

Abstract
Anaplastic lymphoma kinase (ALK), an oncogenic receptor tyrosine kinase, is a therapeutic target in various cancers, including non-small cell lung cancer. Although several ALK inhibitors, including crizotinib, ceritinib, and alectinib, are approved for cancer treatment, their long-term benefit is often limited by the cancer’s acquisition of resistance owing to secondary point mutations in ALK. Importantly, some ALK inhibitors cannot cross the blood-brain barrier (BBB) and thus have little or no efficacy against brain metastases. The introduction of a lipophilic moiety, such as a fluoroethyl group may improve the drug’s BBB penetration. Herein, we report the synthesis of fluoroethyl analogues of crizotinib 1, alectinib 4, and ceritinib 9, and their radiolabeling with 18F for pharmacokinetic studies. The fluoroethyl derivatives and their radioactive analogues were obtained in good yields with high purity and good molar activity. A cytotoxicity screen in ALK-expressing H2228 lung cancer cells showed that the analogues had up to nanomolar potency and the addition of the fluorinated moiety had minimal impact overall on the potency of the original drugs. Positron emission tomography in healthy mice showed that the analogues had enhanced BBB penetration, suggesting that they have therapeutic potential against central nervous system metastases.
Keywords: Lung cancer, anaplastic lymphoma kinase, brain metastasis, targeted therapeutics, PET
Graphical Abstract:
1. Introduction
Anaplastic lymphoma kinase (ALK), a receptor tyrosine kinase and member of the insulin receptor superfamily, is a component of the nucleophosmin (NPM1)-ALK fusion gene, which is expressed by 60% of anaplastic large-cell lymphomas. ALK is also part of the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion gene, which occurs in 3–7% of non-small cell lung cancers (NSCLCs) [1–3]. Thus, ALK is an attractive therapeutic target for cancers that have ALK gene fusions or activating mutations of ALK [4]. Accordingly, much work has been done to develop ALK-inhibiting drugs. Cui et al. [5] were the first to report the structure-based design and synthesis of crizotinib, a potent and selective dual inhibitor of ALK and mesenchymal-epithelial transition factor that was approved by the FDA for the treatment of ALK-mutant NSCLC in 2011 [6, 7]. Although many patients have initial responses to crizotinib, most tend to develop resistance to the drug within 1 or 2 years [8–10], mainly because their disease acquires additional ALK mutations that prevent crizotinib from binding to ALK and inhibiting its activity [11, 12]. Moreover, crizotinib has poor activity against central nervous system (CNS) metastases due to its inability to cross blood brain barrier (BBB) [13]. Compared with crizotinib, the second-generation ALK inhibitor alectinib, initially reported by Kinoshita et al. [14], has much higher potency (1.9 nM) and has selectivity against wild-type ALK. Alectinib also has activity against L1196M, one of the common ALK mutations that lead to crizotinib resistance, and has efficacy against CNS metastases [15, 16]. Ceritinib, another second-generation ALK inhibitor that was first reported by Marsilje et al. [17], elicits high responses in patients with crizotinib-resistant disease and was approved for the treatment of relapsed or refractory NSCLC after crizotinib failure [18]. Another ALK inhibitor is lorlatinib (PF-06463922), a third-generation ALK inhibitor recently approved by the FDA for the treatment of NSCLC [19, 20]. Other potent ALK inhibitors, including X-396, ASP3026, AP26113, PF-06463922, CEP-37440, and TSR-011, some of which have enhanced specificity for ALK, are currently in phase I and II clinical trials [21–25]. The structures of several of these ALK inhibitors are shown in Fig. 1.
Fig. 1.

Structures of several well-known ALK inhibitors.
Although crizotinib has high clinical efficacy against ALK fusion-positive NSCLC, the brain is a frequent site of initial crizotinib failure in NSCLC patients owing to the drug’s poor penetration of the CNS. On the other hand, [14C]labeled alectinib has been shown to have modest BBB penetration in rodent models. A pharmacokinetic study in rats showed that alectinib had a high brain-to-plasma ratio, and an in vitro drug permeability study in Caco-2 colorectal adenocarcinoma cells showed that alectinib was not transported by the P-glycoprotein efflux transporter, a key factor in BBB function [26]. Lorlatinib, which has moderate brain availability [27] and broad-spectrum ALK inhibitory potency for the treatment of tumors that progress despite crizotinib therapy, overcomes various resistance mutations and has efficacy against brain metastases [28]. Ceritinib, another second generation ALK inhibitor, also suffers from crossing BBB. In mice, only 0.4% of the drug was found in the brain 24 h after it’s oral administration [29]. These findings suggest that most of the ALK-inhibiting drugs have limited or poor BBB penetration.
Despite considerable efforts, developing ALK inhibitors that can effectively penetrate the BBB remains a challenge, and no diagnostic method for assessing molecule-specific pharmacodynamics and target sensitivity to ALK inhibition in vivo has been reported. The restricted repertoire of effective ALK inhibitors that can penetrate the BBB limits the targeted treatment of lung cancer brain metastases, and the lack of effective markers and methods for non-invasively monitoring these drugs’ early efficacy inhibits the selection of optimal settings in which to test and monitor the biological and therapeutic efficacy of these novel compounds. Therefore, there is need for development of an ALK inhibiting drug that have sufficient BBB penetration for treatment of NSCLC brain metastases. The addition of a fluoroethyl moiety to ALK inhibitors could give the drugs a more lipophilic character and enhance their brain penetration ability. Moreover, the replacement of fluorine (F) with 18F would enable the use of these drugs as positron emission tomography (PET) tracers to assess the relative levels and heterogeneity of ALK protein in tumors throughout the body, including the CNS, and thus help shape patient selection criteria for clinical trials. In principle, PET could also be used to assess whether the drug has engaged its target and/or to identify suitable drug dosing regimens [30].
Tyrosine kinase inhibitors (TKIs) have been radiolabeled with 11C and 18F for use with PET [31]. Many FDA-approved TKIs, as well as their derivatives, have been radiolabeled and tested preclinically [32–34]. For the majority of these molecules, however, preclinical findings have not triggered clinical translation. One exception is [11C]erlotinib [35], which is an epidermal growth factor receptor (EGFR) inhibitor used to treat patients with NSCLC and certain types of pancreatic cancer [36, 37]. A clinical trial of [11C]erlotinib in NSCLC patients showed that accumulation of the tracer was correlated with response to erlotinib therapy, especially in patients harboring the EGFR-L858R activating mutation [38, 39].
Few studies have investigated ALK inhibitor analogues for use with PET [27, 40]. We previously reported the synthesis of a ceritinib analogue (fluoroethyl ceritinib; compound 9 in the present study) labeled with 18F [41], and others have synthesized and tested [11C] and [18F]labeled lorlatinib for brain penetration [27, 40]. Analyses of the structure-activity relationship and molecular modeling of ceritinib suggest that substitutions on the drug’s piperidine nitrogen are fairly well-tolerated without compromising the drug’s efficacy [42]; similarly, substitutions on the piperidine nitrogen in crizotinib [5] and on the secondary amine nitrogen in alectinib are also well-tolerated [43]. We hypothesized that crizotinib, alectinib, and ceritinib, minimally modified with the attachment of a lipophilic group and radioisotope, retain their ALK-specific efficacy, have enhanced BBB penetration, and enable facile PET pharmacokinetic analysis. Herein, we report the syntheses of fluorinated analogues of crizotinib ([19F/18F]fluoroethyl crizotinib; [19/18F]1), alectinib ([19F/18F]fluoroethyl alectinib; [19/18F]4), and ceritinib ([19/18F]fluoroethyl ceritinib; [19/18F]9) and the preliminary in vitro and PET-based validation of efficacy and brain permeability in vivo.
2. Results and discussion
2.1. Chemistry and radiochemistry
We used two methods to synthesize fluorinated analogues of ALK inhibitors. Method 1 involved a reaction between the original drug and fluoroethyl tosylate [44], and Method 2 involved introducing the fluorine atom in the last step by displacing a suitable leaving group of a preinstalled ethyl moiety in the original drug. Both methods were suitable for producing [18F]1; however, only Method 2 was suitable for producing [18F]4.
2.1.1. Synthesis of fluoroethyl crizotinib 1
Non-radioactive fluoroethyl crizotinib (compound 1) was prepared using the two methods mentioned above. In Method 1, reaction of fluoroethyl tosylate with crizotinib (Scheme 1, Method 1) afforded compound 1 in 51% yield after chromatographic purification in a single-step reaction. Compound 1 was characterized by spectroscopic methods. 1H nuclear magnetic resonance (NMR) spectroscopy of compound 1 showed two sets of additional peaks: one at 4.55 ppm with a coupling constant of 47.8 Hz (J F-H, geminal), and another at 2.96 ppm with a coupling constant of 28.4 Hz (J F-H, vicinal). 13C NMR spectroscopy showed two additional peaks, and 19F NMR spectroscopy showed one additional peak at −217.1 ppm. High resolution mass spectrometry (HRMS) showed the desired m/z [M+Na]+ as 518.1290, which was consistent with the desired molecule.
Scheme 1.

Synthesis of fluoroethyl crizotinib 1 by Methods 1 and 2.
In Method 2, reaction of the precursor chloroethyl crizotinib (compound 3) with tetrabutylammonium fluoride (Bu4NF; Scheme 1, Method 2) afforded compound 1 in 70% yield. Hydroxyethyl crizotinib (compound 2) was obtained in 74% yield by the reaction of crizotinib with hydroxyethyl tosylate. 1H NMR spectroscopy of compound 2 showed four additional protons, 13C NMR showed two additional peaks, and the mass spectrum showed the desired m/z [M+H]+ as 494.88. Reaction of compound 2 with p-toluenesulfonyl chloride (TsCl) in dichloromethane (CH2Cl) produced compound 3 in 39% yield. 1H NMR spectroscopy showed that compound 3 had the same number of protons as compound 2 in the aliphatic region without any additional aromatic peaks, 13C NMR spectroscopy showed that compound 3 had the same number of carbons as compound 2, and the HRMS m/z [M+H]+ was 512.1204. Upon treatment of compound 2 with TsCl, compound 3 was obtained in place of the expected tosylate analogue. This may have been due to a further nucleophilic attack the chloride perpetrated on the intermediate tosylate either directly or in a two-step process, in which the chloride attacks an aziridine ring formed by anchimeric assistance by a lone pair of electrons in the nitrogen in the piperidyl ring of crizotinib. The treatment of alcohols with tosyl chloride in similar reaction conditions has been reported to yield chloroethyl derivatives [41]. Ding et al. also reported that tosylate does not always form when benzyl alcohols are treated with tosyl chloride [45].
