

Keywords: E-cadherin, necrotizing enterocolitis, ROCK inhibitor
Abstract
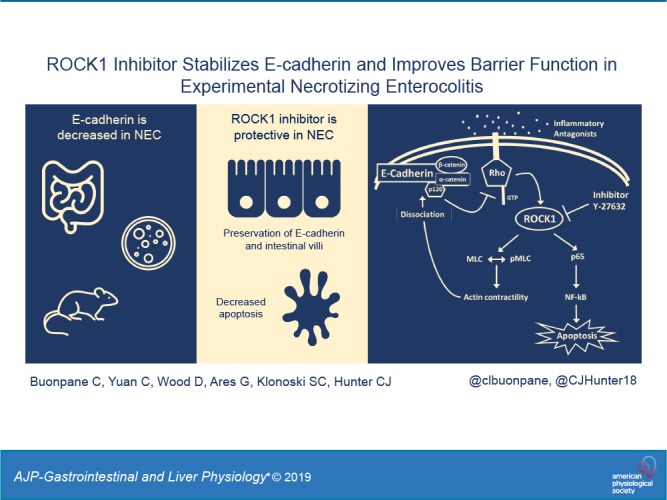
Necrotizing enterocolitis (NEC) is a devastating gastrointestinal disease of newborns. Although incompletely understood, NEC is associated with intestinal barrier dysfunction. E-cadherin, an adherens junction, is a protein complex integral in maintaining normal barrier homeostasis. Rho-associated protein kinase-1 (ROCK1) is a kinase that regulates the E-cadherin complex, and p120-catenin is a subunit of the E-cadherin complex that has been implicated in stabilizing the cadherin complex at the plasma membrane. We hypothesized that E-cadherin is decreased in NEC and that inhibition of ROCK1 would protect against adherens junction disruption. To investigate this, a multimodal approach was used: In vitro Caco-2 model of NEC (LPS/TNFα), rap pup model (hypoxia + bacteria-containing formula), and human intestinal samples. E-cadherin was decreased in NEC compared with controls, with relocalization from the cell border to an intracellular location. ROCK1 exhibited a time-dependent response to disease, with increased early expression in NEC and decreased expression at later time points and disease severity. Administration of ROCK1 inhibitor (RI) resulted in preservation of E-cadherin expression at the cell border, preservation of intestinal villi on histological examination, and decreased apoptosis. ROCK1 upregulation in NEC led to decreased association of E-cadherin to p120 and increased intestinal permeability. RI helped maintain the stability of the E-cadherin-p120 complex, leading to improved barrier integrity and protection from experimental NEC.
NEW & NOTEWORTHY This paper is the first to describe the effect of ROCK1 on E-cadherin expression in the intestinal epithelium and the protective effects of ROCK inhibitor on E-cadherin stability in necrotizing enterocolitis.
INTRODUCTION
Necrotizing enterocolitis (NEC) is a devastating intestinal disease of premature infants (4, 5, 21). Despite decades of research, the complex pathophysiology is incompletely understood, and despite advances in neonatal care, the mortality remains high (21). An intact intestinal epithelial barrier is essential to maintain gastrointestinal homeostasis. Moreover, increased intestinal permeability has been implicated in the pathogenesis of NEC (20). E-cadherin, a type I cadherin cell adhesion molecule, is crucial in maintaining epithelial cell interactions [adherens junctions (AJs)] and the E-cadherin-catenin-cytoskeleton interface. This protein complex serves multiple roles, including control of cell-cell adhesion, barrier function, regulation of the actin cytoskeleton, and intracellular signaling pathways and transcriptional regulation (10, 14, 18). Dysregulation of E-cadherin expression has been associated with a high rate of invasion and metastasis in colorectal cancers (10) and increased intestinal permeability in inflammatory bowel disease (14, 18); however, its role has never been studied in NEC.
The E-cadherin complex is composed of subunits including α-catenin, β-catenin, γ-catenin, and p120-catenin (24). Rho-associated protein kinase (ROCK) is a serine-threonine kinase involved in the regulation of cadherin function. Constitutive activation of ROCK leads to disruption of AJs, whereas pharmacological inhibition of ROCK promotes AJ stability (11, 28). One important mediator of ROCK1/E-cadherin interaction is p120 (24); p120 is implicated in stabilizing the cadherin complex at the plasma membrane (24). However, the interplay among E-cadherin, ROCK1, and p120 is still not well understood. We (11) have previously demonstrated that ROCK inhibition regulates apical tight junction integrity by an occludin-dependent mechanism; however, the effect of ROCK on the AJ was unknown. We hypothesized that inhibition of ROCK1 would promote stabilization of the E-cadherin complex and improve intestinal epithelial barrier function in NEC.
MATERIALS AND METHODS
Bacterial strains and reagents.
Cronobacter sakazakii strain BAA-894 [American Type Culture Collection (ATCC), Manassas, VA] was grown in Luria broth at 37°C overnight for 16 h in a shaker incubator. Bacteria were centrifuged at 3,000 rpm, washed twice, and then reconstituted in 0.9% normal saline (VWR, Radnor, PA). Lipopolysaccharide (LPS) from Escherichia coli, strain 0111:B4 (Sigma, St. Louis, MO) was reconstituted in normal saline at a stock concentration of 5 mg/mL. TNFα (Sigma) was diluted per instructions at a stock concentration of 1 μg/mL. Rho kinase inhibitor Y-27632 (RI) (Sigma, no. Y0503) was suspended in HyClone water (Fisher, Pittsburgh, PA) to a stock concentration of 100 mM for cells.
Cells.
