

Introduction
The concept of the “leaky gut” has received increasing attention in the lay press and also in the scientific literature of late due to its associations with numerous gastrointestinal (GI) and non-GI diseases such as irritable bowel syndrome, Alzheimer’s disease, asthma, type 2 diabetes, hepatic steatosis, and many others [1] to the point that dietary modifications, probiotics, and other interventions intended to increase “gut integrity” are recommended as treatments for a host of diseases [2, 3]. The premise underlying the “leaky gut” hypothesis is that physiologic stressors such as anxiety, intensive exercise, or dietary components such as emulsifiers increase intestinal mucosal paracellular permeability, enhancing entry of pathogenic bacteria and bacterial toxins into the systemic circulation, provoking systemic inflammation and triggering numerous diseases. Although data strongly support the concept that bacterial endtoxin (lipopolysaccharide; LPS) is pro-inflammatory and that inflammation can increase intestinal paracellular permeability, few convincing data obtained form the study of intact intestinal tissue support paracellular transport of bacteria and bacterial toxins from lumen to submucosa. In this perspective, we will review the data regarding intestinal paracellular transport, providing views based on the preponderance of the available data regarding the mechanisms of intestinal transport of solutes, bacteria, and bacterial toxins in relation to paracellular permeability with the primary objective of contrasting the intestinal transport pathways for intact bacteria and bacterial toxins such as LPS with the paracellular pathways by which ions and small organic molecules are transported across the mucosa. In this fashion, we hope to provide data that will support our conclusion that the intestinal paracellular space is a major route of transport of water and small solutes such as ions and small soluble organic molecules between the lumen and submucosal space, and not a means by which large molecules, lipophilic substances, or macromolecular structures such as proteins, particulate matter, or intact bacteria are absorbed. We hope also to further the understanding of the intestinal transport of bacteria and bacterial toxins, in particular since such translocation underlies many important and highly morbid diseases such as sepsis, the systemic inflammatory response syndrome (SIRS) and multiple organ failure (MOF) [4, 5].
Overview of Intestinal Mucosal Structure and Barrier Function
The intestinal mucosal surface is an essential portal of entry of nutrients, ions, and fluids into the body that is comprised of multiple epithelial cell types serving diverse functions, connected by intercellular junctions, a luminal layer composed of glycocalyx, secreted mucus, water, and ions, and a subepithelial layer comprised of subepithelial nerves, vessels, immune cells, and lymphatics. All of these elements coalesce into an actively regulated, dynamic structure whose purpose is to absorb beneficial molecules such as nutrients, vitamins, microbial metabolites, and ions while excluding pathogenic bacteria, bacterial toxins, and other harmful substances [6]. The terms “intestinal barrier function” and “intestinal integrity” are frequently used to describe how the gut prevents harmful substances in its lumen from entering into the bloodstream. Rather than being a monolithic wall-like structure, however, the intestinal barrier consists of numerous specialized components and heterogeneous cell types and intercellular junctions that achieve this function [6, 7]. The surface mucus layer is believed to impede the ingress of intact bacteria and large particulates toward the mucosa [8–10]. Small soluble nutrients such as saccharides, amino acids, vitamins, divalent metals, and organic ions are absorbed by a broad variety of integral membrane transport proteins such as the glucose transporter SGLT1 and the organic ion transporters (OAT)s [11–13]. Ions such as Na+, Cl−, K+, and H+ and metals such as Fe2+ are transported by channels, symporters, and antiporters such as Na+/H+ antiporter NHE1 and the Cl− channel CFTR [14]. Macro-molecules such as intact proteins are transported by a variety of receptor-mediated endocytic mechanisms [15]. Intact bacteria, antigens, and particulate matter are transported by specialized M-cells overlying intestinal lymphoid aggregates (Peyer’s patches) in the distal small intestine [16–18] and are sensed by goblet cell-associated antigen pathways (GAPs) [19], with some data indicating that LPS is absorbed through the lamina propria by GAPs in the small intestine [20].
With the number to date of known intestinal membrane transporters, ion channels, and surface receptors that facilitate transport of the above-named molecules and structures from the intestinal lumen identified directly or indirectly into the systemic circulation estimated at 100 or more, the intestinal barrier is thus composed of numerous components and transport mechanisms, all regulated by bioactive molecules and by neurohormonal signaling in response to physiologic and pathologic stimuli.
Paracellular Transport
Paracellular transport is a term used to describe the movement of water and small solutes between adjacent epithelial cells through intercellular junctions [21, 22]. Although the intercellular junctions comprise ~ 0.01% of the intestinal surface area [23], paracellular transport makes an outsize contribution to the transepithelial movement of water and solutes [24]. The measurement of paracellular permeability is usually quite straightforward since the movement of ions and charged organic molecules can be measured as an electrical current and the movement of neutral molecules can be measured by assaying their concentration in the bloodstream or urine or in a compartment equivalent to the subepithelial space in mounted or perfused tissue or in cultured cells grown on permeable substrates. Although transepithelial electrical resistance and the movement of “marker” solutes is frequently used to measure paracellular permeability, only a small number of publications have directly documented paracellular movement of solutes by autoradiography, in vivo confocal microscopy, or electron microscopy [25–28] whereas for most cases, the differentiation of paracellular from transcellular movement has been indirect [23, 29]. Some of the direct demonstrations of paracellular permeability include autoradiography with 3H polyethylene glycol (PEG) 400 (Da), which reported the presence of the marker molecule in the intercellular spaces, in the enterocytes, and interestingly, in a subset of goblet cells, foreshadowing the discovery of GAPs [26]. Another is in vivo confocal microscopy of fluorescent dextran (MW ~ 10 kDa) that when injected intravenously penetrated the intercellular junctions up the region of the tight junctions. Using confocal endoscopy in humans, Chang et al. visualized leakage of intravenous fluorescein from the intercellular spaces in colitis patients, correlating it with the presence of diarrhea (Fig. 1) [27].
Fig. 1.
