

Macrophages enhance tissue vascularization in different ways depending on their phenotypes.
Abstract
Macrophages are key contributors to vascularization, but the mechanisms behind their actions are not understood. Here, we show that diverse macrophage phenotypes have distinct effects on endothelial cell behavior, with resulting effects on vascularization of engineered tissues. In Transwell coculture, proinflammatory M1 macrophages caused endothelial cells to up-regulate genes associated with sprouting angiogenesis, whereas prohealing (M2a), proremodeling (M2c), and anti-inflammatory (M2f) macrophages promoted up-regulation of genes associated with pericyte cell differentiation. In 3D tissue-engineered human blood vessel networks in vitro, short-term exposure (1 day) to M1 macrophages increased vessel formation, while long-term exposure (3 days) caused regression. When human tissue-engineered blood vessel networks were implanted into athymic mice, macrophages expressing markers of both M1 and M2 phenotypes wrapped around and bridged adjacent vessels and formed vessel-like structures themselves. Last, depletion of host macrophages inhibited remodeling of engineered vessels, infiltration of host vessels, and anastomosis with host vessels.
INTRODUCTION
The underlying goal of tissue engineering is to functionally repair and regenerate complex tissues and organs. One of the major challenges in engineering viable tissues is forming functional and stable blood vessel networks within the tissue, which supply oxygen and nutrients to the cells. The process of blood vessel growth, or angiogenesis, involves a complex cascade of events, including basement membrane degradation, migration and proliferation of endothelial cells (ECs), tube formation, fusion (anastomosis) of newly formed vessels, and stabilization by support cells such as pericytes. Macrophages, the primary cells of the inflammatory response, have long been recognized as crucial regulators of healing, and depletion of macrophages from wounds causes drastically reduced angiogenesis (1), while exogenous addition of macrophages promotes angiogenesis (2). However, macrophages are also highly plastic cells that exist on a spectrum of diverse phenotypes that dynamically respond to changing environmental stimuli. How these changes in phenotypes affect angiogenesis is poorly understood.
In the normal response to injury of multiple tissue types, monocytes infiltrate the wound site and differentiate into macrophages that exhibit a highly proinflammatory phenotype that is often referred to as M1 because of the role of T helper cell 1 (TH1)–derived cytokines in their polarization. Macrophages with an inflammatory phenotype have been shown to stimulate angiogenesis in multiple animal models (3). At later stages, the macrophage population undergoes a dramatic shift in phenotype to a mixed population of diverse phenotypes that is collectively referred to as M2. This change occurs via direct conversion of M1 macrophages (4) as well as differentiation of newly arriving monocytes (5) and proliferation of M2 macrophages at the site of injury (6). The relative contributions of M1- and M2-like macrophages to angiogenesis are not known.
Several of these distinct phenotypes can be modeled in vitro through the addition of relevant stimuli. For example, addition of lipopolysaccharide (LPS) and interferon-γ (IFN-γ) stimulates the M1 phenotype, which secretes the potent initiator of angiogenesis, vascular endothelial growth factor A (VEGFA) (7). M2a macrophages are stimulated in vitro by TH2 cytokines interleukin-4 (IL-4) and IL-13 and secrete platelet-derived growth factor-BB (PDGF-BB), which is important for stabilization of nascent blood vessels (7). M2c macrophages are stimulated by IL-10 and secrete high quantities of matrix metalloproteinases (MMPs) (8), and M2f (or M2eff) macrophages are stimulated by phagocytosis of apoptotic cells (efferocytosis) and secrete anti-inflammatory mediators (9). Despite the fact that M2-like phenotypes have been strongly implicated in angiogenesis in vivo (10), the contributions of particular M2 subtypes have not yet been parsed. Moreover, the majority of reports denote diverse M2 subtypes as simply M2, despite differences among these phenotypes at the transcriptome and proteome levels (7, 8). These differences suggest that M2 subtypes may have diverse functions in promoting angiogenesis and provide an opportunity to refine macrophage characterization to better reflect the function of macrophage phenotypes in tissue repair and regeneration. Also, given the plasticity of macrophages, it is likely that hybrid phenotypes exist in vivo. Recent advancements in single cell analysis are beginning to shed light on the complexity of macrophages in vivo, revealing subtypes beyond those described here; for the purposes of this work, we chose to focus on phenotypes implicated in angiogenesis that can be reliably generated in vitro.
Here, we show that human M1, M2a, M2c, and M2f macrophages differentially affect the angiogenic behavior of human ECs. First, we show that macrophage phenotypes induce distinct changes in EC gene expression via paracrine signaling in a Transwell coculture system, and we identify the biological pathways affected in ECs. We then engineered a three-dimensional (3D) triculture model of in vitro blood vessel network formation incorporating human ECs, mesenchymal stem cell (MSC)–derived support cells, and macrophages and used this system to demonstrate functional changes in vascularization dynamics driven by the M1 and M2a phenotypes. Last, we show that host macrophages facilitate anastomosis of engineered blood vessels with the host vasculature in vivo in a murine model of subcutaneous implantation and that macrophages expressing both M1 and M2 markers interact with engineered and host blood vessels.
RESULTS
Four distinct macrophage phenotypes differentially alter EC behavior
To investigate how macrophages of different phenotypes affect EC behavior in vitro, primary human monocytes were isolated from four healthy donors and differentiated into the M1, M2a, M2c, and M2f phenotypes via administration of cytokines or apoptotic cells, as well as a reference control referred to as M0 (Fig. 1A). We have extensively characterized the gene expression profiles of M1, M2a, and M2c macrophages (7, 8), and here, we show that M2f macrophages up-regulate gene expression of the M2-associated marker CD206 as well as the anti-inflammatory factors IL10 and transforming growth factor–beta 1 (TGFB1) (Fig. 1B), in agreement with previous reports (9). We also observed significantly greater secretion of TGF-β1 by M2a and M2f macrophages relative to the M0, M1, or M2c phenotypes (Fig. 1C), further establishing M2a, M2c, and M2f as unique phenotypes.
Fig. 1. Overview of phenotype-induced changes in EC gene expression via Transwell culture with macrophages.
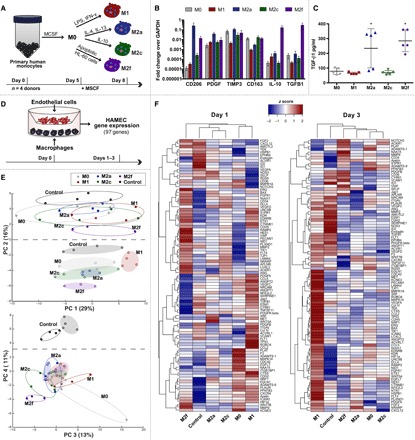
(A) Primary human peripheral blood monocytes were isolated from four human donors, differentiated into macrophages, and polarized into M0, M1, M2a, M2c, or M2f phenotypes for 72 hours. (B) Gene expression of commonly associated M2 markers. (C) Levels of TGFB1 in cell culture media. Data in (B) and (C) represent means ± SEM with n = 3 to 5 technical replicates from one human donor. Differences were determined using one-way ANOVA with Tukey’s multiple comparisons test; *P < 0.05 relative to M0, M1, and M2c phenotypes. (D) Primary HAMECs were Transwell cocultured with M0, M1, M2a, M2c, or M2f macrophages for 1 to 3 days. Changes in HAMEC gene expression were analyzed using a custom CodeSet from NanoString Technologies, inclusive of 97 genes related to angiogenesis. (E) PCA of HAMEC gene count data. To facilitate visualization, color-coded ellipses were drawn to include all samples for each phenotype. Open ellipses represent day 3 data; shaded ellipses represent day 1 data. (F) Gene counts for HAMEC treated with M0, M1, M2a, M2c, or M2f macrophages were z scored across rows and hierarchically clustered on the basis of Euclidean distance.
To assess whether these differences alter EC behavior, M0, M1, M2a, M2c, or M2f macrophages were cocultured with human microvascular ECs (HAMECs) separated by Transwell inserts for 3 days (Fig. 1D). On days 1 and 3, ECs were isolated and analyzed for changes in expression of a custom-curated panel of 97 genes related to various processes in angiogenesis via NanoString (table S1). Principal component analysis (PCA) showed that major changes in EC gene expression occurred over time and that these responses were dependent on macrophage phenotype (Fig. 1E). Distinct effects of the different phenotypes on EC responses were still pronounced even after 3 days in culture.
Consistent with the PCA results, hierarchical clustering revealed that major differences in EC behavior occurred in response to different macrophage phenotypes, with M2f and M1 macrophages inducing the greatest changes at day 1, and M1 and M2c having the strongest effects at day 3 (Fig. 1F). M1 and M2f macrophages also appeared to elicit opposing patterns in EC gene expression. Expression profiles for all genes tested are provided in fig. S1.
We next interrogated the specific changes in ECs that occurred in response to macrophage phenotype. A list of EC genes significantly altered [false discovery rate (FDR) adjusted P < 0.05] in response to each macrophage phenotype was generated (table S2). On day 1, compared with ECs cultured without macrophages, M1 macrophages altered the greatest number of genes (25 total), M2a and M2c macrophages each altered 16 genes, and M2f macrophages altered 24 genes; control M0 macrophages altered 20 genes. Of note, culture with M1 macrophages caused ECs to up-regulate many genes associated with the tip cell phenotype of ECs (ADAMTS-9, CTSS, CXCR4, EDNRB, KDR, NID1, NRCAM, PFKFB3, PLAUR, and UNC5B), which is associated with sprouting behavior and the early stages of angiogenesis (11), although genes associated with vessel stabilization and lumen formation were also up-regulated (APLN, CD34, and SERPINE1) (fig. S1).