Human colon epithelial cells (Caco-2, passages 19–29, ATCC) were grown in F12-DMEM (Fisher) supplemented with 10% fetal bovine serum and 100 U/mL penicillin and streptomycin. Caco-2 cells were grown to confluence for at least 7 days on either 6- or 24-well transwell membrane system (Corning). Establishment of transepithelial resistance (TEER) was determined by measurement of TEER reaching at least 250 Ω/cm2 using a voltohmmeter (EVOM2; World Precision Instruments, Sarasota FL). Y-27632 was applied apically in the transwell system and incubated for 1 h before treatment with LPS and human TNFα (Fisher Scientific, Waltham, MA). 100 μg/mL LPS, and 10 ng/mL human (h)TNFα was applied both apically and basally for a time course postconfluence. Media and reagents were replenished every other day.
Rat pup model.
Timed pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Roanoke, IL) and induced near term at embryonic day (E)21 with subcutaneous injection of oxytocin 0.1–0.3 units. Rat pups were collected from dams before breast feeding and separated into experimental groups. Pups were gavage fed three times daily with 0.2–0.3 mL of formula (15 g Similac 60/40; Ross Pediatrics, Columbus, OH) in 75 mL of Esbilac canine milk replacer (Peg-AG, Hampshire, IL). Pups in the experimental group were exposed twice daily to hypoxia in a 5% O2-95% N2 modular chamber (Billups-Rothenberg, Del Mar, CA). Formula was constituted for each experimental group as follows: clean (control) formula without additional supplementation, NEC-inducing formula was supplemented with 1 × 108 CFU/mL Cronobacter sakazakii (CS) (1 × 107 CFU per feed), RI formula supplemented with 3 mg/kg RI. The clean group was fed standard clean formula three times daily, the NEC group was fed twice daily with CS formula and once with clean formula, the RI group was fed once daily with RI formula and twice with clean formula, and the RI + NEC group was fed once daily with clean formula with RI, and twice with CS formula. Rat pups were euthanized on day of life 4 or if they displayed signs of severe NEC such as abdominal distension, discoloration, and respiratory distress. Intestines were collected for further analysis. NEC was graded histologically by a pathologist blinded to groups. Each intestinal sample was given a grade based on degree of submucosal edema, neutrophil infiltration, epithelial obliteration, necrosis, and perforation. Grades ranged from 0 (normal) to 3 (severe). Injury scores greater or equal to 1.5 were considered as NEC. Animals were housed in the Northwestern University facilities that were fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All procedures were approved by Northwestern University Institutional Animal Care and Use Committee and conducted in accordance with guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Permeability assays.
Permeability was assessed after experimental time points in Caco-2 cells. Briefly, medium was gently aspirated, and cells were washed twice with Hank’s balanced salt solution (HBSS, Thermo Fisher Scientific). After the wash was completed, cells were replenished with 100 μL of HBSS in the apical chamber and 800 μL of HBSS in the basal chamber; 50 μL of 2.5 mg/mL of Lucifer Yellow (Thermo Fisher Scientific) reconstituted in phosphate-buffered saline (PBS) (Thermo Fisher Scientific) was added to the apical chamber, and cells were incubated for 2 h. The basal chamber was collected, and 100 μL of each sample was assayed in triplicate using a GeminiXS fluorescent plate reader (Molecular Devices, San Jose, CA). Concentration of Lucifer Yellow as a measurement of fluorescent value was previously determined by establishment of a standard curve. Final immunofluorescence value as determined by plate reader was compared with the standard curve.
Rat intestinal permeability was measured using 3-kDa FITC-dextran (Thermo Fisher Scientific). Rat pups were gavage fed with 40 mg/100 g body wt of 3-kDa FITC-dextran reconstituted in PBS. Blood serum was collected 2 h post-FITC-dextran feed. Then, 50 μL of EDTA was added to blood as an anticoagulant and was centrifuged at 3,000 rpm for 5 min to separate serum. Serum was measured in triplicate using the GeminiXS fluorescent plate reader.
Human tissue.
Institutional Review Board approval was obtained (IRB no. 2013-15152) for collection of tissue samples from neonates and infants undergoing bowel resection at Ann and Robert H. Lurie Children’s Hospital of Chicago, IL. All procedures were performed in accordance with the relevant institutional guidelines and regulations. Parental consent was obtained before collection of tissue. Tissue samples were classified as either Control or NEC. Patients identified as the NEC group had Bell stage 3 NEC requiring surgical intervention. Control patients were those undergoing bowel resection for other conditions without active NEC, e.g., ileostomy closure, intestinal atresia, or bowel stricture. Tissue obtained was collected in 10% buffered formalin (Cardinal Health, Dublin, OH), preserved in optimal cutting temperature (OCT; Sakura Finetek, Torrance, CA) at −80°C, and flash-frozen in liquid nitrogen.
Protein and Western blot.
Protein from snap-frozen human and rat intestinal tissues were extracted using a dounce homogenizer. Briefly, tissue was homogenized using cell lysis buffer [20 mmol/L Tris·HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L Na2 EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L glycerophosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin; Cell Signaling Technology, Boston MA) supplemented with 1 mmol/L phenylmethylsulfonyl fluoride, protease inhibitors (1.02 mmol/L 4-(2-aminomethyl) benzenesulfonyl fluoride hydrochloride, 0.0008 mmol/L aprotinin, 0.02 mmol/L leupeptin, 0.04 mmol/L bestatin, 0.015 mmol/L pepstatin A, 0.014 mmol/L E-64], and phosphatase inhibitors (sodium vanadate, sodium molybdate, sodium tartrate, and imidazole; Sigma). Lysates were centrifuged at 14,000 g for 2 min at 4°C. Resulting supernatant was either used immediately or stored at −80°C. Protein from cells was also extracted with cell lysis buffer. Cells were gently scraped from the culture dish and centrifuged at 8,000 g for 15 min at 4°C to pellet, and washed in ice-cold PBS. Complete cell lysis buffer was added, and cells were drawn three times through a 27-gauge needle to homogenize. Conical tubes containing cell mixture were then gently mixed on a rotating platform for 30 min at 4°C. Cells were centrifuged at 12,000 rpm for 15 min at 4°C to isolate the supernatant.
