

Abstract
Coronavirus disease 2019 (COVID-19) is caused by SARS-CoV-2 infection and was first reported in central China in December 2019. Extensive molecular surveillance in Guangdong, China’s most populous province, during early 2020 resulted in 1,388 reported RNA-positive cases from 1.6 million tests. In order to understand the molecular epidemiology and genetic diversity of SARS-CoV-2 in China, we generated 53 genomes from infected individuals in Guangdong using a combination of metagenomic sequencing and tiling amplicon approaches. Combined epidemiological and phylogenetic analyses indicate multiple independent introductions to Guangdong, although phylogenetic clustering is uncertain because of low virus genetic variation early in the pandemic. Our results illustrate how the timing, size, and duration of putative local transmission chains were constrained by national travel restrictions and by the province’s large-scale intensive surveillance and intervention measures. Despite these successes, COVID-19 surveillance in Guangdong is still required, because the number of cases imported from other countries has increased.
Keywords: COVID-19, SARS-COV-2, genomic epidemiology, phylogenetics, virus evolution, real-time disease surveillance
Graphical Abstract
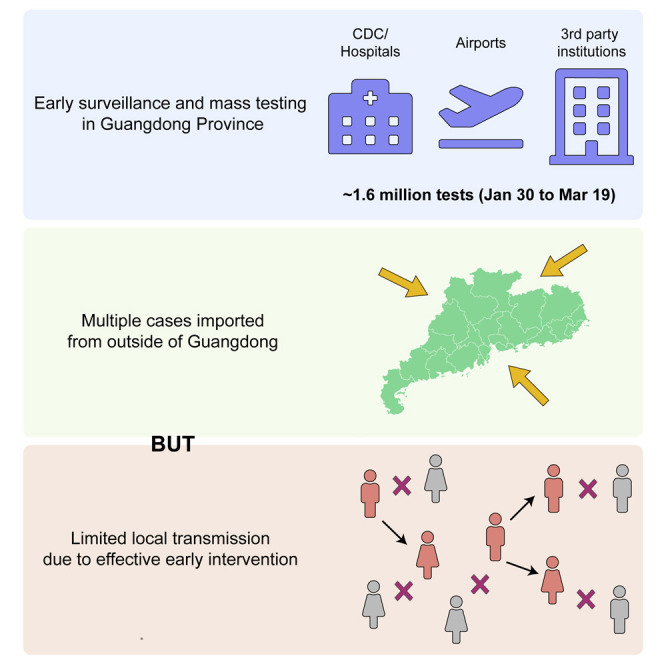
Highlights
-
•
1.6 million tests identified 1,388 SARS-CoV-2 infections in Guangdong by 19 March
-
•
Virus genomes can be recovered using a variety of sequencing approaches
-
•
Analyses reveal multiple viral importations with limited local transmission
-
•
Effective control measures helped reduce and eliminate chains of viral transmission
Genomic and epidemiological analyses provide insights into how COVID-19 was contained in China’s most populous province using a combination of surveillance and travel restriction measures.
Introduction
A new virus-associated disease, coronavirus disease 2019 (COVID-19), was initially reported in China on 30th December 2019 (Wu et al., 2020). The causative agent of COVID-19 is the novel human coronavirus SARS-CoV-2 (Wu et al., 2020, Zhou et al., 2020), and, as of 24th March 2020, there have been 372,757 confirmed infections and 16,231 deaths reported worldwide (World Health Organization, 2020). In China, the COVID-19 epidemic grew exponentially during January 2020, peaking on 12th February 2020 with 15,153 newly confirmed cases per day. One month later, reported COVID-19 cases in China dropped to ~20 per day, indicating the epidemic there was contained. However, the number of cases reported outside of China has risen exponentially since the second half of February 2020. By 11th March 2020, the day that the World Health Organization (WHO) announced COVID-19 to be a new pandemic, 37,371 cases had been reported outside of China (World Health Organization, 2020).
Guangdong Province and the Pearl River Delta Metropolitan Region contain some of the world’s largest and most densely populated urban areas. Guangdong is the most populous province of China (113 m people) and contains many large cities including Guangzhou (12 m), Shenzhen (10 m), Dongguan (8 m), and Foshan (7 m). The province has strong transportation links to Hubei Province, where the first cases of COVID-19 were reported. The Wuhan-Guangzhou high-speed railway has been estimated to transfer 0.1–0.2 million passengers per day during the spring festival period, which started on 10th January 2020. By 19th March 2020, Guangdong had 1,388 confirmed cases of COVID-19, the highest in China outside of Hubei Province.
Understanding the evolution and transmission patterns of a virus after it enters a new population is crucial for designing effective strategies for disease control and prevention (Faria et al., 2018, Grubaugh et al., 2017, Ladner et al., 2019). In this study, we combine genetic and epidemiological data to investigate the genetic diversity, evolution, and epidemiology of SARS-CoV-2 in Guangdong Province. We generated virus genome sequences from 53 patients in Guangdong using both metagenomic sequencing and multiplex PCR amplification followed by nanopore sequencing. Through phylogenetic analysis, interpreted in the context of available epidemiological information, we sought to investigate the timing and relative contributions of imported cases versus local transmission, the nature of genetically distinct transmission chains within Guangdong, and how the emergency response in Guangdong was reflected in the reduction and elimination of these transmission chains. Our results may provide valuable information for implementing and interpreting genomic surveillance of COVID-19 in other regions.