2.1.2. Radiosynthesis of [18F]1.
Radiosynthesis of [18F]1 was also achieved by two different methods. In Method 1 (Scheme 2, Method 1), the prosthetic group [18F]fluoroethyl tosylate was first produced with a mean decay-corrected radiochemical yield of 68% (range, 65–73%; n=15). Compound [18F]1 was produced by reaction of crizotinib with [18F]fluoroethyl tosylate with a mean radiochemical yield of 54% (range, 45–65%; n=8). The average decay-corrected yield of [18F]1 from aqueous [18F]fluoride was 24% (range, 20–28%; n=8). [18F]1 was >99% radiochemically pure and had a mean molar activity of 24 GBq/μmol. The synthesis time was 130–135 min from end of bombardment. High performance liquid chromatography (HPLC) of radioactive [18F]1 co-injected with the non-radioactive standard compound 1 showed co-elution of the radioactive and ultraviolet (UV) peaks, confirming the identity and purity of the radiolabeled tracer. (Supporting Information, Fig. S1).
Scheme 2.

Radiosynthesis of [18F]fluoroethyl crizotinib ([18F]1) by Methods 1 and 2.
Radiosynthesis of [18F]1 by Method 2 was a single-step process (Scheme 2, Method 2), which produced [18F]1 from compound 3 with a 70% decay-corrected yield (n=3). Unfortunately, [18F]1 could not be easily separated from the starting material on the semi-preparative HPLC column or by flash chromatography, which affected the compound’s apparent molar activity and chemical purity. Therefore, Method 1 was preferred when producing [18F]1 for biological studies.
2.1.3. Synthesis of fluoroethyl alectinib 4.
Fluoroethyl alectinib (compound 4) was also prepared using two different methods (Scheme 3).
Scheme 3.

Synthesis of fluoroethyl alectinib 4 by Methods 1 and 2.
In Method 1, the reaction of alectinib with fluoroethyl tosylate in the presence of potassium carbonate (K2CO3) produced compound 4 in 63% yield. The product was fully characterized by spectroscopic methods, including 1H, 13C, and 19F NMR spectroscopy and HRMS. 1H NMR spectra were run in dimethyl sulfoxide (DMSO)-d6 and deuterated chloroform (CDCl3); in DMSO-d6, four additional peaks and two sets of doublets were observed at 5.1 ppm (J=24 Hz) and 4.9 ppm (J=47 Hz), respectively. Interestingly, in CDCl3, one set of protons was observed as a doublet of triplets, and the other peak was a broad singlet. 13C NMR spectroscopy showed 30 peaks, and 19F NMR spectroscopy (decoupled) showed one peak at −218.57 ppm. HRMS revealed the m/z [M+H]+ to be 529.2989, indicating the desired compound.
In Method 2, the precursor methanesulfonylethyl alectinib (compound 7a) was synthesized in multiple steps. Reaction of alectinib with t-butyldimethylsilylethyl tosylate produced an alectinib derivative (t-butyldimethylsilylethyl alectinib; compound 5) in 61% yield. The product was characterized by 1H NMR spectroscopy and mass spectrometry (MS). Hydroxyethyl alectinib (compound 6) was obtained after acidic hydrolysis of compound 5 in 97% (quantitative) yield. This compound was characterized by 1H NMR, MS and HPLC; 13C NMR spectroscopy could not be performed because the compound was sparingly soluble in common NMR solvents. Reaction of compound 6 with methanesulfonyl chloride produced compound 7a in 57% yield. Compound 7a was fully characterized by spectroscopic methods, whose data were consistent with the desired compound. Toluenesulfonylethyl alectinib (compound 7b) was prepared using a process similar to that used to prepare compound 7a and fully characterized by spectroscopic methods. Reaction of compound 7a or 7b with Bu4NF in acetonitrile (MeCN) at 103°C produced compound 4 in 10–12% yield. In this reaction, the undesired vinyl derivative, n-vinyl alectinib (compound 8), formed by elimination, was obtained as the major product.
2.1.4. Radiosynthesis of [18F]4.
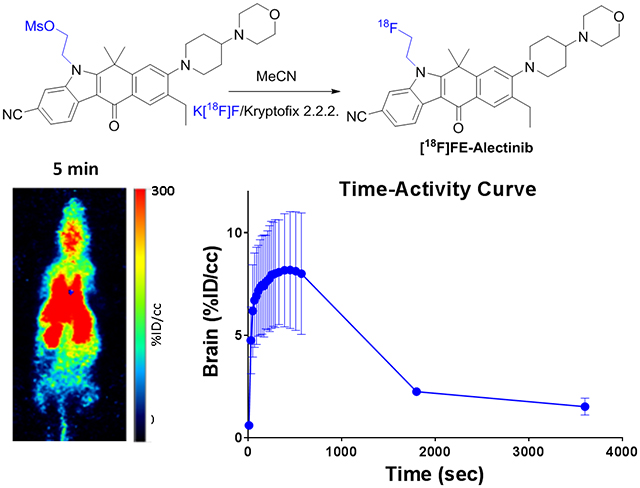
Direct coupling of [18F]fluoroethyl tosylate with alectinib in the presence of K2CO3 (Method 1, Scheme 3) was unsuccessful. Radiosynthesis of [18F]4 could be performed only by Method 2 (Scheme 4). Starting from compound 7a, the mean decay-corrected (d. c.) yield of [18F]4 was 21% (range, 16–26%; n=11). The radiochemical yield of [18F]4 from compound 7b was 18% (d. c.; n=2). [18F]4 was >99% pure and had a mean molar activity of 23 GBq/μmol. The synthesis time was 90–95 min from end of bombardment. The product could be purified by HPLC using a semi-preparative reverse phase amide column. Semi-preparative and analytical HPLC of [18F]4 co-injected with non-radioactive standard compound 4 is presented in Fig. S2 and S3 (Supporting Information).
Scheme 4:

Radiosynthesis of [18F]fluoroethyl alectinib ([18F]4) by Method 2.
2.1.5. Radiosynthesis of [18F]9.
Compound [18F]9 was produced by coupling ceritinib with [18F]fluoroethyl tosylate, as we reported previously [41]. The radiochemical yield, purity, and molar activity of [18F]9 were similar to those we reported previously.
2.1.6. Radiotracer formulation
Because [18F]1, [18F]4, and [18F]9 were lipophilic and insoluble in water or saline, they were dissolved in a small volume of ethanol (0.1–0.2 mL) and then diluted with 10% Tween 80 and phosphate-buffered saline (PBS; pH 7.4). The final formulated radiotracers were filtered through a 0.22-μm Millipore filter before injection into the animals.
2.1.7. LogP values
The LogP values (assessed by octanol/water partition) were 0.73 for [18F]1, 0.87 for [18F]4, and 0.97 for [18F]9. The LogDpH=7.4 values (assessed by octanol/PBS partition) were 1.43 for [18F]1, 1.47 for [18F]4, and 1.51 for [18F]9. Of note, the theoretical LogP values (calculated with Molinspiration software) for [18F]1, [18F]4, and [18F]9 were 4.81, 6.01, and 6.94, respectively, and those of the parent compounds crizotinib, alectinib, and ceritinib were 4.01, 5.28, and 5.91, respectively. Differences in calculated versus experimental LogP values have been previously reported [46, 47], and experimental LogP values of the parent drugs are not reported; therefore, the lipophilicities of the parent compounds and the fluoroethyl derivatives were compared using the calculated LogP values. The higher LogP values of the fluoroethyl derivatives indicated that these compounds have more lipophilicity than their parent compounds; therefore, they are more likely to cross the intact BBB.
2.2. Biology
2.2.1. Cytotoxicity
We assessed the cytotoxicity of the fluorinated ALK inhibitors in ALK-positive H2228 human lung cancer cells, which harbor EML4-ALK variant 3 (86 kDa) [48], and in ALK-negative H441 lung cancer cells. The cell lines’ differential expression of ALK was confirmed by Western blotting (Fig. 2).
Fig. 2.

Western blotting of ALK expression in lung cancer cells. Lane 1, molecular weight markers; lane 2, H2228 cell lysate; lane 3, H441 cell lysate. GAPDH was probed as a loading control.
H2228 cells were treated with the three fluorinated ALK inhibitors, and efficacy was compared with that of their corresponding parent drugs (Table 1, Fig. 3). Compound 1 had a half maximal inhibitory concentration ([IC50]) of 7.5 μM, whereas crizotinib had an IC50 of 2.8 μM, which was less than 3-fold lower than that of the parent drug. Compound 4, which had an IC50 of 10 nM (versus an IC50 of 6.5 nM for alectinib), was the most potent derivative but had lower efficacy. Nonetheless, its potency confirmed compound 4 as a potential PET tracer. Compound 9 had an IC50 of 24.2 μM, indicating that it had nearly 10-fold less potency than ceritinib (IC50, 2.9 μM). Thus, the fluoroethyl moiety impacted the potency of ceritinib, but did not have a detrimental effect on the potency of crizotinib or alectinib. Both alectinib and compound 4 had no substantial effect on ALK-negative H441 cells.
Table 1.
IC50 values of fluoroethyl derivatives and their parent compounds in H2228 cells.
| Compound | Crizotinib | Compound 1 | Alectinib | Compound 4 | Ceritinib | Compound 9 |
|---|---|---|---|---|---|---|
| IC50 value | 2.8 μM | 7.5 μM | 6.5 nM | 10 nM | 2.9 μM | 24.2 μM |
Fig. 3.

Cytotoxicity of compounds 1 (A), 4 (B), and 9 (C) and their parent compounds in ALK-expressing H2228 lung cancer cells, and cytotoxicity of compound 4 and its parent compound in non-ALK-expressing H441 lung cancer cells (D). Data are means ± standard deviations.
2.2.2. Live cell uptake of [18F]1, [18F]4, and [18F]9
The cell-associated radioactivity of [18F]1, [18F]4, and [18F]9 in ALK-positive H2228 and ALK-negative H441 cells is shown in Fig. 4. After 30 min of incubation, both [18F]1 and [18F]9 showed higher uptake in the ALK-positive H2228 cells, as expected. In contrast, [18F]4 showed higher uptake in the ALK-negative H441 cells. For all three analogues, nonspecific binding, measured as the radioactivity retained by the ALK-negative cells, was relatively high. TKI-based PET tracers tend to interact with cell membranes, potentially accounting for the high background binding; they are also often substrates for ATP-binding cassette (ABC) transporters, which could account for the low uptake of [18F]4 in H2228 cells [49]. In fact, P-glycoprotein-mediated ceritinib resistance in ALK-rearranged NSCLC has been reported [50]. Although others have reported that alectinib is not transported by the P-glycoprotein efflux transporter, a key factor in BBB penetration [26], the three [18F]labeled molecules all have theoretical LogP values greater than 3, large surface areas, and molecular weights higher than 400, which may render them susceptible to efflux by other members of the ABC transporter family [51].
Fig. 4.

Uptake of [18F]1 (A), [18F]4 (B), and [18F]9 (C) by ALK-positive H2228 cells and ALK-negative H441 cells. Data are means ± standard deviations.
