

A novel molecular target that signals abnormal involuntary movement in Parkinson’s disease treatment is discovered.
Abstract
The therapeutic effects of l-3,4-dihydroxyphenylalanine (l-DOPA) in patients with Parkinson’s disease (PD) severely diminishes with the onset of abnormal involuntary movement, l-DOPA–induced dyskinesia (LID). However, the molecular mechanisms that promote LID remain unclear. Here, we demonstrated that RasGRP1 [(guanine nucleotide exchange factor (GEF)] controls the development of LID. l-DOPA treatment rapidly up-regulated RasGRP1 in the striatum of mouse and macaque model of PD. The lack of RasGRP1 in mice (RasGRP1−/−) dramatically diminished LID without interfering with the therapeutic effects of l-DOPA. Besides acting as a GEF for Ras homolog enriched in the brain (Rheb), the activator of the mammalian target of rapamycin kinase (mTOR), RasGRP1 promotes l-DOPA–induced extracellular signal-regulated kinase (ERK) and the mTOR signaling in the striatum. High-resolution tandem mass spectrometry analysis revealed multiple RasGRP1 downstream targets linked to LID vulnerability. Collectively, the study demonstrated that RasGRP1 is a critical striatal regulator of LID.
INTRODUCTION
The loss of substantia nigral projections neurons, which results in decreased dopamine levels in the dorsal striatum, is the primary cause of Parkinson’s disease (PD). As a precursor for dopamine, l-DOPA (l-3,4-dihydroxyphenylalanine or levodopa) effectively alleviates motor symptoms in PD; however, its therapeutic benefits are markedly limited by its debilitating dyskinetic side effects, the l-DOPA–induced dyskinesia (LID). Previous studies have shown that LID is mediated by the abnormal activation of dopamine 1 (D1)–dependent cyclic adenosine 3′,5′-monophosphate (cAMP)/protein kinase A (PKA), extracellular signal–regulated kinase (ERK), and mammalian target of rapamycin kinase complex 1 (mTORC1) signaling in the dorsal striatum (1, 2). The inhibitors of these signaling pathways may prevent LID without affecting the beneficial motor effects of l-DOPA (2). Ras–guanine nucleotide–releasing factor 1 (RasGRF1), which is a guanine nucleotide exchange factor (GEF) for Ras guanosine triphosphatase (GTPase), is abundant in the cortex, hippocampus, and striatum and is known to activate ERK in the striatum and regulate LID (3). However, the striatal regulators that modulate both ERK and mTORC1 signaling in LID remain unknown. Previously, we found that Rhes, a striatal-enriched GTPase/SUMO-E3–like protein, binds and activates mTORC1 signaling and promotes LID (4), in agreement with a recent report (5). RasGRP1, a GEF for H-Ras that signals ERK, is highly expressed in hematopoietic cells, is regulated by calcium and diacylglycerol, is known to play a role in T and B cell proliferation, and has been implicated in leukemia and lupus (6–8). Besides blood cells, RasGRP1 is enriched in specific brain regions that control motor and cognitive functions, such as the striatum, but its role in neuronal functions remains less clear (9, 10). Earlier, we found that RasGRP1 can also act as a GEF for Rhes and promote amphetamine-induced hyperactivity via the striatal protein-protein complex known as “Rhesactome” (10). Here, we report a causal role for RasGRP1 in LID. We demonstrate that l-DOPA–increased RasGRP1 level is causally linked to the development of abnormal involuntary movements (AIMs) associated with robust activation of ERK and mTOR pathways in the striatum. Using high-end quantitative proteomic analysis of parkinsonian wild-type (WT) and RasGRP1−/− mice treated with l-DOPA, we have identified multiple striatal targets downstream to RasGRP1 activation that may play critical roles in LID.