Results
Enhanced surveillance was launched in all clinics in Guangdong province following the first reports of patients with undiagnosed pneumonia on 30th December 2019. Initially, screening and sampling for SARS-CoV-2 was targeted toward patients with fever and respiratory symptoms and those who had a history of travel in the 14 days before the date of symptom onset. The first detected case in Guangdong had symptom onset on 1st January and was reported on 19th January 2020 (Kang et al., 2020). COVID-19 cases in Guangdong grew until early February 2020 (peaking at >100 cases per day) and declined thereafter (Figure 1 A). After 22nd February 2020, the daily number of locally infected reported cases in Guangdong did not exceed one. However, since the beginning of March 2020 COVID-19 cases imported into Guangdong from abroad have been detected with increasing frequency. As of 26th March 2020, a total of 102 imported cases were reported from 19 different countries (Figure 1A), highlighting the risk that local COVID-19 transmission could reignite in China.
Figure 1.

Summary of the COVID-19 Epidemic in Guangdong Province, China
(A) Time series of the 1,388 laboratory-confirmed COVID-19 cases in Guangdong until 19th March 2020, by date of onset of illness. Cases are classified according to their likely exposure histories (see inset and main text). The dashed lines indicate the date the first Guangdong case was detected (19th January 2020) and the shutdown of travel from Wuhan (23rd January 2020). An overview of testing and surveillance strategies at different stages of the epidemic is illustrated below the time series, on the same timescale.
(B) Geographic distribution of COVID-19 cases and human population density among the 21 prefecture-level divisions of Guangdong Province.
See also Figure S1.
Different surveillance strategies were applied during the epidemic in Guangdong (Figure 1A; STAR Methods). More intensive surveillance was initiated on 30th January 2020 in response to the Spring Festival period, which results in greater mobility among regions and provinces in China (Kraemer et al., 2020, Tian et al., 2020) and because asymptomatic COVID-19 cases had been reported (Guan et al., 2020). This included monitoring (1) all travelers returning from Hubei or other regions with high epidemic activity, (2) their close contacts, and (3) all hospitalized patients in clinics, including those without fever or respiratory symptoms, regardless of their exposure history. Approximately 1.35 million samples were screened by six third-party institutions between 30th January and 15th March 2020. Surveillance commenced at Guangdong airports in early March, following the growth of COVID-19 outbreaks outside of China. In total, ~1.6 million tests were performed by 19th March 2020, identifying 1,388 SARS-CoV-2 positive cases in 20 of 21 prefectures in Guangdong Province (Figure 1B). Around a quarter of cases (336) were judged to be linked to local transmission and two-thirds (1,014) had a likely exposure history in Hubei (see STAR Methods). For locally infected cases, 181 (53%) were linked to transmission among household members. More than half of the reported cases (60%) were from the cities of Shenzhen and Guangzhou (Figure 1B). We note that the number of detected cases will be less than the true number of infections, although the degree of under-reporting is unknown. Surveillance was targeted toward travelers, hence these data may overestimate the proportion of travel-associated cases.
To understand the genetic structure of the COVID-19 outbreak in Guangdong, we generated near-complete and partial genomes from 53 COVID-19 patients in Guangdong Province. The genomes were generated by a combination of metagenomic sequencing and multiplex PCR amplification followed by nanopore sequencing on a MinION device (see STAR Methods). Sequence sampling dates ranged from 30th January to 28th February 2020 (Figure S1 ).
Figure S1.

Time Series of Reported Cases and Sample Collection Dates, Related to Figure 1
(A) Time series of the 1388 laboratory-confirmed COVID-19 cases in Guangdong until 19th March, by date of onset of illness. Cases are classified according to their likely exposure histories (see inset). The solid line indicates the cumulative number of cases and the dashed lines indicate the date the first case was detected in Guangdong (19th January) and the shutdown of travel from Wuhan (23rd January). (B) Time series of the 53 SARS-CoV-2 genomes we report, by collection date. Genomes are classified according to patients’ likely exposure history. The collection dates of 17 previously released genomes sampled from patients in Guangdong are also shown.
Sequencing was performed on 79 clinical samples (throat swabs, n = 32; anal swabs, n = 24; nasopharyngeal swabs, n = 10; sputum, n = 13) collected from 62 patients with varying disease symptoms, ranging from asymptomatic to very severe (see STAR Methods). Real-time reverse transcription PCR Ct (cycle threshold) values of these samples ranged from 19 to 40.86. Figure 2 A displays the Ct values for the 53 samples with >50% genome coverage for which we report whole and partial genome sequences (see Figure S2 for details of all 79 samples). When Ct values are <30, sequence reads covered ~90% or more of the reference genome (GenBank: MN908947.3) irrespective of the amplification and sequencing approach used (see STAR Methods). However, genome coverage declined for samples with Ct >30 (Figure S2). Using a Kruskal-Wallis rank-sum test we found an association between sample Ct values and sample type (Figure 2B; p < 0.001), and sample Ct values and disease severity (Figure 2C; p = 0.03) (see also Liu et al., 2020), however, we note that sampling was not undertaken with these hypotheses in mind.
Figure 2.

Profile of SARS-CoV-2 Genome Sequences from Guangdong Province, China
(A) Plot of SARS-CoV-2 genome coverage against real-time reverse transcription Ct value for the 53 genome sequences reported here. Each sequence is colored by sequencing approach: blue, BGI metagenomic sequencing; orange, multiplex PCR nanopore sequencing; green, Illumina metagenomic sequencing.
(B) Real-time reverse transcription PCR Ct values for different sample types.
(C) Real-time reverse transcription PCR Ct values for samples from patients with different disease severity; the “mild” category includes 2 asymptomatic cases.
(D) Genome coverage map for the 53 genomes reported here, ordered by % genome coverage. Single nucleotide polymorphisms (with respect to the reference genome MN908947.3) are colored in red. Each genome is colored according to the sequencing approach used.
