. 4, A and B). As expected, tumor cell proliferation was reduced as measured by Ki67 staining in collected xenograft tumors after CIA1 treatment (Fig. 4C). Moreover, the expression of COUP-TFII target genes, including induced FOXM1, CDK1, and repressed p21, was all disturbed in tumor samples upon CIA1 treatment (Fig. 4D). We also observed that CIA1 treatment resulted in a significant reduction of tumor angiogenesis as detected by CD31 immunostaining, consistent with the role of COUP-TFII in promoting angiogenesis (Fig. 4E). All these results indicate that CIA1 can suppress prostate cancer tumor growth in mice. We did not observe apparent body weight loss in mice with CIA1 administration (fig. S6B), indicating that, grossly, CIA1 has limited toxicity in mice.
Fig. 4. Inhibition of prostate cancer tumor growth by COUP-TFII inhibitor.
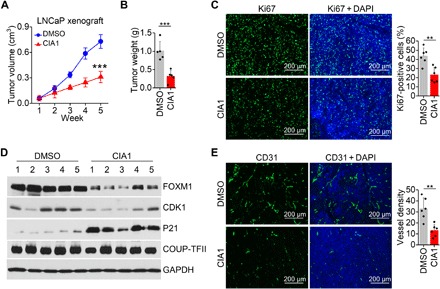
(A) CIA1 treatment has antitumor activity in mice bearing LNCaP xenograft tumors. CIA1 (2.6 mg/kg) was given daily by intraperitoneal injection. Tumor growth curve was shown. n = 5 per group. Two-way ANOVA. Tumor weight was shown in (B). n = 5 per group. t test. (C) Ki67 staining in tumor tissues treated with DMSO and CIA1. n = 6 per group. t test. (D) Indicated protein levels in collected tumor tissue were measured by Western blot assay. (E) CD31 staining in tumor tissues treated with DMSO and CIA1. n = 6 per group. t test. **P < 0.01 and ***P < 0.001. DAPI, 4′,6-diamidino-2-phenylindole.
In addition to androgen-sensitive LNCaP cells, we further tested xenograft tumors of AR-negative PC3 cells, AR-v7–driven CRPC 22Rv1 cells, and wild-type AR CRPC LNCaP-abl cells. Notably, CIA1 also inhibited growth of these CRPC tumors of PC3, 22Rv1, and LNCaP-abl xenografts (Fig. 5, A to F). To reinforce the clinical relevance of CIA inhibitors, we further measured CIA1’s effect on prostate cancer PDX mice. CIA1 treatment also substantially reduced tumor growth in PDX mice (Fig. 5, G and H). Consistently, no obvious body weight loss was observed in these mice (fig. S6C), reinforcing the limited toxicity of CIA1. We next measured COUP-TFII protein levels in prostate cancer cell lines and xenograft tumors (fig. S6, D and E). 22Rv1 cells and xenograft tumors had lower levels of COUP-TFII protein and consequently were less sensitive to inhibitor treatment, suggesting the correlation between inhibitor efficacy and COUP-TFII protein levels. Furthermore, we found that the inhibitor did not obviously affect COUP-TFII protein levels in the tumor (fig. S6F), similar to what we observed in cell lines (fig. S5). Together, these results indicate that COUP-TFII inhibitors can be used for prostate cancer treatment to inhibit tumor growth.
Fig. 5. Inhibition of prostate cancer tumor growth by COUP-TFII inhibitor.
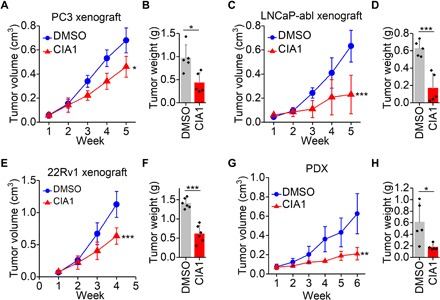
CIA1 treatment reduced tumor growth in the mice that bear PC3 xenograft (A), LNCaP-abl xenograft (C), 22Rv1 xenograft (E), and PDX tumor (G). CIA1 (2.6 mg/kg) was given daily by intraperitoneal injection. n = 5 to 6 per group. Two-way ANOVA. The corresponding tumor weight was shown in (B), (D), (F), and (H). n = 5 to 6 per group. t test. *P < 0.05, **P < 0.01, and ***P < 0.001.
To provide more information for future clinical applications, we performed the drug metabolism and pharmacokinetics (DMPK) assay and measured the tissue distribution of CIA1. After intraperitoneal injection of CIA1 in mice, plasma CIA1 levels reduced about 10-fold within 1 hour (fig. S6G). In contrast, CIA1 was absorbed by tissues and enriched in some tissues including fat, liver, and testis (fig. S6H). These results suggest that CIA1 has a potential to be optimized for improved clearance rate and tissue preference.
