

Abstract
Single-nucleotide polymorphisms (SNPs) are the most abundant form of genomic polymorphisms and are widely used in population genetics research. Here, high-throughput sequencing was used to examine the genome-level diversity, population structure, and relationships of apricot, which are important for germplasm conservation and molecular breeding. Restriction site-associated DNA sequencing (RAD-seq) was adopted to sequence 168 Prunus spp. accessions distributed in five ecological groups, including 74 accessions of cultivated Prunus armeniaca L. and 94 accessions of wild apricots (P. armeniaca L. and Prunus sibirica L.), which generated 417,961 high-quality SNPs. We used cluster, genetic structure, and principal component analyses to examine the genetic diversities and genetic relationships of the 168 accessions. The Dzhungar-Ili ecological group accessions showed the highest genetic diversity in terms of private allele number, observed heterozygosity, and nucleotide diversity. We speculate that the Central Asian ecological group accessions were domesticated from the Dzhungar-Ili ecological group accessions. The population structure and gene flow of the North China and European ecological group accessions suggested a genetic background of P. sibirica. We argue that the two groups should be considered hybrid swarms connected to P. sibirica by continuous and extensive gene flow. P. armeniaca originated in Northwest China (Ili Valley), subsequently spread throughout Central Asia, and eventually spread to Europe. In addition, selective sweep signatures in P. armeniaca during domestication from wild to cultivated apricots, combined with differentially expressed genes, underlie distinct fruit traits, including sugars, aromas, organic acids, and carotenoids. This study provides substantive and valuable genomic resources that will significantly advance apricot improvement and effective utilization.
Subject terms: Comparative genomics, Genome evolution
Introduction
Apricot is a fruit of temperate and subtropical regions and is distributed worldwide. China, Iran, Pakistan, Uzbekistan, Morocco, Algeria, Ukraine, and the USA are the main producers of apricot1. Apricot belongs to section Armeniaca (Lam.) Koch, subgenus Prunophora Focke, and genus Prunus (Rosaceae)2. According to different taxonomic systems, apricots are divided into three to twelve species3,4. However, six species are recognized by most scholars3,5,6: Prunus armeniaca L., Prunus sibirica L., Prunus mandshurica (Maxim.) Skv., Prunus holosericea (Batal.) Kost., Prunus mume Sieb. et Zucc., and Prunus brigantina Vill. Almost all cultivated apricots originated from P. armeniaca7.
Chinese scholars8 divided apricot into six ecological groups based on geographical distribution. The Central Asian ecological group (CAG) accessions are the most abundant, with vigorous tree growth, many slender twigs, and small leaves. These accessions flower late, are self-fruiting, and produce soft, small fruits with a sweet kernel8. The European ecological group (EG) accessions are medium-sized trees with thick branches, weak growth, and a short dormancy period, and mostly produce firm fruits through self-fertilization. The flowers, leaves, and fruits of EG accessions are usually large8. Accessions in the Dzhungar-Ili ecological group (DZG) are small trees that produce small fruits with a pubescent peel and exhibit strong cold, drought, and waterlogging resistance8. The North China ecological group (NCG) accessions are tall trees with vigorous growth. These accessions have intermediate cold and drought resistance and produce fruits via self-fertilization that are large and juicy8. The Northeast Asian ecological group (NAG) accessions are small shrubs that have strong cold resistance. These accessions can tolerate temperatures as low as −50 °C in winter, exhibit early leaf fall, and produce small and dry fruits with bitter flesh that are not suitable for consumption8. The East China ecological group (ECG) accessions include shrubs or medium-sized trees and tall trees and have strong disease resistance. The seed-setting rate of self-pollinated flowers is very low, the colors of the flowers vary, and the fruits are mostly green8. Therefore, the groups exhibit large morphological and physiological differences and wide adaptability to ecological conditions9.
According to Vavilov10, apricots have three origin centers: China, Central Asia, and the Near East. Most scholars5,6,11,12 support the history of ancient apricots in Central Asia and China, regarding these regions as independent centers of domestication. However, the early history of apricot is not completely clear, and the main question of whether the cultivation history in Central Asia is longer or shorter than that in China remains unanswered13. De Candolle et al.14 argued that apricot cultivation in China dates back to the end of 3000 BC. However, there is no archeological evidence for the beginning of apricot domestication and cultivation. In Central Asia, apricot cultivation was recently introduced in ~1000 to 2000 BC15. Many scholars have shown that the genetic diversity of apricots in China is the richest worldwide16–18.
The Kashgar, Hotan, and Kuqa oasis areas around the Tarim Basin in the southern part of the Xinjiang Uygur Autonomous Region of China are the main apricot-producing areas and contain the greatest abundance of apricot cultivars. The wild apricot forest in Ili, Xinjiang, China, is a relic of a broad-leaved forest from the late Tertiary community that played a decisive role in the domestication and cultivation of apricot worldwide18. It is an important part of the deciduous broad-leaved forest under the mountain coniferous forest and above-mountain grassland in Xinjiang8. There is only one mountain between the southern Xinjiang and Ili areas in the northern Tianshan Mountains, and there are several corridors between the northern and southern Tianshan Mountains. Geographically, the apricots cultivated in Xinjiang, southern Tianshan Mountains (CAG), most likely evolved from the spread of wild apricots in the Ili Valley (DZG)8.
Crop domestication not only changes economic and agronomic traits but also retains the genetic characteristics that affect the genetic diversity and population structure of domesticated plants19. Identifying genetic variations in wild and cultivated populations and within populations can provide insights into the general mechanisms of plant domestication and diversification and guide crop genetic improvement in future breeding programs20. There is evidence that domestication can lead to a decrease in the genetic diversity of cultivated crops21. The genetic information present before domestication and artificial selection may be retained in wild populations, which are special resources for studying the effects of variation in the apricot genome. However, the natural apricot population in China is strongly influenced by human activities, such as logging and deforestation. Consequently, protective measures must be taken to prevent further declines in wild apricot resources. Information on genetic diversity and population structure is essential for the development of management and conservation approaches. We studied the genetic diversity and population structure of apricot and identified the representative core germplasm. We can preserve the genetic diversity of the whole population to the greatest extent by using the smallest amount of genetic resources, and further improve the protection efficiency of apricot germplasm resources22. Developing genomic resources, increasing the knowledge of the apricot gene pool, and obtaining information about genetic diversity and population structure can accelerate biological research and genetic improvement.