(E) Genomic structure of SARS-CoV-2 and the genomic location and frequency of single nucleotide polymorphisms (with respect to the reference genome MN908947.3) among our 53 sequences. These mutations correspond to the red lines in (D).
Figure S2.

Plots of SARS-CoV-2 Genome Coverage against RT-PCR Ct Value and the Number of Mapped Reads for 104 Sequencing Runs Performed on 79 Clinical Samples, Related to Figure 2
Plots of SARS-CoV-2 genome coverage against RT-PCR Ct Value (A) and the number of mapped reads (B). Each sequence is colored by sequencing approach: blue = multiplex PCR nanopore sequencing, green = BGI metagenomic sequencing, orange = Illumina metagenomic sequencing. Open circles indicate sequences that were not reported here or used in phylogenetic analyses, either because of insufficient coverage, or because a higher-quality sequence existed for the same patient.
Sequences generated with nanopore sequencing indicate common regions of low coverage (Figure 2D), indicating that the version 1 primer set used here did not amplify some regions efficiently. Efficient primer binding may have been prevented due to genetic divergence from the reference genome (MN908947.3). An alternative explanation is the interaction between two particular primers, resulting in primer dimer formation (Itokawa et al., 2020). After completion of this study, the primers have been redesigned to address these issues and improve coverage (Quick 2020). Shared and unique single nucleotide polymorphisms (SNPs) were observed at 97 sites across the SARS-CoV-2 genome (Figures 2D and 2E), with 77 SNPs present in only one genome. Three SNPs were present in >10 genomes: (C8782T, C21711T, and T28144C). When compared to 49 previously released genomes from Hubei and Guangdong, 118 SNPs are present in only one genome and these three SNPs are still the only variants shared among >10 genomes (Data S1).
To understand the genetic diversity of the SARS-CoV-2 epidemic in Guangdong, we performed phylogenetic analyses using maximum likelihood and Bayesian molecular clock approaches. We added our new virus genomes from Guangdong to 177 publicly available sequences, which includes 73 sequences from China, 17 of which were previously reported Guangdong genomes. The final alignment comprised 250 sequences and increased the number of SARS-CoV-2 sequences from China by ~60% when our data were submitted to GISAID (on 9th March).
The estimated maximum likelihood (ML) phylogeny is shown in Figure 3 A. The SARS-CoV-2 sequences from Guangdong (red) are interspersed with viral lineages sampled from other Chinese provinces and other countries (gray). This pattern agrees with the epidemiological time series in Figure 1A, indicating that most detected cases were linked to travel rather than local community transmission. Despite this, there were a number of instances where sequences from Guangdong appeared to cluster together, sometimes with sequences sampled from other regions. To explore these lineages in more detail, we performed a Bayesian molecular clock analysis that places the phylogenetic history of the genomes on an estimated timescale. A summary visualization of the maximum clade credibility tree from that analysis is shown in Figure 3B and is largely congruent with the ML tree. The current low genetic diversity of SARS-CoV-2 genomes worldwide means that most internal nodes have very low posterior probabilities; we caution that no conclusions should be drawn from these branching events as they will be informed by the phylogenetic prior distribution rather than variable nucleotide sites (Figure 3A). Nevertheless, five clusters (denoted A–E) containing Guangdong sequences had posterior probability support of >80% (i.e., their sequences grouped monophyletically in >80% of trees in the posterior sample; Figure 3B). These clusters were also observed in the ML phylogeny (Figure 3A). Some included only sequences sampled in Guangdong (A, B), others included sequences sampled in other countries and provinces (C, D, E).
Figure 3.

Phylogenetic Analyses of SARS-CoV-2 Genome Sequences from Guangdong Province, China
(A) Estimated maximum likelihood phylogeny of SARS-CoV-2 sequences from Guangdong (red circles) and genomes from other countries and provinces (not circled). The axis is in units of nucleotide changes from the inferred root sequence. A phylogenetic bootstrap analysis was not performed due to the low number of phylogenetically informative sites and the number of missing bases (N) in the alignment. The position of clusters A–E discussed in main text are highlighted with red boxes and labeled.
(B) Visualization of the corresponding time-scaled maximum clade credibility tree. Sequences from Guangdong and their terminal branches are in red and those from other locations in gray. The clusters (A–E) discussed in main text are highlighted with boxes and labeled. All nodes with posterior probabilities <0.5 have been collapsed into polytomies and their range of divergence dates are illustrated as shaded gray expanses.
From the molecular clock analysis, we were able to estimate the times of the most recent common ancestor (tMRCA) of clusters A–E. We found that SARS-CoV-2 lineages were imported multiple times into Guangdong during the second half of January 2020 (Figure 4 ). Three clusters (C, D, E) have earlier tMRCAs that coincide with the start of the Guangdong epidemic and two (A, B) have later tMRCAs, around the time of the epidemic peak in the province (Figure 4). The average time between the tMRCA and the earliest sequence collection date in each cluster was approximately 10.25 days. The observed duration of each phylogenetic cluster (tMRCA to most recently sampled sequence) ranged from 9.49 (cluster B) to 45.2 (cluster D) days. The clusters with earlier tMRCAs contain more sequences from travelers sampled outside of China, possibly reflecting a decrease in air passenger travel from Guangdong after January 2020 (Flightradar24, 2020). The median tMRCA estimate of the COVID-19 pandemic was 1st December 2019 (95% HPD 15th November to 13th December 2019; Figure 4), consistent with previous analyses (Rambaut, 2020).
Figure 4.

Molecular Clock Analysis of the Five Phylogenetic Clusters of Guangdong Sequences that Were Supported with Posterior Probabilities >80% in Bayesian Phylogenetic Analysis
(A) Daily number of local and imported COVID-19 cases in Guangdong province. The first reported case in Guangdong (January 19) and the shutdown of travel from Wuhan (January 23) are indicated by dashed lines.