Disrupted COUP-TFII binding to associated regulators by the inhibitor CIA1
We next investigated the mechanism on how the CIA inhibitor, CIA1, suppresses COUP-TFII activity. First, the CIA1 inhibitor did not affect COUP-TFII protein levels (fig. S7A). Subsequently, we determined whether the CIA1 inhibitor affects COUP-TFII binding to DNA. COUP-TFII binding to the promoter of its target genes FOXM1, CDK1, and CDKN1A (fig. S7B) was measured by chromatin immunoprecipitation (ChIP)–quantitative polymerase chain reaction (qPCR) after CIA1 treatment. CIA1 did not appear to alter COUP-TFII binding to its DNA binding sites on target gene promoters (fig. S7C). This result indicates that CIA inhibitors do not affect COUP-TFII binding to DNA, consistent with undetected interaction between CIA inhibitor and DNA binding domain of COUP-TFII.
COUP-TFII has been shown to regulate gene expression through recruitment of transcription regulators (16, 22–25). Accordingly, we investigated whether CIA inhibitors can disturb COUP-TFII transcription activity through affecting COUP-TFII–cofactor interaction. Through enzyme-catalyzed proximity labeling assay (26), we overexpressed miniTurbo-fused COUP-TFII (fig. S7D) and treated cells with biotin to label COUP-TFII–binding proteins (fig. S7E). MS was further performed to measure interference of COUP-TFII–binding proteins by CIA1 treatment (fig. S7F). Gene Ontology analysis of CIA1-interfered COUP-TFII–binding proteins (table S1) showed that transcription regulators binding to COUP-TFII were disturbed by CIA1 (fig. S7G). Among the ranked transcription regulators (Fig. 6A), we further identified through IP that CIA1 reduced COUP-TFII binding to FOXA1, HOXB13, ZNF148, ZBTB10, and WDR5 (Fig. 6B). To investigate which COUP-TFII–binding regulator plays a similar role in promoting prostate cancer cell growth as COUP-TFII, we suppressed these regulators (fig. S7H) and measured cell growth. The result showed that suppression of FOXA1 reduced prostate cancer cell growth (Fig. 6C), suggesting that COUP-TFII might interact with FOXA1 to regulate gene expression and promote tumor cell growth. CIA1 also reduced FOXA1-induced genes and increased FOXA1-repressed genes (Fig. 6D). In addition, CIA1 did not directly bind to FOXA1 (fig. S7I), and FOXA1 specifically regulated genes were not affected by CIA1 (fig. S7J). These results established that CIA1 disturbs COUP-TFII binding to FOXA1 to regulate gene expression. In addition, COUP-TFII activity on target gene regulation was notably impaired in the absence of FOXA1 (Fig. 6, E and F), indicating that COUP-TFII collaborates with FOXA1 to regulate target gene expression. To determine whether FOXA1 is important for CIA1-induced gene expression change, we investigated whether FOXA1 recruitment to COUP-TFII–binding sites was affected by CIA1. As expected, ChIP-qPCR results showed that the recruitment of FOXA1 to COUP-TFII–binding sites on target gene promoters was reduced by CIA1 treatment (Fig. 6G). Moreover, CIA1-induced gene expression change was largely absent when FOXA1 expression was down-regulated (Fig. 6, H and I). Therefore, the results indicate that CIA1 disturbs COUP-TFII binding to FOXA1 to regulate target gene expression.
Fig. 6. Disrupted COUP-TFII binding to FOXA1 by inhibitor.
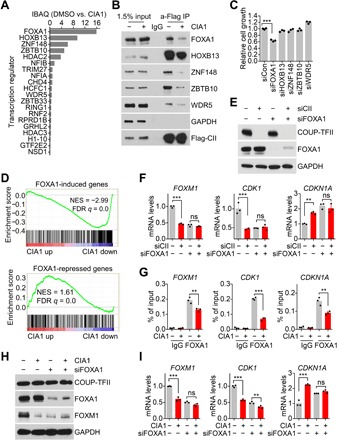
(A) MS analysis of CIA1 reduced COUP-TFII–binding proteins. Cells were treated with 4 μM CIA1 for 3 hours and then with biotin for 4 hours. IBAQ, intensity-based absolute quantification. (B) LNCaP cells with flag-COUP-TFII overexpression were treated with 4 μM CIA1 for 7 hours. IP was performed. (C) LNCaP cell growth was measured 4 days after with siRNA transfection. n = 3 per group. One-way ANOVA. (D) GSEA of FOXA1 signature with CIA1 regulated genes. FOXA1 signature was generated by GSEA37314. (E) LNCaP cells were transfected with siRNA for 72 hours. Western blot was performed, and qPCR was performed in (F). n = 3 per group. Two-way ANOVA. (G) LNCaP cells were treated with 4 μM CIA1 for 12 hours. ChIP-qPCR was performed. n = 3 per group. Two-way ANOVA. IgG, immunoglobulin G. (H) LNCaP cells were transfected with siRNA for 48 hours and then treated with 2 μM CIA1 for 18 hours. Western blot was performed, and qPCR was performed in (I). n = 3 per group. Two-way ANOVA. ns, P > 0.05; **P < 0.01 and ***P < 0.001.