Macrophage phenotypes alter different EC biological pathways
To identify the biological processes with which these genes are associated, Gene Ontology (GO) Enrichment Analysis was performed for each list of differentially expressed genes. All GO terms with fold enrichment >25 were further analyzed using REVIGO, which facilitates identification of the most important pathways by removing redundant terms and plots them in semantic space (fig. S2), such that overlapping ontologies have gene products with similar functions (12). All nonredundant GO terms related to angiogenesis were then grouped, and the top five most highly enriched terms for each phenotype on day 1 (Fig. 2A) and day 3 (fig. S3A) were plotted. Two of the terms highly enriched after 1 day by M1 macrophages were not enriched in response to any other phenotype: Tie signaling pathway and positive regulation of EC chemotaxis. Cell migration involved in sprouting angiogenesis was also highly enriched in response to M1 and, to a lesser extent, M2c macrophages. Notably, EC migration and proliferation are processes that occur early during angiogenesis to facilitate vessel sprouting. In contrast, responses to M2a or M2f macrophages were highly enriched for pericyte cell differentiation and regulation of smooth muscle cell differentiation, processes that occur after endothelial tube formation (13). In addition, EC responses to M2f were uniquely enriched for negative regulation of vascular permeability, which also suggests a later role in vessel stabilization. The most highly enriched GO terms in response to M2c macrophages, including negative regulation of sprouting angiogenesis, EC apoptotic process, and branching involved in blood vessel morphogenesis, were also enriched in response to the M1 and M2f phenotypes. After 3 days, EC responses to M1 macrophages were still highly enriched for EC chemotaxis, as well as basement membrane organization and EC-matrix adhesion. However, there were no unique highly enriched terms by M2a, M2c, or M2f macrophages, which may be due to converging macrophage behaviors in response to EC signals. Collectively, these data suggest that all four phenotypes of macrophages alter different biological processes in ECs, which may be related to specific stages of angiogenesis.
Fig. 2. Macrophage phenotypes differentially influence biological processes in ECs after 1 day of Transwell coculture.
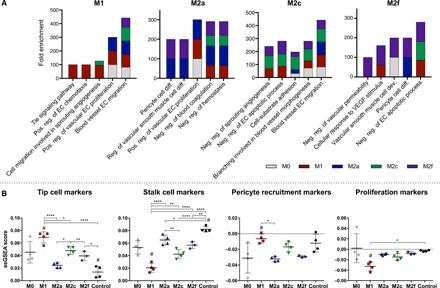
GO enrichment analysis was performed on lists of genes differentially regulated in ECs in response to macrophage phenotype. (A) Top five most highly enriched, nonredundant GO terms for each phenotype. (B) ssGSEA for gene sets related to specific processes of angiogenesis; analysis was not restricted to only differentially expressed genes. One-way ANOVA with Tukey’s post hoc analysis; *P < 0.05; **P < 0.01; ****P < 0.0001, # indicates P < 0.05 relative to the M0 control.
Because we used a targeted panel of genes related to angiogenesis, GO terms include several genes that were not included in our panel. Therefore, to further interrogate phenotype differences, we grouped the genes in our panel into lists based on established functions through a thorough literature search (table S3) and performed single-sample gene set enrichment analysis (ssGSEA) against each gene set. Unlike the analysis described above, which was restricted to only differentially expressed genes, ssGSEA was performed on all 97 genes characterized, regardless of changes in expression level. This enabled us to assess the effects of each phenotype on specific angiogenic processes, such as tip cell activation, stalk cell activation, recruitment of pericytes, and proliferation (Fig. 2B and fig. S3B), as well as others (fig. S4). Consistent with our GO enrichment analysis, ECs cocultured with M1 macrophages were significantly enriched for genes associated with the tip cell phenotype relative to coculture with M2a or M2f or to untreated control ECs (Fig. 2B). Consistent with these findings, ECs cocultured with M1 macrophages were also more enriched for markers of pericyte recruitment, a function associated with tip cells (14). Moreover, compared with M1 and M2c macrophages, coculture with M2a macrophages resulted in a significantly higher enrichment for genes associated with the stalk cell phenotype of ECs. ECs cocultured with M1 macrophages down-regulated genes associated with the stalk cell phenotype as well as proliferation, a major process associated with stalk cells (14). On the basis of the divergent effects of M1 and M2a macrophages on EC gene expression, we chose to further investigate these phenotypes in functional assays of angiogenesis. M1 and M2a macrophages are also the most well mapped to in vivo phenotypes and the most widely characterized in the literature, yet the angiogenic abilities of these phenotypes have not been thoroughly distinguished.
M1 and M2a macrophages display unique interactions with engineered blood vessels in 3D
We next sought to determine whether the phenotype-induced changes in EC gene expression by M1 macrophages translated to functional differences in 3D tissue vascularization. Therefore, we used a tissue-engineered 3D model of human blood vessels (15) to facilitate the in vitro study of macrophage phenotype in angiogenesis. Green fluorescent protein (GFP)–labeled human THP-1 monocyte-derived macrophages were used in these studies to enable visualization of macrophage-EC interactions in real time while obtaining sufficient quantities of macrophages for the studies. Although differences between THP-1 and primary human monocyte-derived macrophages are known to exist (16, 17), THP-1 cells are thought to closely mimic the function of monocytes and macrophages (18). Nevertheless, it should be noted that compared with primary macrophages, THP-1 displays lower expression of CD14, which is involved in apoptotic cell clearance and may affect vascularization. We have also reported differences in gene expression patterns for MMP9, IL-10, TGFB1, and fibroblast growth factor 2 (FGF2) (17), all of which function in angiogenesis.
Our previous work demonstrated that ECs and vascular support cells can be seeded onto poly L-lactic acid (PLLA)/poly(lactic-co-glycolic acid) (PLGA) constructs to generate self-assembled vascular networks in vitro and recapitulate the dynamics of vascular morphogenesis (15, 19). Elongated immature vessels develop within 4 days after seeding, and well-developed vascular networks are formed by day 7 (15). In this study, we adapted this model to use porous gelatin-based sponges, which allowed more uniform incorporation of macrophages at various time points following blood vessel formation.
HAMECs expressing td-Tomato (HAMEC-dTom) and adipose-derived MSCs, which act as pericyte-like support cells to facilitate the formation of stable vessels (15), were seeded together on 8-mm compressed Gelfoam sponges to initiate blood vessel formation. After 3 days, vessel formation was confirmed via confocal microscopy. Human THP-1–derived M0, M1, or M2a macrophages were subsequently seeded onto the constructs, and changes in macrophage-vessel interactions and macrophage morphology were assessed after 1 day of triculture via live cell imaging (Fig. 3A), as changes in macrophage phenotype would be expected after extended culture. Distinct differences in macrophage morphology and interactions with blood vessels were observed between M0, M1, and M2a macrophages (Fig. 3B). In contrast to the primarily rounded morphology of M0 macrophages, the M1 phenotype displayed both rounded and elongated morphologies and was found in close proximity with vessels and at the leading front of sprouts. In contrast, M2a macrophages appeared mostly elongated, clustered together, and physically wrapped around the vessels. Wrapping of macrophages around blood vessels has been described by previous in vivo reports using murine models (20–22). One report did show that the wrapping macrophages expressed the M2 marker CD206 (22), although this marker does not clearly differentiate between different M2 subtypes in mice like it does in humans (17).
Fig. 3. Effects of macrophage phenotype on in vitro tissue vascularization dynamics.
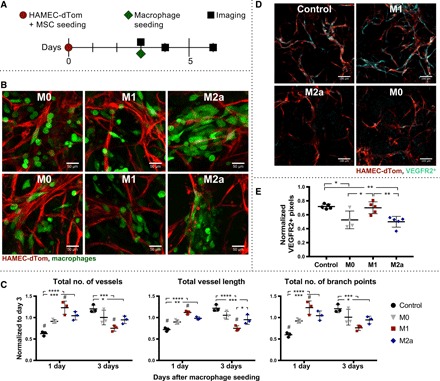
(A) In vitro engineered human blood vessels generated via coculture of HAMEC-dTom and MSCs on Gelfoam constructs. Vessel formation was visualized via live cell confocal imaging after 3 days, before addition of THP-1–derived M0, M1, or M2a macrophages. Changes in network morphology were observed 1 to 3 days after macrophage seeding via confocal microscopy. (B) Representative maximum-intensity projections of M0-, M1-, and M2a-seeded vessel networks illustrating interactions between macrophages (green) and vessels (red) 1 day after macrophage seeding. Scale bars, 50 μm. (C) Quantification of changes in network morphology via MATLAB analysis in 3D, in terms of the total number of vessels, total vessel length, and total number of branch points (junctions between two vessels). All data were normalized to day 3 baseline measurements and represent means ± SD, n = 3. Two-way ANOVA with Tukey’s post hoc analysis; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, and # indicates P < 0.05 relative to the M0 control. Data are representative of two independent experiments. (D) Representative maximum-intensity projections of engineered vascular grafts that were fixed and stained for the tip cell marker KDR (VEGFR2) via whole-mount immunohistochemistry. Scale bars, 100 μm, n = 3. Vessels shown in red; VEGFR2 staining shown in cyan. (E) Quantification of EC VEGFR2 staining, performed on maximum-intensity projections of 2 × 2 tile scans. Pixels colocalized with dTom and VEGFR2 expression were measured and normalized to the total number of dTom+ pixels in each image. Data represent means ± SEM, with n = 4 to 5. One-way ANOVA with Tukey’s post hoc analysis; *P < 0.05 and **P < 0.01.
