

Dosing protein hydrogels with divalent cations allows for programming of complex shapes and shape morphing.
Abstract
Smart materials that are capable of memorizing a temporary shape, and morph in response to a stimulus, have the potential to revolutionize medicine and robotics. Here, we introduce an innovative method to program protein hydrogels and to induce shape changes in aqueous solutions at room temperature. We demonstrate our approach using hydrogels made from serum albumin, the most abundant protein in the blood plasma, which are synthesized in a cylindrical or flower shape. These gels are then programmed into a spring or a ring shape, respectively. The programming is performed through a marked change in stiffness (of up to 17-fold), induced by adsorption of Zn2+ or Cu2+ cations. We show that these programmed biomaterials can then morph back into their original shape, as the cations diffuse outside the hydrogel material. The approach demonstrated here represents an innovative strategy to program protein-based hydrogels to behave as actuators.
INTRODUCTION
Dynamic biomaterials that undergo conformational changes can enable artificial tissue structures, which could experience morphological transformations, and soft robotics, which could react and change in response to their environment. Currently, most common shape-morphing materials are based on polymers and require switching between a stiff and a soft phase (1). These materials typically rely on two or more network skeletons, sharing the same three-dimensional (3D) space (2), or have a chemical response to small ions (3, 4). Programming, defined as the capability of fixing a temporary shape in a material, requires a marked increase in stiffness, in a reversible manner (5). The initial shape recovery entails a switch from a stiff to a soft phase and is typically realized by compromising the integrity of the secondary network, by changing the temperature (1, 6), pH (7), or solvent (8, 9), or through photoswitching (10, 11).
Protein-based hydrogels use a protein as their primary network, in a water-rich environment (12). These hydrogels retain many of the characteristics displayed by the polymer-based materials but can harvest from a much more diverse biofunctional library. Proteins accomplish many life-supporting functions, from structural role to enzymatic reactivity, and their function is, in most cases, directly related to their folded 3D structure. While there is a substantial diversity for the starting material, when compared to polymers, proteins are stable and functional in a much narrower range of temperatures, pH, or salt conditions and require a water-based environment. Since the mechanical response of a material depends directly on the concentration of its constituent network nodes (13–15), the range of obtainable stiffness for protein-based hydrogels is extremely limited: the protein needs to be above the critical gelation concentration to be turned into a biomaterial and below its specific solubility limit (15–17). This narrow range allows for a change in a stiffness of only ~10 to 30%, depending on the starting protein. Furthermore, well-defined cross-linked network connections are critical to ensure a high shape recovery ratio (18). Unlike most linear polymer molecules, which can entangle like spaghetti in a bowl, globular proteins have well-defined 3D structures, acting as hard spheres. This structural integrity provides an excellent control over the cross-linking points and density, while preserving the tertiary structure of the network nodes.
Recently, we have introduced a new method to obtain shape memory in protein-based hydrogels by stiffening them through adsorbed polyelectrolytes (19). Our approach relies on producing protein hydrogels from bovine serum albumin (BSA), which is homologous to human serum albumin, the most abundant protein in blood plasma. BSA solutions can be turned into completely covalently cross-linked hydrogel biomaterials when starting from solutions with protein concentrations above 1 mM (17, 20). Below 1 mM, BSA hydrogels show irreversible plastic deformation under force, indicative of incomplete cross-linking (17). Because of the overall charge of BSA, these protein hydrogels have been programmed by stiffening, induced by the secondary network made from positively charged polyelectrolytes (19). The shape change was induced in this case via the unfolding response of the protein domains in chemical denaturants with complete recovery upon removal of the denaturing solution. In the unfolding phase transition responsible for shape memory, the proteins lose their tertiary structure and show a remarkable decrease in stiffness. This transition is highly repeatable, as protein folding has been tightly controlled by billions of years of evolution. However, the polyelectrolytes adsorption is irreversible, and because of their large size, the loading capacity is relatively limited, resulting in a change in stiffness of up to ~6.5-fold. Here, we explore the viability of using divalent cations to stiffen protein-based hydrogels toward programming them in various shapes. We then manage to morph back the protein materials from the temporary programmed shape to the initial permanent shape through simple diffusion. It has been previously shown, for other protein hydrogels (21) and peptide-based hydrogels (22), that divalent cations can reinforce their network. It is also well known that BSA can bind cations at various exposed amino acids (23, 24). For example, divalent cations such as Ni2+, Cu2+, and Zn2+ were shown to bind histidine (25, 26) and tryptophan (24), and to bridge cysteine amino acids (27). Adsorption of cations was also associated with an increase in the mechanical stability of proteins at the molecular level (28, 29). Here, we explore this mechanical change to induce the increase in stiffness needed to program protein-based biomaterials in various shapes (Fig. 1). The small ions have the advantage of allowing much higher loading of positive charge, leading to a 17-fold stiffening, and the capacity to diffuse outside the programmed material. This novel implementation of programming protein-based hydrogels with small ions is an important step toward obtaining biocompatible materials that can adjust their shape.