RESULTS
RasGRP1 role during LID in a mouse model of PD
We hypothesized that RasGRP1 may be an upstream regulator of LID due to the following reasons: (i) l-DOPA treatment of mice with unilateral 6-hydroxydopamine (6-OHDA) lesions of the nigrostriatal pathway augmented striatal ERK and mTOR signaling (1, 2, 11); (ii) Rhes, a striatal-enriched protein that activates mTOR, is involved in LID (4); (iii) RasGRP1 regulated the synaptic localization of Rhes; and (iv) RasGRP1 and Rhes coexpression strongly activated both ERK and mTORC1 signaling in a striatal cell culture (10). To test our hypothesis, we subjected WT and RasGRP1−/− [RasGRP1 knockout (KO)] mice to the well-established 6-OHDA lesion model of LID, as described in our earlier work (4). Figure 1A shows the timeline of the 6-OHDA lesion and LID analysis. We observed 6-OHDA–induced PD-like symptoms in the drag test, rotarod, and turning test, which were similar between WT and RasGRP1 KO mice (Fig. 1B). The open-field test did not show obvious differences (Fig. 1B). Daily treatment of lesioned mice with l-DOPA (5 mg/kg) (12) induced significantly less AIMs in RasGRP1 KO mice compared to WT controls at every time point (Fig. 1C). All components of dyskinesia appeared to be equally dampened in RasGRP1 KO mice (Fig. 1D). The time course of dyskinesia was similar between WT and RasGRP1 KO mice, suggesting that RasGRP1 did not alter l-DOPA turnover (Fig. 1E). These observations suggest that RasGRP1 is a critical regulator of LID.
Fig. 1. RasGRP1 deletion diminishes LID.
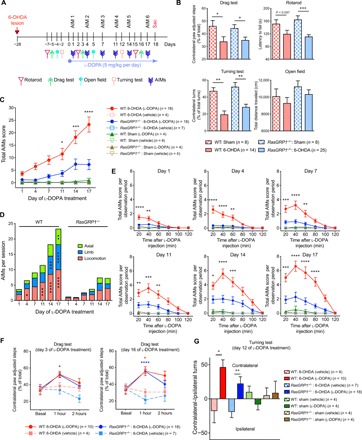
(A) LID scheme. (B) Drag test, rotarod, turning test, and open-field test for the indicated genotypes for sham or 6-OHDA–lesioned mice. Total AIM scores (C) or AIMs per session (D) (axial, limb, or locomotion) for the indicated sham or 6-OHDA–lesioned WT and RasGRP1 KO (RasGRP1−/−) mice, vehicle, or l-DOPA injected. (E) Total AIMs score per observed period (days 1 to 17) after injection of l-DOPA. (F) Drag test on days 3 and 16 after l-DOPA treatment and (G) turning test on day 12 after l-DOPA injection. Error bars represent means ± SEM (n = 4 to 25). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by one-way ANOVA followed by Bonferroni post hoc test and repeated measures two-way ANOVA followed by Bonferroni post hoc test.
RasGRP1 deletion and anti-Parkinson’s effects of l-DOPA
Next, we investigated whether RasGRP1 deletion had any influence on the anti-Parkinson’s effect of l-DOPA. We found that administration of l-DOPA decreased Parkinson’s-like symptoms as measured by the drag test (on days 3 and 16; Fig. 1F) and the turning test (day 12; Fig. 1G) in both WT and RasGRP1 KO–lesioned mice. As expected, sham injections produced no defects in the drag test (fig. S1). The open-field or rotarod test were also used as functional in vivo readouts, but we did not see any difference in total distance traveled or latency to fall between the genotypes and sham treatments (figs. S2 and S3). Thus, RasGRP1 promoted the adverse effects of l-DOPA but did not interfere with its therapeutic motor effects. Moreover, RasGRP1 KO mice displayed no significant changes in basal motor behavior or coordination (figs. S2 and S3) or amphetamine-induced motor activity compared to control mice (10).
Effect of RasGRP1 on striatal signaling during LID
Previous works had showed that dopamine 1 (D1) and dopamine 2 (D2) receptors of medium spiny neurons (MSNs) in the striatum may play different roles in LID. For example, Santini et al. (2) showed that l-DOPA induces ERK and mTOR signaling in D1 MSNs. Consistent with these data, the stimulation of mTOR signaling by l-DOPA has been shown to be abolished by D1 antagonists and unaffected by D2 antagonists (2). Similarly, we found that Rhes, which is predominantly expressed both in D1 and D2 MSNs, promoted mTOR signaling in LID (4). As RasGRP1 KO mice showed diminished dyskinesia, we investigated ERK and mTOR, along with other signaling molecules in the striatal tissue of WT and RasGRP1 KO mice. First, we confirmed that the 6-OHDA lesion procedure produced similar degree of denervation in both WT and RasGRP1 KO mice [assessed by significant loss of tyrosine hydroxylase (TH) protein levels; Fig. 2, A and B]. Intriguingly, we found that RasGRP1 levels were up-regulated after l-DOPA injection (Fig. 2, A and