DISCUSSION
COUP-TFII is an orphan nuclear receptor that regulates gene expression to play important roles in organogenesis. The expression of COUP-TFII is greatly reduced after the completion of organogenesis and differentiation in adulthood. However, recent evidence shows that up-regulation of COUP-TFII occurs under pathological conditions and results in the development of multiple devastating diseases including prostate cancer (7–9), heart failure (14), and muscular dystrophy (15). Studies also demonstrate that reduction of COUP-TFII expression in these disease models greatly reduced disease development, indicating that COUP-TFII can serve as a therapeutic target. In this study, we identified potent and specific COUP-TFII–binding inhibitors and showed their efficacy in targeting COUP-TFII to inhibit prostate cancer tumor growth.
COUP-TFII belongs to an orphan nuclear receptor family in which a ligand has yet to be identified. Accordingly, it is also difficult to find inhibitors for orphan nuclear receptors. However, the x-ray structure of LBD in multiple orphan members, including COUP-TFII, Nurr1, PNR, TR4, and REV-ERB, reveals that they all have a putative hydrophobic ligand-binding pocket with different sizes (16, 27, 28). Recent evidence also shows that small-molecule compounds, which bind to classical binding pockets, surface cavities, and cryptic sites or induce protein conformation changes, may regulate the activity of orphan nuclear receptors, suggesting that the orphan nuclear receptor might be targetable or druggable. Studies on orphan nuclear receptor, Nurr1, showed that the activity of Nurr1 can be regulated by small molecules through its binding to the surface of hydrophobic grooves (29, 30). X-ray structure of the LBD reveals that COUP-TFII also has a putative active ligand-binding pocket and its activity can be promoted by the compound retinoid acid (16). Recent reports also show that COUP-TFII could be inactivated by the 4-methoxynaphthol compound through pocket binding–induced protein instability (17). However, the 4-methoxynaphthol compound needs a high concentration up to 100 μM to reduce COUP-TFII activity. Here, we identified a COUP-TFII direct binding inhibitor that functions in the nanomolar range and shows a promising potential for future clinical treatment.
We chose a large and unselected compound library and used full-length COUP-TFII instead of a truncated domain to increase the yield and ensure that the inhibitor is active on endogenous COUP-TFII. The molecules identified in the screen, CIA1 and CIA2, are closely related for their structure similarity, indicating the reliability of screening. Our subsequent experiments further showed that the inhibitors specifically target COUP-TFII. First, the inhibitors functioned in a COUP-TFII–dependent manner. Second, the inhibitor regulated very similar target genes compared with COUP-TFII knockdown. Furthermore, the inhibitor directly bound to COUP-TFII but not to other tested nuclear receptors, which share the similarity with COUP-TFII in LBD structure or protein sequence.
The COUP-TFII inhibitors were subsequently applied into the treatment of disease models. COUP-TFII has been shown to promote prostate cancer progression through regulating TGFβ signaling and FOXM1/CENPF axis. We found that the inhibitors disturbed the signaling and repressed cell cycle gene expression to reduce tumor growth. The biggest concern in prostate cancer treatment is that androgen deprivation therapy leads to CRPC occurrence due to regained activation of AR such as constitutively active ARv7 and targeting AR lastly results in AR-deficient lethal prostate cancer. Our inhibitor was shown to be potent in repressing growth of AR-sensitive, CRPC, ARv7-dependent, and AR-null tumors, thus indicating its broad potential application in multiple-stage prostate cancer. Besides prostate cancer, COUP-TFII also promotes progression of heart failure (14) and muscular dystrophy (15), which lack effective treatment. Our previous reports have shown that genetic suppression of COUP-TFII can alleviate disease development in these disease models, suggesting that COUP-TFII inhibitors can also have potential applications in muscular dystrophy and heart failure. We will next treat COUP-TFII inhibitors into these two disease models to establish the therapy and expand the application of COUP-TFII inhibitors. In the meantime, we will also modify our lead compound to obtain new inhibitors with more potent activity. Our current tested compound analogs did not succeed with higher activity. However, through structure modification, we learned that the dimethoxybenzene group might be important for activity, as changing this group impaired inhibitor activity. Therefore, further directed modification will likely show improvement.
In addition, potential side effects of an inhibitor are always the major concern on its use in disease treatment. As we have shown previously that COUP-TFII is highly expressed during embryonic stages for its function in organ development and cellular differentiation, the expression level comes down to the basal level after completion of organ development and cellular differentiation. When we deleted the COUP-TFII gene in the adult, we detected no major effect on animal health, suggesting that COUP-TFII is no longer required for adult maintenance. Consequently, we do not expect major side effects coming from inhibition of COUP-TFII activity in healthy tissues, except biological functions that may require new differentiation, such as reproduction and wound healing. We did not observe apparent body weight change or organ abnormality after daily intraperitoneal administration of the inhibitor, indicating the safety of treatment with this inhibitor.