2.2.3. Biodistribution of [18F]1, [18F]4, and [18F]9 in healthy mice
Non-tumor-bearing healthy mice were injected with [18F]1, [18F]4, or [18F]9, and the biodistribution of each of the three compounds was assessed by PET and region of interest analysis. The mean time-activity curves of [18F]1, [18F]4, and [18F]9 demonstrated that the tracers were quickly cleared from the heart, accumulated in both the kidney and liver, and then cleared rapidly from the urinary track (Fig. 5). The tracers steadily accumulated in the brain from 0.5 to 6.0 min, with a peak of 6–8 %ID/cc, followed by a wash out phase from the brain. Within 30 min, most of the activity was cleared from the brain. Most of the radioactivity accumulated in the liver and was slowly excreted into the bowel with significant tracer accumulation in the gallbladder. Tracer accumulation in muscle was low, being somewhat lower than retention in blood.
Fig. 5.

Mean time-activity curves and distributions of [18F]1 (A), [18F]4 (B), and [18F]9 (C) in major organs, including brain, heart, liver, kidney, and muscle, in normal nude mice. Data are means ± standard deviations.
PET images revealed the whole-body distributions of [18F]1, [18F]4, and [18F]9 (Fig. 6–8, respectively), which complemented the compounds’ time-activity curves; there was early accumulation of the tracers in the brain and washout within 30 min. One hour after injection, most of the activity was cleared from the heart (blood), and activity remained in the liver, gallbladder, and intestine. The tracers cleared quickly from the kidneys and accumulated in the bladder.
Fig. 6.

Maximum intensity projection PET images show the biodistribution of [18F]1 in a representative animal at 5, 10, 30, and 60 min after injection. B=brain, K=kidney, H=heart, and L=liver.
Fig. 8.

Maximum intensity projection PET images show the biodistribution of [18F]9 in a representative animal at 5, 10, 30 and 60 min after injection. B=brain, K=kidney, H=heart, and L=liver.
Region of interest analysis confirmed a substantial level of tracer accumulation in the brain at 10 min, with [18F]4 having the greatest accumulation (Table 2).
Table 2.
Biodistribution of [18F]1, [18F]4, and [18F]9 in normal nude mice (n=3 per group) at various times. Data are mean %ID/cc values ± standard deviations.
| Compound | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [18F]1 | [18F]4 | [18F]9 | ||||||||||
| Organ | 5 min | 10 min | 30 min | 60 min | 5 min | 10 min | 30 min | 60 min | 5 min | 10 min | 30 min | 60 min |
| Brain | 6.6±2.6 | 6.5±2.8 | 2.6±0.9 | 1.8±0.6 | 8.1±2.6 | 8.0±2.9 | 2.2±0.1 | 1.5±0.4 | 6.6±1.8 | 6.5±1.6 | 4.6±0.1 | 4.0±0.2 |
| Heart | 5.2±1.0 | 3.6±0.8 | 1.9±0.6 | 1.6±0.4 | 6.1±1.3 | 4.2±1.1 | 1.9±0.3 | 2.0±0.7 | 9.1±2.9 | 7.0±2.6 | 3.1±0.3 | 2.6±0.1 |
| Liver | 6.8±0.8 | 7.1±1.0 | 4.9±1.9 | 3.7±1.2 | 12.7±4.4 | 14.8±4.9 | 9.6±0.7 | 10.2±5.1 | 16.8±2.8 | 17.9±6.4 | 9.5±1.7 | 6.4±0.5 |
| Kidney | 16.9±5.5 | 14.2±4.7 | 3.2±0.9 | 2.2±0.7 | 20.7±6.8 | 17.8±7.6 | 4.3±0.4 | 4.9±1.7 | 20.2±7.9 | 18.2±6.6 | 7.2±1.1 | 5.7±0.2 |
| Muscle | 2.0±0.6 | 2.3±0.8 | 1.6±0.7 | 1.2±0.5 | 2.4±1.2 | 2.5±1.2 | 1.7±0.4 | 2.0±0.6 | 3.6±1.1 | 3.9±1.1 | 2.4±0.3 | 2.1±0.3 |
The fluoroethyl group was incorporated into the parent drugs to increase their lipophilic behavior. As the biodistribution assessments with PET and region of interest analysis show, lipophilicity also increases their brain permeability, as all three newly synthesized compounds crossed the BBB. Based on the vascular volume of the brain (1/30 by gram weight whole brain) [52], the 8.1 % ID/cc of [18F]4 5 min after injection exceeded by 40-fold the value expected for a tracer confined to the vascular compartment (0.2 %ID/g). The compounds quickly washed out of normal brain, except [18F]9, which was retained at a %ID/cc of about 4 at 1 h. Compounds [18F]1 and [18F]4 cleared the brain rapidly (within 30 min), which may reflect a susceptibility of these compounds to be substrates for the ABC transporter family. Nonetheless, initial accumulation of activity in the normal brain suggests that these compounds, especially compound l, have an advantage for therapeutic application in CNS metastasis. For example, therapeutic potential of 1 may exceed the parent compound, crizotinib, which has relatively poor penetration of the BBB [32]. Our results are in line with the findings of Collier et al. [27, 40], who reported brain penetration of [11C]lorlatinib in a rhesus macaque. They reported an activity peak in the cerebellum at 10 min, with a %ID/g value of approximately 0.020 and a standardized uptake value almost equal to 2, and a rapid washout during the subsequent hour. In contrast, the %ID/cc values of our three novel analogues in mice were almost equal to 7. Similar to the present study, the study by Collier et al. in H3122 tumor-bearing mice showed the expected hepatobiliary clearance. The modest degree of blockable tumor uptake in vivo implied a significant component of non-specific binding [40].
3. Conclusion
We synthesized and radiolabeled novel analogues of crizotinib, alectinib, and ceritinib, ([19/18F]fluoroethyl crizotinib, [19/18F]fluoroethyl alectinib, and [19/18F]fluoroethyl ceritinib, respectively). This series of ALK inhibitors was obtained in good yields with high purity and good molar activity. The radiolabeled analogues have potential as PET tracers for quantitative studies in the context of brain metastasis, but this potential is mitigated by these analogues’ high non-specific binding to biological tissues, a trait similar to those of other compounds of this general class. Compared with their parent compounds, which already have proven anticancer efficacy, these analogues may have a better therapeutic effect against brain metastases owing to their enhanced CNS pharmacokinetic properties.
4. Materials and methods
4.1. Reagents and instrumentation
Reagents and solvents were purchased from ThermoFisher Scientific (Waltham, MA) and Sigma-Aldrich (St. Louis, MO) and used without further purification. Crizotinib, alectinib, and ceritinib with greater than 95% purity were purchased from Pure Chemistry Scientific, Inc. (Sugar Land, TX). Silica gel solid-phase extraction cartridges (Sep-Pak, 900 mg) and C18 reverse-phase extraction cartridges (Sep-Pak, 900 mg) were purchased from Alltech Associates (Deerfield, IL). K[18F]F/Kryptofix 2.2.2 as an aqueous solution was purchased from Cyclotope, Inc. (Houston, TX) or from MD Anderson’s Center for Advanced Biomedical Imaging (Houston, TX). Radioactivity was measured using a dose calibrator (model # CRC-15R, Capintec, Inc., Florham Park, NJ), and tracer activity was measured using a gamma counter (model #E500300; Perkin-Elmer, Waltham, MA).
Thin-layer chromatography (TLC) was performed on pre-coated Kieselgel 60 F254 glass plates (Merck, Darmstadt, Germany) and aluminum-backed, pre-coated silica gel plates (Sorbent Technologies, Inc., Norcross, GA). 1H, 13C, and 19F NMR spectroscopy was performed at MD Anderson Cancer Center on a 300-, 500-, or 600-MHz spectrometer (Bruker, Billerica, MA) using tetramethylsilane as an internal reference or hexafluorobenzene as an external reference. HRMS was performed at the University of Minnesota on a Bruker BioTOF II mass spectrometer using electrospray ionization. Low-resolution MS was performed at MD Anderson Cancer Center on a liquid chromatography mass spectrometer with an Acquity TQ detector (Waters Corp., Milford, MA) using electrospray ionization.
HPLC was performed on 1100 Series pumps (Agilent Technologies, Stuttgart, Germany) with built-in ultraviolet detectors and a radioactivity detector with a single-channel analyzer (Bioscan, Washington, DC). Purification of [18F]fluoroethyl tosylate was performed on a reverse-phase C18 semi-prep column (Alltima C18; 10 μ, 10 × 250 mm; Alltech/Grace, Chicago, IL) using a solution of 50% MeCN in water. Purification of [18F]4 was performed on a reverse-phase semiprep amide column (Ascentis-RP-Amide; 10 μm, 25 cm × 10 mm; Supelco, Sigma-Aldrich, St. Louis, MO) using a solution of 38% MeCN in water with 0.1% trifluoroacetic acid (TFA). Quality control analyses of [18F]1 and [18F]9 were performed on an analytical C18 column (Econosil C18; 4.6 × 250 mm; Alltech/Grace) in solvent systems comprising 60% MeCN/40% water/0.1% TFA; [18F]4 was analyzed with 40% MeCN/60% water/0.1% TFA on analytical HPLC.
4.2. Chemistry and Radiochemistry
4.2.1. Ethylene glycol monotosylate and ethylene glycol ditosylate
Ethylene glycol monotosylate and ethylene glycol ditosylate were prepared using a procedure similar to one described previously [53]. Briefly, ethylene glycol (3.5 mL, 62.9 mmol, 8.0 eq.), triethylamine (3.3 mL, 23.6 mmol, 3.0 eq.), and dicholoromethathane (CH2Cl2, 12 mL) were added to a flask and stirred at 0°C for 20 min. A solution of TsCl (1.5 g, 7.9 mmol, 1.0 eq.) in CH2Cl2 (13 mL) was added slowly using a syringe over 1 h. The resulting mixture was warmed to room temperature and stirred for 24 h. TLC indicated complete consumption of TsCl. The reaction mixture was washed with water (2×10 mL), and the aqueous layer was extracted with CH2Cl2. Organic layers were combined and evaporated. The products were purified by flash column chromatography on silica gel using 30% ethyl acetate (EtOAc)/hexanes (Hex) to yield ethylene glycol monotosylate as a colorless oil (970 mg, 57% yield) with >95% purity and ethylene glycol ditosylate as a white crystalline solid (490 mg, 17% yield) with >96% purity. Ethylene glycol monotosylate: Rf = 0.3 in 30% EtOAc/Hex; 1H NMR (CDCl3, 500 MHz) δ: 7.81 (d, J = 7.1, 2H), 7.36 (d, J = 7.1 Hz, 2H), 4.14 (q, J = 2.3 Hz, 2H), 3.82 (t, J = 1.9 Hz, 2H), 2.45 (s, 3H). Ethylene glycol ditosylate: Rf = 0.5 in 30% EtOAc/Hex; 1H NMR (CDCl3, 500 MHz) δ: 7.73 (d, J = 8.1 Hz, 2H), 7.34 (d, J = 7.7 Hz, 2H), 4.18 (s, 2H), 2.46 (s, 3H).