Macrophage phenotypes differentially affect vascularization
Changes in vessel network morphology in response to M0, M1, or M2a macrophages were assessed 1 and 3 days after macrophage seeding via live cell imaging (fig. S5, A and B). A custom computational image analysis code in MATLAB was used to characterize and quantify vessel structures in 3D; changes were normalized to baseline network morphology obtained immediately before macrophage seeding. M1-seeded engineered vascular grafts displayed an increased total number of vessels, vessel length, and number of branch points relative to both the M0 control (P < 0.05) and constructs without macrophages (P < 0.0001) 1 day after seeding (Fig. 3C). In contrast, after 3 days, M1-seeded grafts displayed a significant reduction in the number of vessels and total vessel length relative to control grafts. These data suggest that short-term exposure to M1 enhances vascularization, but long-term exposure causes vessel regression.
M1 macrophages support tip cell behavior
On the basis of the gene expression analysis of macrophage-EC coculture in 2D, which showed significant up-regulation of the VEGF receptor and tip cell marker VEGFR2 (KDR) in ECs cocultured with M1 macrophages, we hypothesized that M1 macrophage–induced sprouting was related to protein-level up-regulation of this receptor. Therefore, we performed whole-mount immunofluorescence staining of macrophage-seeded engineered vascular grafts for VEGFR2 24 hours after macrophage seeding (Fig. 3D and fig. S5C). Colocalization of VEGFR2+ pixels with dTom+ pixels was quantified and normalized to the total number of dTom+ pixels in each image to account for variations in the number of blood vessels present (Fig. 3E). While ECs reduced expression of VEGFR2 in the presence of M0 and M2a macrophages compared with the control, this reduction was obviated in the presence of M1 macrophages.
Temporal effects of macrophage phenotype on vascularization
On the basis of the results that M1 macrophages support tip cell behavior, which function in vessel sprouting, while M2a macrophages may support pericyte cell differentiation to promote stabilization, we reasoned that the sequential addition of M1 and M2a phenotypes would lead to enhanced blood vessel network formation. To this end, M1 and M2a macrophages were added to vessel networks on days 3 and 4, respectively, of blood vessel formation (fig. S6A). This time interval was selected to approximate the temporal profile of M1 and M2 macrophages in vivo (4). Changes in network morphology were assessed on day 5 (1 day after seeding the second macrophage population) by live cell imaging and compared with several controls: the continuous presence of M1 or M2a, M1 followed by M0, or M0 followed by M2a (fig. S6B). While the sequential addition of M1 and M2a macrophages enhanced vessel formation relative to the control without macrophages, these findings did not reach the level of statistical significance (fig. S6C). In addition, this group did not increase the number of vessels, total vessel length, or number of branch points relative to any of the other control groups, suggesting that the presence of macrophages has a greater effect on these parameters than individual phenotypes.
In contrast, sequential seeding on days 3 and 6 of vessel formation resulted in vessel regression by all macrophage-seeded grafts by day 10, irrespective of phenotype or sequence (fig. S7). It is known that vessel networks undergo extensive vascular remodeling processes involving pruning and regression before formation of mature, functional vasculature (23). However, given that vessel regression was observed several days after macrophage seeding when macrophage phenotype was likely altered, we cannot speculate on the role of phenotype in this process.
Host macrophages facilitate blood vessel interactions in vivo
It is well established that both macrophages and vascularization are required for tissue regeneration (24, 25). Therefore, we reasoned that macrophages may play a fundamental role in vascularization of engineered vascular grafts in vivo and anastomosis with host vasculature. To elucidate the in vivo contributions of macrophages to these processes, we first implanted engineered vascular grafts subcutaneously in athymic mice and characterized the interactions of host macrophages with the engineered human vessels.
Engineered vascular grafts were prepared from PLLA and PLGA embedded with GFP-expressing human umbilical vein ECs (HUVEC-GFP) and human neonatal dermal fibroblasts (HNDFs) as support cells in fibrin. We have previously established the efficacy of this system in mice (26) and routinely use these constructs for vascularization studies (27, 28). PLLA/PLGA constructs coseeded with HUVEC-GFP and HNDF were cultured for 14 days in vitro to allow self-assembly into mature vessel networks (Fig. 4A), as previously described (26, 28, 29), and then implanted subcutaneously in athymic nude mice. Confocal images of retrieved grafts, perfused with fluorescent antibodies against murine CD31 to identify host vessels, and stained for the pan macrophage marker F4/80 (30), revealed the presence of macrophages within the grafted tissue 14 days after implantation (Fig. 4B). High-magnification imaging further demonstrated diverse macrophage morphologies and interactions with both host and engineered vessels (Fig. 4, C to E). Some macrophages appeared to form tube-like structures (Fig. 4C and movies S1 and S2). We also observed close associations of macrophages with the vessels; macrophages not only were found lying next to infiltrating host vessels and engineered vessels (Fig. 4D and movies S3 and S4) but also appeared to wrap around the vessels and bridge adjacent sprouts (Fig. 4E and movies S5 and S6). Quantification of host and engineered vessels revealed an increase in host vessel infiltration into the graft and a corresponding decrease in engineered vessels after 7 days, although the number of F4/80+ macrophages did not change over time (Fig. 4F).
Fig. 4. Macrophage interaction with host and engineered vasculature within the graft.
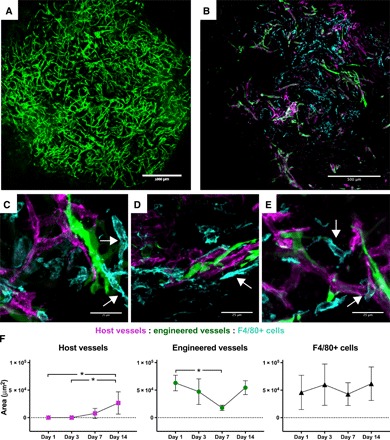
(A) Representative image of an engineered vascular graft bearing GFP-expressing human blood vessels (green) after 14 days of in vitro culture. Scale bar, 1000 μm. (B) F4/80 immunostaining (cyan) of engineered vascular graft explanted 14 days after implantation, indicating interactions between engineered vessel network (green) and penetrating host vessels (red). Scale bar, 500 μm. (C to E) High-magnification images of macrophages (F4/80+, cyan) interacting with engineered (green) and host (magenta) vessels within graft marked with white arrows. Scale bars, 25 μm. (C) Macrophages form vessel-like structures; (D) macrophage adjacent to both engineered and host vessel; (E) macrophage bridging between vessel segments and wrapping around host vessel. (F) Quantification of the area of host vessels, engineered vessels, and F4/80+ cells in sectioned grafts explanted on days 1, 3, 7, and 14. Data represent means ± SD and were analyzed using one-way ANOVA with Tukey’s post hoc multiple comparisons test; *P < 0.05 for n = 6 to 8.
Macrophages facilitate angiogenesis and anastomosis in vivo
We next investigated the importance of macrophages for anastomosis of the engineered vessels with the host vasculature using macrophage depletion via clodronate liposomes, administered 1 day prior and at 4, 8, and 12 days after implantation of the engineered vascular grafts (Fig. 5A). Macrophage depletion was confirmed via staining for the pan macrophage marker F4/80 in the blood and engineered tissues. Flow cytometric analysis demonstrated an increase in macrophage population following graft implantation into control mice and a significant decrease in macrophage population after implantation in clodronate-treated mice (Fig. 5B and fig. S8). Depletion of macrophages in engineered tissues was further confirmed via immunofluorescence 14 days after graft implantation using antibodies against F4/80 (Fig. 5C).
Fig. 5. Consequences of in vivo macrophage depletion on graft revascularization and integration with host vessels.
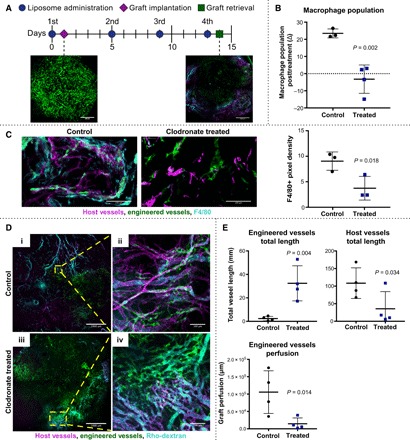
(A) Timeline of clodronate- or PBS-loaded liposome administration, graft implantation, and retrieval. Representative images show engineered vessels (green) in graft before implantation and following retrieval. Host vessels shown in magenta and Rho-dextran perfusion in cyan. Scale bars, 1000 μm. (B) Flow cytometric analysis of whole-blood staining against the pan macrophage marker, murine F4/80, following the second administration of liposomes; n = 3 to 4. (C) Immunofluorescence staining and quantification of pan macrophage marker F4/80 (cyan) in grafts from macrophage-depleted (clodronate-liposome–treated) versus control (PBS-liposome–treated) mice 14 days after implantation. Scale bars, 100 μm. (D) Representative maximum-intensity projections of retrieved grafts 14 days after implantation in (i) control mice and (iii) macrophage-depleted mice. Scale bars, 1000 μm. High-magnification images of (ii) control and (iv) macrophage-depleted grafts. Scale bars, 100 μm. Host vessels shown in magenta, engineered vessels shown in green, and grafts perfused with Rho-dextran shown in cyan. (E) Quantification of total vessel length via AngioTool for engineered vessels and host vessels; engineered vessel perfusion of maximum-intensity projections analyzed using a custom MATLAB code. All data represent means ± SD and were analyzed using an unpaired t test with n = 3 to 4.