Fig. 1. Schematics of the fixation process.
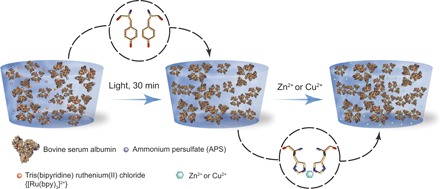
(Left) BSA-based protein hydrogels are fabricated using a light-activated reaction, in the presence of ammonium persulfate (APS) and tris(bipyridine) ruthenium(II) chloride [Ru(bpy)3]2+. (Right) Following synthesis, the protein hydrogels are exposed to Zn2+ or Cu2+, which reversibly increases their stiffness by up to 17-fold. This stiffening effect can be used for shape programming.
RESULTS AND DISCUSSION
Protein-based hydrogels can be obtained using various cross-linking strategies, such as treatment with glutaraldehyde (20) and enzymatic reactions (30), using protein-based lock-and-key ends (31, 32) or photoactivation (12). Here, we use photoactivation via tris(bipyridine) ruthenium(II) chloride ([Ru(bpy)3]2+) to obtain protein-based hydrogels made from BSA (Fig. 1). This reaction was shown to produce covalent carbon─carbon bonds at the exposed tyrosine amino acid sites between adjacent protein domains (12). The advantage of using light to trigger the start of the cross-linking reaction is that it allows us to load the reaction mixture in the desirable shape, without any change in viscosity before light exposure.
Our first step was to explore the range of concentrations of two positively charged ions, which can potentially increase the stiffness of protein hydrogels and allow for shape programming. The change in stiffness of protein-based hydrogels in the presence of positively charged ions was quantified using our force-clamp rheometry apparatus (17, 33). We first produced cylindrical-shaped hydrogels starting from 2 mM BSA, using polytetrafluoroethylene (PTFE) tubes with an inner diameter of 0.56 mm. We chose 2 mM as starting concentration, as our polymerization method produces complete cross-linking for BSA and the hydrogels show reversible behavior without any plastic deformations in the sampled force range (17). Using surgical thread, these gels were then attached in our force-clamp rheometer via two metal hooks connected to a voice-coil motor and force sensor, respectively (Fig. 2, inset, and see also Materials and Methods). Following incubation for 30 min in a phosphate buffer saline with the desired cation concentration, the mechanical response of treated BSA hydrogel was measured in the 0- to 4-kPa range (Fig. 2). The change in stress as a function of strain, as the applied force is increased linearly with time, can be used to assess the stiffness of the material, as the slope in the rising part of the trace directly reports on the dynamic Young’s modulus. Typically, hydrogels made from globular proteins such as BSA also show hysteresis in the stress-stain curves (Fig. 2A). This hysteresis disappears for BSA hydrogels when exposed to chemical denaturants, which break down the tertiary structure of the protein domains forming the hydrogel network (12, 17). This hysteresis was related to the imbalance between the forces where unfolding and refolding take place, with the unfolding transition occurring at much higher forces than the refolding (34), and can allow for large energy dissipation before failure (35, 36). BSA hydrogels show an up to 5-fold increase in stiffness when treated with Cu2+ (50 kPa in 1.5 M Cu2+ from 11 kPa) and a 17-fold increase in stiffness in the presence of Zn2+ (191 kPa in 2 M Zn2+) (Fig. 2B). The stiffening of BSA hydrogels in 2 M Zn2+ is several orders of magnitude greater than that reported for the same gels when treated with polyelectrolytes (19) and should allow for more complex programmed shapes. The stiffening effect seems to depend more on the solution concentration of cations rather than their nature (Fig. 2B). The main advantage of Zn2+ over Cu2+ is its higher solubility in water, which allows us to prepare solutions with higher concentrations and observe greater stiffening.