Antibodies.
The following antibodies were used for both Western blots and immunofluorescence: mouse anti-β-actin (1:10,000; Sigma, no. A5441), rabbit anti-villin (Cell Signaling no. R814), rabbit caspase-3 (Cell Signaling no. 9662), mouse anti-E-cadherin (BD Biosciences, San Jose, CA, no. 610182), rabbit anti-ROCK1 (EP786Y; Abcam, Cambridge, MA, no. ab219587), rabbit anti-catenin δ-1 (D7S2M; Cell Signaling, no. 59854), rabbit anti-GAPDH (Cell Signaling, no. 14C10), anti-sodium-potassium-ATPase (Abcam, Cambridge, MA, no. EP1845Y), goat anti-rabbit IgG (H+L) secondary antibody (LifeTechnologies, Carlsbad, CA, no. 31460), goat anti-mouse IgG (H+L) secondary antibody (LifeTechnologies, no. 31430). Alexa fluor 594 goat anti-mouse (Invitrogen, no. A-11032) and Alexa fluor 488 goat anti-rabbit secondary (Invitrogen, no. A-11034) were used as secondary antibodies for immunofluorescence microscopy.
RhoA activation assay.
A RhoA Activation Assay Kit (Cell Biolabs, San Diego, CA, product no. STA-403-A) was used to measure activated RhoA in protein extracts from rat pup tissue. Protein was isolated from quick-frozen tissues as described in the kit protocol with the lysis buffer provided (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP-40, 10 mM MgCl2, 1 mM EDTA, 2% glycerol).
One milligram of protein (determined by Protein Assay Kit II, no. 5000002; Bio-Rad, Hercules, CA) was used in the assay for each experimental condition. Rhotekin RBD agarose beads were added to each sample, rotated at 4°C, centrifuged at 10,000 g for 1 min, and washed three times with the lysis buffer. Forty microliters of 2× sample buffer was added to the washed agarose beads and boiled for 4 min.
Twenty microliters of the supernatant were added to each well of a 15% acrylamide gel. A positive control for RhoA (supplied with the kit), molecular weight markers, the samples from the pull down, and total protein samples for each condition were loaded. The gels were transferred to nitrocellulose, blocked with 5% milk, probed overnight with anti-RhoA (1:200, Bell Biolabs Mouse Monoclonal, part no. 240302) in 2.5% milk, probed with anti-mouse (1:1,000, goat anti-mouse IgG (H+L) secondary antibody, horseradish peroxidase; LIFE, Carlsbad, CA). Clarity Max Western ECL Substrate (Bio-Rad product no. 1705062) was used to detect the RhoA bands at 21 kDa.
Subcellular fractionation and coimmunoprecipitation.
Posttreatment, Caco-2 cells were collected for subcellular fractionation. Cells were gently washed with PBS and were extracted using a ProteoExtract Subcellular Proteome Extraction Kit (Millipore, no. 539791) following the kit’s protocol. The cytosolic and membrane fractions were analyzed by Western blot and probed for E-cadherin. Cytosol and membrane markers were determined by GAPDH and sodium-potassium-ATPase.
Whole Caco-2 cells were cross-linked using a technique adapted from Smith et al. (25). Briefly, cells were washed three times in cold PBS. Cells were then incubated in cross-link solution consisting of 0.5 mM DSP and 0.5 mM DTME and incubated for 30 min while rotating at room temperature. After incubation, 2 mL of quenching solution consisting of 4 mM cysteine in 20 mM Tris (pH 7.4) was added and incubated for 10 min. Cells were then collected and centrifuged at 8,000 rpm for 10 min. Following cross-linking, cells were extracted using RIPA lysis buffer. Protein concentration was assayed, and protein was coimmunoprecipitated with ROCK1, p120, and E-cadherin antibodies overnight on a rotator at 4°C. After incubation, PGAgarose was added to protein and incubated for 2 h before centrifuging at 8,000 rpm for 15 min and washed three times with PBS. Cross-linked lysates were incubated with 50 mM DTT in 3× lysis buffer for 15 min before boiling for 3 min to ensure cleavage of disulfide bonds within the cross-linkers. Protein was then analyzed via Western blot.
Quantitative RT-PCR.
Total RNA was extracted from human intestinal tissue, rat pup intestinal tissue, and cells by use of an RNeasy Mini Kit (Qiagen, Venlo, The Netherlands). RNA concentration was determined using Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA was converted to cDNA using GeneAMP RNA PCR Core Kit (Thermo Fisher Scientific). Quantitative RT-PCR was performed and analyzed with Bio-Rad CFX and iQ SYBR Green Supermix. Primer sequences used for human and Caco-2 cells were GAPDH: forward 5′-ACCACAGTCCATGCCATCAC-3′, reverse 5′-TCCACCACCCTGTTGCTGTA-3′; E-cadherin: forward 5′-CTTTGACGCCGAGAGCTACA-3′, reverse 5′-TTTGAATCGGGTGTCGAGGG-3′; and ROCK1: forward 5′-TTGTTTGAACAGGAAGGCGGA-3′, reverse 5′-GCCCGATGGAGACTTAGCAG-3′. For rat they were GAPDH: forward 5′-ATCACCATCTTCCAGGAGCG-3′, reverse 5′-TTCTGAGTGGCAGTGAGGGC-3′; E-cadherin: forward 5′-TTGAGAATGAGGTCGGTGCC-3′, reverse 5′-CAGAATGCCCTCGTTGGTCT-3′; and ROCK1: forward 5′-CATGCAAGCGCAATTGGTAG-3′, reverse 5′-TAAGGAATGCAGGCAGAACC-3′. Results were normalized to GAPDH. Relative fold changes of genes of interest were analyzed with Bio-Rad CFX software version 3.0 using ΔΔCT calculations.