Collectively, we identified a small molecule that directly binds to COUP-TFII and inhibits COUP-TFII activity with a high specificity and potent efficacy. On the basis of the results from its inhibition of prostate cancer tumor growth, it is reasonable to believe that this small molecular inhibitor can potentially serve as an efficient and safe inhibitor for human use. Our finding provides a novel drug that can potentially benefit patients with diseases caused by hyperactivation of COUP-TFII.
MATERIALS AND METHODS
Cell lines, culturing condition, and transfection
293T, LNCaP, 22Rv1, C4-2, PC3, DU145, and RWPE-1 cells were purchased from the American Type Culture Collection and maintained in the Tissue and Cell Culture Core Facility at the Baylor College of Medicine. Cells were cultured in RPMI 1640 (11875093, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS) (F2442, Sigma-Aldrich, Saint Louis, MO). LNCaP-abl cell line was obtained from Z. Culig (Medical University of Innsbruck, Innsbruck, Austria) and maintained in RPMI 1640 supplemented with 10% charcoal-stripped FBS (F6765, Sigma-Aldrich, Saint Louis, MO). The authenticity of all cell lines was verified in the past 6 months. PrEC was purchased form Lonza (CC-2555, Lonza, Basel, Switzerland) and cultured in the medium (C-3165, Lonza, Basel, Switzerland) with supplement (CC-3166, Lonza, Basel, Switzerland). Cell transfection was performed using Lipofectamine 2000 (11668019, Thermo Fisher Scientific, Waltham, MA) for vectors and Lipofectamine RNAiMAX (13778075, Thermo Fisher Scientific, Waltham, MA) for small interfering RNAs (siRNAs) according to the manufacturer’s instructions. siRNA targeting sequences are shown in table S2.
Chemicals
CIA1 was from ChemBridge (7960292, ChemBridge, San Diego, CA). CIA2 was from Enamine (T5234163, Enamine, Kiev, Ukraine). Other CIA analogs were from Enamine (Z31249021, Z31249038, Z31249019, Z94847179, Z645446344, Z31264771, and Z31121599, Enamine, Kiev, Ukraine). Other chemicals were made in H.E.X.’s laboratory. Biotin-CIA was synthesized in KareBay Biochem Inc. (Monmouth Junction, NJ).
High-throughput screening
The high-throughput screening was performed in The Scripps Research Institute, Florida using their fully automated platform. This effort was supported by the National Institutes of Health (NIH) MLPCN initiative. Briefly, the luciferase assay was performed using 293T cells transiently transfected with the luciferase reporter (pXP2-NGFIA-Luc) in combination with expression vectors for COUP-TFII (pcDNA6.2–COUP-TFII). Transfection of p17mer-X4TK-luc and pAB-Gal4-VP16 was set up for counter screening. Compounds were added and incubated for 18 hours. ONE-Glo luciferase assay system (Promega, Madison, WI) was added to all wells, and the ViewLux (PerkinElmer Life Sciences) was used to measure luciferase activity for 5-s exposure.
Luciferase assay
293T cells were transfected with the same amount of luciferase reporter and various expression vectors (pXP2-NGFIA-Luc and pcDNA6.2-COUP-TFII or pGIPZ-shCOUP-TFII; p17mer-X4TK-luc and pAB-Gal4-VP16). Twenty-four hours after transfection, cells were harvested and seeded into 96-well microplate at 4 × 104 cells per well. Compounds were added 5 hours after seeding. Luciferase assay activity was measured 16 to 20 hours after compound treatment using ONE-Glo luciferase assay system (E6110, Promega, Madison, WI).
RNA isolation and quantitative reverse transcriptase real-time PCR
Total RNA was extracted from cells using TRIzol reagent (15596018, Thermo Fisher Scientific, Waltham, MA). Complementary DNA (cDNA) was synthesized using Thermo Fisher Scientific Maxima First Strand cDNA Synthesis Kit (FERK1641, Thermo Fisher Scientific, Waltham, MA). Quantitative real-time PCR was performed with PowerUp SYBR Green PCR Master Mix (A25742, Thermo Fisher Scientific, Hampton, NH) on the StepOnePlus Real-Time PCR System (Applied Biosystems). Relative mRNA levels were normalized to ACTB (Beta-actin). All primers were synthesized by Sigma-Aldrich. The primer sequences are shown in table S2.
RNA sequencing and GSEA
LNCaP cells were transfected with COUP-TFII siRNA for 72 hours or treated with inhibitor (1 μM CIA1) for 18 hours, and total RNA was extracted. The RNA sequencing was performed by Q2 Solutions (Morrisville, USA). The sequencing data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (GSE142475). Up- and down-regulated genes were filtered with fold change log21 as cutoff to generate COUP-TFII–induced or COUP-TFII–repressed gene signatures. GSEA was carried out using the GSEA JAVA program from the Broad Institute.