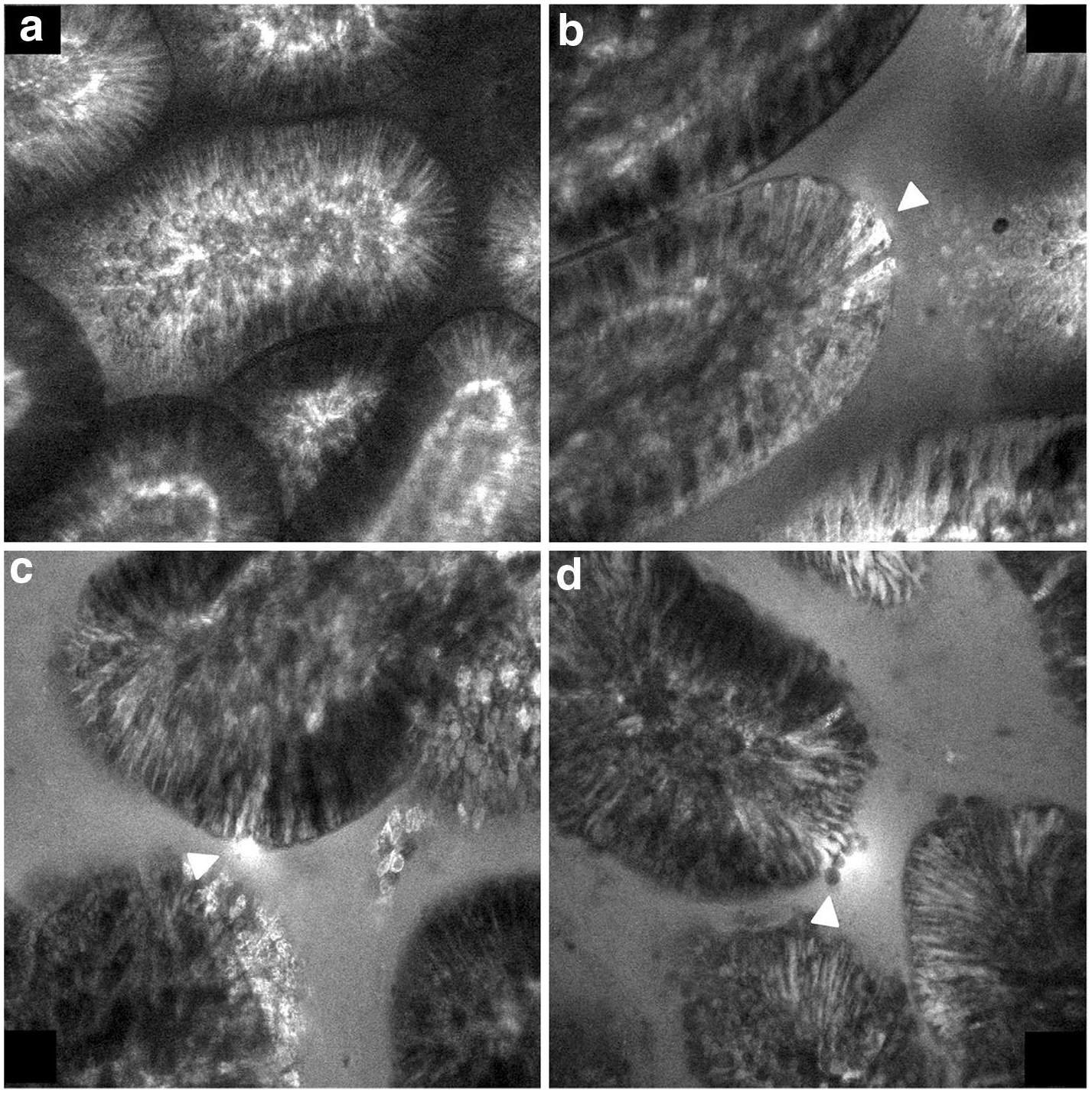
Endoscopic confocal microscopy of grossly normal ileal mucosa in a patient with Crohn’s disease previously injected intravenously with the small solute fluorescent tracer sodium fluorescein (376 Da). Arrowheads: a Normal, b cell junction enhancement, c fluorescein leak, d cell drop out. Reproduced with permission from [27]
Indirect criteria used to identify paracellular transport include the first-order kinetics in which the amount transported plotted as a function of concentration is linear, lacking the “saturation” typical of protein-mediated transport mechanisms [24, 30]. Other supportive evidence is the correlation of the rate of transport of a given substance with transcellular electrical resistance, in which lower transport rates of substances assumed to be transported paracellularly are observed in high-resistance, “tight” epithelia, although electrical resistance and the transport of marker molecules do not always coincide. A third is the lack of known protein-mediated transport pathways for a given substance. A fourth is measurement of transport of a substance in the presence of inhibitors of protein-mediated transport, with the residual transport assumed to be paracellular. Finally, increasing solute flux with tight junction disruptors such as calcium chelators is also presumptive evidence of paracellular movement [31]. Although somewhat unsatisfying due to their indirectness, these criteria have held over time even as more and more transport proteins have been molecularly identified and physiologically characterized.
The transport of ions, small water-soluble compounds, and water itself through the paracellular spaces is usually referred to as “intestinal permeability.” It is measured experimentally or clinically by the use of non- or poorly metabolized, water-soluble marker compounds administered orally and usually measured in the urine in order to calculate the permeability coefficient. Since the amount of the marker compounds that have been administered orally is known and the markers are not substantially metabolized during the study period, their urinary excretion is expressed as a percentage of the administered molecule ingested. The most commonly used marker compounds used clinically are the carbohydrates mannitol and lactulose. Mannitol, with a cross-sectional hydrated diameter of 6.5 Å is a small solute that is used as a marker of total intestinal surface area available for absorption, whereas lactulose (with a larger cross-sectional diameter of 9.5 Å) is used to assess the paracellular permeability through the tight junctions normalized to the total available intestinal surface [32], with results expressed as a ratio of the permeability of mannitol to lactulose. These spaces between intestinal cells vary in size depending on their position along the villi, with larger openings at the base of the villi or the crypts, and smaller openings at the tips of the villi, with recent data supporting the concept that the permeability and composition of the junctions between Paneth cells, goblet cells, stem cells, and other cell types populating the villus-crypt axis may vary widely [7, 32]. Proteins, including tight junction proteins such as the claudins and occludins control paracellular permeability, defined as the conductance of water and small solutes through the spaces between adjacent epithelial cells [33, 34].
The concept of paracellular permeability has recently received considerable attention due to multiple scientific breakthroughs [2]. Although intestinal paracellular junctions have been observed histologically and their function measured for decades, they were initially thought to be static structures. Numerous studies have validated that the paracellular permeability ratio is greater in patients with Crohn’s disease and some of their clinically normal relatives [35, 36] and in patients with celiac disease [37] as compared with people unaffected by or unrelated to these disease states. Another advance was the cloning of the intercellular proteins such as zonulins, claudins, occludins, tricellulins, and the many other protein components that comprise the tight junctions, facilitating the understanding of their contributions toward junctional structure and paracellular permeability [33, 38]. Another was the discovery that tight junctional permeability is dynamic, altered by systemic inflammation and other factors such as enterotoxins and the volume of fluid absorption [23, 39], initially reported in the early 1980s in clinical studies of intestinal permeability in arthritis and celiac disease patients [37, 40]. The lactulose:mannitol permeability ratio is increased in patients ingesting nonsteroidal anti-inflammatory drugs (NSAIDs), alcohol and some chemotherapeutic compounds [2].
More recent work by Jerry Turner and others [29, 41] has provided substantial data supporting the presence of two distinct paracellular pathways: the “pore” pathways that transports ions and small uncharged molecules, and the “leak” pathway that transports larger molecules regardless of charge. Since only the leak pathway varies with inflammation, this concept provides a solid foundation for the use of the above-described ratio measurements, in that the pore pathways transport the small molecules such as mannitol or 400 (Da) fluorescein isothiocyanate (FITC) 400 whereas larger molecules such as lactulose and FITC 4000 are transported by the variable leak pathway. In this fashion, the invariant transport rate through the pore pathway normalizes the variable transport through the leak pathway. Further work has supported the concept that specific members of the claudin family are responsible for charge and water selectivity of the pores [38]. Some of the most convincing human data supporting the pore and leak pathways were generated in normal humans given a low pro-inflammatory intravenous dose of bacterial endotoxin and PEGs of MW 400–10,000 Da. Notably, baseline absorption was inversely related to MW, with no change after LPS injection for the absorption of PEG 400, increased absorption after LPS for PEG 1500 and PEG 4000, and no detectable absorption of PEG 10,000 [42]. This study documented in humans the variability of the leak pathway in response to systemic inflammation versus the constancy of the pore pathway, and that very large solutes cannot penetrate the paracellular barrier, even in the presence of inflammation.