Immunofluorescence microscopy.
Caco-2 cells were grown on a 24-well transwell system membrane. Animal and human tissues were embedded in paraffin and sectioned at 4 μm. After the slides were baked at 65°C for 1 h, paraffin was removed using SafeClear (Fisher Scientific). Antigen retrieval was performed by steaming slides with eBioscience IHC Antigen Retrieval Solution (Fisher Scientific) for 1 h. Both cells and tissue sections were processed similarly for staining. Slides were gently washed in cold PBS and blocked with 5% normal goat serum in PBS with 0.1% Triton for 30 min at room temperature. Slides were incubated in primary antibody at 1:300 in PBS at 4°C overnight. After incubation, slides were washed with PBS and incubated with secondary antibody at 1:1,000 in PBS for 1 h at room temperature. Slides were then mounted with DAPI fluoroshield (Sigma) and covered with a glass coverslip. An ApopTag Red in Situ Apoptosis Detection Kit (Millipore, Burlington, MA) was used for apoptotic cell labeling; the protocol was performed according to kit instructions. All fluorescent microscopy was performed on a Nikon A1R multiphoton confocal microscope. All images were analyzed using ImageJ version 1.51n.
Statistical analysis.
All experiments were performed in biological triplicates or greater. Graphs and statistical analyses were generated using GraphPad Prism 8 (La Jolla, CA). Statistical analyses of data were performed using parametric and nonparametric tests as appropriate. Differences among groups were considered significant at P < 0.05.
RESULTS
E-cadherin is decreased in NEC.
Both total E-cadherin gene and protein expressions were significantly decreased in both human and experimental (rat) NEC compared with controls (Fig. 1, A–D ). To assess the movement of E-cadherin in the cell during NEC, we assayed E-cadherin expression in the Caco-2 cells. Less E-cadherin was located in the membrane during NEC states than under control conditions (Fig. 1E). Immunofluorescence revealed abundant E-cadherin at the basolateral cell border of healthy human intestinal tissue. However, in NEC, there was a decrease in the amount of E-cadherin present at the cell-cell junction that was associated with a spiculated appearance characteristic of internalization (Fig. 1, F and G). In experimental NEC, Caco-2 cells exhibited a decrease in E-cadherin following 12 h of exposure to LPS (Fig. 1H). Consistent with the reported in vivo findings, immunofluorescence of control (healthy) Caco-2 cells demonstrated a similar pattern to that of control human tissue, characterized by a crisp pattern of E-cadherin staining at the cell-cell border. In contrast, the in vitro NEC model revealed that E-cadherin relocalized from the cell border to an intracellular location (Fig. 1I). Epithelial sloughing is a characteristic feature of advanced NEC, and to control for loss of the epithelium, Western blots were performed with villin (a marker of the epithelium) and β-actin (Fig. 1D).
Fig. 1.

Decreased E-cadherin in necrotizing enterocolitis (NEC). A: qRT-PCR showed lower E-cadherin gene expression in human NEC intestinal samples (n = 5) than in healthy controls (n = 10). B: qRT-PCR showed lower E-cadherin gene expression in experimental rat pup NEC intestinal samples (n = 6) than in controls (n = 8). C: Western blot analysis showing that E-cadherin protein expression is decreased in human NEC vs. controls. Bottom: representative immunoblots probed with antibodies against E-cadherin; blots were normalized to β-actin and analyzed in at least 3 separate experiments. D: Western blot analysis showing E-cadherin protein expression in experimental NEC in rats is decreased compared with controls. Bottom: representative immunoblots probed with antibodies against E-cadherin; blots were normalized to β-actin and villin and repeated in triplicate. E: Caco-2 cells treated with LPS for 24 h were fractionated into subcellular compartments and analyzed with Western blot, demonstrating decreased E-cadherin in the membrane during NEC compared with control. Bottom: representative immunoblots probed with antibodies against E-cadherin; blots were reprobed with antibodies against Na-K-ATPase as a marker for the cell membrane. F: representative immunofluorescence micrographs of human intestinal villi stained for E-cadherin (green) show focal expression at the basolateral cell border in control vs. decreased, spiculated pattern of expression in NEC; nuclei stained with DAPI (blue), scale bar, 10 µm. G: mean fluorescent intensity measured in 15 × 30-µm boxes (2 cells wide) showed decreased E-cadherin fluorescence in human NEC compared with controls. H: qRT-PCR showed lower E-cadherin gene expression in Caco-2 cells treated with LPS for 12 h to 5 days. I: representative immunofluorescence micrographs of Caco-2 cells stained for E-cadherin (green) show high-intensity focal expression at basolateral cell borders in control groups but diminished expression in cells treated with LPS for 24 h; nuclei stained with DAPI (blue). Scale bar, 20 µm. All values are means ± SE (*P < 0.05, **P < 0.01, ***P < 0.001).
ROCK1 is increased early in NEC.