Cell viability, proliferation, and colony formation assay
Cells were seeded in 96-well plate at 1000 to 3000 cells per well. Inhibitor was added at the following day. After 96 hours of incubation, cell viability was assessed by CellTiter-Glo assay (G7570, Promega, Madison, WI). Cell proliferation was measured by CellTiter 96 AQueous One Solution Cell Proliferation Assay (G3582, Promega, Madison, WI). The optical density at 490 nm was measured by the Multiskan FC Microplate Photometer (Thermo Fisher Scientific, Waltham, MA) and normalized. For the colony formation assay, PC3 cells were seeded at 300 cells per well in six-well plate and treated with CIA1 or CIA2 for about 10 days. Colony was fixed with methanol, stained with crystal violet, and counted.
Invasion assay
Cells were treated with inhibitor for 48 hours. PC3 cells (0.3 × 105) were seeded with serum-free medium in a transwell chamber precoated with Matrigel (354483, BD Biosciences, Franklin Lakes, NJ). Medium with 10% FBS was added in the lower chamber. Inhibitor was added to both upper and lower chambers. After 24 hours, cells were fixed with methanol. The noninvading cells were gently removed, and invaded cells on the lower side of the chamber were stained with crystal violet, photographed, and counted.
Sprouting assay
Sprouting assay was performed as described (31). Cytodex 3 beads were purchased from GE Healthcare (17-0485-01, GE Healthcare, Chicago, IL). Human umbilical cord endothelial cell culture medium was from Lonza (CC-3162, Lonza, Basel, Switzerland). CIA1 or CIA2 (0.2 μM) was treated. Cumulative sprout length per bead was calculated by ImageJ software.
Western blot assay
Total proteins were extracted from cells following standard protocol. Protein concentration was measured using the bicinchoninic acid protein assay kit (23229, Thermo Fisher Scientific, Waltham, MA). Protein samples were separated by SDS–polyacrylamide gel electrophoresis and transferred onto nitrocellular membrane (162-0112, Bio-Rad, Hercules, CA). The membrane was incubated for 30 min in blocking buffer (tris-buffered saline with Tween 20 with 5% nonfat dry milk), followed by overnight incubation at 4°C with the primary antibody. Membrane was then incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies (7074 or 7076, Cell Signaling Technology, Danvers, MA) for 1 hour. The signals were visualized with SuperSignal Chemiluminescent Substrate (34577, Thermo Fisher Scientific, Waltham, MA). The primary antibodies used in this study were as follows: Flag-HRP (A8592, Sigma-Aldrich, Saint Louis, MO), glyceraldehyde-3-phosphate dehydrogenase (SC-25778 HRP, Santa Cruz Biotechnology, Dallas, TX), Streptavidin-HRP (N100, Thermo Fisher Scientific, Waltham, MA), GST (A190-122A, Bethyl, Montgomery, TX), hemagglutinin (11666606001, Roche, Basel, Switzerland), COUP-TFII (6434, Cell Signaling Technology, Danvers, MA), AR (5153, Cell Signaling Technology, Danvers, MA), PR (SC-7208, Santa Cruz Biotechnology, Dallas, TX), GR (24050-1-AP, Proteintech, Wuhan, Hubei, P.R.C.), ER (SC-542, Santa Cruz Biotechnology, Dallas, TX), RXRa (21218-1-AP, Proteintech, Wuhan, Hubei, P.R. China), PPARa (15540-1-AP, Proteintech, Wuhan, Hubei, P.R. China), HNF4A (3113, Cell Signaling Technology, Danvers, MA), FOXM1 (20459, Cell Signaling Technology, Danvers, MA), CDK1 (A303-663A-T, Bethyl, Montgomery, TX), and P21 (SC-6246, Santa Cruz Biotechnology, Dallas, TX). FOXA1 (58613, Cell Signaling Technology, Danvers, MA), HOXB13 (90944, Cell Signaling Technology, Danvers, MA), ZNF148 (A303-116A-T, Bethyl, Montgomery, TX), ZBTB10 (A303-257A-T, Bethyl, Montgomery, TX), WDR5 (13105, Cell Signaling, Danvers, MA), and RORγ (ab113434, Abcam, Cambridge, UK).
ChIP-qPCR assay
LNCaP cells were treated with 4 μM CIA1 for 12 hours. ChIP assay was performed using Magna ChIP A/G kit from Millipore according to the manufacturer’s protocol. COUP-TFII (PP-H7147-00, R&D Systems, Minneapolis, MN), FOXA1 (ab23738, Abcam, Cambridge, UK), and corresponding control immunoglobulin G antibodies were used. The qPCR assays were carried out on chromatin samples prepared above. Primer sequences are shown in table S2.