(B) Posterior distributions of the tMRCAs of the five phylogenetic clusters (A–E) from the molecular clock analysis (Figure 3B). Distributions are truncated at the upper and lower limits of the 95% HPD intervals; the vertical red lines indicate median estimates. Blue shading and horizontal red lines indicate the sampling period over which genomes in each cluster were collected. Dots indicate the collection dates of genomes, colored by sampling location (red, Guangdong; gray, other).
See also Table S1.
The apparent clusters of Guangdong sequences require careful interpretation because of the relative undersampling of SARS-CoV-2 genomes from other Chinese provinces, including Hubei. Specifically, it is known that undersampling of regions with high incidence can lead to phylogenetic analyses underestimating the number of introductions into recipient locations and overestimating the size and duration of transmission chains in those recipient locations (Grubaugh et al., 2017, Kraemer et al., 2018). For example, the largest Guangdong phylogenetic cluster (denoted A in Figures 3 and 4) comprises 8 sequences, none of which are placed at the root of the cluster, and it is tempting to conclude that the entire cluster derived from community transmission within Guangdong. However, 6 of the 8 genomes reported travel from Hubei and therefore the cluster in fact represents multiple SARS-CoV-2 introductions into Guangdong, with dates of symptom onset around or shortly after the shutdown of travel from Wuhan (Figure 4).
Discussion
Our analyses of the genomic epidemiology of SARS-CoV-2 in Guangdong province indicate that, following the first COVID-19 case detected in early January, most infections were the result of virus importation from elsewhere, and that chains of local transmission were limited in size and duration. This suggests that the large-scale surveillance and intervention measures implemented in Guangdong were effective in interrupting community transmission in a densely populated urban region, ultimately containing the epidemic and limiting the potential for dissemination to other regions (Leung et al., 2020). However, vigilance is still required as there remains a risk that SARS-CoV-2 transmission could reignite in Guangdong following the recent increase in the number of COVID-19 cases imported to China from other countries.
The results also suggest that analyses of phylogenetic structure during the early phase of the pandemic should be interpreted carefully. The number of mutations that define phylogenetic lineages are small (often one), and may be similar to the number of sequence differences arising from errors introduced during reverse transcription, PCR amplification, or sequencing. Bayesian estimates of divergence times (Rannala and Yang, 1996), such as the tMRCA of the pandemic, are based on aggregate numbers of mutations and informed by dense sampling through time, and are thus expected to be more robust. Further, the low and variable sampling of COVID-19 cases among different regions makes it challenging to evaluate phylogenetic clusters that comprise cases from a single region; although such clusters could indeed represent local transmission, our results show they can also include multiple introductions from a genomically undersampled location. Therefore, as with all phylogenetic analyses, the SARS-CoV-2 genomes must be interpreted in the context of all available epidemiological information.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Agencourt RNAClean XP beads | Beckman | Cat# A63987 |
| Agencourt AMPure XP | Beckman | Cat# A63881 |
| Critical Commercial Assays | ||
| SpotON sequencing flow cell | Nanopore Technologies | Cat# FLO-MIN106D |
| Ligation Sequencing Kit | Nanopore Technologies | Cat# SQK-LSK109 |
| Native Barcoding Kit 1D 1-12 | Nanopore Technologies | Cat# EXP-NBD104 |
| Native Barcoding Kit 1D 13-24 | Nanopore Technologies | Cat# EXP-NBD114 |
| Q5® Hot Start High-Fidelity 2X Master Mix | New England BioLabs | Cat# M0494L |
| NEBNext Ultra II End repair/dA-tailing Module | New England BioLabs | Cat# E7546L |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | Cat# M0367L |
| NEBNext Quick Ligation Module | New England BioLabs | Cat# E6056L |
| TURBO DNase | Thermo Fisher | Cat# AM2239 |
| MGIEasy RNA Library Prep kit | MGI | Cat# 1000005274 |
| MGIEasy Circularization Module | MGI | Cat# 1000005260 |
| MGIEasy DNA Adapters-16(Tubes) Kit | MGI | Cat# 1000005284 |
| QIAamp Viral RNA Mini Kit | QIAGEN | Cat# 52904 |
| SMARTer Stranded Total RNA-Seq Kit v2 | Clontech | Cat# 634412 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher | Cat# 18090010 |
| Qubit 1X dsDNA HS Assay Kit | Thermo Fisher | Cat# Q33230 |
| Qubit ssDNA Assay Kit | Thermo Fisher | Cat# Q10212 |
| Qubit RNA HS Assay Kit | Thermo Fisher | Cat# Q32852 |
| Deposited Data | ||
| SARs-CoV-2 Genome Sequences | GISAID | EPI_ISL_413850–413902 |
| Software and Algorithms | ||
| Bowtie2 2.3.4.3 | JOHNS HOPKINS University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools 1.3.1 | htslib | http://www.htslib.org |
| Artic | ARTIC network | https://artic.network/ncov-2019 |
| Geneious | Biomatters Limited | https://www.geneious.com |
| MAFFT | Katoh and Standley, 2013 | https://mafft.cbrc.jp/alignment/software/ |
| PhyML v3.3 | Guindon et al., 2010 | http://www.atgc-montpellier.fr/phyml/ |
| figtreejs-react | N/A | https://doi.org/10.5281/zenodo.3761848 |
| Guppy Basecalling Software 3.4.5+fb1fbfb | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| BEAST v1.10.4 | Suchard et al., 2018 | http://beast.community |
| R Statistical Computing Software v3.5.1 | The R Foundation | https://www.r-project.org/ |
Resource Availablity
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Oliver G Pybus (oliver.pybus@zoo.ox.ac.uk).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession numbers for the sequences reported in this paper are GISAID: EPI_ISL_413850–413902. Code for all figures, tree files, BEAST XML file, BEAST log file, and raw data for Figures 1, 2, 3, and 4 are available at https://github.com/laduplessis/SARS-CoV-2_Guangdong_genomic_epidemiology. A live version of Figure 3B can be found at https://laduplessis.github.io/SARS-CoV-2_Guangdong_genomic_epidemiology/.