Transport of Lipophilic Compounds
Penetration of compounds into the body through the intestinal surface should be considered separately for lipid-soluble and for water-soluble compounds. Lipid-soluble compounds can presumably cross the enterocyte apical plasma membrane by diffusion with subsequent transfer through the enterocytes into the portal or lymphatic circulations. Long-chain lipids are predominantly transported out of the intestinal absorptive cells into the lymphatic circulation [43] via a multistep process including micelle formation with bile salts, intracellular de-esterification, re-esterification, packaging into lipoproteins, and extrusion across the enterocyte basolateral membrane into the lymph [44, 45]. Medium chain triglycerides are absorbed directly into the portal vein by a mechanism that has not been extensively studied [43, 46]. The transmembrane rather than paracellular transport of lipophilic compounds was supported by the demonstration of an inverse association between lipid/water partition coefficient and increased transport rate following calcium chelation, i.e., disrupting intercellular junctions increased the transport of hydrophilic but not of lipophilic small organic molecules [31].
Intestinal Absorption of Endotoxins
LPS is present in the outer cell membranes of most Gram-negative bacteria. Endotoxins are amphiphilic molecules with a hydrophilic polysaccharide chain and a lipophilic lipid A tail that in the presence of bile acids in the intestinal lumen can form micellar aggregates. Since the diameter of the micellar aggregates of endotoxins is > 100–200 Å, they are unable to penetrate the normal paracellular spaces [47]. Endotoxin entry into cells occurs via mechanisms dependent on plasma membrane structures such as lipid rafts and clathrin-dependent mechanisms [48]. It is more likely that endotoxins can either be absorbed directly through the enterocyte apical cell membrane or by a receptor-mediated transcytosis followed by exocytosis at the basolateral epithelial cell membrane [47, 49–51] (Fig. 2). Furthermore, endotoxin penetration of cell membranes requires accessory proteins such as LPS-binding protein (LBP) and the cell-surface protein cluster-of-differentiation (CD) 14 [52]. Endotoxin also penetrates GAPs, a newly discovered mechanism for the presentation of large molecules to the immune system, in the intestine but not in the colon [53]. The findings of endotoxin associated with circulating chylomicrons also suggests that endotoxins are also absorbed by uptake pathways similar to those of long-chain triglycerides [54].
Fig. 2.
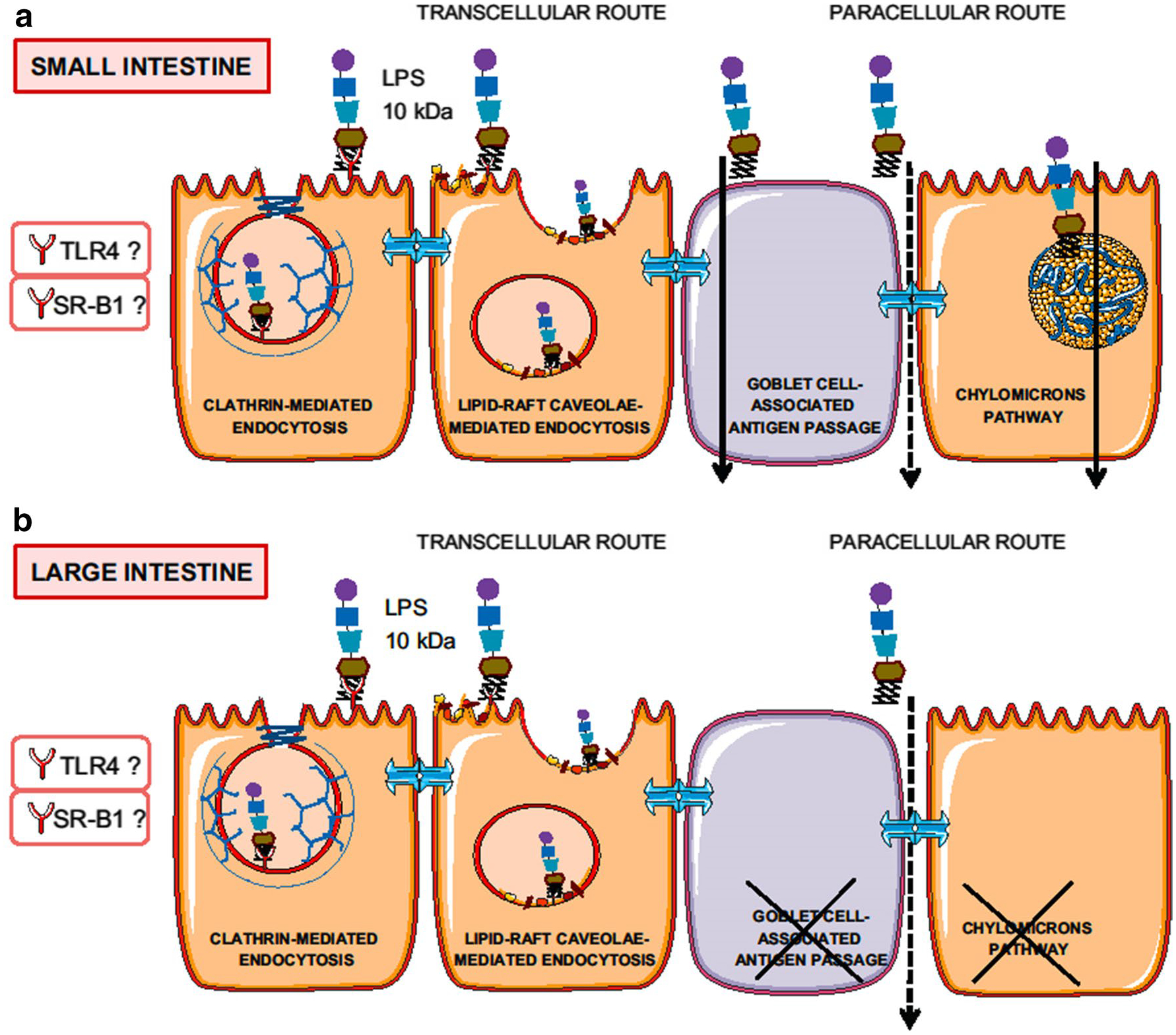
Summary of pathways by which endotoxin is absorbed in the intestine (a) and colon (b). LPS: Lipopolysaccharide. TLR4: Toll-like receptor 4. SR-B1: Scavenger receptor class B, type I. Reproduced from [47] with permission
With the exceptions of exotoxins secreted by enteropathogenic bacteria, such as the clostridial exotoxins or botulinum exotoxins discussed below, most endotoxins of commensal bacteria are not absorbed by the intact intestine of healthy individuals [49, 55]. Nevertheless, when a person whose intestinal barrier has been compromised by trauma, acute inflammatory conditions such as pancreatitis, burns, or surgery, increased endotoxin concentrations are present in the circulation [42, 56, 57]. For example, endotoxins are detected in the circulation of close to 90% of patients with trauma, 78% of patients with pneumonia, and 54% of patients with compromised inflammatory bowel disease [55]. Circulating endotoxins induce the synthesis and release of LBP by the liver. The activated cells limit the toxicity of LPS through a complex series of reactions by LPS-binding protein and receptors [52]. These reactions are part of the body’s protective mechanisms against bacteria-related sepsis.