4.2.2. Fluoroethyl tosylate
Fluoroethyl tosylate was prepared as described previously [41]. Briefly, ethylene glycol ditosylate (100 mg, 1 eq.) was dissolved in dry MeCN (4 mL); then, a solution of Bu4NF in tetrahydrofuran (THF, 300 μL, 1 M, 1.1 eq.) was added, and the reaction mixture was heated at 95°C for 1 h. The crude reaction mixture was concentrated, purified by flash column chromatography, and eluted with 20% EtOAc/Hex. After evaporation of the solvent, 35 mg of fluoroethyl tosylate was obtained as a colorless liquid in a 60% yield. 1H NMR (CDCl3, 600 MHz) δ: 7.81 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 4.57 (dt, J = 47.1 Hz & 4.1 Hz, 2H), 4.26 (dt, J = 27.1 Hz & 4.1 Hz, 2H), 2.46 (s, 3H). 19F NMR decoupled (CDCl3, 300 MHz) δ: −224.65 (s). MS: m/z [M+H]+ 219.03.
4.2.3. 3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(fluoroethylpiperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine (fluoroethyl crizotinib; compound 1)
Method 1:
3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine (crizotinib; 22.5 mg, 0.054 mmol) was dissolved in dry MeCN (2.0 mL) in a v-vial, and triethylamine (300 μL) was added. To this mixture, fluoroethyl tosylate (12 mg, 0.06 mmol, 1.1 eq.) was added, and the reaction mixture was heated for 5.5 h at 95–97°C. TLC showed a higher Rf spot in about 75% intensity. The mixture was concentrated under a stream of air, and then the crude product was purified by flash chromatography on a silica gel column and eluted with 5% methanol (MeOH)/CH2Cl2 to obtain fluoroethyl crizotinib (compound 1) (14.6 mg, 51% yield) with 99% purity as assessed by analytical HPLC. 1: Rf = 0.5 in 5% MeOH/ CH2Cl2;1H NMR (DMSO-d6, 300 MHz) δ: 7.96 (s, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.55–7.60 (m, 1H), 7.53 (s, 1H), 7.45 (t, J = 8.3 Hz, 1H), 6.90 (d, J = 1.6 Hz, 1H), 6.09 (q, J = 6.6 Hz, 1H), 5.65 (s, 2H, NH2), 4.55 (dt, J=47.8 Hz & 4.9 Hz, 2H), 4.05–4.16 (m, 1H), 3.16 (s, 1H), 2.96 (s, 1H), 2.66 (dt, J = 28.4 Hz & 4.9 Hz, 2H), 2.20 (dt, J =11.4 and 2.6 Hz, 2H), 1.90–2.01 (m, 4H), 1.81 (d, J = 6.6 Hz, 3H). 13C NMR decoupled (DMSO-d6, 150 MHz) δ: 149.40, 138.75, 136.80, 135.43, 134.46, 128.72, 123.42, 121.08, 120.95, 119.09, 117.49, 117.38, 117.33, 114.38, 95.38, 82.49, 81.40, 71.91, 58.32, 57.35, 52.17, 32.02, 18.57. 19F NMR, decoupled (DMSO-d6, 300 MHz) δ: −113.2 (s), −217.1 (s). HRMS: m/z [M+Na]+ calculated for C23H25Cl2F2N5ONa, 518.1302; found, 518.1290.
Method 2:
Compound 3 (synthesized from crizotinib as described in sections 4.2.4 and 4.2.5; 3 mg, 0.0058 mmol, 1.0 eq.) was dissolved in dry MeCN (400 μL) in a v-vial, and Bu4NF (15 μL, 0.1 M in THF, 0.0064 mmol, 1.1 eq.) was added. The resulting mixture was heated at 105–107°C for 1 h. The mixture was concentrated under a stream of air, and then the crude product was analyzed by analytical HPLC, which indicated 70% conversion to fluoroethyl crizotinib (compound 1). The product was confirmed by co-injecting it with the reference authentic sample of compound 1; both the product and the authentic sample had the same retention time.
4.2.4. 3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(hydroxyethylpiperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine (hydroxyethyl crizotinib; compound 2)
3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine (crizotinib; 50 mg, 0.11 mmol) was dissolved in dry MeCN (2.0 mL), and then triethylamine (0.6 mL, excess) was added. To this mixture, ethyleneglycol monotosylate (48 mg, 0.2 mmol, 2.0 eq.) was added, and the mixture was refluxed at 95°C for 5 h. The reaction mixture was concentrated under vacuum, and the crude product was purified by flash chromatography on silica gel, eluted with 100% EtOAc to remove unreacted ethylene glycol monotosylate, and then eluted with 10% MeOH/CH2Cl2. After solvent evaporation, hydroxyethyl crizotinib (compound 2) was obtained as an off-white solid (40 mg, 74% yield) with 95% purity. 2: Rf = 0.4 in 10% MeOH/CH2Cl2; 1H NMR (CDCl3, 500 MHz), δ: 7.76 (s, 1H), 7.56 (s, 1H), 7.50 (s, 1H), 7.30 (m, 1H), 7.05 (t, J = 8.0 Hz, 1H), 6.87 (s, 1H), 6.07 (q, J = 6.6 Hz, 1H), 4.79 (s, 2H, NH2), 4.14 (m, J= 4.3 Hz, 1H), 3.65 (t, J= 4.8 Hz, 2H), 3.07 (d, J = 10.9 Hz, 2H), 2.60 (t, J = 4.8 Hz, 2H), 2.28 (t, J = 11.7 Hz, 2H), 2.17 (d, J = 11.4 Hz, 2H), 2.06 (q, J = 11.9 Hz, 2H), 1.85 (d, J = 6.5 Hz, 3H). 13C NMR decoupled (CDCl3, 125 MHz), δ: 148.89, 139.85, 136.95, 135.72, 135.51, 128.96, 128.93, 122.58, 122.13, 121.93, 119.93, 119.21, 116.81, 116.63, 114.96, 72.42, 59.32, 59.07, 58.01, 52.32, 32.54, 18.90. MS: m/z [M+H]+ 494.88.
4.2.5. 3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(chloroethylpiperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine (chloroethyl crizotinib; compound 3)
Compound 2 (40 mg, 0.0811 mmol, 1.0 eq.) was dissolved in dry CH2Cl2 (5.0 mL) and cooled to 0°C under argon, then triethylamine (160 μL, 0.648 mmol, 8.0 eq) was added. The solution was stirred for 10 min, and then a solution of TsCl (62 mg, 0.324 mmol, 4.0 eq.) in dry CH2Cl2 (1.0 mL) was added dropwise over 15 min. This mixture was allowed to slowly warm to room temperature over 3 h, and then it was stirred continuously for another 35 h. The crude reaction mixture was concentrated under vacuum, re-dissolved in CH2Cl2 (5 mL), and washed with an aqueous sodium bicarbonate (NaHCO3) solution (0.1 M, 3×3 mL). The organic layer was isolated, dried under magnesium sulfate (MgSO4), concentrated, and purified by chromatography on silica gel and eluted with 4% MeOH/CH2Cl2. After solvent evaporation, chloroethyl crizotinib (compound 3) was obtained as a semisolid (16 mg, 39% yield) with 97% purity. 3: Rf = 0.5 in 10% MeOH/ CH2Cl2;1H NMR (CDCl3, 600 MHz), δ: 7.68 (s, 1H), 7.54 (s, 1H), 7.48 (s, 1H), 7.31 (m, J = 4.8 Hz, 1H), 7.05 (t, J = 7.92 Hz, 1H), 6.87 (s, 1H), 6.07 (q, J = 6.4 Hz, 1H), 5.26 (s, 2H, NH2), 4.12 (t, J = 11.6 Hz, 1H), 3.61 (t, J = 6.8 Hz, 2H, Cl-CH2), 3.06 (d, J = 11.1 Hz, 2H), 2.79 (t, J = 6.8 Hz, 2H, N-CH2), 2.30 (t, J = 11.7 Hz, 2H), 2.16 (d, J = 12.2 Hz, 2H), 2.06 (m, 2H), 1.85 (d, J = 6.5 Hz, 3H). 13C NMR decoupled (CDCl3, 125 MHz) δ: 175.13, 148.84, 140.02, 136.75, 135.67, 134.07, 128.96, 122.47, 119.70, 119.05, 116.89, 116.70, 115.23, 72.57, 59.49, 52.46, 41.00, 32.25, 18.87. HRMS: m/z [M+H]+ calculated for C23H26Cl3FN5O, 512.1187; found, 512.1204.
4.2.6. [18F]fluoroethyl tosylate
[18F]Fluoroethyl tosylate was prepared as described previously [41]. Briefly, aqueous K[18F]F/kryptofix was dried under a stream of argon at 103–105°C, a solution of ethylene glycol ditosylate (5–6 mg in 0.5 mL dry MeCN) was added to the dry [18F]fluoride, and the reaction mixture was heated at 103°C for 15 min. The crude reaction mixture was passed through a silica gel Sep-Pak cartridge, eluted with CH2Cl2 (2 mL), and evaporated under a stream of argon. The crude product was purified by HPLC on a semi-preparative C18 column (Alltima C18; 10 μ, 10 × 250 mm; Alltech/Grace). The radioactive product [18F]fluoroethyl tosylate was eluted with a mixture of 50% MeCN in water at a flow of 4 mL/min. The radioactive fraction was collected, and an aliquot of the product [18F]fluoroethyl tosylate was co-injected with a non-radioactive authentic sample of fluoroethyl tosylate into an analytical HPLC column to verify the product’s identity and purity. The solvent from the rest of the product was partially evaporated under reduced pressure, and then water (10 mL) was added. The solution was passed through a C18 reverse-phase cartridge; the cartridge was dried by vacuum for 5 min and then flashed with argon for 2 min. The product was eluted with 1.3 mL of dry MeCN and collected into a v-vial containing crizotinib (5.5 mg).
4.2.7. [18F]fluoroethyl crizotinib ([18F]1)
Method 1:
The reaction mixture of crizotinib and [18F]fluoroethyl tosylate in the v-vial described in section 4.2.6 was heated at 103°C for 1 h, and then an aliquot of the reaction mixture was injected into the analytical HPLC system, which showed major consumption of the radioactive [18F]fluoroethyl tosylate. The solvent was evaporated completely under a stream of argon, and the crude product was purified on a Sep-Pak silica gel cartridge and eluted with 4% MeOH/CH2Cl2. Fractions (0.5 mL) were collected, the radioactive fractions were combined, and radioactivity was measured using a dose calibrator (Capintec) to obtain [18F]1. An aliquot of the product, [18F]1, was dissolved in MeCN and then co-injected with the standard compound 1 into an analytical HPLC column to verify the product’s identity and purity. The rest of the product was evaporated and formulated for further biological study.
Method 2:
Compound 3 (3 mg) was placed in a v-vial, and a solution of dry K[18F]F/kryptofix 2.2.2 in anhydrous MeCN (0.5 mL) was added. The reaction mixture was heated at 103°C for 20 min; analysis of an aliquot of the mixture by HPLC showed high intensity of the product with insignificant [18F]fluoride remaining. The crude reaction mixture was passed through a silica gel Sep-Pak cartridge and eluted with 5% MeOH/CH2Cl2 (2 mL). The solvent from the eluent was evaporated, and the product was dissolved in CH2Cl2 and loaded onto a silica Sep-Pak cartridge. The product was eluted with 3% MeOH/CH2Cl2, and 0.5-mL fractions were collected. The radioactive fractions were combined, and the solvent was evaporated at 40°C under a stream of argon. An aliquot of the product, [18F]1, was dissolved in MeCN and co-injected with the nonradioactive authentic sample of compound 1 into an analytical HPLC column to verify the product’s identity and purity.