The rapid development of high-throughput sequencing technology has provided whole-genome information for nonreference crops. The use of this technology has produced large data sets of short sequences within millions of genomes with deep coverage. These methods, including restriction site-associated DNA sequencing (RAD-seq) and genotyping-by-sequencing (GBS), have been successfully applied in population genomics studies23–25. Previous studies used nuclear DNA, mitochondrial DNA, and chloroplast DNA3,26–28 sequence fragments to study the genetic diversity of apricots and the relationships between apricot and other related species29. Compared with genome-level sequencing, plastid and nuclear gene molecular markers have a lower accuracy and resolution. The limited number of polymorphic loci produced by conventional molecular markers hinders genetic research on apricot.
The main purposes of this study were to evaluate the genetic diversity, population structure, and relationships of apricots in different ecological groups using RAD-seq of the whole genome, clarify the phylogenetic relationships between five ecological groups and provide a theoretical basis for further improving and effectively utilizing apricot germplasm resources.
Results
Sequence data quality and processing
A collection of 168 Prunus spp. accessions were successfully sequenced using the HiSeq 2000 system (Illumina Inc., USA) producing a total of 192 Gb of raw reads, with an average of 1.14 Gb for each accession. After filtering low-quality reads, a total of 180 Gb of clean data were retained, with an average of 1.07 Gb per accession and an average success rate of 98.44%. The quality of the sequencing data was high (Q20 > 90%, Q30 > 85%), the GC content was stable at 37.33–41.25%, and all samples were free from contamination (Supplementary Data-Table 2). The enzyme capture rate ranged from 92.84 to 98.92%. The mapping rate ranged from 88.7 to 94.37%. A total of 1,475,632 small nucleotides (SNPs) were obtained from the 168 Prunus spp. accessions using SAMtools software30, and 417,961 SNPs were obtained after filtering, which ensured the accuracy and reliability of subsequent genetic diversity and population structure analyses. At the population level, more than half of the SNPs (with an average of 55.72%) had base transitions, and the transition vs. transversion (ts/tv) ratios for the populations ranged from 1.787 to 1.799. The ts/tv ratios of SNPs in the two wild populations (NAG and DZG) were larger than those in the cultivated populations (Fig. 1; Supplementary Data-Table 3).
Fig. 1. SNP mutation type for the five ecological groups.
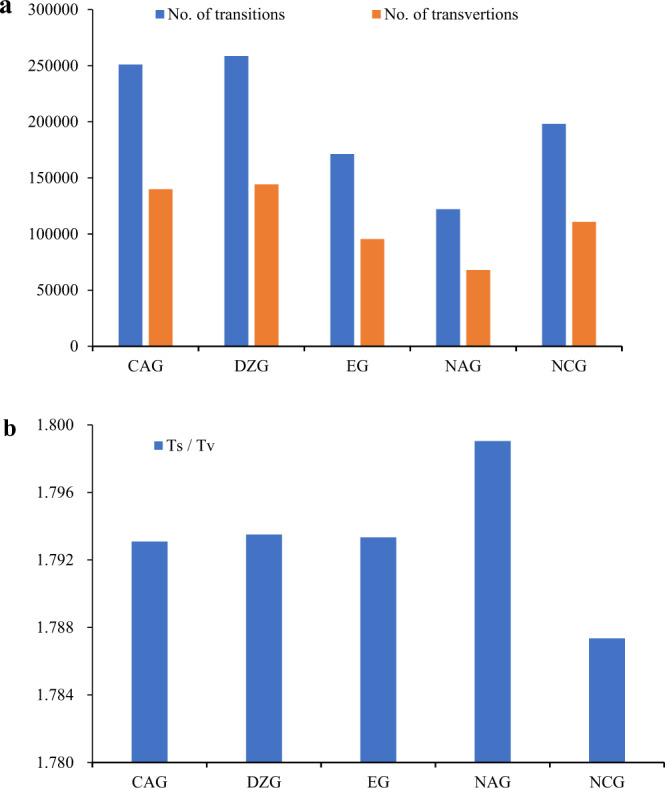
a Number of transitions and transversions for the five ecological groups. b transition/transversion rate
Population phylogenetic relationships
A phylogenetic tree of all 168 accessions was constructed to better understand their relationships (Fig. 2). In the phylogenetic tree, most branches had high bootstrap values, which indicated that the maximum likelihood (ML) tree was highly reliable. The phylogenetic tree divided the collected accessions into five groups. One group included the NAG accessions, located in Northeast China and belonging to P. sibirica. These accessions are adapted to and grown in extremely cold regions in China, and are thus considered very hardy. The other four groups belonged to P. armeniaca. These groups include (1) the DZG (group I), with four subgroups based on the strong geographical distribution pattern among the branches (Yining County (group I-1), Huocheng County (group I-2), Xinyuan County (group I-3), and Gongliu County (group I-4)) in the Ili region, Xinjiang; (2) the CAG (group II), with apricot cultivars mainly from the Kashgar, Hotan, and Kuqa areas in Xinjiang that could not be clearly distinguished based on their geographical distributions, which may be due to the high genetic diversity and frequent gene exchange generated by selection or introduction, reflecting the complex domestication history of cultivated apricots18 (notably, as revealed by the population structure analysis (Fig. 3), the CAG was mixed with the genetic background of DZG; therefore, Xinjiang cultivated apricots originated from Ili wild fruit forests); (3) the EG (group III), with apricot cultivars mainly from North America, Italy, and France; and (4) the NCG (group IV), with apricot cultivars mainly from Northeast China, North China, and Korea.