Cellular thermal shift assay
The CETSA was performed by the standard protocol (32). LNCaP cells were treated with 10 μM CIA1 for 1 hour. Cells were suspended in phosphate-buffered saline with protease inhibitors, heated in the indicated temperature for 3 min, and immediately snap-frozen. Samples were subjected to two freeze-thaw cycles and centrifuged. Supernatants were collected, and Western blot assays were performed.
Tumor xenograft
Six-week-old male nude mice (Nu/J homozygous for Foxn1nu, stock number 002019) were purchased from the Jackson laboratory (Bar Harbor, Maine). A total of 10 × 106 LNCaP cells, 2 × 106 PC3 cells, and 4 × 106 22Rv1 cells mixed with Matrigel (354234, Corning, Corning, NY) were subcutaneously injected into the flank of mice. For LNCaP-abl xenograft, mice were castrated and then injected with 10 × 106 LNCaP-abl cells. PDX mice were purchased from the Jackson laboratory (TM00298). The inhibitor was started to be given daily at 2.6 mg/kg by intraperitoneal injection when tumor diameter reached to about 0.5 cm. The formulation was 10% (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD, 332593, Sigma-Aldrich, Saint Louis, MO) in sterile saline solution. The tumor size was measured by caliper during the entire experimental process. Tumor volume was calculated by the formula: v = 0.5 × a × b2 (v, the tumor volume; a, the major diameter of the tumor; b, the minor diameter). At the end of the experiment, mice were euthanized, and tumor tissue was removed for further examination.
Immunohistochemistry assay
Immunohistochemistry was performed as described previously (9). Primary Ki67 antibody (550609, BD Biosciences, Franklin Lakes, NJ; 1:2000 dilution) and CD31 antibody (AF3628-SP, R&D Systems, Minneapolis, MN; 1:1000 dilution) were incubated overnight at 4°C, and secondary antibodies were incubated for 1 hour at room temperature.
DMPK assay
Mice were fasted 12 hours before treatment and then administered a single dose of CIA1 in 10% HP-β-CD though intraperitoneal injection (2.6 mg/kg). Plasma samples were taken at 0.25, 0.5, 1, 2, 4, 8, and 24 hours after CIA1 injection. For CIA1 tissue distribution analysis, tissue samples were collected at 5, 10, and 30 min and 1 hour after CIA1 injection. Samples were analyzed by LC-MS, using Phoenix WinNonlin software (Certara USA Inc.).
Pulldown assay
Cells were lysed in IP buffer [20 mM tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA with protease inhibitor]. Cell lysate, purified COUP-TFII LBD protein (16), and purified FOXA1 protein (ab98301, Abcam, Cambridge, UK) were incubated for 4 hours at 4°C with streptavidin beads (65601, Thermo Fisher Scientific, Waltham, MA) that were preloaded with biotinylated inhibitor. For the competition assay, cell lysate was preincubated with 20 μM competitor inhibitor for 3 hours. Beads were washed three times by IP buffer and resuspended in loading buffer. Samples were boiled at 95°C for 5 min for separation of the protein and beads and then analyzed by Western blot.
Enzyme-catalyzed proximity labeling and MS
Enzyme-catalyzed proximity labeling was performed by the standard protocol (26). The flag-miniTurbo-13GGGGS-COUP-TFII was overexpressed in LNCaP cells. Cells were treated with 4 μM CIA1 for 3 hours, and 400 μM biotin was added for 4 hours. Cells were washed, collected, and lysed in radioimmunoprecipitation assay (RIPA) lysis buffer [89900, Thermo Fisher Scientific; 25 mM tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS] with protease inhibitor added. After preclearing with Protein G Dynabeads (10003D, Thermo Fisher Scientific, Waltham, MA) for 1 hour, lysates were incubated with streptavidin-coupled magnetic Dynabeads (65601, Thermo Fisher Scientific, Waltham, MA) for 1 hour with rotation at 4°C. The beads were subsequently washed twice with 1 ml of RIPA lysis buffer, once with 1 ml of 1 M KCl, once with 1 ml of 0.1 M Na2CO3, once with 1 ml of 2 M urea in 10 mM tris-HCl (pH 8.0), and twice with 1 ml of RIPA lysis buffer. Beads were collected after wash. MS was performed in the Mass Spectrometry Proteomics Core at the Baylor College of Medicine. For Western blot assay, beads were resuspended in Laemmli sample buffer (1610747, Bio-Rad, Hercules, CA) and boiled at 95°C for 5 min for magnetic separation of the protein and beads.
Co-IP assay
LNCaP cells with flag-COUP-TFII overexpression were treated with 4 μM CIA1 for 7 hours. Cells were collected, and nuclear component was separated by extract buffer [10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 0.5 mM dithiothreitol with protease inhibitor]. Nuclear component was lysed in IP buffer [20 mM tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA with protease inhibitor]. After preclearing with Protein G Dynabeads (10003D, Thermo Fisher Scientific, Waltham, MA) for 1 hour, lysates were incubated with Protein G Dynabeads preloaded with anti-flag antibody (F1804, Sigma-Aldrich, Saint Louis, MO) for 2 hours at 4°C. Beads were washed three times in IP buffer and resuspended in Laemmli sample buffer (1610747, Bio-Rad, Hercules, CA) and boiled at 95°C for 5 min for separation of the protein and beads. Samples were then analyzed by Western blot.