4.2.8. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5(2-fluoroethyl)-benzo[b]carbazole-3-carbonitrile (fluoroethyl alectinib; compound 4)
Method 1:
9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile (alectinib; 14.5 mg, 0.03 mmol, 1.0 eq.) was dissolved in dry MeCN (2.0 mL) in a v-vial, and anhydrous K2CO3 (29 mg, 0.21 mmol, 7 eq.) was added. To this mixture, fluoroethyl tosylate (21.8 mg, 0.1 mmol, 3.3 eq.) was added, and the reaction mixture was heated at 105°C for 5.5 h. TLC showed that the mixture had a higher Rf spot (0.6 in 5% MeOH/CH2Cl2) at about 65% intensity as compared with the starting material (0.5 in 5% MeOH/CH2Cl2). The mixture was filtered and concentrated under a stream of air, and then the crude product was purified by flash chromatography on silica gel and eluted with 3% MeOH/CH2Cl2 to obtain fluoroethyl alectinib (compound 4; 10 mg, 63% yield) with 99% purity as assessed by analytical HPLC. 4: Rf = 0.6 in 5% MeOH/CH2Cl2;1H NMR (DMSO-d6, 600 MHz) δ: 8.49 (d, J=8.0, 1 H), 8.29, (s, 1 H), 8.01 (s, 1 H), 7.64 (d, J=8.0, 1H), 7.34 (s, 1 H), 5.05, (d, JHF=24.1 Hz, 2 H), 4.92 (d, JHF=47.1 Hz, 2 H), 3.61 (s, 5 H), 3.23 (d, J=11.6 Hz, 2 H), 2.79 (t, J=11.5 Hz, 2 H), 2.73 (q, J=7.1 Hz, 2 H), 2.33 (quintet, 1 H), 2.49, 2H, 1.93 (d, J=10.9, 2H), 1.85 (s, 6 H), 1.75 (s, 1 H), 1.61 (q, J= 10.4 Hz, 2 H), 1.29 (t, J=7.2 H, 3H). 1H NMR (CDCl3, 300 MHz) δ: 8.75 (d, J = 8.2 Hz, 1H), 8.22 (s, 1H), 7.74 (s, 1H), 7.60 (d, J = 6.9 Hz, 1H), 7.16 (s, 1H), 4.93 (dt, J = 24.6 Hz, 4.6 Hz, 2H), 4.82 (bs, 2H), 3.77 (t, J = 4.4 Hz, 4H), 3.31 (d, J= 10.7 Hz, 2H), 2.75 (m, 4H), 2.63 (t, J = 4.6 Hz, 4H), 2.36 (m, 1H), 2.01 (d, J = 11.3 Hz, 2H), 1.87 (s, 6H), 1.73 (m, 2H), 1.33 (t, J = 7.5 Hz, 3H). 13C NMR decoupled (CDCl3, 150 MHz) δ: 180.61, 156.85, 156.15, 147.71, 137.71, 136.89, 128.37, 126.76, 125.89, 125.73, 123.43, 120.04, 115.92, 114.45, 114.44, 111.97, 106.44, 81.92, 80.76, 67.33, 61.98, 52.18, 49.99, 45.94, 45.79, 37.59, 29.53, 28.96, 23.20, 14.39. 19F NMR, decoupled (CDCl3, 300 MHz) δ: −218.57 (s). HRMS: m/z [M+H]+ calculated for C32H38FN4O2, 529.2979; found, 529.2989.
Method 2:
Compound 7a or 7b (prepared from alectinib as described in sections 4.2.10 to 4.2.13; 2.4 mg, 0.004 mmol, 1 eq.) was dissolved in dry MeCN (0.4 mL); then a solution of Bu4NF in THF (12 μL, 1M, 0.006 mmol, 1.5 eq.) was added, and the reaction mixture was heated at 100°C for 30 min. Analytical HPLC of an aliquot of the reaction mixture indicated formation of the desired compound as a minor peak and a new product as a major peak. The crude reaction mixture was concentrated, purified by column chromatography, and eluted with 2% MeOH/CH2Cl2 to afford fluoroethyl alectinib (compound 4) in 11% yield (Rf = 0.6 in 3% MeOH/CH2Cl2) with 99% purity as assessed by analytical HPLC. Another off-white solid was obtained and identified as the elimination product 9-ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5-ethenyl-benzo[b]carbazole-3-carbonitrile (n-vinyl alectinib; compound 8; Rf 0.7 in 3% MeOH/CH2Cl2) in 80% yield with 97% purity as assessed by analytical HPLC. 8: 1H NMR (CDCl3, 600 MHz) δ: 8.65 (d, J = 8.1 Hz, 1H), 8.23 (s, 1H), 7.87 (s, 1H), 7.59 (d, J=8.1Hz, 1H), 7.27 (dd, J = 8.0 Hz, 1H), 7.15 (s, 1H), 5.85 (d, J = 8.0 Hz, 1H), 5.72 (d, J = 15.48 Hz, 1H), 3.77 (t, J= 4.4 Hz, 4H), 3.31 (d, J = 12.0 Hz, 2H), 2.79–2.72 (m, 4H), 2.63 (t, J = 4.4 Hz, 4H), 2.35 (m, 1H), 2.01 (d, J = 11.7 Hz, 2H), 1.85 (s, 6H), 1.74 (qd, J = 11.8 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H). 13C NMR decoupled (CDCl3, 75 MHz) δ: 180.90, 156.57, 156.12, 147.54, 137.58, 136.72, 130.91, 128.31, 126.80, 125.96, 125.88, 123.15, 120.11, 117.41, 115.86, 115.59, 111.90, 106.53, 67.33, 61.97, 52.15, 49.98, 37.75, 28.95, 28.50, 23.21, 14.14. MS: m/z [M+H]+ calculated for C32H37N4O5, 509.2811; found, 509.2819. Similar results were obtained when compound 7b was reacted with Bu4NF.
4.2.9. t-Butyldimethylsilylethyl tosylate
t-Butyldimethylsilylethyl tosylate was prepared as described previously [54] with modification. Briefly, hydroxyethyl tosylate (200 mg, 0.924 mmol, 1.0 eq.) and t-butyldimethylsilyl chloride (418 mg, 2.774 mmol, 3.0 eq.) were dissolved in CH2Cl2 (8 mL). Then, triethylamine (0.64 mL, 4.624 mmol, 5.0 eq.) was added at 0°C, and the resulting mixture was stirred for 10 min. The mixture was then warmed to room temperature and stirred for 23 h. The reaction mixture was washed with water (2×5 mL), and the aqueous layer was extracted with CH2Cl2. Combined organic layers were dried over MgSO4, filtered, and evaporated. The crude product was purified by flash column chromatography on silica gel using 12% EtOAc/Hex to obtain t-butyldimethylsilylethyl tosylate as a pale yellow oil (243 mg, 79% yield) with 95% purity. The compound was fully characterized by 1H NMR spectroscopy and MS. Rf = 0.9 in 50% EtOAc/Hex; 1H NMR (CDCl3, 600 MHz) δ: 7.79 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H), 4.06 (t, J = 4.9 Hz, 2H), 3.79 (t, J = 5.2 Hz, 2H), 2.44 (s, 3H), 0.83 (s, 9H), 0.016 (s, 6H). MS: m/z [M+H]+ 331.13. The compound’s NMR spectrum was consistent with that reported previously [55].
4.2.10. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5(2-t-butyldimethylsilylethyl)-benzo[b]carbazole-3-carbonitrile (t-butyldimethylsilylethyl alectinib; compound 5)
Alectinib (50 mg, 0.103 mmol, 1.0 eq.) and anhydrous K2CO3 (100 mg, 0.725 mmol, 7.0 eq.) were suspended in dry MeCN (3.5 mL) in a 5-mL v-vial. To this mixture, t-butyldimethylsilylethyl tosylate (68 mg, 0.206 mmol, 2.0 eq.) was added, and the reaction mixture was heated at 100°C for 23 h. TLC showed a higher Rf spot in about 75% intensity compared to the starting material. The reaction mixture was cooled to room temperature and filtered; the filtrate was washed with 5% MeOH/CH2Cl2 (10 mL). The filtrate was concentrated under a stream of air, and the crude product was purified by flash chromatography on silica gel and eluted with 3% MeOH/CH2Cl2 to obtain t-butyldimethylsilylethyl alectinib (compound 5) (41 mg, 61% yield) with 96% purity. 5: Rf = 0.7 in 5% MeOH/ CH2Cl2; 1H NMR (CDCl3, 600 MHz) δ: 8.64 (d, J = 8.2 Hz, 1H), 8.23 (s, 1H), 7.80 (s, 1H), 7.56 (dd, J = 7.2 Hz, 1H), 7.17 (s, 1H), 4.65 (t, J = 6.3 Hz, 2H), 4.07 (t, J = 6.2 Hz, 2H), 3.77 (t, J = 4.4 Hz, 4H), 3.31 (d, J = 12.1 Hz, 2H), 2.74 (m, J = 7.5 Hz, 4H), 2.63 (t, J = 4.1 Hz, 4H), 2.36 (m, J = 6.7 Hz, 1H), 2.01 (d, J = 11.9 Hz, 2H), 1.88 (s, 6H), 1.74 (q d, J = 3.8 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H), 0.87 (s, 9H), −0.05 (s, 6H). 13C NMR decoupled (CDCl3, 150 MHz) δ: 180.66, 157.00, 156.00, 147.93, 137.57, 137.19, 128.22, 126.69, 125.83, 125.52, 123.17, 120.20, 115.95, 115.17, 111.59, 105.94, 67.32, 62.00, 61.52, 52.19, 49.98, 47.91, 37.63, 29.48, 28.95, 25.77, 23.10, 18.25, 14.41, −5.53. MS: m/z [M+H]+ calculated for C38H53N4O3Si, 641.3887; found, 641.34.