Fig. 2. ML tree created for 168 Prunus spp. accessions.

Red indicates an outgroup (Malus sieversii). Group-0 represents Northeast Asian ecological group (wild P. sibirica), Group I represents Dzhungar-Ili ecological group (wild P. armeniaca), Group II represents Central Asian ecological group (cultivated P. armeniaca), Group III represents European ecological group (cultivated P. armeniaca), and Group IV represents North China ecological group (cultivated P. armeniaca). Node values correspond to bootstrap values
Fig. 3. Population structure of 168 accessions of Prunus spp.

Each column represents an individual, with the length of the different color segments representing the proportion of an ancestor in the individual’s genome. K = 2–8 indicates the number of ancestral groups assumed in this study from 2 to 8; The x-coordinate indicates the name of the sample, and the order of the sample of the same group was specified together. HC Huocheng county, YN Yining county, GL Gongliu county, XY Xinyuan county
Population genetic structure
Population genetic structure analysis was conducted based on high-quality SNPs filtered by VCFtools. Based on the ML, K = 2–8 ancestral populations were assumed in this study. The optimal population K value was determined according to the coefficient of variation (CV), and the optimal value obtained in this study was K = 5 (Supplementary Data-Fig. 1). According to Fig. 4, when the K value was 5, 168 materials were not assigned to the five groups, which was inconsistent with the results of the phylogenetic tree analysis. Due to divergence in the population structure and phylogenetic tree results, it was necessary to analyze population structure under different K values. Therefore, we examined the population structure for K values ranging from 2 to 8 (Fig. 3).
Fig. 4. PCA analysis in 168 accessions of Prunus spp.
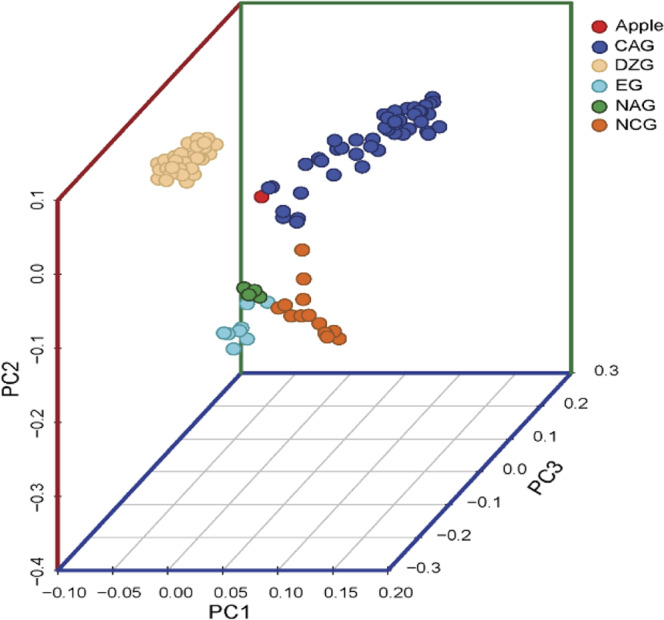
Red indicates an outgroup (Malus sieversii). The CAG, DZG, EG, NAG, and NAG indicate Central Asian ecological group, Dzhungar-ili ecological group, European ecological group, Northeast Asian ecological group, and North China ecological group
When K = 3, the CAG and DZG accessions were distinguished, while the EG, NCG, and NAG accessions could not be separated. When K = 6, in addition to the CAG and DZG accessions, the NCG and EG accessions could be distinguished from each other. When K = 8, the population structure of the CAG, NCG, and EG accessions remained unchanged, and internal differentiation occurred in only the DZG accessions, which were divided into four subgroups: Huocheng, Yining, Gongliu, and Xinyuan. The genetic background of the CAG accessions was complex, but most of the genetic background was found to originate from the DZG accessions, which supports the hypothesis that the cultivated apricots in Xinjiang originated from wild apricots in the Ili region. However, it was interesting to note that the population structure of the EG and NAG accessions changed very little as K increased from 2 to 8. The highly polymorphic and diverse properties of the DZG, CAG, and NCG might have influenced the population structure of the EG and NAG. Combined with the phylogenetic tree, these results clearly divided apricot into five groups: CAG, DZG, NCG, EG, and NAG accessions.
Population genetic relationships
The collection of 168 accessions was analyzed by principal component analysis (PCA) to better understand the relationships between ecological groups (Fig. 4), and the PCA scores were used to evaluate genetic variance. The first three components explained 16.49% of the total genetic variation (8.28, 5.88, and 2.33%, respectively). According to the first three components, the accessions were divided into five groups: the CAG, DZG, NCG, EG, and NAG.
Gene flow analysis
We analyzed the gene flow between the five ecological groups (Supplementary Data-Fig. 2). Allowing just one migration event (m = 1) (Fig. 5a), we observed that gene flow occurred from the NAG to NCG accessions. In terms of geographical location, these two ecological groups were close to each other, which may have caused the germplasm of the NCG accessions to be mixed with that of P. sibirica. Allowing two admixture events (m = 2) (Fig. 5b), the EG, NCG, and CAG accessions exhibited extensive gene flow, which likely reflects their many shared genomic components due to hybridization in their domestication and breeding histories. This result was consistent with that observed on the basis of population structure (Fig. 3).
Fig. 5. Detection of gene flow between five ecological groups accessions.