Bacterial Translocation
The hypothesis of BT originated with clinical observations of sepsis occurring in patients with burns, trauma and circulatory collapse [58]. Since sepsis was usually caused by bacteria of enteric origin, the supposition was made that intestinal bacteria can translocate across the intestinal barrier under stress when the structural or functional integrity of the intestinal barrier is compromised. Due to the inaccessibility of the mesenteric lymphatics and portal vein, BT has not been demonstrated in healthy humans. One of the few reports to support this supposition was the measurement of portal venous endotoxin concentrations and culturable bacteria in patients undergoing elective laparotomy [59]. The authors reported that although 97% of the portal vein samples were positive for endotoxin as measured by a Limulus bioassay, only 9% of the patients had gut flora culturable from the portal vein. Considering that all of the patients had underlying diseases such as inflammatory bowel disease, colon cancer, and chronic cholecystitis, and that they had all undergone the trauma of surgery and bowel manipulation, the probability that BT exists in the absence of disease or trauma is low. The actual mechanism of BT has been investigated in cell cultures or in laboratory animals with induced trauma or burns or circulatory compromise of the intestine. It appears from these studies that physical damage to the intestinal epithelium is an important factor underlying BT. The damage can be caused by a broad variety of conditions that compromise the intestinal mucosa, including vascular or hormonal or vasoactive factors or by bacterial endotoxins [1, 4]. The mechanism by which bacteria traverse the mucosal barrier remains unclear, with the most convincing studies suggesting that a specific transcytotic process involving specialized cells overlying the domes of intestinal lymphoid aggregates (Peyer’s patches) termed M-cells, which are characterized by a sparse glycocalyx, short and irregular microvilli and a “microfold” appearance [16, 17]. M-cells take up large antigens, macromolecular complexes, particulates, and proteins such as ferritin and peroxidase and also intact viruses, bacteria, and protozoa through a specific and regulated process termed receptor-mediated endocytosis in which cell-surface receptors such as GP2 recognize specific antigens expressed on microorganisms that initiate transit through the cell, exocytosis, and engulfment by phagocytic subepithelial dendritic cells (Fig. 3) [16, 47, 60, 61]. This situation is vastly different from normal permeability of water-soluble small molecules with diameters < 10–12 Å that usually traverse the paracellular spaces and are absorbed into the circulation [32]. Another recently discovered mechanism by which proteins and particulates and possibly bacteria can penetrate the mucosa is via GAPs, in which large solutes such as 10 kDa dextran and proteins such as albumin traverse the mucosa where they are presented to subepithelial dendritic cells [19, 53, 62].
Fig. 3.
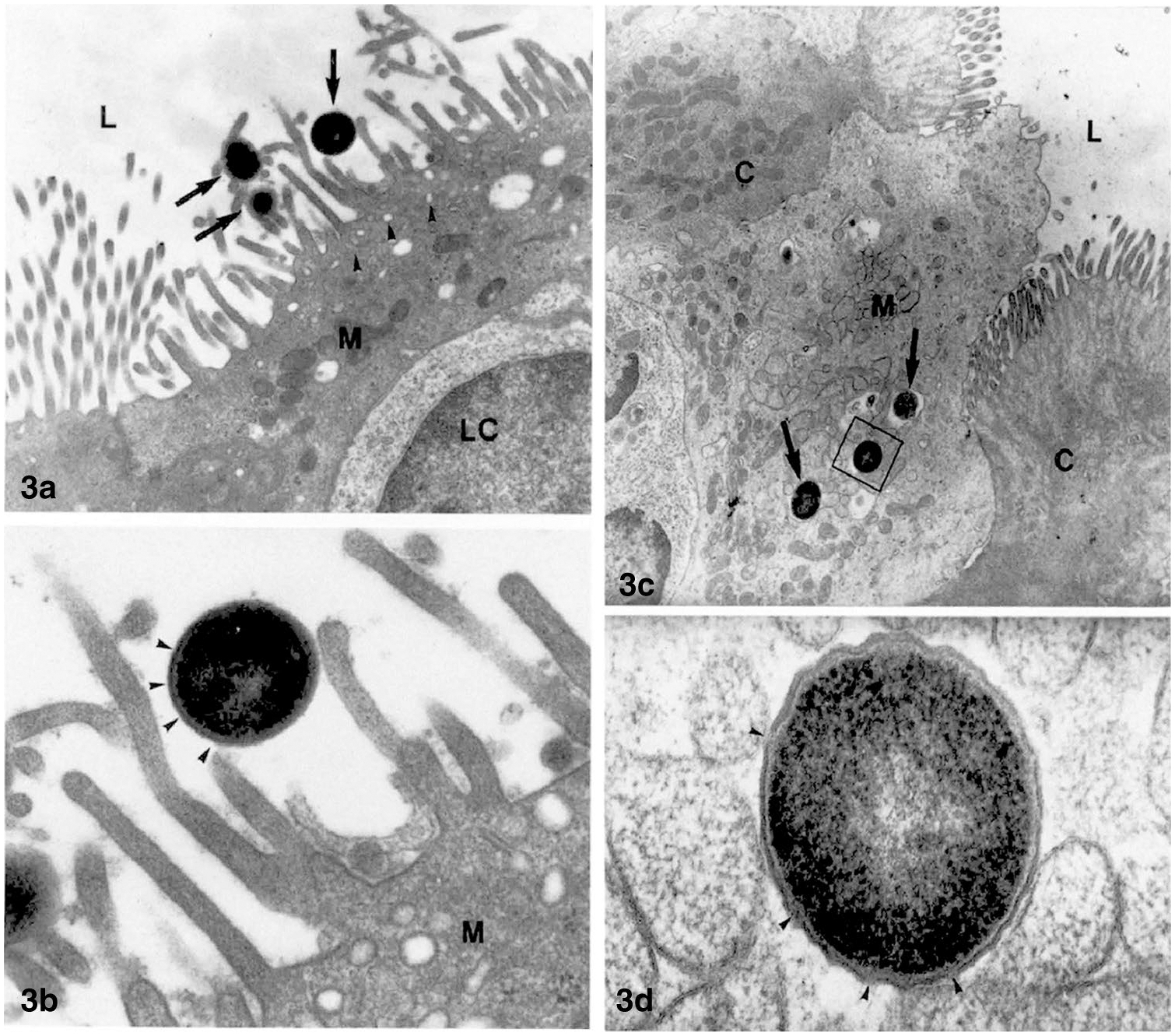
Transmission electron microscopic images of the entry of intact bacteria into M-cells overlying the Peyer’s patches in rabbit ileum. Isolated loops of ileum were incubated with a suspension of S. pneumoniæ R36a prior to harvesting the tissue. Caption from 61 “a TEM image of a M-cell (M) from 30 min treated PP. The cell shows the typical morphology: short microvilli, many pinocytotic vesicles (arrowheads) in the apical cytoplasm and a lymphoid cell (LC) close to the gut lumen (L). Among the microvilli three S. pneumoniae (arrows) are present. × 1000. b Detail of a. The bacterial wall (arrowheads) appears intact without damaged areas. M = M-cell. × 36,360. c Apical portion of a M-cell (M) with some streptococci inside endosomes (arrows), from a 60 min treated PP. The M-cell protrudes into the intestinal lumen (L) and joins the adjacent columnar cells (C). × 7870. d Enlargement of the square in c. The pneumococcus shows some broken areas in its wall (arrowheads). × 77,780.” Reproduced from [61] with permission
Pathologic Consequences of BT and Endotoxin Transport
The gut lumen contains 103–1011 bacteria/ml, [63] that in aggregate contain 1 g or more of endotoxin, that if purified and intravenously injected could be fatal to hundreds or even thousands of humans [64]. Endotoxins activate specific membrane receptors termed Toll-like receptors that are members of a class of pro-inflammatory receptors hat recognize pro-inflammatory molecules termed “pattern-associated molecular patterns” or PAMPS that in turn can activate genes that initiate inflammatory cascades in the host [49, 65]. Parenteral administration of minute amounts of endotoxins to humans elicits an inflammatory reaction typical of SIRS (fever or hypothermia, tachycardia, hyperpnea, leukocytosis, and hypotension), whereas larger amounts of administered endotoxins typically produce sepsis and shock [66, 67]. In Gram-negative bacterial infections, serum endotoxin measurements provide earlier detection of sepsis than the culture of bacterial pathogens themselves [55, 68].