Fig. 2. Cation-based stiffening of protein-based hydrogels.
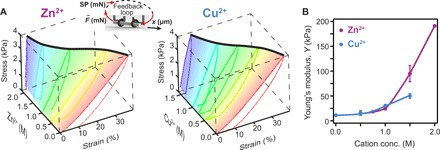
(A) Chemomechanical changes induced by adsorption of various concentrations of Zn2+ (left) and Cu2+ (right) by protein hydrogels made from 2 mM BSA. The mesh highlights the force-loading part, used to assess the change in stiffness, and the thick, black curve follows the final strain at 4-kPa stress. Inset: Schematics of a hydrogel tube pulled under a feedback-controlled force, where the set point (SP) was increased and decreased linearly with 40 Pa/s. (B) Change in measured Young’s modulus as a function of cation concentrations. Both Zn2+ and Cu2+ induce stiffening when adsorbing to BSA-based hydrogels. Lines between points are eye guides. Error bars are SD (n = 3).
Apart from the stiffening effect, incubation of protein hydrogels in solutions with high concentrations of cations improves their mechanical failure properties (22). For these tests, we use a typical bone shape, where the BSA hydrogels were extended until failure or maximum force range of our force sensor (Fig. 3A). BSA-based hydrogels show an increase in both toughness and failure stress with increasing cation concentration. The measured toughness, which represents the ability of a material to absorb energy and deform without fracturing and is derived from the area under the pulling stress-strain curve (Fig. 3B and 3C, left), increased from 1 to 2.8 kJ/mol with cation concentration. The failure stress increased from 15 to 33 kPa. The maximum elongation did not show a substantial variation with cation concentration. This behavior suggests that, while the stiffness is probably given by the increase in the mechanical stability of protein domains (27) and noncovalent bridging (22) at ~120%, the primary network of the BSA hydrogels is the one starting to fail, experiencing irreversible breaking of covalent bonds. Hence, the cross-linking geometry is the limiting factor for extensions, and for further improvements in maximum elongation, the primary hydrogel network would require refinement.
Fig. 3. Mechanical characterization of BSA-hydrogels immersed in cations of various concentrations.
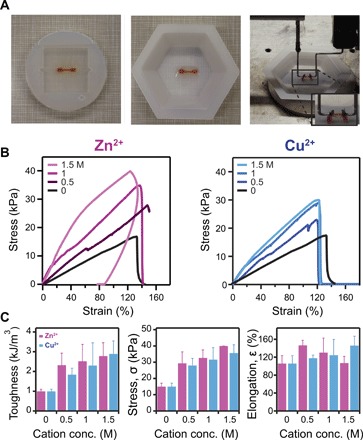
(A) Picture of a gel casted using a bone-shaped silicone mold (left and middle) and attached in the force-clamp rheometer (right). (B) Stress versus strain of BSA hydrogels immersed in Zn2+ (left) and Cu2+ (right) and pulled until breaking. (C) Toughness (left), failure stress (middle), and maximum elongation (right) of BSA hydrogels incubated with increasing concentrations of Zn2+ (magenta) and Cu2+(blue). Toughness increased from 1.0 ± 0.1 to 2.8 ± 0.7 kJ/mol in the presence of 1.5 M Zn2+ and to 2.9 ± 0.7 kJ/mol in the presence of 1.5 M Cu2+. The breaking stress increases from 15 ± 2 to 33 ± 5 kPa when BSA hydrogels were treated with 1.5 M Zn2+ and to 36 ± 5 kPa when treated with 1.5 M Cu2+. The failing strain shows little variation (106 ± 18% versus 107 ± 15% in 1.5 M Zn2+ and 146 ± 21% in 1.5 M Cu2+). Error bars are SD (n = 3). (Photo credit: Luai R. Khoury, UWM; Marina Slawinski, UWM).