Lines represent gene flow; arrows indicate the direction of ene flow. The scale bar shows a tenfold average standard error of the entries in the sample covariance matrix. The color bar shows the migration weight: a red color denotes a strong gene flow, while a yellow color denotes a weak gene flow
Population genetic diversity
The expected heterozygosity (He) of the P. armeniaca populations ranged from 0.378 to 0.407; the observed heterozygosity (Ho) of the P. armeniaca populations ranged from 0.218 to 0.260; Wright’s F-statistic (FIS) of the P. armeniaca populations ranged from 0.030 to 0.078; and the number of private alleles (AP) in the populations of P. armeniaca ranged from 11–11,069. The average nucleotide diversity (π) and polymorphism (Waterson’s theta, θw) estimates of the P. armeniaca population mutation parameters were 0.0027 and 0.0016 at the genome level, respectively (Table 1, Fig. 6). The mean nucleotide variation of P. armeniaca was higher than that of other perennial crops, such as peach (π = 0.0015)31 and cassava (π = 0.0026)32, but lower than that of date palm (π = 0.0092)33.
Table 1.
The statistical values of genetic diversity within populations from different ecological groups
| Taxon | Apricot accessions groups | Private allele number (AP) | Effective site | SNP number | Ho | He | FIS | θw | Tajima’s D |
|---|---|---|---|---|---|---|---|---|---|
| Wild P. armeniaca | DZG | 11,069 | 417,961 | 402,703 | 0.260 | 0.378 | 0.071 | 2.02E-03 | 1.035 |
| Cultivated P. armeniaca | CAG | 3784 | 417,961 | 390,892 | 0.251 | 0.405 | 0.078 | 1.52E-03 | 0.844 |
| NCG | 202 | 416,708 | 308,930 | 0.223 | 0.407 | 0.033 | 2.20E-03 | 0.468 | |
| EG | 11 | 411,449 | 266,769 | 0.218 | 0.406 | 0.030 | 2.05E-03 | 0.510 | |
| Wild P. sibirica | NAG | 0 | 385,166 | 189,976 | 0.155 | 0.391 | 0.031 | 1.52E-03 | 0.220 |
Ho observed heterozygosity, He expected heterozygosity, π nucleotide diversity, θw Watterson’s theta, FIS inbreeding coefficient of an individual relative to the population
Fig. 6. Summary of nucleotide diversity and population divergence across the five ecological groups.

Values in parentheses represent measures of nucleotide diversity for the group, and values between pairs indicate population divergence (Fst). The thickness of the lines is proportional to the value of Fst. The circle size is proportional to the value of π
Further analysis of the DZG accessions (Table 2) showed that the observation heterozygosity (Ho) ranged from 0.373 to 0.383, the expected heterozygosity (He) ranged from 0.256 to 0.268, the inbreeding coefficient (FIS) ranged from 0.015 to 0.055, and the private allele number (AP) ranged from 2026 to 8847. The Ho and AP in Xinyuan County were higher than those in Gongliu, Yining, and Huocheng Counties.
Table 2.
The statistical values of genetic diversity within groups from wild apricots
| Apricot accessions groups | Population | Private allele number (Ap) | Polymorphic loci, % | Ho | He | FIS |
|---|---|---|---|---|---|---|
| DZG | Xinyuan County | 3217 | 88.1983 | 0.3828 | 0.2608 | 0.0219 |
| Yining County | 2026 | 85.7091 | 0.37906 | 0.26767 | 0.0152 | |
| Huocheng County | 3046 | 86.4664 | 0.37277 | 0.2583 | 0.037 | |
| Gongliu County | 8847 | 95.0875 | 0.37768 | 0.2558 | 0.0553 |
Notably, the Ho and π (0.260 and 0.274, respectively) of the DZG accessions were higher than those of the CAG accessions (0.251 and 0.265, respectively), NCG accessions (0.223 and 0.229, respectively) and EG accessions (0.218 and 0.222, respectively) (Fig. 6). Therefore, some of the genetic diversity in apricots was lost during the process of domestication from the DZG to the EG. The genetic diversity index of P. armeniaca was larger than that of P. sibirica.
Tajima’s D represents intraspecific polymorphism based on locus variation. The Tajima’s D values of the five groups were positive, and significantly different from zero (Table 1). Thus, the null hypothesis of neutral evolution could be rejected. Furthermore, numerous intermediate-frequency alleles were detected in the populations, which may have been caused by a bottleneck effect, population structure, or balancing selection.
Genetic differentiation and AMOVA
The genetic differentiation index (Fst) between populations ranged from 0.1080 to 0.1946 (Fig. 6), and the degree of differentiation among populations was low. The Fst values of the CAG and DZG accessions were the smallest among all the groups analyzed (Fig. 6), indicating that the genetic differences within populations were greater than those between populations and the occurrence of gene flow between populations34,35. The Fst values of the EG and NAG accessions were the largest among all the groups analyzed (Fig. 6), which may have been due to geographical isolation, low gene flow between populations, and large genetic differences34,35.
Assessment of genetic differences amDNA was extracted from young leavesong the five geographical groups by analysis of molecular variance (AMOVA) revealed that 12.02% of the genetic variation occurred between populations and 87.98% occurred within populations (Table 3). Although the variation among the five geographical groups of accessions mainly occurred within populations, the differences between populations were also large.
Table 3.
Results of the analyses of molecular variance from five ecological groups
| Source of variation | Degree of freedom | Sum of squares | Variance components | Percentage of variation |
|---|---|---|---|---|
| Among populations | 5 | 82466.52 | 340.03 Va | 12.02 |
| Within populations | 332 | 826059.33 | 2488.13 Vb | 87.98 |
| Total | 337 | 908525.85 | 2828.16 |
Scans for selective sweeps
Separate selective sweeps driven by artificial selection were detected in the CAG, EG, and NCG accessions. For CAG accessions, selective sweep signatures were detected for 0.27 Mb of the genome sequence, containing 53 putative genes (Fig. 7a; Supplementary Data-Table 5). For EG accessions, selective sweep signatures were detected for 1.22 Mb of the genome sequence, containing 278 putative genes (Fig. 7b; Supplementary Data-Table 7). For NCG accessions, selective sweep signatures were detected for 1.19 Mb of the genome sequence, containing 290 putative genes (Fig. 7c; Supplementary Data-Table 9).
Fig. 7. Distribution of Fst values of selective sweeps of P. armeniaca during domestication from wild to cultivated apricots.