BT from the intestine has been proposed by numerous authors in order to explain sepsis and endotoxemia observed in patients with severe trauma, burns, intestinal obstruction, pancreatitis, and acute liver diseases [4, 58]. Under these circumstances, the bacteria translocated from the intestine into either the portal or lymphatic circulations may be transported to multiple organs. There are also numerous animal studies demonstrating BT following different models of trauma and burn injuries. In these studies, sepsis is usually detected by the culture of enteric bacteria in the systemic circulation, in the mesenteric lymphatics, or in a variety of extra-intestinal tissues and organs. Usually, the portal vein of such laboratory animals does not have culturable bacteria despite the presence of systemic sepsis, suggesting that BT takes place by transfer of intestinal bacteria predominantly into the lymphatic circulation via the intestinal lymph nodes, likely via the specialized M-cells overlying lymphoid aggregates [69, 70].
Are Bacteria and Endotoxin Transported via the Paracellular Route?
Given the marked deleterious effects of circulating endotoxins, the main issues at the center of this discussion are the examination of evidence for the mechanisms of BT, i.e., how do bacteria and their endotoxins translocate across the intestine (transcellular vs. paracellular) and does BT or uptake of endotoxins take place at baseline or only under stressed conditions? Can bacteria or their endotoxins penetrate the intact intestinal epithelium in normal humans [59, 71] or animals through the intestinal cells themselves or through the intercellular junctions, or can BT occur only through damaged intestinal epithelium? Furthermore, the authors would like to clarify the difference between the concepts of BT and “intestinal permeability,” “intestinal integrity,” or “barrier function” that are often used interchangeably in the literature although they refer to vastly different mechanisms.
Generally, most water-soluble compounds of cross-sectional hydrated diameter < 15 Å that are not transported by membrane transporters or channels are transported across the intestinal mucosa through the intercellular spaces that are not of sufficient dimension to allow the passage of larger molecules, particulates, or microorganisms, including bacteria, with cross-sectional diameters of more than 1000 Å [42, 72]. There are rare reports of bacteria crossing gastrointestinal epithelia such as a demonstration of the pathogen H. pylori visualized in the intercellular spaces between gastric epithelial cells [73]. In vitro studies indicate that intact bacteria can penetrate monolayers of cancer-derived intestinal cell lines such as Caco-2 cells grown on permeable supports [74, 75]. It is not entirely clear if the bacteria penetrate the cells themselves or translocate through the paracellular spaces in these in vitro cultured cell monolayers [76]. These in vitro studies in monolayers with cancer-derived cell lines differ radically from the situation in vivo in the normal intact intestinal epithelium with numerous specialized cell types, intact blood and lymphatic circulations, and other structures and factors such as the glycocalyx, secreted mucus, reticuloendothelial cells, other immune cells, and mesenteric nerves. Although useful for studies of protein-mediated transport, for helping describe the contributions of specific proteins toward transcellular and paracellular solute and ion transport, and for high-throughput drug screening, [77, 78] extrapolation of data supporting the transport of microorganisms or large lipophilic molecules from cultured cells to the intact epithelium is at best hazardous. These studies should be interpreted with great caution and reservation as to their applicability to the normal intestine in individuals or damaged intestine in patients with trauma, severe burns or patients with obstructed bowel or circulatory collapse [79].
Substantiation of the hypothesis of BT as a mechanism of sepsis in humans is difficult to demonstrate once trauma or burns have occurred, or when patients are septic, have disrupted circulation to the intestine, or have a damaged intestinal epithelial surface itself. Nevertheless, several investigators have studied BT by studying mesenteric lymph node tissue at the onset of laparotomy in patients without sepsis or trauma, which is cultured with viable bacteria categorized and compared to the existing intestinal luminal bacteria. One of the largest series involved culturing of mesenteric lymph node tissue at the start of a laparotomy in patients with a variety of acute and elective surgical procedures [80]. In over 900 patients, 11% of patients undergoing elective surgery had viable bacteria cultured from the mesenteric lymph at onset of laparotomy indicating that laparotomy itself is associated with bacterial translocation in some patients. Patients with emergency surgery had 25.4% whereas patients with intestinal obstruction had 21–29% positive cultures of their intestinal lymph nodes depending on the site of obstruction. The demonstration of increased incidence of culturable bacteria in the mesenteric lymph nodes of patients with intestinal obstruction supports the concept of BT in patients with severe physiologic stress and mucosal injury and would suggest that BT does not take place without mucosal damage and/or inflammation or trauma. These studies, although confirming the concept of BT in humans, do not identify the mechanism whereby the bacteria are transported from lumen to the lymphatics. Given that M-cells have well-accepted BT capability and overlie intestinal lymphoid aggregates, M-cell mediated BT is one plausible mechanistic explanation [16, 17].