Overall vascularization and anastomosis between engineered and host vessels were assessed 14 days after implantation via perfusion with fluorescently tagged antibody against murine CD31, which labels host blood vessels, and rhodamine-dextran, which allows quantification of vessel perfusion. Retrieved grafts were imaged using confocal microscopy to visualize host-graft vessel interactions (Fig. 5D). The depletion of macrophages caused a significant increase in the number of engineered vessels remaining in the graft and a decrease in the number of infiltrating host blood vessels (Fig. 5E). A significant reduction in perfusion of engineered vessels was also observed in mice with depleted macrophages (Fig. 5D), providing evidence that macrophages are important for anastomosis between host and graft vessels. Overall, these data suggest that macrophages are indispensable for vascularized tissue integration.
Diverse macrophage phenotypes facilitate vessel integration
To further characterize the contributions of macrophage phenotypes to graft integration, we retrieved the engineered vascular grafts 1, 3, 7 and 14 days after implantation in healthy mice. Before graft retrieval, fluorescently tagged anti-murine CD31 antibody was injected through the mouse tail vein for vessel staining and visualization. Upon retrieval, grafts were fixed and stained for the pan macrophage marker F4/80. Macrophage subsets were evaluated via costaining to specific macrophage phenotype markers, including iNos and Arg1, which are expressed at high levels by M1 and M2 macrophages, respectively (immunohistochemical markers of specific M2 subsets have not yet been delineated in the mouse like they have been in human macrophages) (Fig. 6) (31). Analysis of retrieved graft sections showed that as early as 1 day after implantation, macrophages infiltrated into the engineered vascular grafts and were positive for markers of both M1 and M2 phenotypes (Fig. 6, A and B). The presence of iNos colocalized with F4/80 increased slightly over time, although not significantly (P > 0.05), whereas the presence of Arg1 colocalized with F4/80 remained relatively unchanged (Fig. 6C). It is not clear whether the F4/80+ macrophages map to particular M1 or M2 phenotypes, or hybrid phenotypes, as coexpression of multiple phenotypic markers was not assessed.
Fig. 6. Macrophage phenotype subsets during graft integration.
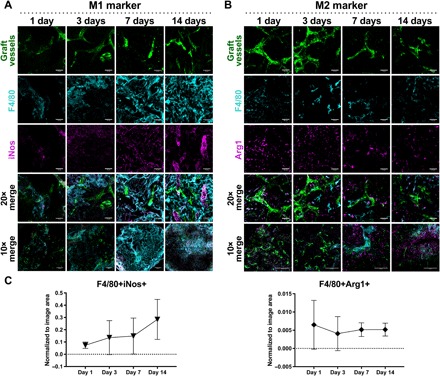
Representative maximum-intensity projections of engineered vascular grafts bearing GFP-expressing vasculature (green) extracted on days 1, 3, 7, and 14 after implantation and sectioned. Graft sections were stained for pan macrophage marker F4/80 (cyan) and costained for either iNos [magenta, (A)] or Arg1 [magenta, (B)] to determine presence of macrophages expressing M1 (A) and M2 (B) markers. Scale bars, 50 μm. (C) Quantification of F4/80+iNos+ staining and F4/80+Arg1+ staining. Data represent means ± SD and were assessed via one-way ANOVA with Tukey’s post hoc analysis; n = 3 to 4.
DISCUSSION
Macrophages play important and diverse roles during vascularization. Prior work has shown that macrophages physically interact with blood vessels and contribute to fusion of sprouting vessels (20, 21) and can form functional vascular channels that mimic blood vessels but lack ECs (32). However, there are conflicting reports regarding the angiogenic potential of different macrophage phenotypes. For instance, M1 macrophages have been implicated in both the enhancement (3, 7) and the inhibition (10, 33) of angiogenesis. For example, proinflammatory CCR2+ macrophages have been reported to induce vascular sprouting by VEGF secretion at early stages of healing in murine skin wounds (3), but angiogenesis is impaired in chronic wounds, which are characterized by the sustained presence of proinflammatory (M1) macrophages (34). In vitro, M1 macrophages enhanced tube formation of HUVECs on Matrigel (7) but inhibited tubule formation by aortic ECs in a 3D hydrogel (35). In the present study, short-term presence (1 day) of M1 macrophages enhanced angiogenesis, but long-term presence (3 days) caused vessel regression, suggesting that a potential reason for the conflicting reports may be differences in timing. On the other hand, vessel regression is an important part of healthy angiogenesis (23), so it is possible that M1 macrophages support angiogenesis over time by changing behaviors.
Gene expression analyses of ECs cocultured with macrophages allowed investigation into potential pathways affected by the different phenotypes. Most notably, we found that M1 macrophages promoted expression of genes associated with vessel sprouting, including those indicative of tip cell activation, EC proliferation, and migration. These findings are consistent with prior work demonstrating that the M1 macrophage–conditioned media promote vascular tube formation in vitro (7). These proangiogenic effects may be attributed to M1 secretion of proinflammatory factors such as VEGF (7), a known regulator of angiogenesis, and tumor necrosis factor (TNF), which induces endothelial tip cell differentiation (36). Proinflammatory mediators can also play a role in modulating vascular morphogenesis, which may explain the observed promotion of genes associated with vessel stabilization and lumen formation in the presence of M1 macrophages.
While M2 macrophages are often characterized as proangiogenic, the differences between multiple M2 subtypes have not been delineated in vivo, and few studies have examined the differences in angiogenic potential of each M2 subtype. Our results suggest that three distinct phenotypes, which have all been described as “M2,” may contribute to angiogenesis in different ways. In particular, gene expression analysis of ECs cocultured with the different phenotypes suggests that M2a macrophages may mediate EC-pericyte interactions, M2c macrophages may regulate sprouting, and M2f macrophages may regulate vessel maturation. We have previously demonstrated M2a production of PDGFBB (7), a recruiting factor for pericytes, and M2c secretion of MMPs (8), which facilitate degradation of the basement membrane and surrounding extracellular matrix to create space for lumen formation, and support cell migration and proliferation and vascular remodeling. M2c macrophages also produce osteopontin, a known immunomodulator that enhances VEGF expression in ECs and promotes proliferation, migration, and tube formation of ECs in vitro. M2f macrophages up-regulate TGFB1, which is implicated in endothelial cross-talk with support cells to modulate vessel stabilization and has been shown to promote EC migration in vitro and vessel formation in vivo (37). Together, these data implicate multiple M2 phenotypes in diverse aspects of angiogenesis, including vessel stabilization, remodeling and maturation, and strongly support a need not only to characterize the functional effects of these phenotypes, as well as others not described here, but also to thoroughly phenotype M2-like macrophages participating in angiogenesis in vivo. Single cell analysis conducted over time will likely be important for this analysis, considering that these diverse phenotypes may act simultaneously or in sequence.
Our findings of proangiogenic contributions of both M1 and M2 macrophages are in line with previous reports that both M1 and M2 phenotypes are present in vascularizing engineered tissue (21). We observed greater physical interaction in vitro between vessels and M2a macrophages compared with the M1 phenotype, which appeared at vascular fronts, corroborating previous observations that macrophages expressing CD206, an M2 marker, physically wrap around remodeling vessels (22). In vivo interactions of macrophages with engineered vessels were also consistent with prior findings that macrophages can act as bridging cells to support anastomosis (20), as well as guide sprouting vessels by forming tunnels within the local extracellular matrix (38).
The use of engineered vascular grafts in combination with clodronate-loaded liposomes demonstrated the in vivo contribution of macrophages to integration of human engineered blood vessels with host tissue. This analysis showed the critical role of macrophages in the remodeling of engineered vessels and anastomosis with host vasculature. These findings are consistent with recent data showing that macrophages augment blood vessel ingrowth, cell survival, and tissue regeneration of engineered skeletal muscle tissue grafts (39). Our findings that macrophages express markers of both M1 and M2 phenotypes support the concept that multiple phenotypes contribute to vascularization in unique ways. At present, it is not clear whether the phenotype(s) of the infiltrating macrophages were affected by interactions with the host vessels or with the PLLA/PLGA scaffolds, or both.
In summary, we showed that macrophages of distinct phenotypes differentially affect EC behavior, with M1 macrophages enhancing angiogenesis of human engineered blood vessels in vitro to a greater extent than M0 and M2a. Furthermore, macrophages of both M1 and M2 phenotypes were critical for anastomosis between engineered blood vessels and host vasculature in vivo. The results highlight the need for in-depth studies investigating the effects of different phenotypes on multiple processes involved in angiogenesis and tissue regeneration.