Activation of ROCK1 is associated with loss of cell-to-cell adhesion and increased apoptosis (6). Whereas the Rho signal pathway has been previously identified as a critical player in the development of heart disease, pancreatic fibrosis, and pulmonary hypertension, the effects of ROCK1 in the intestine are less well described. However, ROCK1 was found to induce intestinal barrier damage and mucosal permeability impairment in inflammatory bowel disease (13). On the basis of these data and our prior work on ROCK, we aimed to define the timing of ROCK activation during NEC. In human NEC intestinal samples, we found that ROCK1 was decreased in NEC (Fig. 2C). In addition, immunofluorescence demonstrated that ROCK1 was internalized and scattered within the cytosol of the enterocyte in NEC. This contrasted with the crisp, organized pattern of ROCK1 staining at the cell-cell border in healthy controls (Fig. 2, A and B). Following exposure to NEC-inducing conditions, rat pup intestine was collected, stained, and scored for the degree of intestinal injury and NEC. In rat pups with mild to moderate villous necrosis (injury severity score of 1.5–2), an increase in ROCK1 expression was seen compared with healthy controls. However, pups with severe NEC (injury severity score of 2.5–3) demonstrated ROCK1 expression similar to the baseline levels of controls (Fig. 2, E and F). To examine whether this phenomenon was a result of a time-dependent response to disease, we assayed ROCK1 gene expression at different time points in the in vitro model of NEC. Indeed, we identified that, during earlier time points (4 h), ROCK1 expression was increased in NEC; however, later, at 24 h, ROCK1 gene expression was decreased in NEC (Fig. 2D). We additionally assayed activation of ROCK by using an assay for GTP-bound RhoA. We found increased levels of bound RhoA in rat pups with histologically confirmed NEC versus healthy controls (Fig. 2E).
Fig. 2.

Increased Rho-associated protein kinase-1 (ROCK1) early in necrotizing enterocolitis (NEC). A: immunofluorescence micrographs of human intestinal villi stained for ROCK1 (red) show crisp focal expression at apical and basolateral cell borders in control vs. decreased, disorganized pattern of expression in NEC; nuclei stained with DAPI (blue); ×20 scale bar,100 µm, ×100 scale bar, 20 µm. B: mean fluorescent intensity measured in 15 × 30-µm boxes (2 cells wide) demonstrate decreased ROCK1 fluorescence in human NEC compared with controls. C: qRT-PCR showed lower ROCK1 gene expression in human NEC intestinal samples (n = 5) than in healthy controls (n = 10). D: qRT-PCR showed increased ROCK1 gene expression in Caco-2 cells treated with LPS at 4 h and decreased expression at 24 h. E: pull-down assay (RhoA Activation Assay, Cell Biolabs) of activated RhoA demonstrates an increase in RhoA activation in rat pups with NEC (n = 4). Total RhoA is similar between groups, and all protein was normalized to β-actin. F: qRT-PCR showed increased ROCK1 gene expression in rats with injury severity (IS) scores of 1.5–2 (n = 6) compared with controls (n = 5), and rats with ISscores of 2.5–3 (n = 7) showed lower ROCK1 gene expression than rats with IS scores of 1.5–2. G: representative immunofluorescence micrographs of rat intestinal villi stained for ROCK1 (red) show increased expression in rats with IS scores of 1.5–2; nuclei stained with DAPI (blue), scale bar, 20 µm. All values are means ± SE (*P < 0.05, **P < 0.01, ***P < 0.001).
ROCK1 inhibitor is protective in NEC.
The early activation of ROCK in experimental NEC suggests that ROCK may be an early activator in the cascade. To test whether early administration of RI could protect against experimental NEC, RI was administered to rat pups after induction of NEC on the first experimental day. When RI was given to the experimental NEC rat pups, E-cadherin gene and protein expressions were markedly preserved (Fig. 3, A and B). Immunofluorescence revealed that the RI-treated group had preservation of the crisp E-cadherin staining at the basolateral domain, similar to that of controls (Fig. 3, C and D). Mean fluorescence imaging (MFI) analysis demonstrated significant differences in the intensity of E-cadherin staining. However, MFI alone has its limitations, and these results are best interpreted in conjunction with the IF images themselves.
Fig. 3.

Rho-associated protein kinase-1 (ROCK1) inhibitor (RI; Y-27632) preserves E-cadherin in necrotizing enterocolitis (NEC). A: qRT-PCR showed decreased E-cadherin gene expression in rats with NEC compared with controls. E-cadherin gene expression was higher in rats with RI+NEC than those with NEC (P = 0.056; control, n = 8; RI, n = 9; RI+NEC, n = 9; NEC, n = 6). B: Western blot analysis showing E-cadherin protein expression in experimental NEC in rats is decreased compared with controls. E-cadherin protein expression was higher in rats with RI+NEC than in those with NEC. Bottom: representative immunoblots probed with antibodies against E-cadherin, balanced by protein concentration; all groups with n = 3. C: representative immunofluorescence micrographs of rat intestinal villi stained for E-cadherin (green) show crisp focal expression at basolateral cell borders in control vs. decreased spiculated pattern of expression in NEC. E-cadherin signal and location preserved in RI+NEC; nuclei stained with DAPI (blue), ×20 scale bar, 100 µm, ×100 scale bar, 20 µm, ×500 scale bar, 5 µm. D: mean fluorescent intensity (MFI) measured in 15 × 30-µm boxes (2 cells wide) showed decreased E-cadherin fluorescence in NEC compared with controls, an increase in E-cadherin in RI compared with control, and decreased E-cadherin fluorescence in NEC compared with RI+NEC. All values are means ± SE (*P < 0.05, **P < 0.01, ***P < 0.001).
Rat pups treated with RI, despite exposure to disease-triggers, showed preservation of the intestinal villi on histological examination, whereas experimental NEC pups had severe villous necrosis (Fig. 4A). As anticipated, rat intestinal injury was greater as measured by injury severity scores in NEC pups than in healthy controls. Furthermore, the RI + NEC group had lower injury scores than the NEC group (Fig. 4B). Rat intestinal permeability was significantly increased in NEC. RI treatment corresponded to a significant preservation of intestinal permeability (Fig. 4C). Apoptosis was also significantly increased in NEC; however, RI treatment led to less apoptosis (Fig. 4, D and E).
Fig. 4.