Toxins of Pathogenic Bacteria
Thus far, we have limited our discussion to the generic endotoxins that comprise part of the outer cell wall of Gram-negative bacteria. Pathogenic bacteria, such as Clostridium perfringens, Salmonella typhimurium, Shigella flexneri, Vibrio cholerae, toxigenic E. coli, and others have specific exotoxins that have characteristic host interactions that differ from the host interaction with LPS. Though the normal intestinal microbiome does not include large amounts of pathogenic organisms, as an example, increased proliferation of the pathogen Clostridium botulinum is associated with release of botulinum neurotoxins, damaging the intestinal barrier and fostering absorption of the neurotoxin into the systemic circulation and the nervous system, causing botulism [49]. Furthermore, the exotoxins of diarrheagenic toxin-producing bacteria such as Clostridium perfringens, Salmonella typhimurium, Shigella flexneri, Vibrio cholerae, and toxigenic E. coli specifically bind to and alter the function of tight junctions, with consequent massive fluid and electrolyte secretion [81, 82].
Pathogenic exotoxins have a variety of means of penetrating through the intestinal barrier in otherwise healthy individuals. For example, botulinum neurotoxin is associated with a hemagglutinin complex or other protein complexes that either bind to specific receptors on the enterocyte luminal cell membrane or disrupt the junctional protein E-cadherin to open the paracellular spaces for the absorption of the toxin [83]. In some specific experiments, the intestinal M-cells serve as a portal of entry of the hemagglutinin-botulinum toxin complex [49, 70]. Different exotoxins use a wide variety of strategies to traverse the intestinal barrier via complex interactions with the enterocytes or their surface receptors or even the protein complex that controls the opening size of the paracellular pathways [84, 85]. These mechanisms, however, appear to be different than the means by which LPS and systemic inflammation increase paracellular permeability and should not be interpreted as equivalent.
Given that diarrhea and not endotoxemia is one of the principal clinical consequences of infection with enteric toxigenic bacteria, which demonstrably affect tight junction function, one must differentiate the modest increase in paracellular permeability reported in inflammatory conditions with no known over acute clinical consequences from the marked increase in paracellular permeability induced by enteric exotoxins with consequent massive fluid and electrolyte secretion and diarrhea.
Gut Barrier During Inflammation and Injury
As stated, BT from the intestinal microbiome through the intestinal mucosa does not take place in healthy individuals. Yet, patients with severe trauma or burn injury lose the normal mucosal protection barrier due to inflammatory mediators affecting paracellular and transcellular transport processes [4, 71, 80]. The mechanisms by which inflammation affects tight junction structure and function and transcellular mechanisms such as endocytosis are largely unknown, although reproducible correlations have been observed between the severity of experimental and clinical disease and on the one hand and transepithelial electrical resistance, transport of paracellular markers, and systemic LPS concentrations and on the other [42, 86]. Moreover, concentrations of circulating endotoxin and the number of bacteria in the intestinal lymph nodes and the lymphatic circulation also correlate with disease severity [80]. These correlations have been cited in support of the concept of the “leaky gut” wherein inflammatory states increase paracellular permeability to endotoxins and bacteria. Despite these assumptions, no direct evidence exist supporting bacterial or endotoxin transport through the paracellular pathway; rather, the translocation of bacteria appears to take place only when the intestinal structure is altered [1]. Furthermore, as mentioned previously, bacteria are far too large and lipophilic to be transported via the paracellular pathway, even in the presence of inflammation [42, 72]. The finding of bacteria in subepithelial lymph nodes and intestinal lymph supports the possibility of enhanced bacterial uptake via the well-described M-cell mediated pathway. BT, therefore, should be considered as a distinct process that is quite different from the normal paracellular intestinal permeability of small water-soluble compounds [29, 32].
Conclusions
The concept of the “leaky gut,” although used to explain the pathogenesis of many common diseases, should be rethought in terms of the available data that support the many means by which luminal substances cross the gut mucosal barrier. In terms of bacteria and bacterial products, the data strongly support transcellular uptake mechanisms in all but the most extreme situations. That is to say that environmental pressures such as acute inflammation and ischemia do increase transmucosal transport of bacteria and bacterial products in humans, but not likely by the paracellular route. Accordingly, the terms “gut permeability,” “leaky gut,” and “gut integrity” should probably be discarded in favor of more meaningful though cumbersome nomenclature such as “gut solute paracellular permeability” so as to imply organ, route, and mechanism. Furthermore, the associations between gut solute paracellular permeability and circulating endotoxin concentrations should not be regarded as cause and effect, but rather increased gut paracellular permeability should be regarded as a useful biomarker for systemic inflammation.
Increased knowledge of how bacteria and bacterial products such as endotoxin cross the epithelial barrier should yield data that facilitate the discovery of treatments aimed at abrogating the most feared complications of systemic endotoxemia, namely SIRS and MOF, whereas further study of paracellular permeability could likely lead to breakthroughs in the prevention of toxigenic diarrhea, a cause of substantial morbidity and mortality worldwide [87]. Furthermore, the study of the non-transport-related functions of junctional proteins should yield promising insights into cancer pathogenesis, immune function, and identifying biomarkers [88]. It is hoped that this review will convince scientists and clinicians to look past paracellular permeability to other mechanisms by which endotoxin is taken into the body in the hope that interventions can be devised that prevent endotoxin uptake and that mechanistic interpretations of processes that increased paracellular permeability can be made.
Key Messages.