MATERIALS AND METHODS
Cell culture
Induction of neutrophil apoptosis
Human promyelocytic leukemia (HL-60) cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and expanded in Iscove’s modified Dulbecco’s medium (ATCC) supplemented with 25% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified chamber at 37°C and 5% CO2. Neutrophils were maintained at a density of 1 × 105 cells/ml via media exchange every 3 days, and trypan blue exclusion was performed to assess cell viability, which was routinely >85%. To induce apoptosis, HL-60 cells were first resuspended in culture media without FBS for 2 hours for cell synchronization. Following serum deprivation, 800 μM H2O2 (Sigma-Aldrich) was added to the cells and incubated for an additional 3 hours. Apoptosis was confirmed via flow cytometry using an APO–BrdU (bromo deoxyuridine) TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions, and compared to healthy neutrophil controls. All apoptotic HL-60 cells were washed in 1X PBS before incubation with M0 macrophages.
Primary human monocyte-derived macrophages
Primary human monocytes from four healthy donors were purchased from the University of Pennsylvania Human Immunology Core (Philadelphia, PA). Monocytes were cultured at a concentration of 1 × 106 cells/ml in ultralow attachment well plates for 8 days in RPMI 1640 media, supplemented with 10% heat-inactivated human serum, 1% penicillin/streptomycin, and MCSF (macrophage colony stimulating factor, 20 ng/ml) as previously described (17). Media were replenished on days 3 and 5. On day 5, macrophages were stimulated into the M0, M1, M2a, and M2c phenotypes by addition of IFN-γ (100 ng/ml) and LPS (100 ng/ml) for M1 activation, IL-4 (40 ng/ml) and IL-13 (20 ng/ml) for M2a activation, and IL-10 (40 ng/ml) for M2c activation. For M2f activation, macrophages were cocultured with apoptotic HL-60 cells in a 1:5 ratio of macrophages to neutrophils. On day 8, all macrophages were washed to remove polarizing stimuli.
Monocyte cell line
THP-1 cells used in 3D angiogenesis assays were provided by the Admon Lab (Technion, Israel) and expanded in suspension flasks at 200,000 to 400,000 cells/ml in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin solution. Media were replenished every 2 to 3 days by centrifugation of the cells at 200g for 5 min. Following expansion, THP-1 cells were transferred to ultralow attachment culture flasks and stimulated with phorbol 12-myristate-13-acetate (PMA; 320 nM final concentration) for 16 to 24 hours at 37°C and 5% CO2, to induce differentiation to M0 macrophages as previously described (17). Following PMA treatment, M0 macrophages were washed in 1X PBS. For differentiation into the M1 and M2a phenotypes, the cells were incubated for an additional 48 hours in media supplemented with IFN-γ (100 ng/ml) and LPS (100 ng/ml) for M1 activation, or IL-4 (40 ng/ml) and IL-13 (20 ng/ml) for M2a activation. After differentiation, the cells were gently scraped and collected for use, as indicated. To visualize macrophages, THP-1 cells expressing GFP (GFP–THP-1) were purchased from Angio-Proteomie (Boston, MA). GFP–THP-1 were expanded in ultralow attachment flasks at 200,000 to 400,000 cells/ml in RPMI 1640 media, supplemented with 10% FBS and 1% penicillin/streptomycin solution. Media were replenished every 2 to 3 days by centrifugation of the cells at 200g for 7 min. Differentiation into the M0, M1, and M2a phenotypes was accomplished using the same procedure as described for the unlabeled THP-1 cells. All cells were cultured in a humidified chamber at 37°C and 5% CO2.
Endothelial cells
HAMECs were transfected to express td-Tomato (HAMEC-dTom). HAMEC-dTom were cultured at 5000 to 7000 cells/cm2 in EC medium (ECM, ScienCell, #1001) supplemented with 5% FBS, 1% EC Growth Supplement, and 1% penicillin/streptomycin solution. HUVECs expressing GFP (HUVEC-GFP) were purchased from Angio-Proteomie (Boston, MA) and cultured at 5000 to 7000 cells/cm2 in EC Growth Medium (EGM-2-BulletKit, Lonza, #CC-3162), supplemented with EGM-2 SingleQuots and 3% FBS (HyClone, Thermo Fisher Scientific). All ECs were maintained in a humidified chamber at 37°C and 5% CO2 and routinely subcultured via trypsinization at 90% confluency; ECs were used within nine passages.
Lentivirus packaging and transduction with td-Tomato fluorescent protein
Production of lentiviruses was performed according to the manufacturer’s instructions. Briefly, recombinant, replication-incompetent, VSV-G pseudotyped lentiviruses were generated using a Lenti-X HTX Packaging System [Clontech, fourth generation (five vectors split packaging technology and inducible promoters)]. The expression vector (pLVX-tdTomato-C1, Clontech) was transfected, in a Lenti-X HTX Packaging Mix, into the Lenti-X 293T packaging cells (Clontech). Following 72 hours, supernatants containing the lentiviral produced by the transfected packaging cells were filtered through a 45-μm filter to remove cellular debris. HAMECs were seeded at a density of 5000 cells/cm2 and allowed to adhere overnight. Cells were transduced with td-Tomato lentivirus particles in the presence of polybrene (1.5 μg/ml; Sigma-Aldrich) per well. Transduction media were replaced after 24 hours, and the cells were incubated for up to 72 hours to allow for gene products to accumulate within the target cells. Selection for stably expressing cells was performed using puromycin (3 μg/ml; Takara Bio Company).
Vascular support cells
Human adipose-derived MSCs were purchased from Lonza and cultured at 5000 cells/cm2 in Human Adipose Derived Stem Cell Growth BulletKit medium (Lonza, #PT-4503) according to the manufacturer’s instructions. MSCs were routinely subcultured via trypsinization at 90% confluency and used within seven passages. HNDFs (Lonza) were cultured in Dulbecco’s modified Eagle’s medium (Gibco Life Technologies) supplemented with 10% FBS, 1% nonessential amino acids (Biological Industries, Israel), 1% penicillin/streptomycin solution (Biological Industries, Israel), and 0.2% β-mercaptoethanol (Gibco, Carlsbad, CA). HNDFs were routinely subcultured via trypsination at 90% confluency and used within eight passages.
In vitro Transwell coculture
HAMEC-dTom (25,000) were seeded in the apical chamber of Transwell inserts (0.4-μm pore size and 6.5-mm diameter) in 100 μl of ECM and allowed to attach for 1 hour at 37°C and 5% CO2. Media were removed from all macrophage-seeded wells and replaced with 100 μl of coculture media (1:1 ratio of ECM to cRPMI 1640 with MCSF) to support cell survival. Transwell inserts containing HAMEC-dTom were transferred to macrophage-seeded wells (n = 4 per phenotype). An additional 500 and 100 μl of coculture media were added to the basolateral and apical chambers, respectively. All samples were incubated for 1 to 3 days at 37°C and 5% CO2. HAMEC-dTom cultured in Transwell inserts in the absence of macrophages served as a control. Controls were cultured in equal volumes of coculture media to account for changes in cell behavior induced by media supplements.
On days 1 and 3, Transwell inserts were transferred to clean 24-well plates and stored on ice. Conditioned media were collected from the basolateral and apical chambers and stored at −80°C, and all samples were washed in 1X PBS to remove residual media. HAMEC-dTom were lysed directly in the inserts in 200 μl of Buffer RLT (Qiagen). All lysates were transferred to 1.5-ml polymerase chain reaction (PCR) clean tubes and stored at −80°C until RNA extraction.
RNA extraction and purification
HAMEC-dTom lysates were thawed on ice and vortexed briefly. An additional 150 μl of Buffer RLT was added to HAMEC-dTom lysates to bring the final volume to 350 μl. Samples were mixed with an equal volume of 70% ethanol and directly loaded onto RNeasy mini-spin columns (RNeasy Micro Kit, Qiagen) for purification, according to the manufacturer’s instructions. RNA was eluted in a final volume of 14 μl of ribonuclease-free water and stored at −80°C until multiplex gene expression analysis.
cDNA synthesis and quantitative reverse transcription PCR
To characterize differences in macrophage phenotypes, purified RNA was quantified on a NanoDrop 1000 (Thermo Scientific) and treated with deoxyribonuclease (DNase) I, Amplification Grade (Invitrogen) for DNA removal, according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from 1 μg of RNA (A260/280 > 1.8) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and stored at −80°C. Quantitative reverse transcription PCR (qRT-PCR) was performed using 20 ng of cDNA and Fast SYBR Green Master Mix (Applied Biosystems) according to the manufacturer’s instructions. Mean quantification cycle, Cq, values were calculated from technical replicates. The expression of target genes was normalized to the reference gene, glyceraldehyde phosphate dehydrogenase (GAPDH). All primers were synthesized by Life Technologies: CD163, 5′-TTTGTCAACTTGAGTCCCTTCAC-3′ (forward) and 5′-TCCCGCTACACTTGTTTTCAC-3′ (reverse); CD206 (MRC1), 5′-AAGGCGGTGACCTCACAAG-3′ (forward) and 5′-AAAGTCCAATTCCTCGATGGTG-3′ (reverse); GAPDH, 5′-AAGGTGAAGGTCGGAGTCAAC-3′ (forward) and 5′-GGGGTCATTGATGGCAACAATA-3′ (reverse); IL-10, 5′-TCAAGGCGCATGTGAACTCC-3′ (forward) and 5′-GATGTCAAACTCACTCATGGCT-3′ (reverse); PDGF, 5′-CTCGATCCGCTCCTTTGATGA-3′ (forward) and 5′-CGTTGGTGCGGTCTATGAG-3′ (reverse); TGFB1, 5′-CAATTCCTGGCGATACCTCAG-3′ (forward) and 5′-GCACAACTCCGGTGACATCAA-3′ (reverse); TIMP3, 5′-ACCGAGGCTTCACCAAGATG-3′ (forward) and 5′-CATCATAGACGCGACCTGTCA-3′ (reverse).