Study approval
All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the Baylor College of Medicine, and all experiments were performed in compliance with the institutional guidelines of the Baylor College of Medicine.
Statistical analysis
The standard statistical test analyses were completed and performed using Origin 2017 (Northampton, USA), and data were presented as individual data points with means ± SD. Unpaired two-tailed Student’s t test was used for two-group comparison. One-way analysis of variance (ANOVA) was used for comparison among multiple groups with one independent variable, followed by post hoc Tukey’s multiple comparisons test. Two-way ANOVA was used for comparison among multiple groups with two independent variables, followed by Sidak’s multiple comparisons test. The difference was regarded as significant when P < 0.05. “ns” indicates not significant (P > 0.05). *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank J. R. Hebert for editorial assistance. We appreciate the technical support of P. Li and G. Xu. We also thank the Tissue Culture Core of Molecular and Cellular Biology Department, Genetically Engineered Mouse Core, and Mass Spectrometry Proteomics Core for their assistance. We thank P. Baillargeon and L. DeLuca (Lead Identification, Scripps Research Institute Molecular Screening Center, Scripps Florida) for assistance with compound management. We thank L. Scampavia (Lead Identification, Scripps Research Institute Molecular Screening Center, Scripps Florida) for LC-MS analysis of the presented compounds. Funding: This work was supported by grants from NIH DK45641 to M.-J.T., NIH HL114539 to S.Y.T., and NIH DK071662 to H.E.X. This work was supported by the NIH National Institute of Mental Health (grant U54-MH084512). Author contributions: L.W., S.Y.T., and M.-J.T. conceived and designed the experimental approaches and prepared the manuscript. L.W. performed most of the experiments. C.-M.C., J.Q., and M.X. performed a subset of experiments. C.-Y.K. contributed to the statistical analysis. J.S., E.Y., W.G., and H.E.X. performed the chemical synthesis, protein purification, and pharmacology efforts. C.-M.C., L.P.R., P.C., L.S., F.M., T.S., and P.H. performed the high-throughput screening and medicinal chemistry efforts. L.W. wrote the original draft. L.W., S.Y.T., and M.-J.T. reviewed and edited the manuscript. Competing interests: H.E.X. is a founder and a stakeholder of Cascade Pharmaceutical, a start-up interested in the development of COUP-TFII ligands. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The Escherichia coli–expressed COUP-TFII protein used in this paper is obtained from H.E.X. of the Van Andel Research Institute. Request of this protein should be addressed to H.E.X. (eric.xu@simm.ac.cn) pending scientific review and a completed material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8031/DC1
REFERENCES AND NOTES
- 1.Overington J. P., Al-Lazikani B., Hopkins A. L., How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Xie X., Tang K., Yu C.-T., Tsai S. Y., Tsai M.-J., Regulatory potential of COUP-TFs in development: Stem/progenitor cells. Semin. Cell Dev. Biol. 24, 687–693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira F. A., Tsai M. J., Tsai S. Y., COUP-TF orphan nuclear receptors in development and differentiation. Cell. Mol. Life Sci. 57, 1388–1398 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin F. J., Qin J., Tang K., Tsai S. Y., Tsai M. J., Coup d’Etat: An orphan takes control. Endocr. Rev. 32, 404–421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin J., Tsai S. Y., Tsai M.-J., The critical roles of COUP-TFII in tumor progression and metastasis. Cell Biosci. 4, 58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie X., Wu S.-P., Tsai M.-J., Tsai S., The role of COUP-TFII in striated muscle development and disease. Curr. Top. Dev. Biol. 125, 375–403 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Qin J., Wu S.-P., Creighton C. J., Dai F., Xie X., Cheng C.-M., Frolov A., Ayala G., Lin X., Feng X.-H., Ittmann M. M., Tsai S.-J., Tsai M.-J., Tsai S. Y., COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 493, 236–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin S.-C., Kao C.-Y., Lee H.-J., Creighton C. J., Ittmann M. M., Tsai S.-J., Tsai S. Y., Tsai M.-J., Dysregulation of miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of prostate cancer. Nat. Commun. 7, 11418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L., Xu M., Qin J., Lin S.-C., Lee H.-J., Tsai S. Y., Tsai M.-J., MPC1, a key gene in cancer metabolism, is regulated by COUPTFII in human prostate cancer. Oncotarget 7, 14673–14683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navab R., Gonzalez-Santos J. M., Johnston M. R., Liu J., Brodt P., Tsao M.-S., Hu J., Expression of chicken ovalbumin upstream promoter-transcription factor II enhances invasiveness of human lung carcinoma cells. Cancer Res. 64, 5097–5105 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Bao Y., Gu D., Feng W., Sun X., Wang X., Zhang X., Shi Q., Cui G., Yu H., Tang C., Deng A., COUP-TFII regulates metastasis of colorectal adenocarcinoma cells by modulating Snail1. Br. J. Cancer 111, 933–943 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin J., Chen X., Xie X., Tsai M.