a Distribution of Fst values of selective sweeps in DZG and CAG accessions during domestication. b Distribution of Fst values of selective sweeps in DZG and EG accessions during domestication. c Distribution of Fst values of selective sweeps in DZG and NCG accessions during domestication. The horizontal line indicates the threshold of Fst (5%)
One carotenoid biosynthesis gene, one cellulose biosynthesis gene, two serine and threonine metabolism genes, and two GTPase regulatory genes were identified in the selective sweep regions of the CAG accessions (Supplementary Data-Table 4). Eight serine and threonine metabolism genes, three ethylene biosynthesis genes, two aromatic metabolism genes, two organic acid metabolism genes, one high-affinity transport system gene, two stress tolerance-related genes, one pectin degradation gene, one pathogen-related gene, and one salicylic acid biosynthesis gene were detected in the EG accessions (Supplementary Data-Table 6). One auxin-related gene, thirteen serine and threonine metabolism genes, five GTPase regulatory genes, five ethylene biosynthesis genes, one organic acid metabolism gene, one stress tolerance-related gene, one pathogen-related gene, one salicylic acid biosynthesis gene, three sugar metabolism genes, one auxin-related gene, one alanine metabolism gene, one aspartate metabolism gene, one glutamate metabolism gene, and one glucuronoxylan biosynthesis gene were detected in the NCG accessions (Supplementary Data-Table 8).
The candidate genes identified in selective sweeps of P. armeniaca during domestication from wild to cultivated apricots differ greatly in function, indicating that different ecological groups of P. armeniaca adapted to different selection pressures for desired agronomic characteristics in cultivated apricot. Thus, the domestication of cultivated apricot was a process of differential selection.
Discussion
Genomic data provide a novel perspective for studying the genetic diversity and domestication of apricot. In this study, we collected 168 accessions from five geographical groups and performed whole-genome restriction enzyme digestion to quickly obtain highly accurate variation information based on markers. The sample sizes averaged 1.14 Gb, ensuring the accuracy of the population genetic analysis. We used RAD-seq to detect 417,961 SNPs, which exceeded the numbers of amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) markers.
Substitution between purine and purine or between pyrimidine and pyrimidine is called transition, and substitution between purine and pyrimidine is called transversion. Point mutations are common in organisms and cause a wide variety of phenotypic changes. This nucleotide mutation pattern is also observed in other plants, such as peanut36, maize37, Amorphophallus paeoniifolius38, and Arabidopsis39. Due to the biased mutation process in the plant genome, the predicted number of transitions is much larger than the predicted number of transversions38. Consistent with these predictions, we found that the base transition values were higher than the base transversion values and that the ts/tv ratios of the five geographical groups ranged from 1.787 to 1.799 (Fig. 1), indicating strong transition bias. Further analysis showed that the ts/tv ratio of the cultivated population was lower than that of the wild population, which may have been due to long-term, strict artificial selection, and the loss of evolutionary potential.
The genetic diversity index of the cultivated population was much lower than that of the wild population (P. armeniaca) (Table 1, Fig. 6), and the genetic diversity of the EG accessions was lower than that of the other groups of accessions. The AP was most significant in the wild population (DZG), measuring 11,069, indicating a large pool of genetic variation, which could guide the genetic improvement of crops in future breeding programs. In contrast, AP values of only 11–3784 (Table 1) were found in the domesticated population (P. armeniaca). These differences may be due to inbreeding or continuous directional selection during the domestication process, which narrows the genetic basis of germplasm, reduces genetic diversity, and increases the possibility of genetic drift during the domestication process. Similar results were obtained by Li et al.26.
According to Vavilov10, China and Central Asia were the two main centers of apricot domestication. Most cultivated apricots belong to P. armeniaca. Among the four main ecological groups of P. armeniaca species, Kostina40 argued that DZG accessions are the most primitive and that CAG accessions have the longest cultivation history. The cultivation of EG accessions has a relatively recent history, being traced back to ~2000 years ago16. In Central Asia, local cultivars are most likely to have originated from wild apricots, moving southward from the Kazakhstan–China border (Dzhungar-Ili) to Kashmir and westward into the mountains of Afghanistan16. In our study, 168 accessions were divided into five ecological groups based on population structure, PCA and ML tree analysis. The CAG and DZG accessions were tightly clustered, indicating that the DZG (wild apricot) may be the ancestor of the CAG. Zhebentyayeva et al.18 and Li et al.26 reached similar conclusions. The genetic background of the CAG was most similar to that of the DZG (Fig. 4). The genetic diversity indexes (Ho, AP, and π) of the DZG accessions were higher than those of accessions in the other ecological groups (Table 1, Fig. 6), which showed the lowest genetic differentiation for CAG accessions (Fig. 6), further supporting the CAG accessions as having originated from the DZG accessions. We speculate that the DZG accessions are the common ancestors of the CAG, NCG, and EG accessions. P. armeniaca originated in Northwest China (Ili Valley), subsequently dispersed throughout Central Asia, and eventually spread to Europe. These findings are reasonable from a historical perspective, as there was extensive cultural contact along the Silk Road from 207 BCE to 220 CE41. Therefore, historical and commercial influences may have contributed to the development of this unique species of cultivated apricot.
Further analysis of the DZG (Fig. 2) clustered the Xinyuan County and Gongliu County accessions into one group and the Yining County and Huocheng County accessions into another group, providing further geographical support for the relationships among these accessions. According to the genetic diversity indexes (Table 2), the Xinyuan County accessions were the most diverse. He et al.42 also found that wild apricots in the Yili Valley maintained a high level of diversity and that genetic variation mainly occurred within populations.
The population structure and gene flow analyses of the NCG and EG accessions suggested a genetic background of P. sibirica. We argue that the two groups should be considered hybrid swarms connected to P. sibirica by continuous and extensive gene flow.