Enzyme-linked immunosorbent assay
Supernatants from macrophages were collected after 2 days of stimulation and analyzed for the presence of transforming growth factor-beta 1 (TGFB1). Protein secretion was measured using a commercially available enzyme-linked immunosorbent assay (ELISA) Development Kit (R&D Systems, USA) according to the manufacturer’s instructions.
Multiplex gene expression analysis
NanoString
RNA was thawed on ice and quantified using a NanoDrop 1000 (Thermo Scientific). Multiplex gene expression analysis was performed via NanoString using a custom nCounter XT CodeSet (NanoString Technologies, Seattle, WA), inclusive of 97 endogenous genes for HAMEC-dTom RNA, 3 housekeeping genes, 8 External RNA Control Consortium (ERCC) negative controls, and 6 ERCC positive controls. All genes included in the CodeSet are listed in table S1. These genes were selected on the basis of literature demonstrating their association with various processes involved in angiogenesis, including regulation of EC differentiation into tip and stalk phenotypes, regulation of the potent angiogenic stimulator, VEGF, tube formation, vessel stabilization and maturation, hypoxia, and apoptosis. Hybridization reactions were prepared with 100 ng of RNA for all samples according to the manufacturer’s instructions. Raw count data were extracted using nSolver Analysis Software 4.0 (NanoString Technologies, Seattle, WA).
NanoString data normalization
Before data normalization, a value of 1 was added to all raw gene counts, including positive and negative controls. The data were then normalized to the ERCC positive controls, as recommended by the manufacturer, to normalize all platform-associated sources of variation. The geometric mean of the positive controls was calculated for each sample and averaged across all samples. This average was divided by the geometric mean of the positive controls for each sample, yielding a sample-specific scaling factor. All gene counts for each sample were then multiplied by the corresponding sample-specific scaling factor. The data were subsequently normalized to the ERCC negative controls. The sample-specific scaling factor was set as the average plus the SD of the negative controls for each sample. This background threshold was subtracted from all counts, and all negative count data were set to 1. The corrected data were then log transformed. Where indicated, the data were further normalized to the day 1 and day 3 EC-only controls by subtracting the average of log-normalized EC data from the log-normalized sample data.
Principal components analysis
To visualize the global representation of gene count data and emphasize differences in EC responses, PCA was implemented using MATLAB software (MathWorks, Natick, MA). PCA is a multivariate data analysis approach that reduces the dimensionality of a dataset by capturing most of the variation in the dataset into new variables known as principal components. Samples can then be plotted using a few of these uncorrelated principal components to detect the hidden phenomena in the dataset, including similarities and dissimilarities among samples. Before analysis, the data were standardized using z scores to enable comparison across the dataset.
Data analysis and heat map clustering in R
Log-transformed gene count data were hierarchically clustered using RStudio Software 1.1.453 with the Bioconductor package. The data were imported into RStudio, and replicates were averaged. z scores were generated by scaling the data across genes using the scale function; NaN values were removed from downstream analysis. The z score data were first clustered between samples using a Euclidean distance matrix with complete linkage and subsequently clustered between genes using a Pearson distance matrix with complete linkage using the get_dist and hclust functions. Heat maps of clustered z scores were generated using the heatmap.2 function.
GO enrichment analysis
GO Enrichment Analysis was performed on genes up-regulated or down-regulated in ECs by macrophage phenotype. GO Enrichment was performed via the PANTHER Overrepresentation Test (released 5 December 2017) with the GO Ontology database released 3 July 2018. The default Homo sapiens reference list, including all genes in the database, was used with the GO biological process complete dataset. Overrepresented genes were determined using Fisher’s exact with FDR multiple test correction for P < 0.05.
REVIGO
All GO terms with fold enrichment >25 were analyzed using REVIGO, which summarizes and visualizes long lists of GO terms by removing redundant terms (12). Nonredundant or nondispensable terms were identified as those with dispensability <0.25. These terms were visualized in semantic space as bubble plots and demarcated based on P value and fold enrichment using RStudio with the ggplot2, scales, and ggrepel packages. As an extension of this analysis, nonredundant terms for each phenotype were then compiled, and the variance across phenotypes was calculated for each GO term; for the purposes of sorting GO terms by variance, phenotypes that were not associated with a specific GO term were given a fold enrichment value of 0.
Single-sample gene set enrichment analysis
All genes were classified into one of 17 gene sets based on related biological function. Normalized and log-transformed gene expression data were imported into RStudio Version 1.2.1335 running R version 3.6.0. Enrichment analyses were performed for all gene sets using the ssGSEA method of the GSVA function, resulting in a total of 17 enrichment scores per experimental sample. For each gene set, GraphPad Prism was used for visualization of the distribution of scores per phenotype, and statistical analysis was performed using one-way analysis of variance (ANOVA) with Tukey’s post hoc multiple comparisons test.
In vitro 3D engineered vascular grafts
Effects of phenotype
Commercially available size 100 compressed Gelfoam sponge was purchased from Pfizer (New York, NY), cut using a sterile 8-mm-diameter biopsy punch, and prewet in sterile 1X PBS or media. Vascular networks were formed on Gelfoam constructs by seeding a 5:1 ratio of HAMEC-dTom to MSCs directly onto the constructs in 10 μl of EC growth media. The cell-seeded constructs were incubated for at least 30 min in a humidified chamber at 37°C and 5% CO2 to facilitate cell attachment; this process was repeated to achieve seeding on both sides of the constructs. After seeding, all constructs were transferred to clean tissue culture dishes, immersed in a 1:1 solution of EC and MSC coculture media, and incubated to initiate vessel formation. On day 3, the engineered vascular grafts were imaged via confocal microscopy as a baseline measurement of vessel growth. After imaging, 100,000 THP-1–derived M0, M1, or M2a macrophages were seeded directly onto the grafts in 10 μl of coculture media. Control grafts without macrophages were seeded with 10 μl of media alone. All samples were incubated for 30 min to allow cell attachment and subsequently transferred to clean 24-well tissue culture plastic for continued incubation in coculture media, which was previously verified to support THP-1 viability (>99.5%) after 3 days in vitro via trypan blue exclusion. Macrophage media were omitted to prevent changes in vessel development. Changes in vascular network formation were monitored over 10 days in vitro using confocal microscopy. For all studies, n ≥ 3 grafts were included per group.
Effects of temporally modulating phenotype
Gelfoam constructs were preseeded with a 5:1 ratio of HAMEC-dTom and MSCs to initiate vessel formation, and on day 3, the grafts were imaged via confocal microscopy as a baseline measurement of vessel growth. After imaging, 50,000 M1 or M2a macrophages were seeded directly onto the grafts in 10 μl of coculture media. All samples were incubated for 30 min to allow cell attachment and subsequently transferred to clean 24-well tissue culture plastic for continued incubation in coculture media. Grafts were imaged on days 4 and 6. After imaging on day 6, 50,000 M1 or M2a macrophages were again seeded directly onto the grafts in 10 μl of coculture media and incubated for cell attachment. The effects of this sequential seeding were measured on days 7 and 10 using confocal microscopy. Experimental groups included the following: control grafts without macrophages, M1-to-M0, M1-to-M1, M1-to-M2a, M0-to-M2a, M2a-to-M2a, and M1+M2a-to-M1+M2a; n ≥ 3 per group.
In vivo contribution of macrophages to anastomosis
Macrophage depletion
Systemic macrophage depletion was achieved by intraperitoneal injections of clodronate-loaded liposomes (0.1 ml per 10 g mouse body weight) as per the manufacturer’s protocol (ClodLip BV, ClodronateLiposomes.org); PBS-loaded liposomes served as a control. Clodronate- or PBS-loaded liposomes were administered 1 day before engineered vascular graft implantation and every 3 to 4 days thereafter for continued macrophage depletion throughout the duration of the study.
Flow cytometry
To assess basal macrophage presence, blood was drained from the tail vein of mice using EDTA-coated capillaries before graft implantation and liposome administration. To assess the macrophage population in periphery blood upon clodronate-loaded liposome administration and graft implantation, blood was collected again after the second liposome administration at day 6 of the experiment. Blood was immediately transferred into a 5-ml tube and incubated with AlexaFlour647-conjugated anti-mouse F4/80 (1 μg/ml; BioLegend) antibody, a pan mouse macrophage marker, for 30 min. After staining, red blood cells (RBCs) were lysed using RBC lysis buffer (BD FACS Lysing Solution; BD Biosciences) following the manufacturer’s protocol and washed several times. Cells were then resuspended in staining buffer [5% (w/v) bovine serum albumin (BSA) in PBS]. Data were acquired on either a BD LSRII or BD FACSCalibur (BD Biosciences) flow cytometer using BD FACSDiva or BD CellQuest Pro software (BD Biosciences). Data analyses were performed using FCS Express software (De Novo software, version 5). Cell populations were identified using a sequential gating strategy; the expression of markers is presented as the percentage of positive cells for the specified markers (fig. S8).