Rho-associated protein kinase-1 (ROCK1) inhibitor (RI; Y-27632) preserves barrier function and prevents apoptosis in necrotizing enterocolitis (NEC). A: hypoxia exposure and gavage feeding of rats with bacteria-inoculated formula led to histological development of NEC compared with control clean-fed rats; rats fed RI, and those treated with both RI and experimental NEC (RI+NEC) showed no histological changes compared with controls; ×20 scale bar, 50 µm. B: intestinal injury was graded histologically based on severity of mucosal erosion, submucosal edema, and derangement of villous architecture; NEC was defined as an injury severity (IS) score of >1.5. Rats exposed to experimental NEC had higher IS scores than control and RI+NEC rats (control, n = 60; RI, n = 19; RI+NEC, n = 18; NEC, n = 70). Values are median ± interquartile range (*P < 0.05, **P < 0.01, ***P < 0.001). C: intestinal permeability was increased in rats with NEC compared with controls and rats in the RI+NEC treatment group. Rats with RI alone had increased intestinal permeability compared with controls (control, n = 8; RI, n = 4; RI+NEC, n = 4; NEC, n = 6). D: representative rat Apoptag immunofluorescence images show minimal apoptosis (red) in control, RI, or RI+NEC rats. Increased apoptosis was seen in NEC rats; nuclei stained with DAPI (blue), ×10 scale bar, 200 µm, ×40 scale bar, 50 µm. E: rat apoptotic cell count was increased in NEC (n = 3 for all groups). Unless otherwise specified, all values are means ± SE (*P < 0.05).
Interaction of E-cadherin, actin cytoskeleton, ROCK1, and p120 in NEC.
E-cadherin is anchored to the actin cytoskeleton, which aids in both stabilization and trafficking of the protein within the intestinal epithelium. Immunofluorescence demonstrated an organized characteristic pattern of both E-cadherin and actin at the cell-cell junctions of healthy cells (Fig. 5A). Conversely in NEC-like states, the actin cytoskeleton was broken down, and E-cadherin appeared to be accumulated in endocytic vesicles. Administration of RI led to preservation of E-cadherin and actin at the cell-cell border.
Fig. 5.

Relationship of actin cytoskeleton, Rho-associated protein kinase-1 (ROCK1), and p120 to E-cadherin in necrotizing enterocolitis (NEC). A: immunofluorescence micrographs of Caco-2 cells stained for E-cadherin (green, top) and actin (red, bottom) show matching expression in all groups; nuclei stained with DAPI (blue), ×100 scale bar, 20 µm. B: representative immunofluorescence micrographs of rat intestinal epithelium stained for ROCK1 (red, top), E-cadherin (green, middle), and composite (yellow, bottom). White arrows indicate areas of colocalization of E-cadherin and ROCK1; nuclei stained with DAPI (blue), ×500 scale bar, 5 µm. C: Western blot analysis showing coimmunoprecipitation of E-cadherin and ROCK1 with ROCK1 and p120 in all 4 experimental groups [control, ROCK1 inhibitor (RI; Y-27632), RI+NEC, NEC]. E-cadherin is associated with p120 but not with ROCK1. ROCK1 is not associated with E-cadherin or p120.
The subcellular location of ROCK1 and E-cadherin were determined in our experimental and control groups by immunofluorescence (Fig. 5B). E-cadherin was found to be well organized along the basolateral border in healthy control intestine, and this was associated with a low ROCK1 signal at the same location. In experimental NEC, ROCK1 epithelial staining was no longer prominently seen at the basolateral region. Instead, an intracellular increase is noted. With administration of RI (without NEC-induction), more ROCK1 was seen intracellularly, and E-cadherin appeared unchanged. When RI was administered to the experimental NEC group, ROCK1 presence at the basolateral border was preserved, as well as the strong E-cadherin presence at the junction, suggesting a stabilization of the AJ.
Coimmunoprecipitation of E-cadherin, ROCK1, and p120 in Caco-2 cells was used to evaluate the interaction of the AJ during NEC conditions. Under control conditions, E-cadherin was strongly associated with p120 but not with ROCK1 (Fig. 5C). RI increased E-cadherin complex stability even in the setting of experimental NEC. ROCK1 did not associate with either E-cadherin or p120, suggesting that ROCK1 stimulates actin contraction causing E-cadherin dissociation without, however, direct binding to E-cadherin or p120.
DISCUSSION
The intestinal epithelium stands as a crucial component of the body’s protection against the millions of microorganisms in the luminal contents of the intestine. Breakdown of that barrier leads to bacterial translocation and extensive inflammatory reaction in the lamina propria (17). Tight junctions and adherens junctions serve to establish an intact intestinal barrier, preventing hyperpermeability. Disruption of tight junctions predisposes the host to bacterial translocation, which has been implicated in the pathogenesis of NEC (1, 4, 7). However, the role of E-cadherin has not yet been studied in NEC. Through investigation of the role of E-cadherin in NEC, we found E-cadherin to be significantly decreased in NEC, resulting in increased intestinal permeability.
E-cadherin serves as a potent cell adhesion molecule and determinant of cell polarity. In normal intestinal epithelium, E-cadherin has a strong, uniform basolateral expression (10). Studies looking at intestinal reconstitution after mucosal injury found that E-cadherin is decreased in regenerating epithelium (12). It is believed that loss of expression and/or function of E-cadherin allows regenerating cells to dedifferentiate and lose cell-cell cohesiveness, allowing for epithelial regeneration (12). However, this loss of cell-cell adhesion further impairs the integrity of the mucosal barrier, allowing exposure of luminal contents to the immune system (18). Our results match those of studies done in inflammatory bowel disease and colon cancer, showing decreased E-cadherin at the basolateral cell border in the diseased (NEC) state.