Human intervention via the artificial selection of favorable phenotypic traits to enhance production and improve desirable agronomic traits can both reduce the levels of genetic variability and skew allele frequencies43. The different genes identified in selective sweeps of CAG, EG, and NCG apricots were found to be enriched in biological processes, including the metabolism of sugars, aromas, organic acids, and carotenoids. These genes may have been involved in different domestication pathways, leading to differences in traits among apricot ecological groups. Some sugar-related genes have been found in selective sweeps, suggesting a preference for sweet fruit during domestication. For the NCG accessions (Supplementary Data-Table 8), a total of three sugar-related genes were identified in the selected regions, including two genes that encode enzymes (vacuolar invertase and neutral invertase). Two organic acid genes were identified in the EG accessions (Supplementary Data-Table 6), and only one was identified in the NCG accessions, indicating that the dominant acid components of apricot differ among ecological groups. Fewer genes involved in carotenoid biosynthesis were identified in selective sweeps. One carotenoid biosynthesis gene was identified in the CAG accessions (Supplementary Data-Table 4), indicating a preference for a certain color during domestication. For volatile compounds in apricot fruit, two genes related to aromatic metabolism (fatty acid desaturase and cytochrome P450) were identified in selective sweeps of the EG accessions (Supplementary Data-Table 6), indicating that aroma in these groups of apricots is regulated by different genes in the metabolic pathway.
Conclusion
In this study, we report genomic information for 168 wild and cultivated apricot accessions. Our findings provide insights into the dissemination of P. armeniaca. Selective sweep signatures in P. armeniaca during domestication from wild to cultivated apricots, combined with differentially expressed genes, underlie distinct fruit traits, including sugars, aromas, organic acids, and carotenoids. Population structure analysis provided new evidence for the mixed genetic background of apricot in different ecological groups. Our study provides abundant genomic resources for wild and cultivated apricots and makes important contributions to the genetic improvement and molecular breeding of apricot.
Materials and methods
Plant material
A diverse collection of 168 Prunus spp. accessions encompassing five ecological groups, namely, the CAG (53 domesticated landraces or cultivars), NCG (13 cultivars), EG (8 cultivars), DZG (90 cultivars), and NAG (4 wild accessions), were considered for RAD-seq.
Seventy-four of the accessions were provided by the Luntai National Fruit Germplasm Resources Garden of the Xinjiang Academy of Agricultural Sciences, the Yingjisha County Apricot National Forest Germplasm Bank, and the Xiongyue National Germplasm Resources Garden of the Liaoning Institute of Pomology. Ninety wild apricots (P. armeniaca) were collected from the natural distribution areas of wild fruit forests in Xinyuan County, Gongliu County, Yining County and Huocheng County in Ili, Xinjiang. Four wild apricots (P. sibirica) were provided by the Xiongyue National Germplasm Resources Garden of the Liaoning Institute of Pomology (Supplementary Data-Table 1).
DNA extraction, library preparation, and sequencing
DNA was extracted from young leaves using a Plant Genomic DNA Kit (Tiangen, Beijing, China)44. The purity and integrity of DNA were analyzed by 1% agarose gel electrophoresis and a NanoDrop® spectrophotometer (ND-1000, Thermo Fisher Scientific, USA). The DNA concentration was accurately quantified with a QubitTM 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA), after which the DNA was stored at −20 °C34. RAD library preparation and sequencing were conducted by Novogene Bioinformatics Technology Co. Ltd., Beijing. The DNA concentration used for library preparation was 75 ng/μl, and the qualified DNA was digested by the EcoRI (GAATTC) restriction enzyme. Each sample was physically randomly fragmented by the Covaris S220 system (Covaris, Woburn, MA, USA), and then subjected to barcode ligation, gel fragment selection, adapter connection and PCR fragment amplification, and 300–500 bp sequences were recovered38. Paired-end sequencing was performed on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA), and each sample generated ~2.32 Gb of raw data. The clean sequencing data have been submitted to the NCBI Short Read Achieve with accession number PRJNA592274.
Mapping and SNP calling
Qualified sequencing data were aligned to the CAG 09 reference genome (P. armeniaca cv. “Cuijianali”; NCBI accession number PRJNA592274) by Burrows-Wheeler Aligner (version 0.7.8-r455)45. We used SAMtools software30 to detect SNPs in the population. The final product was converted to variant call format (VCF).
Sequencing and SNP-calling statistics
To obtain high-quality data, the raw data were sequenced, compared, assessed for mutations, screened, and analyzed. After the reads were successfully aligned to the reference genome through quality control, SAMtools30 software was used to calculate read lengths. The ts/tv ratio was obtained with BCFtools (version 1.8)30.
Population structure analysis
After filtering 1,475,632 SNPs (filter conditions: depth < 5, proportion missing > 0.5, and minor allele frequency (maf) < 0.05), 417,961 SNPs were obtained for PCA, population structure analysis and phylogenetic tree construction.
To clarify the genetic relationships among the five ecological groups, phylogenetic trees were constructed. We used the published RAD-seq data of M. sieversii46 (NCBI accession number SRX2934932) as an outgroup. TreeBeST (version 1.9.2)47 was used to calculate the distance matrix, RAxML (version 8.2.12)48 was used to construct the ML phylogenetic tree, and 1000 bootstrap replicates were used.
Population structure was analyzed by the frappe program49 by setting the number of genetic groups from 2 to 8 and using 1000 bootstrap replicates. To determine the optimal group structure classification, the optimal group number was identified as that with the minimum CV50. Group structure was analyzed by PCA using GCTA51 software, and the significance level of feature vectors was determined by using a Tracy–Widom test. Genome-wide complex trait analysis (GCTA) software integrates the layered correction method of Eigenstart52,53, which can clearly distinguish cases and controls.
Gene flow analysis
Based on the SNPs, we used TreeMix (version 1.13) to evaluate the gene flow between the five ecological groups with the “-se-bootstrap-k 1000 -m” setting, where the number (-m) varied from 1 to 3.