4.2.11. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5(2-hydroxyethyl)-benzo[b]carbazole-3-carbonitrile (hydroxyethyl alectinib; compound 6)
Compound 5 (50 mg, 0.0778 mmol) was suspended in MeOH (9 mL); 15 drops of 1N hydrochloric acid (HCl) was added, and the mixture was heated under reflux for 1.5 h. TLC showed the full consumption of the starting material and formation of a new lower Rf spot (Rf = 0.3 in 5% MeOH/CH2Cl2). Most of the MeOH and aqueous HCl were evaporated under vacuum azeotropically with MeCN (4×5 mL) and dried under high vacuum to yield hydroxyethyl alectinib (compound 6) as a pale yellow solid (40 mg, 97% yield) with >95% purity. 6: Rf = 0.3 in 5% MeOH/ CH2Cl2; 1H NMR (DMSO-d6, 500 MHz) δ: 10.97 (br, 1H), 8.45 (d, J = 7.3 Hz, 1H), 8.29 (s, 1H), 8.03 (s, 1H), 7.64 (d, J = 7.2 Hz, 1H), 7.39 (s, 1H), 4.70 (s, 2H), 4.01 (d, J = 10.85 Hz, 3H), 3.87 (s, 6H), 3.75 (bs, 2H), 3.33 (d, J = 8.8 Hz, 2H), 3.14 (br, 1H), 2.88 (m, J = 10 Hz, 1H), 2.70 (d, J = 6.6 Hz, 1H), 2.24 (d, J = 10 Hz, 2H), 2.06 (s, 1H), 1.88 (s, 7H), 1.28 (s, 3H). MS: m/z [M+H]+ calculated for C32H39N4O3, 527.30; found, 527.64. This compound was used for the next step for the synthesis of compound 7a without further characterization or purification.
4.2.12. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5(2-methanesulfonylethyl)-benzo[b]carbazole-3-carbonitrile (methanesulfonylethyl alectinib; compound 7a)
Compound 6 (41 mg, 0.077 mmol, 1.0 eq.) was suspended in anhydrous CH2Cl2 (6.5 mL), and triethylamine (250 μL, excess) was added; the mixture was stirred for 15 min, and the solution became a pale-yellow suspension. The suspended material was cooled to 0°C, and methane sulfonyl chloride (38 μL, 0.5 mmol, 6.0 eq.) was added. The reaction mixture was warmed to room temperature and stirred for 3 h. Reaction progress was monitored by TLC, which showed a higher Rf (0.6 in 5% MeOH/CH2Cl2) as compared with the starting material (0.3 in 5% MeOH/CH2Cl2). The reaction mixture was diluted with 5 mL of CH2Cl2 and washed with saturated NaHCO3, and the organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude product was purified by flash column chromatography on silica gel and eluted with 2% MeOH/CH2Cl2 to yield methanesulfonyl ethyl alectinib (compound 7a) as a pale yellow solid (27 mg, 57% yield) with 98% purity as assessed by analytical HPLC. 7a: Rf = 0.6 in 5% MeOH/CH2Cl2;1H NMR (CDCl3, 600 MHz) δ: 8.67 (d, J = 8.1 Hz, 1H), 8.22 (s, 1H), 7.78 (s, 1H), 7.61 (d, J = 8.1 Hz, 1H), 7.17 (s, 1H), 4.91 (t, J = 6.9 Hz, 2H), 4.60 (t, J = 6.8 Hz, 2H), 3.77 (t, J = 4.1 Hz, 4H), 3.31 (d, J = 11.7 Hz, 2H), 3.01 (s, 3H), 2.75 (m, J = 6.8 Hz, 4H), 2.63 (t, J = 4.1 Hz, 4H), 2.36 (m, J = 17.2 Hz, 1H), 2.01 (d, J = 11.7 Hz, 2H), 1.89 (s, 6H), 1.74 (dq, J = 3.0 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H). 13C NMR decoupled (CDCl3, 150 MHz) δ: 180.57, 156.72, 156.26, 147.57, 137.77, 136.64, 128.37, 126.77, 126.08, 125.62, 123.64, 119.86, 115.92, 113.99, 112.15, 106.65, 67.30, 65.16, 61.95, 52.14, 49.98, 44.53, 37.82, 37.60, 29.44, 28.93, 23.20, 14.39. HRMS: m/z [M+H]+ calculated for C33H41N4O5S, 605.2798; found, 605.2802.
4.2.13. 9-Ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5(2-p-toluenesulfonylethyl)-benzo[b]carbazole-3-carbonitrile (p-toluenesulfonylethyl alectinib; compound 7b)
Alectinib (30 mg, 0.621 mmol, 1.0 eq.) was added to dry MeCN (3.0 mL) in a v-vial, and then anhydrous K2CO3 (62 mg, 0.4 mmol, 7.0 eq.) was added. To this mixture, ethylene glycol ditosylate (32 mg, 0.09 mmol, 1.4 eq.) was added; the suspension became clear after heating at 105°C for 10 min. The reaction mixture was heated at 105°C for 20 h. TLC (5% MeOH/CH2Cl2) showed a higher Rf (0.6) spot at about 65% intensity as compared with the starting material (0.3 in 5% MeOH/CH2Cl2). The reaction mixture was filtered and washed with 10% MeOH/CH2Cl2 (10 mL), and the filtrate was evaporated under vacuum. The crude product was purified by flash chromatography on silica gel and eluted with 2% MeOH/CH2Cl2 to obtain p-toluenesulfonylethyl alectinib (compound 7b) as a pale-yellow solid (24 mg, 57% yield). 7b: Rf = 0.6 in 5% MeOH/CH2Cl2;1H NMR (CDCl3, 600 MHz) δ: 8.61 (d, J = 8.1 Hz, 1H), 8.21 (s, 1H), 7.52 (t, J = 8.1 Hz, 3H), 7.45 (s, 1H), 7.16 (d, J = 7.9 Hz, 3H), 4.80 (t, J = 6.4 Hz, 2H), 4.42 (t, J = 6.3 Hz, 2H), 3.77 (t, J = 3.9 Hz, 4H), 3.31 (d, J = 11.6 Hz, 2H), 2.75 (m, J = 7.6 Hz, 4H), 2.63 (t, J = 4.2 Hz, 4H), 2.38 (s, 3H), 2.01 (d, J = 11.8 Hz, 2H), 1.85 (s, 6H), 1.74 (dq, J = 11.8 Hz, 2H), 1.64 (s, 1H), 1.34 (t, J = 7.5 Hz, 3H). 13C NMR decoupled (CDCl3, 150 MHz) δ: 180.55, 156.80, 156.26, 147.66, 145.78, 137.74, 136.25, 131.29, 130.03, 128.34, 127.57, 126.71, 125.77, 123.50, 119.72, 115.94, 113.74, 112.01, 106.39, 67.34, 65.55, 61.95, 52.16, 49.99, 44.19, 37.60, 29.57, 28.96, 23.20, 21.69, 14.40. MS: m/z [M+H]+ calculated for C39H45N4O5S, 681.31; found, 681.31.
4.2.14. [18F]fluoroethyl alectinib ([18F]4)
Method 2: An aqueous solution of K[18F]F/Kryptofix 2.2.2 (110 mCi in 0.5 mL) was transferred into a v-vial. Water was removed by azeotropic evaporation with MeCN (1.0 mL) at 103°C under a stream of argon, and the dry K[18F]F/Kryptofix 2.2.2 was dissolved in 0.5 mL of anhydrous MeCN and then transferred to a v-vial containing compound 7a (2 mg) or compound 7b (2.9 mg). The reaction mixture in the v-vial was heated at 103°C for 30 min, and an aliquot was injected into the analytical HPLC system, which showed the presence of radioactive [18F]4 as the major product, with undetectable [18F]fluoride. The solvent was completely evaporated by heating under a stream of argon, and the crude product was dissolved in CH2Cl2 (0.2 mL) and passed through a silica Sep-Pak cartridge to remove unreacted fluoride. The cartridge was eluted with 10% MeOH/CH2Cl2 (2 mL). The solvent was evaporated, and the crude product was purified by HPLC using a reverse-phase amide column and eluted with 38% MeCN/water at 4 mL/min; the radioactive fraction was collected. An aliquot of the purified product was co-injected with the standard cold reference compound 4 into an analytical HPLC column to verify the product’s identity and purity. The solvent was evaporated under reduced pressure, and the product was dissolved in a small volume of ethanol and then diluted with 10% Tween 80 in water.
4.2.15. 5-Chloro-N2–(2-isoprpoxy-5-methyl-4-(1-fluoroethylpiperidin-4-yl)phenyl)-N4-[2-(propane-2-sulfonyl)phenyl]pyrimidine-2,4-diamine ([18F]fluoroethyl ceritinib; ([18F]9)
The title compound was prepared as we described previously [38]. Briefly, [18F]fluoroethyl tosylate was produced and reacted with ceritinib and then purified by chromatography. The product was assessed by analytical HPLC to determine its identity and purity. The solvent was evaporated, and the product was dissolved in a small volume of ethanol and then diluted with 10% Tween 80 solution for formulation and animal injection.
4.3. Formulation
Because compounds 1, 4, and 9 were not soluble in saline or PBS solutions, they were dissolved in DMSO prior to their use in the cytotoxic assay. The radiolabeled analogues, after evaporation of the chromatographic solvent (MeOH/CH2Cl2 or MeCN), were dissolved in a minimum volume of ethanol (0.1–0.2 mL, depending on the amount of activity). The alcoholic solutions were diluted with a 10% solution of aqueous Tween 80 to make a stock solution with a final concentration of 10% ethanol. The stock solution was then diluted with PBS prior to injection into animals.
4.4. LogP measurements
LogP values were determined using a standard method [46]. Briefly, [18F]1, [18F]4, or [18F]9 (10 μL, 1 μCi) in ethanol was added to a mixture of 1-octanol (1.0 mL) and water (1.0 mL) in test tubes in triplicate. The mixtures in the test tubes were subjected to vortexing for 5 min, which was repeated four times; the mixtures were then subjected to centrifugation (2×5 min, 4000 rpm at 4°C). From each test tube, three aliquots of the 1-octanol layer (50 μL each) were transferred into three gamma-counting tubes (nine tubes total). Similarly, 50-μL aliquots of the aqueous layer were transferred into nine separate test tubes. The amount of radioactivity in each tube was counted using a gamma counter. The partition coefficient (P) was calculated as the ratio of activity in octanol to activity in water. The LogD value was also measured in octanol/PBS (pH 7.4) using the same methodology. In addition, Cheminformatics software (Molinspiration, Slovak Republic) was used to calculate theoretical LogP values for [18F]1 (4.81), [18F]4 (6.01), and [18F]9 (6.94).
4.5. Biology
4.5.1. Cell lines and ALK expression
The human lung cancer cell lines H2228 (ALK-positive, EML4-ALK v3) and H441 (ALK-negative) were purchased from ATCC (Manassas, VA). Cells were grown in RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS). For Western blot analysis, the cells were lysed with mammalian cell lysis buffer (50 mM Tris HCl, pH 8.0, 100 mM NaCl, 2 mM dithiothreitol, 5 mM EDTA, 0.5% NP-40, 1 mM microcystin, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 0.15 μg/ml aprotinin, 20 mM leupeptin, and 20 mM pepstatin). Each sample (15 μg total protein lysate) was loaded and fractionated on a 4–20% Mini-PROTEAN TGX gel (Bio-Rad, Hercules, CA). Then, the gel was transferred onto a polyvinylidene fluoride membrane (Trans-Blot Turbo Transfer Pack, Bio-Rad) and blocked with 5% milk in a mixture of Tris-buffered saline solution and polysorbate 20 (Tween 20). The blot was incubated overnight with conjugated primary antibody (Alk D5F3 CP rabbit monoclonal antibody–horseradish peroxidase conjugate; Cell Signaling Technology, Beverly, MA) at a 1:3000 dilution.