Experimental Model and Subject Details
Ethics
This study was approved by ethics committee of the Center for Disease Control and Prevention of Guangdong Province. Written consent was obtained from patients or their guardian(s) when samples were collected. Patients were informed about the surveillance before providing written consent, and data directly related to disease control were collected and anonymized for analysis.
Sample collection, clinical surveillance and epidemiological data
After reports of hospitalized cases with undiagnosed, severe pneumonia on December 30th 2019, enhanced surveillance was initiated in Guangdong Province to detect suspected infections, especially among cases with recent travel history to Hubei or other epidemic regions over the last 14 days. Suspected COVID-19 cases were screened by 31 designated hospitals, local CDCs in 21 prefecture cities, and 6 third-party detection institutions with commercial real-time reverse transcription PCR (RT-PCR) kits (see below for further details). A subset of positive samples was sent to Guangdong Provincial CDC for verification and further sequencing (see below for further details). Imported infections were defined when confirmed cases had travel history from Hubei or other epidemic regions and did not have close contact with local positive cases in 14 days preceding illness onset. The severity of the disease was classified into mild, moderate, severe, or critical (see below for further details). Further details of clinical case definitions are provided in STAR Methods. Demographic information, date of illness onset, and clinical outcomes of sequenced cases were collected from medical records. The exposure history for each case was obtained through an interview. Information regarding the demographic and geographic distribution of SARS-CoV-2 cases can be found at the website of Health Commission of Guangdong Province (http://wsjkw.gd.gov.cn/xxgzbdfk/yqtb/).
Further details of surveillance of COVID-19 in Guangdong, China
The surveillance scheme in Guangdong included 3 main components:
-
a
Twenty-one prefecture CDCs and 31 designated hospitals. These are responsible for the suspected cases diagnoses launched on 30th December 2019. A suspected case was defined if he/she met one of the following criteria: (i) epidemic history and (ii) fever or respiratory symptoms (see below for further details). Epidemic history included: (i) a history of travel to Wuhan or a person who lived in Wuhan or another region where sustained local transmission existed in the 14 days prior to symptom onset; (ii) contact with a patient with fever/respiratory symptoms from Wuhan or another region where sustained local transmission existed in the 14 days prior to symptom onset; (iii) originated from a cluster of COVID-19 cases or is epidemiologically linked to other COVID-19 cases. As of 15th March, 1152 cases were identified through local CDCs and hospitals.
-
b
Six third-party detection institutions. More intense surveillance was initiated on 30th January 2020 in response to the Spring Festival period. This included monitoring (i) all healthy travelers returning from Hubei or other regions with high epidemic activity, (ii) their close contacts, and (iii) all hospitalized patients in clinics, including those without fever or respiratory symptoms, regardless of their exposure history. Approximately 1.35 million samples were screened by six third-party institutions between 30th January and 15th March 2020 and 199 SARS-CoV-2 positive cases were identified from travelers from Hubei without clinical symptoms (76 in 316,214 or 0.02%), fever clinics (99 in 475,949 or 0.02%), non-fever clinics (3 in 447,702) and their close contacts (14 in 70,509 or 0.02%).
-
c
Airport enhanced surveillance. Surveillance commenced at Guangdong airports on 9th March. As of 15th, 3 positive cases were identified from 7,909 diagnoses in Guangzhou Baiyun Airport and a total of 92 imported COVID-19 cases were confirmed as of 26th March.
Further details of sequence sample collection
We collected 58 samples for sequencing from 44 patients (some patients were sampled more than once) in four sentinel hospitals in Guangzhou, Shenzhen and Foshan (Guangzhou Eighth People’s Hospital, 9 samples from 9 patients, collected on 30th January 2020; Guangdong Second Provincial General Hospital, 33 samples from 19 patients, collected between 31st January and 9th January 2020; The Third People’s Hospital of Shenzhen, 11 samples from 11 patients, collected on 5th February 2020; Foshan First People’s Hospital, 5 samples from 5 patients, collected between 10th February and 12th February 2020). These cities recorded the highest number of COVID-19 cases (Figure 1B). We collected a further 21 samples from 18 patients from a screening project of hospitalized COVID-19 cases in Guangdong, which was launched on 28th February. We therefore attempted sequencing on 79 samples from 62 patients (Table S1.1). Because this study focuses on epidemiological questions, we retained only one sequence per patient (the highest quality sequence) and we retained only genomes with > 50% coverage (our quality threshold). This resulted in a final total of 53 genomes.
Clinical classification of COVID-19 cases
Cases were diagnosed and the severity status was categorized as mild, moderate, severe, and critical according to the Diagnosis and Treatment Scheme for Covid-19 released by the National Health Commission of China (Version 7).
Mild cases
The clinical symptoms were mild, and there was no sign of pneumonia on imaging.
Moderate cases
Showing fever and respiratory symptoms with radiological findings of pneumonia.
Severe cases
Adult cases meeting any of the following criteria:
-
i)
Respiratory distress (≧30 breaths/ min);
-
ii)
Oxygen saturation ≤ 93% at rest;
-
iii)
Arterial partial pressure of oxygen (PaO2)/ fraction of inspired oxygen (FiO2) ≦ 300mmHg (l mmHg = 0.133kPa).