Population genetic diversity and differentiation analysis
The genetic diversity indexes included AP, Ho, He, π43, θw54, FIS, and Tajima’s D43. Sliding windows with a 5 kb step size and 10 kb size were used to calculate Ho, He, π, θw, and Tajima’s D. For each window, these values were run using the Bio::PopGen package with a custom Perl script55. Paired Fst values56 were calculated in the same window to measure the difference between populations. FIS29 was calculated using the POPULATION program in the STACKS pipeline. The statistical values were all calculated using VCFtools (version 0.1.14)57.
Using AMOVA in Arlequin (version 3.5.1)58, different genetic structures were defined and statistically tested by classifying and dividing the populations at different levels.
Identification of selective sweeps
Qualified sequencing data were aligned to the P. mume L59. reference genome to identify selective sweeps in the 168 Prunus spp. accessions distributed in five ecological groups. The results were plotted using the ggplot2 package in R software.
Supplementary information
Table S1,Table S2,Table S3,Table S4,Table S5,Table S6,Table S8,Fig. 1,Fig. 2
Acknowledgements
This work was supported by the National Key Research and Development Program (grant number 2016YFC0501504), the Xinjiang Uygur Autonomous Region Key Research and Development Program (grant number 2016B01005–1), the Xinjiang Uygur Autonomous Region Horticulture Key Discipline Fund (grant number 2016–10758–3), and the Xinjiang Uygur Autonomous Region Graduate Innovation Project (grant number XJ2019G134). The authors would like to thank the different research institutions, scientists, and breeders involved in this work, Shanghai Majorbio Bio-pharm Technology Co., Ltd. for providing the analysis data and American Journal Experts (AJE) for providing English editorial assistance.
Author contributions
Conception and design of the study: K.L. Sample preparation and processing: W.W.L., Y.N.W., L.Q.L., Q.P.Z., Y.T.W., S.K.Z., and G.Q.F. Data and result analyses: W.W.L. and K.L. Paper writing: W.W.L. and K.L. All authors read and approved the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-0284-6).
References
- 1.Romero C, Pedryc A, Munoz V, Llacer G, Badenes ML. Genetic diversity of different apricot geographical groups determined by SSR markers. Genome. 2003;46:244–252. doi: 10.1139/g02-128. [DOI] [PubMed] [Google Scholar]
- 2.Rehder, A. Manual of Cultivated Trees and Shrubs Hardy in North America, Exclusive of the Subtropical and Warmer Temperate Regions (Macmillan, New York, 1940).
- 3.Bortiri E, et al. Phylogeny and systematics of Prunus (Rosaceae) as determined by sequence analysis of ITS and the chloroplast trnL-trnF Spacer DNA. Syst. Bot. 2001;26:797–807. [Google Scholar]
- 4.Folta, K. M. & Gardiner, S. E. in Genetics and Genomics of Rosaceae, Genomics-Based Opportunities in Apricot (eds Folta, K. M. & Gardiner, S. E.) Ch. 10–13, 315–335 (Springer, New York, 2009).
- 5.Kryukova, I. in Botanical Classification and Geographical Distribution, Apricot (ed. Smykov, V. K.) 9–23 (Agropromizdat, Moscow, 1989).
- 6.Faust M, Surányi D, Nyujtó F. Origin and dissemination of apricot. Hortic. Rev. 1998;22:225–266. [Google Scholar]
- 7.Zhebentyayeva, T. N, Ledbetter, C, Burgos, L. & Llácer, G. in Fruit Breeding, Apricot (eds Badenes, M. L. & Byrne, D. H.) 415–458 (Springer Science+Business Media: New York, 2012).
- 8.Zhang, J. Y. & Zhang, Z. in Flora of Chinese Fruit Trees (eds Zhang, J. Y. & Zhang, Z.) 42–46 (China Forestry Press: Beijing, 2003).
- 9.Zhang Q, Liu W. Advances of the apricot resources collection, evaluation and germplasm enhancement. Acta Hortic. Sin. 2018;45:1642–1660. [Google Scholar]
- 10.Vavilov NI. The origin, variation, immunity and breeding of cultivated plants. Science. 1951;115:433–434. [Google Scholar]
- 11.Bailey, C. H. & Hough, L. F. in Advances in Fruit Breeding, Apricots. (eds Janick, J. et al.) 367–383 (Purdue University Press, West Lafayette, Indiana, 1975).
- 12.Hormaza, J., Yamane, H. & Rodrigo, J. in Genome Mapping and Molecular Breeding in Plants, Pulses, Sugar and Tuber Crops (ed Kole C.) 123–132 (Springer-Verlag Press: Berlin Heidelberg, 2007).
- 13.Zohary, D., Hopf, M.. & Weiss, E. in Domestication of Plants in the Old World, third edn (eds Zohary, D., Hopf, M. & Weiss, E.) Domestication of Plants in the Old World. 334 (Oxford University Press: Oxford, 2000).
- 14.Candolle AD. Origin of Cultivated Plants. New York, USA: Appleton and Company; 1959. [Google Scholar]
- 15.Sinskaya, E. N. Historical Geography of Cultivated Floras (at the Dawn of Agriculture) (Kolos, Leningrad, 1937).
- 16.Mehlenbacher, S., Cociu, V. & Hough, L. in Genetic Resources of Temperate Fruit and Nut Crops, Apricot (Prunus) (eds Moore, J. N. & Ballington, J. R.) 63–108 (International Society for Horticultural Science, Wageningen, 1990).