While stiffening through a secondary network is necessary for programing in-shape of the hydrogel material, the dynamics of morphing from the programmed to the initial shape will directly depend on the diffusion of the cations outside the hydrogel. To monitor this effect, we first programmed a cylindrical hydrogel in a U-shape (Fig. 4). BSA hydrogels were mounted in a U-shape configuration, corresponding to a bending angle of 180° (as defined in Fig. 4, inset), and incubated for 30 min in three different concentrations of Zn2+ (8, 37). The fixity, Rf, which reports on the degree of the programmed hydrogel to maintain its shape when taken out of the mold used during the programming phase, varied from 96 ± 3% in 2 M Zn2+ to 58 ± 15% in 1 M Zn2+ (fig. S2). The fixity of protein hydrogels depends on the amount of stiffening that can be induced when dosing with cations. Hence, the ~17-fold increase in stiffness when incubating BSA hydrogels with 2 M Zn2+ produces a programmed bending degree closer to the U-shape mold than the equivalent ~2-fold stiffening in 1 M Zn2+ (Fig. 4 and fig. S2). These values are comparable with those measured for BSA hydrogels programmed with polyethylenimine (19) and with other polymeric materials using double network or polymer-ion interactions (37). As shown in Fig. 4, when a programmed hydrogel is immersed in regular Tris buffer, the bending angle decreases to a final value of ~45° in ~3 min. The mechanism behind obtaining the temporary shape involves a combination of ionic cross-linking (4) and the stabilization due to divalent cations of BSA domains (38). The shape morphing results from the diffusion of these divalent ions in the surrounding medium (Fig. 4).
Fig. 4. Changes in the hydrogel shape due to cation diffusing outside the hydrogel.
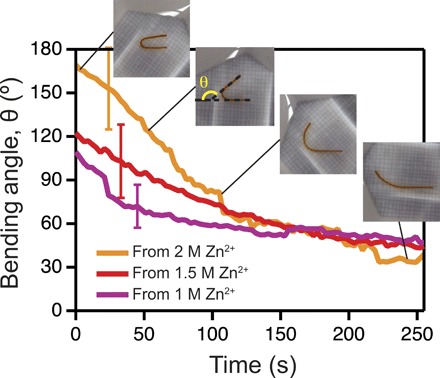
Measured programmed angle of a U-shape gel, θ, as a function of time, upon immersion from Zn2+ into regular Tris buffer. Inset: Pictures of the hydrogel recovering from a U-shape at four different time points. Second inset from the left shows how the angle is measured. The error bars represent SD (n = 3). Movie S3 accompanies this figure. (Photo credit: Luai R. Khoury, UWM; Marina Slawinski, UWM).
Using the large change in stiffness of BSA hydrogels, induced by immersion in Zn2+ and Cu2+ solutions, we programmed cylindrically casted biomaterials into a spring shape and flower-casted materials into a ring shape (Fig. 5, top, and fig. S3). As shown with polyelectrolytes, a ~6.5-fold increase in stiffness already suffices to program BSA hydrogels into a spring shape, and both Zn2+ and Cu2+ induce a strong enough stiffening (up to ~17-fold). The main advantage of the small ions over polyelectrolytes is their diffusion, which happens relatively fast (<5 min). Furthermore, in the present case, the shape morphing is driven by simple diffusion and does not require compromising the primary protein network with the help of a chemical denaturant. Another advantage of the increase in Young’s modulus of BSA hydrogels in the presence of cations is that more complex shapes can be obtained. For example, we demonstrate the morphing from a ring to a flower shape (Fig. 5, bottom, and fig. S3). Compared to the spring shape, in this case, there are no free ends that can release any torsional tension. To obtain this complex shape morphing, we first casted the hydrogel in a flower-like shape using a silicone mold. Following the light-activated cross-linking reaction, we then programmed the hydrogel into a ring shape by mounting it onto a plastic tube, which was then immersed in the cation solution for 30 min. When removed from the plastic tube into a Zn2+ solution with the same concentration, the hydrogel maintains the ring shape (Fig. 5B, bottom left). However, when immersed into a regular Tris buffer, the ring shape quickly morphs into the original flower shape (Fig. 5B, bottom left, and movie S1).
Fig. 5. Programming of protein hydrogels with cations and morphing via chemomechanical changes.