In high-altitude areas (at an altitude of over 1,000 m above the sea level), PaO2/ FiO2 shall be corrected by the following formula:
Cases with chest imaging that showed obvious lesion progression within 24-48 hours > 50% shall be managed as severe cases.
Child cases meeting any of the following criteria:
-
i)
Tachypnea (RR ≥ 60 breaths/min for infants aged below 2 months; RR ≥ 50 BPM for infants aged 2-12 months; RR ≥ 40 BPM for children aged 1-5 years, and RR ≥ 30 BPM for children above 5 ye PaO2/ FiO2 x [Atmospheric pressure (mmHg)/760]ars old) independent of fever and crying
-
ii)
Oxygen saturation ≤ 92% on finger pulse oximeter taken at rest
-
iii)
Labored breathing (moaning, nasal fluttering, and infrasternal, supraclavicular and intercostal retraction), cyanosis, and intermittent apnea
-
iv)
Lethargy and convulsion
-
v)
Difficulty feeding and signs of dehydration
Critical cases
Cases meeting any of the following criteria:
-
i)
Respiratory failure and requiring mechanical ventilation
-
ii)
Shock
-
iii)
With other organ failure that requires ICU care
Method Details
Virus amplification and sequencing
Virus genomes were generated by two different approaches, (i) untargeted metagenomic sequencing on the BGI MGISEQ-2000 (n = 63) and Illumina NextSeq (n = 4) sequencing platforms, and (ii) using version 1 of the ARTIC COVID-19 multiplex PCR primers (https://artic.network/ncov-2019), followed by nanopore sequencing on an ONT MinION (n = 45). Untargeted metagenomic sequencing was initially attempted as it is well suited to the characterization of a previously unknown virus. Subsequently, a protocol for sequencing SARS-CoV-2 using multiplex PCR with nanopore sequencing was made available, which showed good performance on samples with higher Ct values (as described below and in Table S1.3). Thereafter, most clinical samples were sequenced using this latter approach. We report only those genomes for which we were able to generate > 50% genome coverage, and report only one genome per patient.
For metatranscriptomics, total RNAs were extracted from different types of samples by using QIAamp Viral RNA Mini Kit, followed by DNase treatment and purification with TURBO DNase and Agencourt RNAClean XP beads. Both the concentration and the quality of all isolated RNA samples were measured and checked with the Agilent Bioanalyzer 2100 and Qubit. For Illumina sequencing, libraries were prepared using the SMARTer Stranded Total RNA-Seq Kit v2 (according to the manufacturer’s protocol starting with 10 ng total RNA. Briefly, purified RNA was first fragmented and converted to cDNA using reverse transcriptase. The ribosome cDNA was depleted by using ZapRv2 (mammalian-specific). The remaining cDNA was converted to double stranded DNA and subjected to end-repair, A-tailing, and adaptor ligation. The constructed libraries were amplified using 9-16 PCR cycles. Sequencing of metatranscriptome libraries was conducted on the Illumina NextSeq 550 SE 75 platform. For BGI sequencing, DNA-depleted and purified RNA was used to construct the single-stranded circular DNA library with MGIEasy RNA Library preparation reagent set following manufacturer’s protocol. Finally, 60fmol of PCR products were Unique Dual Indexed (UDI), circularized, and amplified by rolling circle replication (RCR) to generate DNA nanoball (DNBs)-based libraries. DNBs preps of clinical samples were sequenced on the MGISEQ-2000 platform.
For the multiplex PCR approach, we followed the general method of multiplex PCR as described in (https://artic.network/ncov-2019) (Quick et al., 2017). Briefly, the multiplex PCR was performed with two pooled primer mixture and the cDNA reverse transcribed with random primers was used as a template. After 35 rounds of amplification, the PCR products were collected and quantified, followed with end-repairing and barcoding ligation. Around 50 fmol of final library DNA was loaded onto the MinION. The nanopore sequencing platform takes less than 24 hours to obtain 10Gb of sequencing data, achieving between 0.3–0.6 million reads per sample. The ARTIC bioinformatics pipeline for COVID (https://artic.network/ncov-2019) was used to generate consensus sequences and call single nucleotide changes relative to the reference sequence. SNP differences were sometimes observed when the same sample was sequenced using different sequencing approaches. These differences were random and not platform specific and, upon close inspection of the reads, most likely resulted from low coverage regions in the metagenomics data. Only the single highest-quality genome was retained per patient.
To test the precision and threshold of the multiplex PCR and nanopore sequencing method, we undertook a serial dilution experiment. Viral RNA was extracted from a cell strain of SARS-CoV-2. To mimic clinical samples with different viral loads, we diluted this viral RNA with SARS-CoV-2-negative RNA extracted from nasopharyngeal swab specimens. Viral loads were estimated using RT-PCR with serial diluted plasmid as a standard. At each dilution level we performed multiplex PCR and nanopore sequencing and assembly as per the approach above, except that reads were assembled against the consensus genome obtained from the original sample using metagenomic sequencing. As expected, relative virus load, % genome coverage, and average read depth decreased at higher dilutions. Genome coverage exceeded 75% for all except the final dilution (Table S1.3).
Quantification and Statistical Analysis
Virus genome assembly
Reference-based assembly of the metagenomic raw data was performed as follows. Illumina adaptors were removed, and reads were filtered for quality (q30 threshold and read length > 15nt) using Cutadapt 1.18 (Martin, 2011). The mapping of cleaned reads was performed against GenBank reference strain MN908947.3 using Bowtie2 (Langmead and Salzberg, 2012). Consensus sequences were generated using samtools 1.2 (Li et al., 2009). Sites were called at depth > = 3 if they matched the reference strain, or depth > = 5 if they differed from the reference, otherwise sites were denoted N. Ambiguity nucleotide codes were used if (i) the minor variant is observed at > 30% frequency and (ii) the minor variant is represented by 5 or more reads. Assembly of the nanopore raw data was performed using the ARTIC bioinformatic pipeline for COVID-19 with minimap2 (Li, 2018) and medaka (https://github.com/nanoporetech/medaka) for consensus sequence generation. For patient samples that were sequenced using both metagenomics and nanopore sequencing, we retained only the sequence with the highest genome coverage.