B). This increase was dependent on l-DOPA administration, as we found no striatal increase of RasGRP1 in 6-OHDA lesion vehicle control (fig. S4). We observed an up-regulation of mTORC1 activity as measured by the levels of phosphorylation of ribosomal protein S6 kinase (S6K) at T389, phosphorylation of S6 at S235/236, and phosphorylation of eukaryotic translation initiation factor 4E (eIF4E)–binding protein 1 (4EBP1; T37/46) at a site that primes p4EBP1 for subsequent activity phosphorylation at S65 in l-DOPA–treated 6-OHDA–lesioned WT mice but not in the striatum of RasGRP1 KO mice (Fig. 2, A and B). We were unable to detect p4EBP1 S65, as the antibodies for this site did not work for brain lysate. We also found that mTORC2 activity, as measured by phosphorylation of Akt (S473), was up-regulated in 6-OHDA–lesioned WT mice but not in the striatum of RasGRP1 KO mice treated with l-DOPA (Fig. 2, A and B). Previous findings have shown that phosphorylated Akt (S473) was up-regulated in the putamen of a monkey model of LID (13). In addition, D1/cAMP/PKA-dependent pGlur1 S845 levels along with pERK (T202/Y204), and the phosphatidylinositol 3-kinase (PI3K) target [pAkt (T308)], were also highly up-regulated in the striatum of WT but not RasGRP1 KO mice after LID (Fig. 2, A and B). On the other hand, we found that Rheb and Rhes levels were significantly down-regulated in the lesion side of RasGRP1 KO mice (Fig. 2, A and B), consistent with our earlier report (10), suggesting that RasGRP1 may physiologically stabilize these proteins in the striatum. RasGRP1 up-regulation was also observed in the striatum of rat under LID (14, 15), but whether it is a cause or consequence of dyskinesia was unknown. Together, these biochemical studies indicated that (i) striatal RasGRP1 is up-regulated in a l-DOPA–dependent manner and is causally linked to the development of LID; (ii) RasGRP1 deletion prevents the up-regulation of l-DOPA–induced cAMP/PKA, mTOR, ERK, and Akt signaling in the striatum; and (iii) Rhes and Rheb, activators of mTOR, are down-regulated in the striatum of RasGRP1 KO mice compared to WT mice during LID.
Fig. 2. RasGRP1 mediates l-DOPA–induced mTOR, ERK, and GluR1 signaling in the striatum.
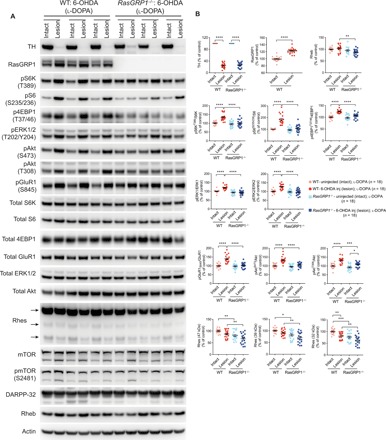
(A) Western blot analysis of intact and 6-OHDA–lesioned striatum of WT and RasGRP1 KO mice after l-DOPA treatment. (B) Quantification of the indicated proteins in WT or RasGRP1 KO intact side or lesioned side of the striatum. Protein levels were normalized to actin. Phosphorylated proteins were normalized against the total protein levels. Error bars represent means ± SEM (n = 18). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test.
Up-regulation of RasGRP1 in D1 MSNs by l-DOPA in 6-OHDA–lesioned striatum
We next wondered whether RasGRP1 up-regulation occurs within D1 MSNs. We made serial brain sections from the WT mice that were 6-OHDA lesioned and treated with l-DOPA. The parallel brain sections were immunostained for RasGRP1/D1R/4′,6-diamidino-2-phenylindole (DAPI) or TH using immunohistochemistry (IHC) protocol. As shown in the Fig. 3A, we found RasGRP1 up-regulation in the lesioned side of the dorsal striatum, coincided with TH loss (Fig. 3B). In 6-OHDA–lesioned mice, RasGRP1 up-regulation is predominantly seen in the dorsolateral region of the striatum (Asterix; Fig. 3A). Unexpectedly, we also observed enhanced signal for RasGRP1 in the ipsilateral cortex when compared to nonlesioned contralateral hemisphere, indicating that striatal 6-OHDA lesion can also promote RasGRP1 expression in the cortex (white arrow; Fig. 3A). Magnified and orthogonal confocal images show RasGRP1 basal expression (Fig. 3C) and its up-regulation in the dorsal striatum within the D1R+ MSNs of lesioned mice (Fig. 3, D and E), consistent with Western blot analysis (Fig. 2, A and B). Earlier, we found biochemically that RasGRP1 is predominantly enriched in the cytoplasmic fractions compared to synaptic fractions in the striatum (10). Consistent with this data, the IHC data reveal that RasGRP1 is predominantly perinuclear and colocalizes partially with D1R staining (arrow; Fig. 3E). Thus, RasGRP1 is clearly up-regulated in the dorsal striatum of 6-OHDA–lesioned animals treated with l-DOPA and colocalized with D1R+ neurons located on the dorsolateral part of striatum.