- 17.Gu M. Apricot cultivars in China. Acta Hortic. 1979;209:63–67. [Google Scholar]
- 18.Zhebentyayeva TN, Reighard GL, Gorina VM, Abbott AG. Simple sequence repeat (SSR) analysis for assessment of genetic variability in apricot germplasm. Theor. Appl. Genet. 2003;106:435–444. doi: 10.1007/s00122-002-1069-z. [DOI] [PubMed] [Google Scholar]
- 19.Abbo S, et al. Plant domestication versus crop evolution: a conceptual framework for cereals and grain legumes. Trends Plant Sci. 2014;19:351–360. doi: 10.1016/j.tplants.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Diamond J. Evolution, consequences and future of plant and animal domestication. Nature. 2002;418:700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, et al. Effect of domestication on the genetic diversity and structure of Saccharina japonica populations in China. Sci. Rep. 2017;7:42158. doi: 10.1038/srep42158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, et al. Study on the main meteorological characteristics and artificial regeneration rain-collection and weeds-controlling techniques of wild fruit forest in Xinyuan. Non-wood. Res. 2018;36:127–135. [Google Scholar]
- 23.Hipp AL, et al. Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. N. Phytol. 2018;217:439–452. doi: 10.1111/nph.14773. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, et al. Genetic relationships among Eriobotrya species revealed by genome-wide RAD sequence data. Ecol. Evol. 2017;7:2861–2867. doi: 10.1002/ece3.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Shahid MQ, Lin S, Chen C, Hu C. Footprints of domestication revealed by RAD-tag resequencing in loquat: SNP data reveals a non-significant domestication bottleneck and a single domestication event. BMC Genomics. 2017;18:354. doi: 10.1186/s12864-017-3738-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, Zhao Z, Miao XJ. Genetic variability of wild apricot (Prunus armeniaca L.) populations in the Ili Valley as revealed by ISSR markers. Genet. Resour. Crop Evol. 2013;60:2293–2302. doi: 10.1007/s10722-013-9996-x. [DOI] [Google Scholar]
- 27.Yuan ZH, et al. Population genetic structure in apricot (Prunus armeniaca L.) cultivars revealed by fluorescent-AFLP markers in southern Xinjiang, China. J. Genet. Genomics. 2007;34:1037–1047. doi: 10.1016/S1673-8527(07)60117-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang QP, et al. The genetic relationship and structure of some natural interspecific hybrids in Prunus subgenus Prunophora, based on nuclear and chloroplast simple sequence repeats. Genet. Resour. Crop Evol. 2017;65:625–636. doi: 10.1007/s10722-017-0559-4. [DOI] [Google Scholar]
- 29.Ren G, et al. Genetic consequences of quaternary climatic oscillations in the Himalayas: Primula tibetica as a case study based on restriction site-associated DNA sequencing. N. Phytol. 2017;213:1500–1512. doi: 10.1111/nph.14221. [DOI] [PubMed] [Google Scholar]
- 30.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verde I, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013;45:487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 32.Kawuki RS, Ferguson M, Labuschagne M, Herselman L, Kim D-J. Identification, characterisation and application of single nucleotide polymorphisms for diversity assessment in cassava (Manihot esculenta Crantz) Mol. Breed. 2009;23:669–684. doi: 10.1007/s11032-009-9264-0. [DOI] [Google Scholar]
- 33.Hazzouri KM, et al. Whole genome re-sequencing of date palms yields insights into diversification of a fruit tree crop. Nat. Commun. 2015;6:8824. doi: 10.1038/ncomms9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pereira-Dias L, Vilanova S, Fita A, Prohens J, Rodriguez-Burruezo A. Genetic diversity, population structure, and relationships in a collection of pepper (Capsicum spp.) landraces from the Spanish centre of diversity revealed by genotyping-by-sequencing (GBS) Hortic. Res. 2019;6:54. doi: 10.1038/s41438-019-0132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta, S. K., Baek, J., Carrasquilla-Garcia, N. & Penmetsa, R. V. Genome-wide polymorphism detection in peanut using next-generation restriction-site-associated DNA (RAD) sequencing. Mol. Breed. 35, 145 (2015).
- 37.Jiao Y, et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2012;44:812–815. doi: 10.1038/ng.2312. [DOI] [PubMed] [Google Scholar]
- 38.Gao Y, et al. Genetic diversity and structure of wild and cultivated Amorphophallus paeoniifolius populations in southwestern China as revealed by RAD-seq. Sci. Rep. 2017;7:14183. doi: 10.1038/s41598-017-14738-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kostina KF. The cultivation of apricots in Fergana Valley (in Russian) Bull. Appl. Bot. Genet. Plant Breed. 1931;27:153–158. [Google Scholar]
- 41.Boulnois L. Silk Road: Monks, Warriors & Merchants on the Silk Road. New York: WW Norton & Co Inc; 2004. [Google Scholar]
- 42.He TM, et al. Using SSR markers to determine the population genetic structure of wild apricot (Prunus armeniaca L.) in the Ily Valley of West China. Genet. Resour. Crop Evol. 2007;54:563–572. doi: 10.1007/s10722-006-9117-1. [DOI] [Google Scholar]
- 43.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Z, et al. Population genomics of pearl millet (Pennisetum glaucum (L.) R. Br.): comparative analysis of global accessions and Senegalese landraces. BMC Genomics. 2015;16:1048. doi: 10.1186/s12864-015-2255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang HX, Li HY, Li YX. Identifying evolutionarily significant units for conservation of the endangered Malus sieversii using genome-wide RADseq data. Nord. J. Bot. 2018;36:e01733. doi: 10.1111/njb.01733. [DOI] [Google Scholar]
- 47.Vilella AJ, et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Q, et al. Genome-wide SNP markers based on SLAF-seq uncover breeding traces in rapeseed (Brassica napus L.) Front. Plant Sci. 2017;8:648. doi: 10.3389/fpls.2017.00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan N, et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 2017;8:249. doi: 10.1038/s41467-017-00336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watterson GA. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 55.Hudson RR, Slatkint M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 57.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, et al. Genomic analyses yield markers for Identifying agronomically Important genes in potato. Mol. Plant. 2018;11:473–484. doi: 10.1016/j.molp.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Q, et al. The genome of Prunus mume. Nat. Commun. 2012;3:1318. doi: 10.1038/ncomms2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1,Table S2,Table S3,Table S4,Table S5,Table S6,Table S8,Fig. 1,Fig. 2