Phylogenetic analysis
All available SARS-CoV-2 sequences (n = 323) on GISAID (gisaid.org) on 13th March 2020 were downloaded. Sequences from GISAID that were error-rich, those which represented multiple sequences from the same patient, and those without a date of sampling were removed. Finally, the dataset was reduced by only retaining the earliest and most recently sampled sequences from epidemiologically linked outbreaks (e.g., the Diamond Princess cruise ship). The resulting dataset of 250 sequences therefore represents the global diversity of the virus while minimizing the impact of sampling bias. Sequences were aligned using MAFFT v7.4 (Katoh and Standley, 2013) and manually inspected in Geneious v11.0.3 (https://www.geneious.com). The final alignment length was 29,923 nucleotides.
We used both the maximum likelihood (ML) and Bayesian coalescent methods to explore the phylogenetic structure of SARS-CoV-2. The ML phylogeny was estimated with PhyML (Guindon et al., 2010) using the HKY+⌈4 substitution model (Hasegawa et al., 1985) with gamma-distributed rate variation (Yang, 1994). Linear regression of root-to-tip genetic distance against sampling date indicated that the SARS-CoV-2 sequences evolve in clock-like manner (r = 0.592) (Figure S3 ). The Bayesian coalescent tree analysis was undertaken with BEAST v1.10.4 (Ayres et al., 2012, Suchard et al., 2018), also using the HKY+⌈4 substitution model with gamma-distributed rate variation with an exponential population growth tree prior and a strict molecular clock, under a non-informative continuous-time Markov chain (CTMC) reference prior (Ferreira and Suchard 2008). Taxon sets were defined and used to estimate the posterior probability of monophyly and the posterior distribution of the tMRCA of observed phylogenetic clusters A-E (Table S1.2). Two independent chains were run for 100 million states and parameters and trees were sampled every 10,000 states. Upon completion, chains were combined using LogCombiner after removing 10% of states as burn-in and convergence was assessed with Tracer (Rambaut et al., 2018). The maximum clade credibility (MCC) tree was inferred from the Bayesian posterior tree distribution using TreeAnnotator, and visualized with figtreejs-react (https://github.com/jtmccr1/figtreejs-react). Monophyly and tMRCA (time to the most recent common ancestor) statistics were calculated for each taxon set from the posterior tree distribution.
Figure S4.

Details of the Clusters (A–E) of Guangdong Genome Sequences, Related to Figure 3
(A–E) Details of the clusters of Guangdong genome sequences. Extracts from the maximum-likelihood phylogeny are shown on the left and extracts from the maximum clade credibility (MCC) tree are shown on the right. Tip labels show GISAID accession number; those in red are from Guangdong and those in black are from other locations. Node bars on the MCC extracts indicate the 95% HPD interval of node ages. Nodes with posterior probability > 0.8 are labeled with a number and gray circle.
Figure S5.

Screenshots of the Online Tree Visualization Tool, Related to Figure 3
The top image shows the 5 clusters A-E highlighted. The bottom image shows the genomes from Guangdong highlighted.
Figure S3.

Root-to-Tip Genetic Distance for 250 Sequences in the Maximum Likelihood Tree Plotted against Collection Date, Related to Figure 3
The Pearson correlation coefficient between root-to-tip distance and collection date is displayed in the top-right corner (r = 0.592). Sequences are colored by sampling location (Guangdong = red, other location = gray).
Acknowledgments
We gratefully acknowledge the efforts of local CDCs, hospitals, and the third-party detection institutions in epidemiological investigations, sample collection, and detection. We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found in Table S1.4. This work was supported by grants from Guangdong Provincial Novel Coronavirus Scientific and Technological Project (2020111107001), Science and Technology Planning Project of Guangdong (2018B020207006), and the Key Research and Development Program of Guangdong Province (2019B111103001). O.G.P., M.U.G.K., and L.d.P. were supported by the Oxford Martin School. V.H. is supported by the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/M010996/1). N.R.F. acknowledges funding from a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (204311/Z/16/Z) and from a Medical Research Council and FAPESP award (MR/S0195/1). J.Q. is funded by a UK Research and Innovation Future Leaders Fellowship. A.R. is supported by the European Research Council (725422-ReservoirDOCS). This work was supported by the Wellcome Trust ARTIC network (Collaborators Award206298/Z/17/Z).
Author Contributions
J. Lu., O.G.P., and C.K. designed the study. J. Lu, Z.L., H.L., J. Sun, J.P., Q.X., R.Y., L.Z., P.Z., C.L., W.L., J. Su, H.Z., B.P., S.F., W.S., K.L., R.S., R.B., X.T., M.L., and T.S. undertook fieldwork and experiments. L.d.P., J. Lu, Z.L., V.H., S.F., J.T.M., L.Y., J.Q., A.R., N.L., J.R., and O.G.P. performed genetic analyses. L.d.P., M.K., M.U.G.K., J. Liu, J.X., J.H., T.L., and W.M. performed epidemiological analyses. J.R., J. Lu, L.d.P., V.H., N.R.F., and O.G.P. wrote the manuscript. L.d.P., J. Lu, V.H., M.U.G.K., A.R., J.R., and O.G.P. edited the manuscript. All authors were involved in coordination, collection, processing, sequencing, and/or bioinformatics of clinical samples. All authors read and approved the contents of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 30, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.04.023.