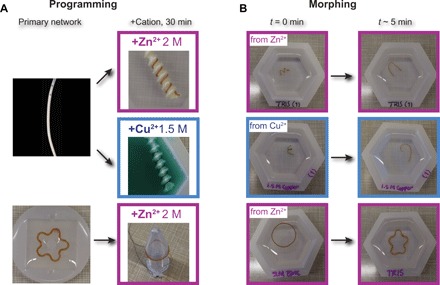
(A) BSA hydrogels were casted in cylindrical shape in PTFE tubes (top left) and programmed in a spring shape using a negative cast, by immersion in 2 M Zn2+ solution (top right) or 1.5 M Cu2+ solution (middle right) for 30 min; BSA hydrogels were produced in a flower shape using a silicone mold (bottom left) and programmed into a ring, by immersion in 2 M Zn2+ solution for 30 min (bottom right). (B) Morphing from the programmed shape into the casted shape of BSA hydrogel upon immersion in regular Tris buffer at time 0 (left) and 5 min (right) for the hydrogels from (A). Movies S1 and S2 accompany this figure. (Photo credit: Luai R. Khoury, UWM; Marina Slawinski, UWM).
CONCLUSIONS
Polymer-based hydrogels have found various applications for shape memory and shape morphing. While these approaches can be made biocompatible, polymers cannot reach the same diversity and control over the sequence and structure as proteins. The approach demonstrated here enables shape morphing in protein-based hydrogels, which could harvest the best of both worlds. This approach relies on the stiffening induced by Zn2+ and Cu2+ to program a permanent shape into a new temporary configuration, and the diffusion of these ions outside the material enables the recovery of the original shape. While we demonstrate this approach with both Zn2+ and Cu2+, we envision that Zn2+ will enable more biologically relevant applications, as it is substantially less toxic than Cu2+.The main advantages of cation-programmed protein hydrogels are that the attainable stiffness is much higher than in regular buffer (~17-fold), enabling programming in complex shapes, and that the fast diffusion of the small ions leads to fast irreversible morphing (<5 min). Furthermore, the shape change is taking place in an aqueous environment at room temperature, which is compatible with conditions present in the human body. Permanent shape morphing based on protein hydrogels and cations could find applications for various implants (39) and injectable hydrogels (40). In addition, the shape morphing method demonstrated here, from a temporary to a permanent profile, does not require denaturation of the tertiary structure of protein domains inside the hydrogels. Hence, this approach allows the preservation of the functionality of proteins forming the skeleton of the hydrogel. In conclusion, the approach presented here provides a remarkable combination of biological diversity and programming capability.
MATERIALS AND METHODS
Materials
BSA [molecular weight (MW), ~66.5 kDa) was purchased from Rocky Mountain Biologicals. Sodium phosphate monobasic anhydrous (NaHPO4) (MW, 119.98 g/mol) was obtained from Research Products International. Sodium chloride (NaCl) (MW, 58.44 g/mol) was purchased from Thermo Fisher Scientific. Ammonium persulfate (APS) [(NH4)2S2O8] (MW, 228.20 g/mol; 1 M) solution, tris(bipyridine) ruthenium(II) chloride {[Ru(bpy)3]2+} (MW, 748.62 g/mol; 6.67 mM) solution, Trizma base NH2C(CH2OH)3 (MW, 121.14 g/mol), hydrochloric acid (HCl) (37%), zinc sulfate monohydrate (ZnSO4·H2O; purity, ≥99.9%) (MW, 179.47 g/mol), copper(ii) chloride dihydrate (CuCl2·2H2O; purity, ≥99.99%) (MW, 170.48 g/mol), and Sigmacote were purchased from Sigma-Aldrich. Tris [20 mM Tris-NaCl and 150 mM NaCl (pH ~7.4)] and phosphate-buffered saline [10 mM NaHPO4 and 150 mM NaCl (pH ~3)] were used as buffers. Double distilled H2O was used in all solution preparations.
Synthesis of BSA-based hydrogels
Three different shapes of BSA-based hydrogels were prepared in this study. First, a BSA-based hydrogel mixture was prepared by mixing 2 mM BSA solution with 1 M APS and 1 M [Ru(bpy)3]2+ in a volume ratio of 15:1:1. To prepare the hydrogels with the cylindrical shape, we followed the same procedure as described in our previous studies (17, 33). Briefly, PTFE tubes (inner diameter, 0.56 mm; Cole-Parmer) were passivated with Sigmacote for 5 to 10 min and dried thoroughly. The solution mix was then injected, and the tube was placed under a light source for 30 min. The bone- and flower-like shape hydrogels were prepared starting from a custom-made silicone rubber mold made of Dragon Skin 30 (purchased from Smooth-On) (fig. S1). The bone-shape hydrogel samples had an overall length of 9 mm. The gauge length and width are 5 and 1 mm, respectively, and the thickness is 1 mm (fig. S1A). The flower-like shape sample has a width of 0.8 mm and a thickness of 1 mm (fig. S1B). Thereafter, the molds were passivated with Sigmacote for 10 min. The hydrogel mixture was casted into the slot and covered with a glass coverslip to reduce evaporation. The loaded molds were then placed under a 100-W mercury lamp for 30 min, after which the hydrogel samples were removed from the molds and immersed in Tris solution.