Intestinal paracellular permeability accounts for the movement of ions and small solutes between the intestinal lumen and the circulation through intercellular tight junctions
Studies of the intestinal paracellular transport of large molecules, particulates, and bacteria investigated in cultured epithelially derived cells have to a large extent not been replicated in living organisms
“Intestinal permeability” implying transport through intercellular tight junctions is only one component of the “intestinal barrier.” The two terms should be used precisely and not interchangeably
Many exotoxigenic pathogenic enteric bacteria disrupt the structure and function of the tight junctions, resulting in massive fluid secretion and diarrhea
It is unlikely that endotoxin enters the circulation via the paracellular route in undamaged intestinal mucosa
M-cells are the most probable entry point of luminal bacteria into the submucosal lymphatics
The common assumption that bacterial endotoxins or intact bacteria are transported through the intestine via the paracellular pathway in health or disease is not supported by data obtained in vivo or in vitro in intact mucosa
The concept of the “leaky gut” should be broadened to encompass pathways other than paracellular permeability in the genesis of diseases attributed to endotoxemia
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nagpal R, Yadav H. Bacterial translocation from the gut to the distant organs: an overview. Ann Nutr Metab. 2017;71:11–16. [DOI] [PubMed] [Google Scholar]
- 2.Camilleri M Leaky gut: mechanisms, measurement and clinical implications in humans. Gut. 2019;68:1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quigley EM. Leaky gut—concept or clinical entity? Curr Opin Gastroenterol. 2016;32:74–79. [DOI] [PubMed] [Google Scholar]
- 4.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. 2012;10:350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatia M, He M, Zhang H, et al. Sepsis as a model of SIRS. Front Biosci (Landmark. Ed). 2009;14:4703–4711. [DOI] [PubMed] [Google Scholar]
- 6.Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearce SC, Al-Jawadi A, Kishida K, et al. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol. 2018;16:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer-Hoffert U, Hornef MW, Henriques-Normark B, et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut. 2008;57:764–771. [DOI] [PubMed] [Google Scholar]
- 10.Johansson ME, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105:15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karasov WH. Integrative physiology of transcellular and paracellular intestinal absorption. J Exp Biol. 2017;220:2495–2501. [DOI] [PubMed] [Google Scholar]
- 12.Lameris AL, Nevalainen PI, Reijnen D, et al. Segmental transport of Ca2 + and Mg2 + along the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2015;308:G206–G216. [DOI] [PubMed] [Google Scholar]
- 13.Ganapathy V, Thangaraju M, Gopal E, et al. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008;10:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh AK, Liu Y, Riederer B, et al. Molecular transport machinery involved in orchestrating luminal acid-induced duodenal bicarbonate secretion in vivo. J Physiol. 2013;591:5377–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haq S, Grondin J, Banskota S, et al. Autophagy: roles in intestinal mucosal homeostasis and inflammation. J Biomed Sci. 2019;26:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno H Intestinal M cells. J Biochem. 2016;159:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams IR, Owen RL. M Cells: Specialized Antigen Sampling Cells in the Follicle-Associated Epithelium, vol. 1 4th ed. Amsterdam: Elsevier Inc; 2015:211–229. [Google Scholar]
- 18.Kucharzik T, Lügering N, Rautenberg K, et al. Role of M cells in intestinal barrier function. Ann N Y Acad Sci. 2000;915:171–183. [DOI] [PubMed] [Google Scholar]
- 19.Knoop KA, Newberry RD. Goblet cells: multifaceted players in immunity at mucosal surfaces. Mucosal Immunol. 2018;11:1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howe SE, Lickteig DJ, Plunkett KN, et al. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS One. 2014;9:e86656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond JM, Wright EM. Biological membranes: the physical basis of ion and nonelectrolyte selectivity. Annu Rev Physiol. 1969;31:581–646. [DOI] [PubMed] [Google Scholar]
- 22.Madara JL. Warner-Lambert/Parke-Davis award lecture: pathobiology of the intestinal epithelial barrier. Am J Pathol. 1990;137:1273–1281. [PMC free article] [PubMed] [Google Scholar]
- 23.Pappenheimer JR. Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J Membr Biol. 1987;100:137–148. [DOI] [PubMed] [Google Scholar]
- 24.Pappenheimer JR, Reiss KZ. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membr Biol. 1987;100:123–136. [DOI] [PubMed] [Google Scholar]
- 25.Soler AP, Miller RD, Laughlin KV, et al. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis. 1999;20:1425–1431. [DOI] [PubMed] [Google Scholar]
- 26.Ma TY, Hollander D, Riga R, et al. Autoradiographic determination of permeation pathway of permeability probes across intestinal and tracheal epithelia. J Lab Clin Med. 1993;122:590–600. [PubMed] [Google Scholar]
- 27.Chang J, Leong RW, Wasinger VC, et al. Impaired intestinal permeability contributes to ongoing bowel symptoms in patients with inflammatory bowel disease and mucosal healing. Gastroenterology. 2017;153:e1. [DOI] [PubMed] [Google Scholar]
- 28.Watson AJ, Chu S, Sieck L, et al. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–912. [DOI] [PubMed] [Google Scholar]
- 29.Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb Perspect Biol. 2018;10:a029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrmann JR, Turner JR. Beyond Ussing’s chambers: contemporary thoughts on integration of transepithelial transport. Am J Physiol Cell Physiol. 2016;310:C423–C431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Artursson P, Magnusson C. Epithelial transport of drugs in cell culture. II: effect of extracellular calcium concentration on the paracellular transport of drugs of different lipophilicities across monolayers of intestinal epithelial (Caco-2) cells. J Pharm Sci. 1990;79:595–600. [DOI] [PubMed] [Google Scholar]
- 32.Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. 1995;108:1566–1581. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. 2017;1397:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luettig J, Rosenthal R, Barmeyer C, et al. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers. 2015;3:e977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives: a possible etiologic factor. Ann Intern Med. 1986;105:883–885. [DOI] [PubMed] [Google Scholar]
- 36.Buhner S, Buning C, Genschel J, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjarnason I, Peters TJ, Veall N. Intestinal permeability defect in coeliac disease. Lancet. 1983;1:1284–1285. [DOI] [PubMed] [Google Scholar]
- 38.Krug SM, Schulzke JD, Fromm M. Tight junction, selective permeability, and related diseases. Semin Cell Dev Biol. 2014;36:166–176. [DOI] [PubMed] [Google Scholar]
- 39.Dubreuil JD. Enterotoxigenic Escherichia coli targeting intestinal epithelial tight junctions: an effective way to alter the barrier integrity. Microb Pathog. 2017;113:129–134. [DOI] [PubMed] [Google Scholar]
- 40.Bjarnason I, Williams P, So A, et al. Intestinal permeability and inflammation in rheumatoid arthritis: effects of non-steroidal anti-inflammatory drugs. Lancet. 1984;2:1171–1174. [DOI] [PubMed] [Google Scholar]
- 41.Shen L, Su L, Turner JR. Mechanisms and functional implications of intestinal barrier defects. Dig Dis. 2009;27:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hietbrink F, Besselink MG, Renooij W, et al. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock. 2009;32:374–378. [DOI] [PubMed] [Google Scholar]
- 43.Hyun SA, Vahouny V, Treadwell CR. Portal absorption of fatty acids in lymphand portal vein-cannulated rats. Biochim Biophys Acta. 1967;137:296–305. [DOI] [PubMed] [Google Scholar]