Engineered vascular graft preparation
Porous sponges composed of 50% PLLA (Polysciences, Warrington) and 50% PLGA (Boehringer Ingelheim) were fabricated using a particle leaching technique to achieve pore sizes of 212 to 600 μm and 93% porosity. Briefly, PLLA and PLGA were dissolved 1:1 in chloroform to yield a 5% (w/v) polymer solution, and 0.24 ml of this solution was loaded into molds packed with 0.4 g of sodium chloride particles. Following solvent evaporation overnight, sponges were subsequently immersed in distilled water (changed every hour) for 8 hours to facilitate salt leaching, resulting in an interconnected pore structure. The ensuing sponges were roughly 0.8 to 1 mm thick and punched into disks using a 6-mm biopsy punch (Integra Miltex). The sponges were sterilized in 70% (v/v) ethanol for at least 2 hours and washed in triplicate with PBS before cell seeding.
Engineered vascular grafts were obtained by seeding 0.5 × 106 HUVEC-GFP (Angio-Proteomie, USA) together with 0.1 × 106 HNDF (Lonza, USA) in 7 μl of a 1:1 mixture of fibrinogen (15 U/ml; Sigma-Aldrich) and thrombin (50 mg/ml; Sigma-Aldrich). The fibrinogen solution was prepared by diluting lyophilized human fibrinogen (Sigma-Aldrich Chemical, St Louis, MO) in 40 mM glycine-tris buffer. The thrombin solution was prepared by diluting thrombin (Sigma-Aldrich) in 40 mM calcium chloride. The suspended coculture was then embedded into the scaffolds and allowed to solidify in non–tissue culture–treated six-well plates for 30 to 45 min at 37°C. Following solidification, a mixture of 1:1 modified EGM-2:HNDF culture medium (total volume of 2 ml) was added to each well; medium was exchanged every other day.
Subcutaneous implantation, perfusion, and retrieval
All surgical procedures and animal studies were performed under protocols reviewed and approved by the animal ethics committee at the Technion, and all efforts were made to minimize animal suffering. Athymic nude mice (Harlan Laboratories) 7 to 9 weeks old were anesthetized via intraperitoneal injection using a 30-gauge needle with a mixture of ketamine-xylazine at a dose of 100 and 10 mg/kg, respectively, or with 2% isoflurane. Every 12 hours for 2 days, buprenorphine (0.05 mg/kg) was subcutaneously injected. The dorsal region of the mouse torso and the entire tail were cleaned with chlorhexidine solution and iodine to establish an aseptic working field. A 12- to 15-mm incision was made in the dorsal-lateral portion of the mouse, and the engineered vascular graft was inserted subcutaneously. After implantation, the incision was sutured with 4.0 silk surgical sutures. At the end of each experiment, mice were anesthetized with a mixture of ketamine-xylazine, as described above, and euthanized by CO2 overdose; grafts were retrieved and fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences).
To visualize host vessel infiltration within implanted grafts, AlexaFlour647-conjugated anti-mouse CD31 antibody (BioLegend, #102416) was injected into the mouse tail vein 30 min before euthanasia. Following graft retrieval and fixation, GFP-expressing engineered vessels and penetrating host vessels were imaged via confocal microscopy. Vascular networks were characterized on maximum-intensity projections of z-stack images for total vessel length using AngioTool software (National Cancer Institute). Whole-mount immunohistochemistry with anti-murine F4/80, a pan macrophage marker, was also performed to visualize host macrophage interactions with native and engineered blood vessels.
To detect the presence of blood flow within engineered and host blood vessels, mice were anesthetized and intravenously injected with tetramethylrhodamine isothiocyanate–dextran (average molecular weight 155,000, Sigma-Aldrich) via the tail vein. Mice were immediately euthanized with CO2 overdose. Grafts were retrieved, fixed with 4% PFA (Electron Microscopy Sciences), and imaged immediately via confocal microscopy. Image processing and further analyses were performed using Fiji Software. Graft perfusion was quantified in 2D using a custom MATLAB code developed by the Levenberg laboratory; analysis was performed on maximum-intensity projections of z-stack images.
Immunofluorescence staining of graft tissue for macrophage phenotype
Grafts retrieved at days 1, 3, 7, and 14 after transplantation were fixed with 4% PFA, incubated overnight in a 30% (w/v) sucrose solution, embedded in optimal cutting temperature compound (Tissue-Tec), and frozen for subsequent cryosectioning to 10-, 20-, and 60-μm-thick sections. For immunofluorescence staining, the sections were incubated in 0.5% Tween solution for 15 min, rinsed with PBS, and then blocked with 5% (w/v) BSA (Sigma-Aldrich) for an additional 30 min. The sections were subsequently incubated with 1:100 anti-F4/80 antibody and with either 1:400 anti-iNos (Cell Signaling Technology) or 1:100 anti-Arg1 (Santa Cruz Biotechnology) in blocking solution overnight at 4°C. The sections were washed in PBS in triplicate and labeled with either 1:100 Cy3-conjugated immunoglobuin G (IgG) (Jackson ImmunoResearch, #712-166-153), 1:100 Dylight 405–conjugated IgG (Jackson ImmunoResearch, #705-475-147), 1:200 AF647-conjugated IgG (Invitrogen, #A-21247), or 1:400 Cy3-conjugated IgG (Jackson ImmunoResearch, #711-165-152) for 30 min at room temperature. Ten- and 20-μm sections were then mounted with Vectamount (Vector Laboratories) and covered with coverslip; 60-μm sections were resuspended in PBS. Sections were then examined under a Zeiss LSM700 confocal microscope (Carl Zeiss) using the Zen software (Zeiss) or imaged using the Pannoramic 250 Flash III automatic slide scanner (3DHISTECH). Image processing and further analyses were performed using Pannoramic Viewer software (3DHISTECH) or with Fiji software.
Confocal microscopy
Network development within the engineered vascular grafts was monitored over 14 days in vitro using confocal imaging. Before imaging, each graft was rinsed in 1X PBS and transferred to a sterile glass-bottom dish. Images were captured using a Zeiss LSM700 inverted laser scanning confocal microscope (Carl Zeiss, Germany) equipped with 10×/0.3 and 20×/0.3 objectives. The pinhole was set to 1 Airy unit, and the z-stack was defined to capture the maximum field of view in the z axis. Tiled z-stack images were acquired and stitched using Zen 2010 software (Zeiss, Germany). Grafts retrieved from in vivo studies were imaged using a similar approach; images were acquired using 5×/0.16, 10×/0.3, 20×/0.8, and 63×/1.4 objectives. For 2D analysis using AngioTool software, maximum-intensity projections were generated using Zen software (Carl Zeiss, Germany).
Whole-mount immunofluorescence
Engineered vascular grafts were washed in 1X PBS and fixed in 10% formalin (4% formaldehyde) for 15 min at room temperature, protected from light. The grafts were washed three times for 5 min in 1X PBS and subsequently treated with 0.3% Triton X-100 for 10 min at room temperature. The grafts were again triple washed and blocked overnight at 4°C in blocking buffer (10% donkey serum and 0.1% Triton X-100 in 1X PBS). After blocking, primary antibody was diluted in blocking buffer; primary antibody or blocking buffer alone (primary delete) was added to each graft and incubated overnight at 4°C with gentle rocking. The grafts were then triple washed in 1X PBS and incubated with secondary antibody, diluted in 1X PBS, and incubated for 3 hours at room temperature, protected from light. After staining, the grafts were washed and stored in 1X PBS at 4°C until imaging.
Quantitative image analysis
Analysis of KDR (VEGFR2) staining
Positive KDR staining of macrophage-seeded engineered vascular grafts was visualized using confocal microscopy. Z-stack images were processed using Fiji Software to adjust brightness and contrast, with gamma set to 0.65. Processed images were analyzed in 3D in MATLAB (MathWorks, Natick, MA); images were binarized, and the total number of pixels colocalized with dTom (expressed by HAMEC-dTom cells) and KDR staining was quantified. To account for effects of macrophages on vessel number, the number of colocalized pixels was normalized to the total number of ECs (pixels containing dTom signal) for each image.
3D in vitro vascular analysis
Z-stack images acquired via confocal microscopy were processed and analyzed in MATLAB as described previously (40), with Phi Max tools. Briefly, metadata from z-stack images were extracted, and images were binarized to generate a grayscale image. Median filtering was applied to remove background noise, followed by Otsu thresholding in 3D. The images were skeletonized in 3D using the bwskel function, with a minimum branch length set to 15 pixels to remove prunes, and the skeletonized structures were graphed using the Skel2Graph3D function. Original and skeletonized images were concatenated to inspect the accuracy of the skeleton in 3D. Vessel structures were characterized in terms of number of vessels, vessel length, the number of vessel endpoints, and the number of branch points or nodes. 3D reconstructions were prepared using LEVER 3D (41).
In vivo vascular perfusion analysis
Rhodamine staining within the graft perimeter was visualized using confocal microscopy. Z-stack images were processed using Fiji Software to adjust brightness and contrast, and maximum-intensity projections were generated. The processed images were analyzed a custom MATLAB (MathWorks, Natick, MA) code. Briefly, images were binarized, and the total number of pixels was quantified and converted into micrometers.