Mechanical studies
The mechanical investigation of hydrogel samples was carried out using a force-clamp machine, as described in previous studies (17, 33). A force-ramp protocol with a rate of 0.04 kPa/s was applied on the hydrogel sample while immersing in Tris solution at room temperature. Afterward, the hydrogel sample was immersed for 30 min at room temperature into one of two cations solutions: Zn2+ and Cu2+, which were dissolved in phosphate buffer saline at different concentrations. We chose a 30-min incubation time, as for the gels used here (with cylindrical shape and a diameter of 0.56 mm or with a square cross section of 0.5 mm by 0.5 mm), the material fully equilibrates with the solvent environment. Then, same force protocol was applied on the treated hydrogel sample. The Young’s modulus, toughness, and breaking stress and strain were calculated from stress-strain curves.
Shape programming and morphing
The shape programming and morphing of the BSA-based hydrogel samples were performed by starting from the tube or the flower shapes. First, a 2 mM BSA-based hydrogel was synthesized inside the plastic tube or the silicone mold. Then, the hydrogels obtained in the tubes were fixed in a 3D spiral-like shape or U-shape, while the flower-shaped hydrogels were fixed in a circular shape around a 5-ml plastic tube. Following treatment with Zn2+ or Cu2+ for 30 min at room temperature, at various concentrations, the gels were removed from the fixing mold and placed into the same solution buffer. The shape morphing was then induced by transferring the fixed gels from the Zn2+ or Cu2+ solution into a regular tris buffer. Recordings were performed with a Panasonic digital camera, in the time-lapse video mode, with a frame saved every 3 s.
Characterizations
All data acquisition and analysis of the mechanical behavior of hydrogels was performed in Igor Pro (WaveMetrics). All image processing for measuring bending angle of the U-shape gels was accomplished in ImageJ. The stress values were calculated by dividing the measured force (millinewtons) to the cross-section area, while the strain values were obtained from the measured extension, in respect to the tethered gel length.
To quantify the ability of our protein hydrogels to memorize their temporary shape and the dynamics of recovery from the temporary programmed shape into the initial permanent shape, we used the U-shape recovery method (37, 41). In this approach, the hydrogel is programmed into a U-shape, and a bending angle θ is defined as the difference between the original orientation (in our case, 180°) and the programmed orientation (the angle between the arms of the programmed shape). The shape fixity, Rf, reports on how well the temporary programmed shape can memorize the geometry of the casting mold in the presence of cations and was calculated as
where θ0 was the measured bending angle at time zero, right after the gel was removed from the programming process. θp is the expected programmed angle (which, for the U-shape, is 180°). The dynamics of switching from the temporary programmed shape into the initial permanent shape was also quantified by monitoring θ(t), the bending angle of the U-shape hydrogel in tris buffer, at various times t.