4.5.2. Cytotoxicity assay
H2228 and H441 cells were seeded in 96-well clear-bottom black-wall plates at a density of 8,000 cells per well in RPMI-1640 medium (100 μL) supplemented with 10% FBS. The next day, the cells were treated with various concentrations of crizotinib, alectinib, and ceritinib or their corresponding fluoroethyl derivatives for 72 h. Subsequently, cell viability was measured by adding 100 μL of Alamar Blue reagent (1:10 dilution) in phenol red-free RPMI medium with 10% FBS to each well. Cells were incubated for 4 h, and fluorescence was measured with a Synergy H4 microplate reader (BioTek, Winooski, VT) at the excitation wavelength of 530 nm and the emission wavelength of 580 nm. IC50 values were calculated with GraphPad Prism 7.01. Experiments were performed in triplicate in three independent sets, and results are reported as means with standard deviations.
4.5.3. Tracer uptake assays
H2228 and H441 cells were seeded in 6-well clear-bottom plates at a density of 106 cells per well in RPMI-1640 medium (2 mL) supplemented with 10% FBS. Aliquots of tracer (1 μCi in 30 μl PBS) were added to each well, and the cells were incubated for 30, 60, or 120 min. At the designated times, the medium was removed, the wells were washed twice with ice-cold PBS (1 mL), the cells were trypsinized for 5 min at 37°C, and the cell suspension was transferred to gamma-counting tubes and assessed for [18F]radioactivity. Experiments were performed in triplicate. Radioactivity uptake was calculated as counts per minute per million cells.
4.5.4. In vivo biodistribution and PET imaging
All animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of MD Anderson Cancer Center. PET imaging and image analysis were performed on an Albira trimodality rodent scanner (Bruker). In brief, 8- to 10- week-old female nude mice were injected intravenously with approximately 100 μCi (3.7 MBq) of one of the PET tracers under anesthesia (2% isoflurane in oxygen) and then imaged on an Albira trimodality rodent scanner (Bruker). Dynamic PET scans were acquired 0–10 min after injection under anesthesia. Static PET scans (10 min) were acquired under anesthesia at 30 and 60 min after injection. For image analysis, PMOD software (PMOD Technologies Ltd., Zurich, Switzerland) was used to draw regions of interest over the major organs on decay-corrected whole-body coronal images. The radioactivity concentration was obtained from the mean values within multiple regions of interest and then converted to percent injected dose per cubic centimeter (%ID/cc) of tissue. Results are reported as means with standard deviations.
Supplementary Material
Fig. S1. HPLC chromatogram of [18F]fluoroethyl crizotinib [18F]1, co-injected with the non-radioactive standard 1. Analytical C18 column; 60% MeCN/40% H2O/0.1% TFA, flow 1 mL/min.
Fig. S2. Purification of [18F]fluoroethyl alectinib [18F]4 by HPLC. Semi-preparative reverse phase amide column; 38% MeCN/62% H2O/0.1% TFA, flow 4 mL/min.
Fig. S3. HPLC chromatogram of [18F]fluoroethyl alectinib [18F]4, co-injected with the non-radioactive standard 4. Analytical C18 column; 40% MeCN/60% H2O/0.1% TFA, flow 1 mL/min.
Fig. 7.

Maximum intensity projection PET images show the biodistribution of [18F]4 of in a representative animal at 5, 10, 30, and 60 min after injection. B=brain, K=kidney, H=heart, and L=liver.
Highlights:
Anaplastic lymphoma kinase (ALK) is a therapeutic target in lung cancer
Many ALK inhibitors are routinely used for therapy of lung cancer
The blood-brain barrier remains a challenge for treatment of brain metastasis
[19F/18F]Fluoroethyl analogues of ALK inhibitors have been developed and tested
Preliminary in vitro and in vivo PET imaging pharmacokinetic results were encouraging, showing significant brain penetration
Acknowledgments
The authors thank Dr. Seth T. Gammon for editing the figures and for providing helpful discussion. The authors also thank Kathryn Hale and Joe Munch in Scientific Publication Services in the Research Medical Library at MD Anderson for editing the manuscript. This work was supported by the U.S. National Institutes of Health through the Washington University-MD Anderson Cancer Center Inter-Institutional Molecular Imaging Center Grant (NCI P50 CA94056; DP-W) and through MD Anderson’s Cancer Center Support Grant (P30CA016672), which supports the NMR Facility and Small Animal Imaging Facility used in the study; and by an MD Anderson Cancer Center Institutional Research Grant (MMA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A: Supplementary data
Supplementary data to this article can be found online.
References
- [1].Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, Look A, Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma, Science, 263 (1994) 1281–1284. 10.1126/science.8122112 [DOI] [PubMed] [Google Scholar]
- [2].Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H, Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer, Nature, 448 (2007) 561–566. 10.1038/nature05945 [DOI] [PubMed] [Google Scholar]
- [3].Palmer RH, Vernersson E, Grabbe C, Hallberg B, Anaplastic lymphoma kinase: signalling in development and disease, Biochemical Journal, 420 (2009) 345–361. 10.1042/BJ20090387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grande E, Bolós M, Arriola E, Targeting oncogenic ALK: a promising strategy for cancer treatment, Mol Cancer Ther 10 (2011) 569–579._ 10.1158/1535-7163.MCT-10-0615 [DOI] [PubMed] [Google Scholar]
- [5].Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, McTigue M, Grodsky N, Ryan K, Padrique E, Alton G, Timofeevski S, Yamazaki S, Li Q, Zou H, Christensen J, Mroczkowski B, Bender S, Kania RS, Edwards MP, Structure based drug design of Crizotinib (PF-02341066), a potent and selective dual inhibitor of Mesenchymal-Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK), J Med Chem, 54 (2011) 6342–6363. 10.1021/jm2007613 [DOI] [PubMed] [Google Scholar]
- [6].Ou SI, Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond, Drug Des Devel Ther, 5 (2011) 471–485. 10.2147/DDDT.S19045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kazandjian D, Blumenthal GM, Chen H, He K, Patel M, Justice R, Keegan P, Pazdur R, FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements, The Oncologist, 19 (2014) e5–e11. https://doi:10.1634/theoncologist.2014-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, Sequist LV, Iafrate AJ, Engelman JA, Mechanisms of acquired Crizotinib resistance in ALK-rearranged lung cancers, Science Translational Medicine, 4 (2012) 120ra117–120ra117._ 10.1126/scitranslmed.3003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dagogo-Jack I, Shaw AT, Crizotinib resistance: implications for therapeutic strategies, Ann Oncol, 27 (2016) iii42–iii50._ 10.1093/annonc/mdw305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Crizotinib resistance arises through multiple mechanisms, Cancer Discovery, 2 (2012) 204–204. 10.1158/2159-8290.CD-RW2012-018 [DOI] [Google Scholar]
- [11].Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang Y, Narasimhan NI, Dalgarno D, Shakespeare WC, Miret JJ, Clackson T, Rivera VM, Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen, Chem Biol Drug Des, 78 (2011) 999–1005. 10.1111/j.1747-0285.2011.01239.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C, Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated ALK inhibitors, Mol Cancer Res, 11 (2013) 122–132._ 10.1158/1541-7786.MCR-12-0569 [DOI] [PubMed] [Google Scholar]
- [13].Frampton JE, Crizotinib: a review of its use in the treatment of anaplastic lymphoma kinase-positive, advanced non-small cell lung cancer, Drugs, 73 (2013) 2031–2051. 10.1007/s40265-013-0142-z [DOI] [PubMed] [Google Scholar]
- [14].Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S, Ohwada J, Miyagi T, Kobayashi T, Takanashi K, Tsukaguchi T, Sakamoto H, Tsukuda T, Oikawa N, Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802), Bioorg Med Chem, 20 (2012) 1271–1280. 10.1016/j-bmc.2011.12.021 [DOI] [PubMed] [Google Scholar]
- [15].Wu J, Savooji J, Liu D, Second- and third-generation ALK inhibitors for non-small cell lung cancer, Journal of Hematology & Oncology, 9 (2016) 19 10.1186/s13045-016-0251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, Gieser G, Stephens O, Zahalka E, Ringgold K, Helms W, Shord S, Yu J, Zhao H, Davis G, McKee AE, Keegan P, Pazdur R, FDA Approval: Alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following Crizotinib, Clin Cancer Res, 22 (2016) 5171–5176._ 10.1158/1078-0432.CCR-16-1293 [DOI] [PubMed] [Google Scholar]
- [17].Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, Sarkisova Y, Sun F, Steffy A, Pferdekamper AC, Li AG, Joseph SB, Kim Y, Liu B, Tuntland T, Cui X, Gray NS, Steensma R, Wan Y, Jiang J, Chopiuk G, Li J, Gordon WP, Richmond W, Johnson K, Chang J, Groessl T, He Y, Phimister A, Aycinena A, Lee CC, Bursulaya B, Karanewsky DS, Seidel HM, Harris JL, Michellys P, Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective Anaplastic Lymphoma Kinase (ALK) inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials, J Med Chem, 56 (2013) 5675–5690._ 10.1021/jm400402q [DOI] [PubMed] [Google Scholar]
- [18].Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, Liu Q, Zhao P, Zhao H, Lu D, Tang Z, Al Hakim A, Boyd K, Keegan P, Justice R, Pazdur R, FDA approval: Ceritinib for the treatment of metastatic Anaplastic Lymphoma Kinase-positive non-small cell lung cancer, Clin Cancer Res, 21 (2015) 2436–2439._ 10.1158/1078-0432.CCR-14-3157 [DOI] [PubMed] [Google Scholar]
- [19].Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng Y, Liu W, Brooun A, Timofeevski S, McDonnell SRP, Jiang P, Falk MD, Lappin PB, Affolter T, Nichols T, Hu W, Lam J, Johnson TW, Smeal T, Charest A, Fantin VR, PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations, Proc Natl Acad Sci U S A, 112 (2015) 3493–3498. 10.1073/pnas.1420785112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, Li Y, Zou HY, Lee NV, Smeal T, Lemmon MA, Mossé YP, The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to Crizotinib in ALK-driven neuroblastoma, Cancer Discovery, 6 (2016) 96–107. 10.1158/2159-8290.CD-15-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jang WD, Kim J, Son HY, Park SY, Cho YS, Koo T, Lee H, Kang NS, Discovery of Tyk2 inhibitors via the virtual site-directed fragment-based drug design, Bioorg Med Chem Lett 25 (2015) 3947–3952. https://doi.org/10.10167j.bmcl.2015.07.037 [DOI] [PubMed] [Google Scholar]