Statistical analysis
Statistical analysis was performed in GraphPad Prism (GraphPad Software Inc., La Jolla, CA). All data are represented as means ± SEM. To determine the effects of macrophage phenotype on ECs in Transwell coculture, all HAMEC-dTom data were analyzed using a two-way ANOVA, and the FDR was controlled for post hoc analysis. The two-stage linear step-up method of Benjamini, Krieger, and Yekutieli was used with Q = 0.05 to control from the number of false positives resulting from multiple comparisons. To assess the effects of macrophage phenotype on in vitro 3D tissue vascularization, one-way ANOVA or two-way repeated-measures ANOVA was performed with Tukey’s post hoc multiple comparisons test. Group differences between clodronate- or PBS-treated mice were determined by one- or two-tailed Student’s t tests. Where appropriate, data were analyzed by two-way ANOVA with Tukey’s post hoc multiple comparisons test or by one-way ANOVA with Mann-Whitney’s post hoc comparisons test. For all analyses, P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We acknowledge the Cell Imaging Center at Drexel for use of resources. We also acknowledge the Lorry I. Lokey Interdisciplinary Center for Life Sciences and Engineering at Technion for help and resources, and the Technion Pre-Clinical Research Authority for providing assistance with in vivo animal studies. We wish to thank J. Zavin for assistance with cryosectioning, O. Katovitz for assistance with data quantification, and I. Michael for assistance with viral transfection. We also thank E. Cardenas for providing guidance on the 3D image analysis. Funding: This work was supported by the U.S. NIH NHLBI R01 HL130037, a gift from the Senel family, and a Drexel Career Development Award awarded to K.L.S. P.L.G. was also supported by a U.S.-Israel Binational Science Foundation Prof. Rahamimoff Travel, a Louis and Bessie Stein Family Foundation Fellowship Award, and the Koerner Family Foundation. This work was supported by funding from the FP7 European Research Council Grant 281501, ENGVASC (S.L.). Author contributions: S.Le. and K.L.S. conceived the research. P.L.G. and S.B.-S. designed the research, performed all experiments, and wrote the manuscript. S.La. assisted with immunohistochemistry, image acquisition, and image analysis for in vivo studies. A.B. prepared and characterized M2f macrophages. B.S. assisted with image analysis. A.C. assisted with development of the custom 3D computational image analysis code. P.L.G., S.B.-S., S.Le., and K.L.S. interpreted the data and edited the manuscript. All authors approved the final manuscript for publication. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data and custom MATLAB and R codes are available upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaay6391/DC1
REFERENCES AND NOTES
- 1.Kubota Y., Takubo K., Shimizu T., Ohno H., Kishi K., Shibuya M., Saya H., Suda T., M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 206, 1089–1102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirose N., Maeda H., Yamamoto M., Hayashi Y., Lee G. H., Chen L., Radhakrishnan G., Rao P., Sasaguri S., The local injection of peritoneal macrophages induces neovascularization in rat ischemic hind limb muscles. Cell Transplant. 17, 211–222 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Willenborg S., Lucas T., van Loo G., Knipper J. A., Krieg T., Haase I., Brachvogel B., Hammerschmidt M., Nagy A., Ferrara N., Pasparakis M., Eming S. A., CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120, 613–625 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., Gherardi R. K., Chazaud B., Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nahrendorf M., Swirski F. K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J. L., Libby P., Weissleder R., Pittet M. J., The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenkins S. J., Ruckerl D., Cook P. C., Jones L. H., Finkelman F. D., van Rooijen N., MacDonald A. S., Allen J. E., Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spiller K. L., Anfang R. R., Spiller K. J., Ng J., Nakazawa K. R., Daulton J. W., Vunjak-Novakovic G., The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 35, 4477–4488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lurier E. B., Dalton D., Dampier W., Raman P., Nassiri S., Ferraro N. M., Rajagopalan R., Sarmady M., Spiller K. L., Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology 222, 847–856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M., Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jetten N., Verbruggen S., Gijbels M. J., Post M. J., De Winther M. P. J., Donners M. M. P. C., Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17, 109–118 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Jakobsson L., Franco C. A., Bentley K., Collins R. T., Ponsioen B., Aspalter I. M., Rosewell I., Busse M., Thurston G., Medvinsky A., Schulte-Merker S., Gerhardt H., Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943–953 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Supek F., Bošnjak M., Škunca N., Šmuc T., REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benjamin L. E., Hemo I., Keshet E., A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 125, 1591–1598 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Betsholtz C., Cell-cell signaling in blood vessel development and function. EMBO Mol. Med. 10, e8610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freiman A., Shandalov Y., Rozenfeld D., Shor E., Segal S., Ben-David D., Meretzki S., Egozi D., Levenberg S., Adipose-derived endothelial and mesenchymal stem cells enhance vascular network formation on three-dimensional constructs in vitro. Stem Cell Res. Ther. 7, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosshart H., Heinzelmann M., THP-1 cells as a model for human monocytes. Ann. Transl. Med. 4, 438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spiller K. L., Wrona E. A., Romero-Torres S., Pallotta I., Graney P. L., Witherel C. E., Panicker L. M., Feldman R. A., Urbanska A. M., Santambrogio L., Vunjak-Novakovic G., Freytes D. O., Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp. Cell Res. 347, 1–13 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Qin Z., The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 221, 2–11 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Blinder Y. J., Freiman A., Raindel N., Mooney D. J., Levenberg S., Vasculogenic dynamics in 3D engineered tissue constructs. Sci. Rep. 5, 17840 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fantin A., Vieira J. M., Gestri G., Denti L., Schwarz Q., Prykhozhij S., Peri F., Wilson S. W., Ruhrberg C., Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116, 829–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu C. W., Poché R. A., Saik J. E., Ali S., Wang S., Yosef N., Calderon G. A., Scott L. Jr., Vadakkan T. J., Larina I. V., West J. L., Dickinson M. E., Improved angiogenesis in response to localized delivery of macrophage-recruiting molecules. PLOS ONE 10, e0131643 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Awojoodu A. O., Ogle M. E., Sefcik L. S., Bowers D. T., Martin K., Brayman K. L., Lynch K. R., Peirce-Cottler S. M., Botchwey E., Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, 13785–13790 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korn C., Augustin H. G., Mechanisms of vessel pruning and regression. Dev. Cell 34, 5–17 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Godwin J. W., Pinto A. R., Rosenthal N. A., Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 9415–9420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Amerongen M. J., Harmsen M. C., van Rooijen N., Petersen A. H., van Luyn M. J., Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 170, 818–829 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesman A., Koffler J., Atlas R., Blinder Y. J., Kam Z., Levenberg S., Engineering vessel-like networks within multicellular fibrin-based constructs. Biomaterials 32, 7856–7869 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Shandalov Y., Egozi D., Koffler J., Dado-Rosenfeld D., Ben-Shimol D., Freiman A., Shor E., Kabala A., Levenberg S., An engineered muscle flap for reconstruction of large soft tissue defects. Proc. Natl. Acad. Sci. U.S.A. 111, 6010–6015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Shaul S., Landau S., Merdler U., Levenberg S., Mature vessel networks in engineered tissue promote graft-host anastomosis and prevent graft thrombosis. Proc. Natl. Acad. Sci. U.S.A. 116, 2955–2960 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koffler J., Kaufman-Francis K., Shandalov Y., Egozi D., Amiad Pavlov D., Landesberg A., Levenberg S., Improved vascular organization enhances functional integration of engineered skeletal muscle grafts. Proc. Natl. Acad. Sci. U.S.A. 108, 14789–14794 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S. H., Starkey P. M., Gordon S., Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J. Exp. Med. 161, 475–489 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez F. O., Gordon S., The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 6, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnett F. H., Rosenfeld M., Wood M., Kiosses W. B., Usui Y., Marchetti V., Aguilar E., Friedlander M., Macrophages form functional vascular mimicry channels in vivo. Sci. Rep. 6, 36659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zajac E., Schweighofer B., Kupriyanova T. A., Juncker-Jensen A., Minder P., Quigley J. P., Deryugina E. I., Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 122, 4054–4067 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sindrilaru A., Peters T., Wieschalka S., Baican C., Baican A., Peter H., Hainzl A., Schatz S., Qi Y., Schlecht A., Weiss J. M., Wlaschek M., Sunderkötter C., Scharffetter-Kochanek K., An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 121, 985–997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore E. M., Suresh V., Ying G., West J. L., M0 and M2 macrophages enhance vascularization of tissue engineering scaffolds. Reg. Eng. Transl. Med. 4, 51–61 (2018). [Google Scholar]
- 36.Sainson R. C. A., Johnston D. A., Chu H. C., Holderfield M. T., Nakatsu M. N., Crampton S. P., Davis J., Conn E., Hughes C. C. W., TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 111, 4997–5007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brecht K., Weigert A., Hu J., Popp R., Fisslthaler B., Korff T., Fleming I., Geisslinger G., Brüne B., Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 25, 2408–2417 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Anghelina M., Krishnan P., Moldovan L., Moldovan N. I., Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: Conversion of cell columns into fibrovascular bundles. Am. J. Pathol. 168, 529–541 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juhas M., Abutaleb N., Wang J. T., Ye J., Shaikh Z., Sriworarat C., Qian Y., Bursac N., Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat. Biomed. Eng. 2, 942–954 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen A. R., Extracting meaning from biological imaging data. Mol. Biol. Cell 25, 3470–3473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wait E., Winter M., Bjornsson C., Kokovay E., Wang Y., Goderie S., Temple S., Cohen A. R., Visualization and correction of automated segmentation, tracking and lineaging from 5-D stem cell image sequences. BMC bioinformatics 15, 328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaay6391/DC1