Supplementary Material
Acknowledgments
Funding: This research was funded by the National Science Foundation (grant numbers MCB-1846143 and DBI-1919670), the Greater Milwaukee Foundation (Shaw Award), and the University of Wisconsin system (RGI 101X396). M.S. and D.R.C. also acknowledge support from SURF and UR@UWM. Author contributions: L.R.K. and I.P. designed the research. L.R.K., M.S., and D.R.C. performed the research. L.R.K. and I.P. analyzed the data. I.P. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaba6112/DC1
REFERENCES AND NOTES
- 1.Lendlein A., Langer R., Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 296, 1673–1676 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Zhao Q., Behl M., Lendlein A., Shape-memory polymers with multiple transitions: Complex actively moving polymers. Soft Matter 9, 1744–1755 (2013). [Google Scholar]
- 3.Nan W., Wang W., Gao H., Liu W., Fabrication of a shape memory hydrogel based on imidazole-zinc ion coordination for potential cell-encapsulating tubular scaffold application. Soft Matter 9, 132–137 (2013). [Google Scholar]
- 4.Du X., Cui H., Zhao Q., Wang J., Chen H., Wang Y., Inside-out 3D reversible ion-triggered shape-morphing hydrogels. Research SPJ 2019, 6398296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun J.-Y., Zhao X., Illeperuma W. R. K., Chaudhuri O., Oh K. H., Mooney D. J., Vlassak J. J., Suo Z., Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu H., Nan K., Bai W., Huang W., Bai K., Lu L., Zhou C., Liu Y., Liu F., Wang J., Han M., Yan Z., Luan H., Zhang Y., Zhang Y., Zhao J., Cheng X., Li M., Lee J. W., Liu Y., Fang D., Li X., Huang Y., Zhang Y., Rogers J. A., Morphable 3D mesostructures and microelectronic devices by multistable buckling mechanics. Nat. Mater. 17, 268–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ju H. K., Kim S. Y., Lee Y. M., pH/temperature-responsive behaviors of semi-IPN and comb-type graft hydrogels composed of alginate and poly (N-isopropylacrylamide). Polymer 42, 6851–6857 (2001). [Google Scholar]
- 8.Chen F., Yang K., Zhao D., Yang H., Thermal- and salt-activated shape memory hydrogels based on a gelatin/polyacrylamide double network. RSC Adv. 9, 18619–18626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomes C. M., Liu C., Paten J. A., Felton S. M., Deravi L. F., Protein-based hydrogels that actuate self-folding systems. Adv. Funct. Mater. 29, 1805777 (2019). [Google Scholar]
- 10.Lendlein A., Jiang H., Jünger O., Langer R., Light-induced shape-memory polymers. Nature 434, 879–882 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Chen H., Yang F., Chen Q., Zheng J., A novel design of multi-mechanoresponsive and mechanically strong hydrogels. Adv. Mater. 29, 1606900 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Lv S., Dudek D. M., Cao Y., Balamurali M. M., Gosline J., Li H., Designed biomaterials to mimic the mechanical properties of muscles. Nature 465, 69–73 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Fang J., Mehlich A., Koga N., Huang J., Koga R., Gao X., Hu C., Jin C., Rief M., Kast J., Baker D., Li H., Forced protein unfolding leads to highly elastic and tough protein hydrogels. Nat. Commun. 4, 2974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shmilovich K., Popa I., Modeling protein-based hydrogels under force. Phys. Rev. Lett. 121, 168101 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Wu J., Li P., Dong C., Jiang H., Xue B., Gao X., Qin M., Wang W., Chen B., Cao Y., Rationally designed synthetic protein hydrogels with predictable mechanical properties. Nat. Commun. 9, 620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang J., Li H., A facile way to tune mechanical properties of artificial elastomeric proteins-based hydrogels. Langmuir 28, 8260–8265 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Khoury L. R., Nowitzke J., Shmilovich K., Popa I., Study of biomechanical properties of protein-based hydrogels using force-clamp rheometry. Macromolecules 51, 1441–1452 (2018). [Google Scholar]
- 18.Huang W. M., Zhao Y., Wang C. C., Ding Z., Purnawali H., Tang C., Zhang J. L., Thermo/chemo-responsive shape memory effect in polymers: A sketch of working mechanisms, fundamentals and optimization. J Polym. Res. 19, 9952 (2012). [Google Scholar]