ROCK (Rho-associated kinase) is an important signal molecule downstream of the Rho kinase signal pathway. Inflammatory agonists activate Rho kinases, which in turn signals ROCK to phosphorylate the myosin light chain, causing contraction of the actin cytoskeleton. This actin polymerization leads to dissociation of the E-cadherin complex (16). Therefore, we hypothesized that ROCK1 would be increased in NEC. We first examined human NEC samples and found ROCK1 expression levels to be significantly decreased compared with healthy controls. However, in our rat pups with NEC, ROCK1 expression was increased. Intrigued by this discrepancy, we analyzed ROCK1 expression by rat pup severity score. Rat pups with injury severity scores (IS) of 1.5–2 (mild villous necrosis) showed high expression of ROCK1, whereas pups with IS of 2.5–3 (severe villous necrosis) had decreased ROCK1 expression. Additionally, we found the same pattern in Caco-2 cells, with an early increase in ROCK1 at 4 h and then a significant decrease in the expression by 24 h. As a result of these findings, we believe that ROCK1 plays a role in the early phases and development of NEC, as an early surge, as inflammatory mediators activate the Rho kinase pathway. However, ROCK1 is likely an important early mediator in NEC but not a key factor in the continued propagation and progression of the disease.
Knowing that ROCK1 is increased early in NEC, we sought to understand the effects of RI in NEC. To do so, we administered RI in our rat pups subjected to experimental NEC conditions. We found that E-cadherin expression and localization were preserved with early administration of RI. Additionally, intestinal villous architecture was preserved, barrier function was maintained, and levels of apoptosis were significantly decreased with administration of RI to our experimental NEC pups. We know that ROCK1 activates p65 through phosphorylation, leading to upregulation of NF-κB and increased apoptosis (6). It should be noted that ROCK1 is a substrate of caspase-3 and that apoptotic singling may itself be responsible, at least in part, for ROCK1 activation (9). Furthermore, inhibition of ROCK1 disrupts the upregulation of p65 and decreases NF-κB activity, leading to decreased apoptosis and preservation of E-cadherin complexes in the membrane. Since 1995, RI has been used in Japan to prevent vasospasm associated with subarachnoid hemorrhage (23). Additional human trials have since been carried out using the drug for several cardiovascular indications. Many recent studies are evaluating RI as a potential target for treatment of a wide range of diseases, including cancer, neuronal degeneration, kidney failure, asthma, glaucoma, osteoporosis, erectile dysfunction, insulin resistance, corneal wound healing, and hepatic fibrosis (15, 22, 23, 26, 29). Given the safety and efficacy of this drug in humans, and favorable experimental animal studies, RI is a promising target for prevention and early treatment of NEC.
Although our data show that E-cadherin is decreased in NEC, the complex regulation of E-cadherin and its dynamic turnover at the cell surface are yet to be fully elucidated. We know that E-cadherin leads a dynamic existence, with constant turnover via both endocytic and exocytic pathways (8). This junctional turnover is required for cell-cell adhesion, and lack of proper trafficking can lead to barrier breakdown. Additionally, E-cadherin is anchored to the actin cytoskeleton through catenins, allowing for epithelial stabilization or further trafficking (27). In NEC, it is likely that the E-cadherin complex is destabilized, downregulated, and trafficked within the cell during NEC (24). This destabilization is due in part to activation of the RhoA pathway, leading to activation of ROCK1, causing actin contraction resulting in the dissociation of E-cadherin from the membrane and subunits (Fig. 6). From our coimmunoprecipitation data, we do not believe that E-cadherin and ROCK1 are bound to one another, rather that ROCK1 activation leads to E-cadherin dissociation via actin cytoskeleton contraction. On the basis of the presented coimmunoprecipitation data, we examined the role of p120 in the regulation of E-cadherin stability. We found that E-cadherin and p120 were structurally associated and that administration of RI alone led to a significant increase in this association. Inhibition of ROCK1 activation would result in less E-cadherin dissociation at the membrane, causing the bond between E-cadherin and p120 to remain strong. When the E-cadherin complex does dissociate, as described above, p120 splits away from the complex. Unbound cytosolic p120 then directly or indirectly inhibits RhoA activity (2, 3, 19). Therefore, regulation of p120 could also serve as a potential target for NEC therapy. Further research is needed to unfold the complex interactions between the E-cadherin complex, ROCK1, the actin cytoskeleton, and p120 in NEC.
Fig. 6.

Proposed mechanism of Rho-associated protein kinase-1 (ROCK1) regulation of E-cadherin. Inflammatory antagonists (such as LPS and TNFα) are recognized by the cell and activate Rho kinases, which in turn activate ROCK1. ROCK1 stimulates myosin light chain (MLC) phosphorylation and causes actin contraction, leading to E-cadherin complex dissociation. p120 splits from the E-cadherin complex and inhibits Rho kinase activity. ROCK1 inhibitor (RI; Y-27632), inhibits ROCK1, disrupting upregulation of p65, leading to decreased NF-κB activity and decreased apoptosis. Inhibition of ROCK1 also leads to preservation of the E-cadherin complex stability in the membrane.
GRANTS
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Disease Grants K08 DK-106450 and R03 DK-117216 and the Jay Grosfeld Award from the American Pediatric Surgical Association to C. Hunter. Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by National Cancer Institute Cancer Center Support Grant P30 CA-060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Multiphoton microscopy was performed on a Nikon A1R multiphoton microscope, acquired through the support of National Institutes of Health Grant 1S10 OD-010398–01.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.B., C.Y.Y., and C.J.H. conceived and designed research; C.B., C.Y.Y., D.R.W., G.A., and S.C.K. performed experiments; C.B., C.Y.Y., D.R.W., and S.C.K. analyzed data; C.B., C.Y.Y., D.R.W., G.A., S.C.K., and C.J.H. interpreted results of experiments; C.B., C.Y.Y., S.C.K., and C.J.H. prepared figures; C.B. and C.Y.Y. drafted manuscript; C.B., C.Y.Y., and C.J.H. edited and revised manuscript; C.B., C.Y.Y., D.R.W., G.A., and C.J.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Nikon A1R: https://cam.facilities.northwestern.edu.