-J., Tsai S. Y., COUP-TFII regulates tumor growth and metastasis by modulating tumor angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 3687–3692 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin J., Chen X., Yu-Lee L.-y., Tsai M.-J., Tsai S. Y., Nuclear receptor COUP-TFII controls pancreatic islet tumor angiogenesis by regulating vascular endothelial growth factor/vascular endothelial growth factor receptor-2 signaling. Cancer Res. 70, 8812–8821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu S.-P., Kao C.-Y., Wang L., Creighton C. J., Yang J., Donti T. R., Harmancey R., Vasquez H. G., Graham B. H., Bellen H. J., Taegtmeyer H., Chang C.-P., Tsai M.-J., Tsai S. Y., Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat. Commun. 6, 8245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X., Tsai S. Y., Tsai M.-J., COUP-TFII regulates satellite cell function and muscular dystrophy. J. Clin. Invest. 126, 3929–3941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruse S. W., Suino-Powell K., Zhou X. E., Kretschman J. E., Reynolds R., Vonrhein C., Xu Y., Wang L., Tsai S. Y., Tsai M.-J., Xu H. E., Identification of COUP-TFII orphan nuclear receptor as a retinoic acid–activated receptor. PLOS Biol. 6, e227 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Guével R., Oger F., Martinez-Jimenez C. P., Bizot M., Gheeraert C., Firmin F., Ploton M., Kretova M., Palierne G., Staels B., Barath P., Talianidis I., Lefebvre P., Eeckhoute J., Salbert G., Inactivation of the nuclear orphan receptor COUP-TFII by small chemicals. ACS Chem. Biol. 12, 654–663 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Crawford E. D., Schellhammer P. F., McLeod D. G., Moul J. W., Higano C. S., Shore N., Denis L., Iversen P., Eisenberger M. A., Labrie F., Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J. Urol. 200, 956–966 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Katzenwadel A., Wolf P., Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer Lett. 367, 12–17 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Rathkopf D., Scher H. I., Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 19, 43–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song C.-H., Lee H. J., Park E., Lee K., The chicken ovalbumin upstream promoter-transcription factor II negatively regulates the transactivation of androgen receptor in prostate cancer cells. PLOS ONE 7, e49026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litchfield L. M., Klinge C. M., Multiple roles of COUP-TFII in cancer initiation and progression. J. Mol. Endocrinol. 49, R135–R148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey P., Sartorelli V., Hamamori Y., Muscat G. E., The orphan nuclear receptor, COUP-TF II, inhibits myogenesis by post-transcriptional regulation of MyoD function: COUP-TF II directly interacts with p300 and myoD. Nucleic Acids Res. 26, 5501–5510 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey P. J., Dowhan D. H., Franke K., Burke L. J., Downes M., Muscat G. E. O., Transcriptional repression by COUP-TF II is dependent on the C-terminal domain and involves the N-CoR variant, RIP13δ1. J. Steroid Biochem. Mol. Biol. 63, 165–174 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Smirnov D. A., Hou S., Ricciardi R. P., Association of histone deacetylase with COUP-TF in tumorigenic Ad12-transformed cells and its potential role in shut-off of MHC class I transcription. Virology 268, 319–328 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Branon T. C., Bosch J. A., Sanchez A. D., Udeshi N. D., Svinkina T., Carr S. A., Feldman J. L., Perrimon N., Ting A. Y., Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhi X., Zhou X. E., Melcher K., Xu H. E., Structures and regulation of non-X orphan nuclear receptors: A retinoid hypothesis. J. Steroid Biochem. Mol. Biol. 157, 27–40 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ingraham H. A., Redinbo M. R., Orphan nuclear receptors adopted by crystallography. Curr. Opin. Struct. Biol. 15, 708–715 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Hintermann S., Chiesi M., von Krosigk U., Mathé D., Felber R., Hengerer B., Identification of a series of highly potent activators of the Nurr1 signaling pathway. Bioorg. Med. Chem. Lett. 17, 193–196 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Poppe L., Harvey T. S., Mohr C., Zondlo J., Tegley C. M., Nuanmanee O., Cheetham J., Discovery of ligands for Nurr1 by combined use of NMR screening with different isotopic and spin-labeling strategies. J. Biomol. Screen. 12, 301–311 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Yung Y. C., Chae J., Buehler M. J., Hunter C. P., Mooney D. J., Cyclic tensile strain triggers a sequence of autocrine and paracrine signaling to regulate angiogenic sprouting in human vascular cells. Proc. Natl. Acad. Sci. U.S.A. 106, 15279–15284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., Molina D. M., The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8031/DC1