- [22].Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, Pao W, Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors, Cancer Res, 71 (2011) 4920–4931._ 10.1158/0008-5472.CAN-10-3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, Sakagami H, Furutani T, Doihara H, Kudoh M, Kuromitsu S, The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice, Mol Cancer Ther 13 (2014) 329–340._ 10.1158/1535-7163.MCT-13-0395 [DOI] [PubMed] [Google Scholar]
- [24].Ceccon M, Mologni L, Giudici G, Piazza R, Pirola A, Fontana D, Gambacorti-Passerini C, Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor AP26113 in human NPM-ALK-positive anaplastic large cell lymphoma, Mole Cancer Res, 13 (2015) 775–783. 10.1158/1541-7786.MCR-14-0157 [DOI] [PubMed] [Google Scholar]
- [25].Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D, Novel ALK inhibitors in clinical use and development, J Hematol Oncol, 8 (2015) 17 10.1186/s13045-015-0122-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H, Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases, Cancer Chemothe Pharmacol, 74 (2014) 1023–1028. 10.1007/s00280-014-2578-6 [DOI] [PubMed] [Google Scholar]
- [27].Collier TL, Maresca KP, Normandin MD, Richardson P, McCarthy TJ, Liang SH, Waterhouse RN, Vasdev N, Brain penetration of the ROS1/ALK inhibitor Lorlatinib confirmed by PET, Molecular Imaging, 16 (2017) 1–3._ 10.1177/1536012117736669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, Burke BJ, Deng Y, Liu W, Dardaei L, Frias RL, Schultz KR, Logan J, James LP, Smeal T, Timofeevski S, Katayama R, lafrate AJ, Le L, McTigue M, Getz G, Johnson TW, Engelman JA, Resensitization to Crizotinib by the Lorlatinib ALK resistance mutation L1198F, N Engl J Med, 374 (2016) 54–61. 10.1056/NEJMoa1508887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kort A, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH, Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2), Pharmacological Research, 102 (2015) 200–207. 10.1016/j.phrs.2015.09.003 [DOI] [PubMed] [Google Scholar]
- [30].Sharma R, Aboagye E, Development of radiotracers for oncology--the interface with pharmacology, Br J Pharmacol, 163 (2011) 1565–1585. 10.1111/jM476-5381.2010.01160.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Slobbe P, Poot AJ, Windhorst AD, van Dongen GAMS, PET imaging with small-molecule tyrosine kinase inhibitors: TKI-PET, Drug Discovery Today, 17 (2012) 1175–1187. 10.1016/j.drudis.2012.06.016 [DOI] [PubMed] [Google Scholar]
- [32].Peng Z, Maxwell DS, Sun D, Bhanu Prasad BA, Pal A, Wang S, Balatoni J, Ghosh P, Lim ST, Volgin A, Shavrin A, Alauddin MM, Gelovani JG, Bornmann WG, Imatinib analogs as potential agents for PET imaging of Bcr-Abl and c-KIT expression at a kinase level, Bioorg Med Chem, 22 (2014) 623–632. 10.1016/j.bmc.2013.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Veach DR, Namavari M, Pillarsetty N, Santos EB, Beresten-Kochetkov T, Lambek C, Punzalan BJ, Antczak C, Smith-Jones PM, Djaballah H, Clarkson B, Larson SM, Synthesis and biological evaluation of a fluorine-18 derivative of Dasatinib, J Med Chem, 50 (2007) 5853–5857. 10.1021/jm070342g [DOI] [PubMed] [Google Scholar]
- [34].Pisaneschi F, Nguyen Q, Shamsaei E, Glaser M, Robins E, Kaliszczak M, Smith G, Spivey AC, Aboagye EO, Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: Synthesis and biological evaluation, Bioorg Med Chem 18 (2010) 6634–6645._ 10.1016/j.bmc.2010.08.004 [DOI] [PubMed] [Google Scholar]
- [35].Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E, Positron emission tomography (PET) imaging with [11C]-labeled Erlotinib: a micro-PET study on mice with lung tumor xenografts, Cancer Research, 69 (2009) 873–878._ 10.1158/0008-5472.CAN-08-3118 [DOI] [PubMed] [Google Scholar]
- [36].Rocha-Lima CM, Raez LE, Erlotinib (tarceva) for the treatment of non-small-cell lung cancer and pancreatic cancer, P & T : a peer-reviewed journal for formulary management, 34 (2009) 554–564. https://www.ptcommunity.com/journal/article/archives/2009/10/554/erlotinib-tarceva-treatment-non-small-cell-lung-cancer-and [PMC free article] [PubMed] [Google Scholar]
- [37].Gridelli C, Bareschino MA, Schettino C, Rossi A, Maione P, Ciardiello F, Erlotinib in Non-Small Cell Lung Cancer Treatment: Current Status and Future Development, The Oncologist, 12 (2007) 840–849. 10.1634/theoncologist.12-7-840 [DOI] [PubMed] [Google Scholar]
- [38].Bahce I, Smit EF, Lubberink M, van der Veldt AAM, Yaqub M, Windhorst AD, Schuit RC, Thunnissen E, Heideman DAM, Postmus PE, Lammertsma AA, Hendrikse NH, Development of [11C]-Erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status, Clin Cancer Res, 19 (2013) 183–193._ 10.1158/1078-0432.CCR-12-0289 [DOI] [PubMed] [Google Scholar]
- [39].Memon AA, Weber B, Winterdahl M, Jakobsen S, Meldgaard P, Madsen HHT, Keiding S, Nexo E, Sorensen BS, PET imaging of patients with non-small cell lung cancer employing an EGF receptor targeting drug as tracer, Brit J Cancer, 105 (2011) 1850–1855. 10.1038/bjc.2011.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Collier TL, Normandin MD, Stephenson NA, Livni E, Liang SH, Wooten DW, Esfahani SA, Stabin MG, Mahmood U, Chen J, Wang W, Maresca K, Waterhouse RN, El Fakhri G, Richardson P, Vasdev N, Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib, Nat Commun, 8 (2017) 15761_ 10.1038/ncomms15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Perera S, Piwnica-Worms D, Alauddin MM, Synthesis of a [18F]-labeled Ceritinib analogue for positron emission tomography of anaplastic lymphoma kinase, a receptor tyrosine kinase, in lung cancer, J Labelled Compd Radiopharm, 59 (2016) 103–108. 10.1002/jlcr.3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang P, Cai J, Chen J, Ji M, Synthesis and anticancer activities of ceritinib analogs modified in the terminal piperidine ring, Eur J Med Chem, 93 (2015) 1–8. 10.1016/j.ejmech.2015.01.056 [DOI] [PubMed] [Google Scholar]
- [43].Roskoski R, Anaplastic lymphoma kinase (ALK) inhibitors in the treatment of ALK-driven lung cancers, Pharmacol Res 117 (2017) 343–356._ 10.1016/j.phrs.2017.01.007 [DOI] [PubMed] [Google Scholar]
- [44].Kniess T, Laube M, Brust P, Steinbach J, 2-[18F]Fluoroethyl tosylate - a versatile tool for building 18F-based radiotracers for positron emission tomography, MedChemComm, 6 (2015) 1714–1754. 10.1039/C5MD00303B [DOI] [Google Scholar]
- [45].Ding R, He Y, Wang X, Xu J, Chen Y, Feng M, Qi C, Treatment of Alcohols with Tosyl Chloride Does Not always Lead to the Formation of Tosylates, Molecules, 16 (2011) 5665–5673. 10.3390/molecules16075665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S, An admonition when measuring the lipophilicity of radiotracers using counting techniques, Appl Radiat Isotopes, 54 (2001) 203–208. 10.1016/S0969-8043(00)00269-4 [DOI] [PubMed] [Google Scholar]
- [47].Vraka C, Nics L, Wagner K, Hacker M, Wadsak W, Mitterhauser M, LogP, a yesterday’s value?, Nucl Med Biol 50 (2017) 1–10. 10.1016/j.nucmedbio.2017.03.003 [DOI] [PubMed] [Google Scholar]
- [48].Martelli MP, Sozzi G, Hernandez L, Pettirossi V, Navarro A, Conte D, Gasparini P, Perrone F, Modena P, Pastorino U, Carbone A, Fabbri A, Sidoni A, Nakamura S, Gambacorta M, Fernández PL, Ramirez J, Chan JKC, Grigioni WF, Campo E, Pileri SA, Falini B, EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues, Am J Pathol, 174 (2009) 661–670. 10.2353/ajpath.2009.080755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim HJ, Lee KY, Kim YW, Choi YJ, Lee J, Choi CM, Baek I, Rho JK, Lee JC, P-glycoprotein confers acquired resistance to 17-DMAG in lung cancers with an ALK rearrangement, BMC cancer, 15 (2015) 553–553._ 10.1186/s12885-015-1543-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Katayama R, Sakashita T, Yanagitani N, Ninomiya H, Horiike A, Friboulet L, Gainor JF, Motoi N, Dobashi A, Sakata S, Tambo Y, Kitazono S, Sato S, Koike S, John lafrate A, Mino-Kenudson M, Ishikawa Y, Shaw AT, Engelman JA, Takeuchi K, Nishio M, Fujita N, P-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancer, EBioMedicine, 3 (2016) 54–66. 10.1016/j.ebiom.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Slade RL, Pisaneschi F, Nguyen Q, Smith G, Carroll L, Beckley A, Kaliszczak MA, Aboagye EO, Identification of ABC transporter interaction of a novel cyanoquinoline radiotracer and implications for tumour imaging by positron emission tomography, PLOS ONE, 11 (2016) e0161427 10.1371/journal.pone.0161427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Leonidova A, Foerster C, Zarschler K, Schubert M, Pietzsch HJ, Steinbach J, Bergmann R, Metzler-Nolte N, Stephan H, Gasser G, In vivo demonstration of an active tumor pretargeting approach with peptide nucleic acid bioconjugates as complementary system, Chem Sci, 6 (2015) 5601–5616. 10.1039/c5sc00951k [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Saha S, Copic D, Bhaskar S, Clay N, Donini A, Hart AJ, Lahann J, Chemically controlled bending of compositionally anisotropic microcylinders, Angew Chemie Int Ed 51 (2012) 660–665. 10.1002/anie.201105387 [DOI] [PubMed] [Google Scholar]
- [54].Alauddin MM, Conti PS, Selective alkylation of pyrimidyl dianions II: synthesis, characterization, and comparative reactivity of 3’, 5’-o-bis- tetrahydropyranyl, trimethylsilyl and tert-butyldimethylsilyl derivatives of 5-bromo-2’-deoxyuridine, Tetrahedron, 50 (1994) 1699–1706. 10.1016/S0040-4020(01)80845-0 [DOI] [Google Scholar]
- [55].Darmanin T, Guittard F, One-pot method for build-up nanoporous super oil-repellent films, Journal of Colloid and Interface Science, 335 (2009) 146–149. 10.1016/j.jcis.2009.03.073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. HPLC chromatogram of [18F]fluoroethyl crizotinib [18F]1, co-injected with the non-radioactive standard 1. Analytical C18 column; 60% MeCN/40% H2O/0.1% TFA, flow 1 mL/min.
Fig. S2. Purification of [18F]fluoroethyl alectinib [18F]4 by HPLC. Semi-preparative reverse phase amide column; 38% MeCN/62% H2O/0.1% TFA, flow 4 mL/min.
Fig. S3. HPLC chromatogram of [18F]fluoroethyl alectinib [18F]4, co-injected with the non-radioactive standard 4. Analytical C18 column; 40% MeCN/60% H2O/0.1% TFA, flow 1 mL/min.