- 19.Khoury L. R., Popa I., Chemical unfolding of protein domains induces shape change in programmed protein hydrogels. Nat. Commun. 10, 5439 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma X., Sun X., Hargrove D., Chen J., Song D., Dong Q., Lu X., Fan T.-H., Fu Y., Lei Y., A biocompatible and biodegradable protein hydrogel with green and red autofluorescence: Preparation, characterization and in vivo biodegradation tracking and modeling. Sci. Rep. 6, 19370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong N., Fu L., Peng Q., Li H., Metal chelation dynamically regulates the mechanical properties of engineered protein hydrogels. ACS Biomater. Sci. Eng. 3, 742–749 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez M. A., Simon J. R., Ghoorchian A., Scholl Z., Lin S. T., Rubinstein M., Marszalek P., Chilkoti A., López G. P., Zhao X., Strong, tough, stretchable, and self-adhesive hydrogels from intrinsically unstructured proteins. Adv. Mater. 29, 1604743 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Liang H., Tu C.-Q., Zhang H.-Z., Shen X.-C., Zhou Y.-Q., Shen P.-W., Binding equilibrium study between Mn(II) and HSA or BSA. Chinese J. Chem. 18, 35–41 (2000). [Google Scholar]
- 24.Butkus J. M., O'Riley S., Chohan B. S., Basu S., Interaction of small zinc complexes with globular proteins and free tryptophan. Int. J. Spectrosc. 2016, 12 (2016). [Google Scholar]
- 25.Shen T., Cao Y., Zhuang S., Li H., Engineered bi-histidine metal chelation sites map the structure of the mechanical unfolding transition state of an elastomeric protein domain GB1. Biophys. J. 103, 807–816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beedle A. E. M., Lezamiz A., Stirnemann G., Garcia-Manyes S., The mechanochemistry of copper reports on the directionality of unfolding in model cupredoxin proteins. Nat. Commun. 6, 7894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perales-Calvo J., Lezamiz A., Garcia-Manyes S., The mechanochemistry of a structural zinc finger. J. Phys. Chem. Lett. 6, 3335–3340 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Park P. S.-H., Sapra K. T., Koliński M., Filipek S., Palczewski K., Muller D. J., Stabilizing effect of Zn2+ in native bovine rhodopsin. J. Biol. Chem. 282, 11377–11385 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramanujam V., Kotamarthi H. C., Ainavarapu S. R. K., Ca2+ binding enhanced mechanical stability of an archaeal crystallin. PLOS ONE 9, e94513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanella R., Bazin A., Ta D. T., Nash M. A., Genetically encoded stimuli-responsive cytoprotective hydrogel capsules for single cells provide novel genotype-phenotype linkage. Chem. Mater. 31, 1899–1907 (2019). [Google Scholar]
- 31.Sun F., Zhang W.-B., Mahdavi A., Arnold F. H., Tirrell D. A., Synthesis of bioactive protein hydrogels by genetically encoded SpyTag-SpyCatcher chemistry. Proc. Natl. Acad. Sci. U.S.A. 111, 11269–11274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X., Fang J., Xue B., Fu L., Li H., Engineering protein hydrogels using SpyCatcher-SpyTag chemistry. Biomacromolecules 17, 2812–2819 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Khoury L. R., Nowitzke J., Dahal N., Shmilovich K., Eis A., Popa I., Force-clamp rheometry for characterizing protein-based hydrogels. J. Vis. Exp. 2018, 58280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valle-Orero J., Rivas-Pardo J. A., Popa I., Multidomain proteins under force. Nanotechnology 28, 174003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown A. E. X., Litvinov R. I., Discher D. E., Purohit P. K., Weisel J. W., Multiscale mechanics of fibrin polymer: Gel stretching with protein unfolding and loss of water. Science 325, 741–744 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao X., Multi-scale multi-mechanism design of tough hydrogels: Building dissipation into stretchy networks. Soft Matter 10, 672–687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiao C., Chen Y., Liu T., Peng X., Zhao Y., Zhang J., Wu Y., Wang H., Rigid and strong thermoresponsive shape memory hydrogels transformed from poly(vinylpyrrolidone-co-acryloxy acetophenone) organogels. Acs Appl. Mater. Interfaces 10, 32707–32716 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Liu X., Zhang W., Liu J., Pearce R., Zhang Y., Zhang K., Ruan Q., Yu Y., Liu B., Mg2+ inhibits heat-induced aggregation of BSA: The mechanism and its binding site. Food Hydrocoll. 101, 105450 (2020). [Google Scholar]
- 39.Annabi N., Zhang Y.-N., Assmann A., Sani E. S., Cheng G., Lassaletta A. D., Vegh A., Dehghani B., Ruiz-Esparza G. U., Wang X., Gangadharan S., Weiss A. S., Khademhosseini A., Engineering a highly elastic human protein-based sealant for surgical applications. Sci. Transl. Med. 9, eaai7466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun W., Duan T., Cao Y., Li H., An injectable self-healing protein hydrogel with multiple dissipation modes and tunable dynamic response. Biomacromolecules 20, 4199–4207 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Lendlein A., Kelch S., Shape-memory polymers. Angew. Chem. Int. Ed. Engl. 41, 2035–2057 (2002). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaba6112/DC1