REFERENCES
- 1.Anand RJ, Leaphart CL, Mollen KP, Hackam DJ. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock 27: 124–133, 2007. doi: 10.1097/01.shk.0000239774.02904.65. [DOI] [PubMed] [Google Scholar]
- 2.Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120 catenin. Nat Cell Biol 2: 637–644, 2000. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- 3.Anastasiadis PZ, Reynolds AB. Regulation of Rho GTPases by p120-catenin. Curr Opin Cell Biol 13: 604–610, 2001. doi: 10.1016/S0955-0674(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 4.Ares G, Buonpane C, Sincavage J, Yuan C, Wood DR, Hunter CJ. Caveolin 1 is associated with upregulated claudin 2 in necrotizing enterocolitis. Sci Rep 9: 4982, 2019. doi: 10.1038/s41598-019-41442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ares GJ, Buonpane C, Yuan C, Wood D, Hunter CJ. A novel human epithelial enteroid model of necrotizing enterocolitis. J Vis Exp 146: e59194, 2019. doi: 10.3791/59194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aznar S, Lacal JC. Rho signals to cell growth and apoptosis. Cancer Lett 165: 1–10, 2001. doi: 10.1016/S0304-3835(01)00412-8. [DOI] [PubMed] [Google Scholar]
- 7.Bergmann KR, Liu SX, Tian R, Kushnir A, Turner JR, Li HL, Chou PM, Weber CR, De Plaen IG. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am J Pathol 182: 1595–1606, 2013. doi: 10.1016/j.ajpath.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends Cell Biol 14: 427–434, 2004. doi: 10.1016/j.tcb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci USA 103: 14495–14500, 2006. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doğan A, Wang ZD, Spencer J. E-cadherin expression in intestinal epithelium. J Clin Pathol 48: 143–146, 1995. doi: 10.1136/jcp.48.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grothaus JS, Ares G, Yuan C, Wood DR, Hunter CJ. Rho kinase inhibition maintains intestinal and vascular barrier function by upregulation of occludin in experimental necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 315: G514–G528, 2018. doi: 10.1152/ajpgi.00357.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanby AM, Chinery R, Poulsom R, Playford RJ, Pignatelli M. Downregulation of E-cadherin in the reparative epithelium of the human gastrointestinal tract. Am J Pathol 148: 723–729, 1996. [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Xiao S, Jiang Q. Role of Rho kinase signal pathway in inflammatory bowel disease. Int J Clin Exp Med 8: 3089–3097, 2015. [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang S, Zimmerman NP, Agle KA, Turner JR, Kumar SN, Dwinell MB. E-cadherin is critical for collective sheet migration and is regulated by the chemokine CXCL12 protein during restitution. J Biol Chem 287: 22227–22240, 2012. doi: 10.1074/jbc.M112.367979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lane J, Martin TA, Watkins G, Mansel RE, Jiang WG. The expression and prognostic value of ROCK I and ROCK II and their role in human breast cancer. Int J Oncol 33: 585–593, 2008. [PubMed] [Google Scholar]
- 16.Lecuit T, Yap AS. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol 17: 533–539, 2015. doi: 10.1038/ncb3136. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Li N, Neu J. Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr 94: 386–393, 2005. doi: 10.1111/j.1651-2227.2005.tb01904.x. [DOI] [PubMed] [Google Scholar]
- 18.Mehta S, Nijhuis A, Kumagai T, Lindsay J, Silver A. Defects in the adherens junction complex (E-cadherin/β-catenin) in inflammatory bowel disease. Cell Tissue Res 360: 749–760, 2015. doi: 10.1007/s00441-014-1994-6. [DOI] [PubMed] [Google Scholar]
- 19.Menke A, Giehl K. Regulation of adherens junctions by Rho GTPases and p120-catenin. Arch Biochem Biophys 524: 48–55, 2012. doi: 10.1016/j.abb.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 20.Moore SA, Nighot P, Reyes C, Rawat M, McKee J, Lemon D, Hanson J, Ma TY. Intestinal barrier dysfunction in human necrotizing enterocolitis. J Pediatr Surg 51: 1907–1913, 2016. doi: 10.1016/j.jpedsurg.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niño DF, Sodhi CP, Hackam DJ. Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol 13: 590–600, 2016. doi: 10.1038/nrgastro.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okumura N, Koizumi N, Kay EP, Ueno M, Sakamoto Y, Nakamura S, Hamuro J, Kinoshita S. The ROCK inhibitor eye drop accelerates corneal endothelium wound healing. Invest Ophthalmol Vis Sci 54: 2493–2502, 2013. doi: 10.1167/iovs.12-11320. [DOI] [PubMed] [Google Scholar]
- 23.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol 20: 242–248, 2008. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith AL, Dohn MR, Brown MV, Reynolds AB. Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol Biol Cell 23: 99–110, 2012. doi: 10.1091/mbc.e11-06-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith AL, Friedman DB, Yu H, Carnahan RH, Reynolds AB. ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PLoS One 6: e16206, 2011. doi: 10.1371/journal.pone.0016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tada S, Iwamoto H, Nakamuta M, Sugimoto R, Enjoji M, Nakashima Y, Nawata H. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in rats. J Hepatol 34: 529–536, 2001. doi: 10.1016/S0168-8278(00)00059-3. [DOI] [PubMed] [Google Scholar]
- 27.Vasioukhin V, Fuchs E. Actin dynamics and cell-cell adhesion in epithelia. Curr Opin Cell Biol 13: 76–84, 2001. doi: 10.1016/S0955-0674(00)00177-0. [DOI] [PubMed] [Google Scholar]
- 28.Wójciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 114: 1343–1355, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita K, Kotani Y, Nakajima Y, Shimazawa M, Yoshimura S, Nakashima S, Iwama T, Hara H. Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res 1154: 215–224, 2007. doi: 10.1016/j.brainres.2007.04.013. [DOI] [PubMed] [Google Scholar]