- 44.Iqbal J, Hussain MM. Intestinal lipid absorption. Am J Physiol Endocrinol Metab. 2009;296:E1183–E1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernier-Latmani J, Petrova TV. Intestinal lymphatic vasculature: structure, mechanisms and functions. Nat Rev Gastroenterol Hepatol. 2017;14:510–526. [DOI] [PubMed] [Google Scholar]
- 46.Sivaprakasam S, Bhutia YD, Yang S, et al. Short-chain fatty acid transporters: role in colonic homeostasis. Compr Physiol. 2017;8:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guerville M, Boudry G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointest Liver Physiol. 2016;311:G1–G15. [DOI] [PubMed] [Google Scholar]
- 48.Hansen GH, Dalskov SM, Rasmussen CR, et al. Cholera toxin entry into pig enterocytes occurs via a lipid raft- and clathrin-dependent mechanism. Biochemistry. 2005;44:873–882. [DOI] [PubMed] [Google Scholar]
- 49.Connan C, Popoff MR. Uptake of clostridial neurotoxins into cells and dissemination. Curr Top Microbiol Immunol. 2017;406:39–78. [DOI] [PubMed] [Google Scholar]
- 50.Lacy DB, Stevens RC. Sequence homology and structural analysis of the clostridial neurotoxins. J Mol Biol. 1999;291:1091–1104. [DOI] [PubMed] [Google Scholar]
- 51.Benoit R, Rowe S, Watkins SC, et al. Pure endotoxin does not pass across the intestinal epithelium in vitro. Shock. 1998;10:43–48. [DOI] [PubMed] [Google Scholar]
- 52.Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leukoc Biol. 1998;64:25–32. [DOI] [PubMed] [Google Scholar]
- 53.Knoop KA, Gustafsson JK, McDonald KG, et al. Antibiotics promote the sampling of luminal antigens and bacteria via colonic goblet cell associated antigen passages. Gut Microbes. 2017;8:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghoshal S, Witta J, Zhong J, et al. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–97. [DOI] [PubMed] [Google Scholar]
- 55.Opal SM, Scannon PJ, Vincent JL, et al. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis. 1999;180:1584–1589. [DOI] [PubMed] [Google Scholar]
- 56.Ammori BJ, Fitzgerald P, Hawkey P, et al. The early increase in intestinal permeability and systemic endotoxin exposure in patients with severe acute pancreatitis is not associated with systemic bacterial translocation: molecular investigation of microbial DNA in the blood. Pancreas. 2003;26:18–22. [DOI] [PubMed] [Google Scholar]
- 57.Ammori BJ, Leeder PC, King RF, et al. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–262. [DOI] [PubMed] [Google Scholar]
- 58.Deitch EA, Rutan R, Waymack JP. Trauma, shock, and gut translocation. New Horiz. 1996;4:289–299. [PubMed] [Google Scholar]
- 59.Jacob AI, Goldberg PK, Bloom N, et al. Endotoxin and bacteria in portal blood. Gastroenterology. 1977;72:1268–1270. [PubMed] [Google Scholar]
- 60.Da Silva C, Wagner C, Bonnardel J, et al. The Peyer’s patch mono-nuclear phagocyte system at steady state and during infection. Front Immunol. 2017;8:1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Regoli M, Borghesi C, Bertelli E, et al. Uptake of a gram-positive bacterium (Streptococcus pneumoniae R36a) by the M cells of rabbit Peyer’s patches. Ann Anat. 1995;177:119–124. [DOI] [PubMed] [Google Scholar]
- 62.Knoop KA, McDonald KG, McCrate S, et al. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 2015;8:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wassenaar TM, Zimmermann K. Lipopolysaccharides in food, food supplements, and probiotics: should we be worried? Eur J Microbiol Immunol (Bp). 2018;8:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pérez-Reytor D, Jaña V, Pavez L, et al. Accessory toxins of Vibrio pathogens and their role in epithelial disruption during infection. Front Microbiol. 2018;9:2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fullerton JN, Segre E, De Maeyer RP, et al. Intravenous endotoxin challenge in healthy humans: an experimental platform to investigate and modulate systemic inflammation. J Vis Exp. 2016;111:e53913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Engelhardt R, Mackensen A, Galanos C, et al. Biological response to intravenously administered endotoxin in patients with advanced cancer. J Biol Response Mod. 1990;9:480–491. [PubMed] [Google Scholar]
- 68.Adamik B, Smiechowicz J, Kübler A. The importance of early detection of endotoxemia. Innate Immun. 2016;22:503–509. [DOI] [PubMed] [Google Scholar]
- 69.Sugii S, Ohishi I, Sakaguchi G. Intestinal absorption of botulinum toxins of different molecular sizes in rats. Infect Immun. 1977;17:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujinaga Y, Popoff MR. Translocation and dissemination of botulinum neurotoxin from the intestinal tract. Toxicon. 2018;147:13–18. [DOI] [PubMed] [Google Scholar]
- 71.Gnauck A, Lentle RG, Kruger MC. Aspirin-induced increase in intestinal paracellular permeability does not affect the levels of LPS in venous blood of healthy women. Innate Immun. 2015;21:537–545. [DOI] [PubMed] [Google Scholar]
- 72.Richter W, Vogel V, Howe J, et al. Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate Immun. 2011;17:427–438. [DOI] [PubMed] [Google Scholar]
- 73.Necchi V, Candusso ME, Tava F, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–1023. [DOI] [PubMed] [Google Scholar]
- 74.Wang B, Chen J, Wang S, et al. Lactobacillus plantarum L9 but not Lactobacillus acidophilus LA reduces tumour necrosis factor induced bacterial translocation in Caco-2 cells. Benef Microbes. 2017;8:497–505. [DOI] [PubMed] [Google Scholar]
- 75.Yasuda M, Nagata S, Yamane S, et al. Pseudomonas aeruginosa serA gene is required for bacterial translocation through Caco-2 cell monolayers. PLoS One. 2017;12:e0169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Backert S, Boehm M, Wessler S, et al. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: paracellular, transcellular or both? Cell Commun Signal. 2013;11:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun H, Chow EC, Liu S, et al. The Caco-2 cell monolayer: usefulness and limitations. Expert Opin Drug Metab Toxicol. 2008;4:395–411. [DOI] [PubMed] [Google Scholar]
- 78.van Breemen RB, Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol. 2005;1:175–185. [DOI] [PubMed] [Google Scholar]
- 79.Cruz N, Qi L, Alvarez X, et al. The Caco-2 cell monolayer system as an in vitro model for studying bacterial-enterocyte interactions and bacterial translocation. J Burn Care Rehabil. 1994;15:207–212. [DOI] [PubMed] [Google Scholar]
- 80.MacFie J, Reddy BS, Gatt M, et al. Bacterial translocation studied in 927 patients over 13 years. Br J Surg. 2006;93:87–93. [DOI] [PubMed] [Google Scholar]
- 81.Eichner M, Augustin C, Fromm A, et al. In colon epithelia, clostridium perfringens enterotoxin causes focal leaks by targeting claudins which are apically accessible due to tight junction derangement. J Infect Dis. 2017;217:147–157. [DOI] [PubMed] [Google Scholar]
- 82.Eichner M, Protze J, Piontek A, et al. Targeting and alteration of tight junctions by bacteria and their virulence factors such as Clostridium perfringens enterotoxin. Pflugers Arch. 2017;469:77–90. [DOI] [PubMed] [Google Scholar]
- 83.Yoseph BP, Klingensmith NJ, Liang Z, et al. Mechanisms of intestinal barrier dysfunction in sepsis. Shock. 2016;46:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Connan C, Voillequin M, Chavez CV, et al. Botulinum neurotoxin type B uses a distinct entry pathway mediated by CDC42 into intestinal cells versus neuronal cells. Cell Microbiol. 2017;19:e12738. [DOI] [PubMed] [Google Scholar]
- 85.Fujinaga Y, Sugawara Y, Matsumura T. Uptake of botulinum neurotoxin in the intestine. Curr Top Microbiol Immunol. 2013;364:45–59. [DOI] [PubMed] [Google Scholar]
- 86.Schietroma M, Pessia B, Carlei F, et al. Intestinal permeability and systemic endotoxemia in patients with acute pancreatitis. Ann Ital Chir. 2016;87:138–144. [PubMed] [Google Scholar]
- 87.Kotloff KL. The burden and etiology of diarrheal illness in developing countries. Pediatr Clin North Am. 2017;64:799–814. [DOI] [PubMed] [Google Scholar]
- 88.Sawada N Tight junction-related human diseases. Pathol Int. 2013;63:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]