Supplemental Information
Table S1.1: Sample and sequencing information. Table S1.2: Taxon sets assessed for monophyly. Table S1.3. Serial dilution experiment to test the precision and threshold of the multiplex PCR and nanopore sequencing method for SARS-CoV-2. Table S1.4: GISAID Sequence Acknowledgments.
Data S1.1: Detailed summary of sequencing information and case data for the 53 genomes reported here. Data S1.2: Genome coverage map for genomes reported here as well as 49 previously reported genomes from Hubei and Guangdong.
References
- Ayres D.L., Darling A., Zwickl D.J., Beerli P., Holder M.T., Lewis P.O., Huelsenbeck J.P., Ronquist F., Swofford D.L., Cummings M.P. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012;61:170–173. doi: 10.1093/sysbio/syr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N.R., Kraemer M.U.G., Hill S.C., Goes de Jesus J., Aguiar R.S., Iani F.C.M., Xavier J., Quick J., du Plessis L., Dellicour S. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science. 2018;361:894–899. doi: 10.1126/science.aat7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M.A., Suchard M.A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008;36:355–368. [Google Scholar]
- Flightradar24 . 2020. Air traffic at China’s busiest airports down 80% since the beginning of the year.https://www.flightradar24.com/blog/air-traffic-at-chinas-busiest-airports-down-80-since-the-beginning-of-the-year/ [Google Scholar]
- Grubaugh N.D., Ladner J.T., Kraemer M.U.G., Dudas G., Tan A.L., Gangavarapu K., Wiley M.R., White S., Thézé J., Magnani D.M. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature. 2017;546:401–405. doi: 10.1038/nature22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2002032. Published online February 28, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Kishino H., Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Itokawa K., Sekizuka T., Hashino M., Tanaka R., Kuroda M. A proposal of an alternative primer for the ARTIC Network’s multiplex PCR to improve coverage of SARS-CoV-2 genome sequencing. bioRxiv. 2020 doi: 10.1101/2020.03.10.985150. [DOI] [Google Scholar]
- Kang M., Wu Jie, Ma Wenjun, He Jianfeng, Lu Jing, Liu Tao, Li Baisheng, Mei Shujiang, Ruan Feng, Lin Lifeng. Evidence and characteristics of human-to-human transmission of SARS-CoV-2. medRxiv. 2020 doi: 10.1101/2020.02.03.20019141. Published online February 17, 2020. [DOI] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer M.U.G., Cummings D.A.T., Funk S., Reiner R.C., Faria N.R., Pybus O.G., Cauchemez S. Reconstruction and prediction of viral disease epidemics. Epidemiol. Infect. 2018;Nov 5:1–7. doi: 10.1017/S0950268818002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer M.U., Yang C.-H., Gutierrez B., Wu C.-H., Klein B., Pigott D.M., Open COVID-19 Data Working Group. Plessis P., Faria N.R., Li R. The effect of human mobility and control measures on the COVID-19 epidemic in China. Science. 2020:eabb4218. doi: 10.1126/science.abb4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner J.T., Grubaugh N.D., Pybus O.G., Andersen K.G. Precision epidemiology for infectious disease control. Nat. Med. 2019;25:206–211. doi: 10.1038/s41591-019-0345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K., Wu J.T., Liu D., Leung G.M. First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: a modelling impact assessment. Lancet. 2020 doi: 10.1016/S0140-6736(20)30746-7. Published online April 8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Yan L.M., Wan L., Xiang T.X., Le A., Liu J.M., Peiris M., Poon L.L.M., Zhang W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis. 2020 doi: 10.1016/S1473-3099(20)30232-2. Published online March 19, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
- Quick J. 2020. Artic-ncov2019 primer schemes.https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019/V3 [Google Scholar]
- Quick J., Grubaugh N.D., Pullan S.T., Claro I.M., Smith A.D., Gangavarapu K., Oliveira G., Robles-Sikisaka R., Rogers T.F., Beutler N.A. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017;12:1261–1276. doi: 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. 2020. Phylodynamic Analysis, 176 genomes.http://virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356 [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala B., Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J. Mol. Evol. 1996;43:304–311. doi: 10.1007/BF02338839. [DOI] [PubMed] [Google Scholar]
- Suchard M.A., Lemey P., Baele G., Ayres D.L., Drummond A.J., Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Liu Y., Li Y., Wu C.H., Chen B., Kraemer M.U.G., Li B., Cai J., Xu B., Yang Q. An investigation of transmission control measures during the first 50 days of the COVID-19 epidemic in China. Science. 2020:eabb6105. doi: 10.1126/science.abb6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . 2020. Coronavirus disease (COVID-2019) situation reports.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.1: Sample and sequencing information. Table S1.2: Taxon sets assessed for monophyly. Table S1.3. Serial dilution experiment to test the precision and threshold of the multiplex PCR and nanopore sequencing method for SARS-CoV-2. Table S1.4: GISAID Sequence Acknowledgments.
Data S1.1: Detailed summary of sequencing information and case data for the 53 genomes reported here. Data S1.2: Genome coverage map for genomes reported here as well as 49 previously reported genomes from Hubei and Guangdong.
Data Availability Statement
The accession numbers for the sequences reported in this paper are GISAID: EPI_ISL_413850–413902. Code for all figures, tree files, BEAST XML file, BEAST log file, and raw data for Figures 1, 2, 3, and 4 are available at https://github.com/laduplessis/SARS-CoV-2_Guangdong_genomic_epidemiology. A live version of Figure 3B can be found at https://laduplessis.github.io/SARS-CoV-2_Guangdong_genomic_epidemiology/.