Fig. 3. RasGRP1 is up-regulated in the dorsal side of striatum in LID.
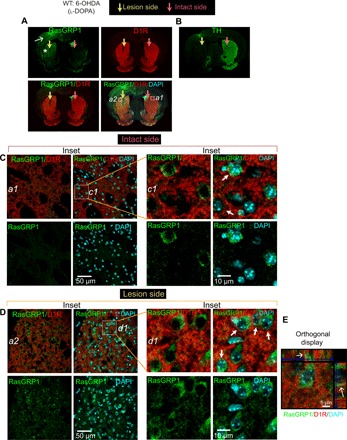
(A) Representative images of brain sections showing immunostaining for (A) RasGRP1 (green) and D1R (red), costained with DAPI (blue) in hemi-parkinsonian WT mice after 10 days of l-DOPA treatment. Image is representative of three independent experiments. Asterisk indicates dorsal striatum. White arrow indicates cortical area. Yellow arrow indicates the 6-OHDA–lesioned side, and pink arrow indicates intact side of the striatum. (B) Representative brain sections stained for TH (green) (C) Magnified inset from intact side (a1) and its magnification (c1). Arrow indicates partial colocalization (D) Magnified inset from lesion side (a2) and its magnification (d1). Arrow indicates partial colocalization. (E) Confocal orthogonal display of RasGRP1 and D1R in the striatum. Arrow indicates partial colocalization. (A) 10× objective; zoom, 0.6. (C to E) 63× objective.
RasGRP1 and mTOR signaling crosstalk
Next, we examined the potential mechanisms by which RasGRP1 up-regulation in LID promoted mTOR and ERK signaling. To this aim, first, we transiently overexpressed RasGRP1 in human embryonic kidney–293 (HEK293) cells, which were transfected with a control vector (His) or His-RasGRP1 complementary DNA (cDNA). Then, we incubated the transfected cells in a serum-free F12 medium supplemented with full amino acids (AA+) or F12 medium that lacked l-leucine (AA−), which is a potent inducer of mTORC1 (16). After 2 hours, cells in AA− medium were restimulated by addition of l-leucine (3 mM). In AA+ conditions, RasGRP1-expressing cells, compared to control cells, had two to three times as much mTORC1 activity as measured by the phosphorylated levels of S6K at T389 (pS6K-T389) and 4EBP1 at S65 (p4EBP1-S65; Fig. 4, A and B). In AA− medium, mTORC1 activity was 50% lower than AA+ medium in RasGRP1-expressing cells yet was higher than the control cells. Upon restimulation with l-leucine for 15 min, mTORC1 activity rapidly returned to levels comparable with those in AA+ conditions. This indicated that RasGRP1 promoted amino acid–mediated mTORC1 activity. RasGRP1 expression increased the constitutive phosphorylation of ERK, which is not sensitive to amino acids (Fig. 4C).
Fig. 4. RasGRP1 regulates mTOR activation, independent of ERK signaling.
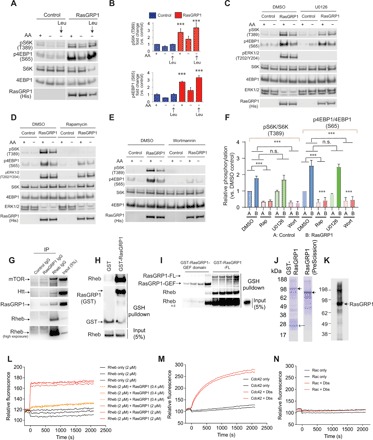
(A) RasGRP1 mediates amino acid–induced mTORC1 activity. HEK293 cells (grown in DMEM and serum) were transfected with His (control) or His-RasGRP1 constructs (0.5 μg each), and after 36 to 48 hours, the cells were exposed to serum-free media (F12+) containing all amino acids (AA+) or serum free media (F12–) that lacks l-leucine (AA–) for 2 hours, and wherever indicated, F12– media was stimulated with l-leucine (3 mM) for 15 min. Cell lysates were probed for pS6K (T389), p4EBP1 (S65), and other indicated proteins by Western blotting. (B) Displays quantification of (A). Error bars represent means ± SEM (n = 3 independent experiments). ***P ≤ 0.001 by unpaired Student’s t test. (C) RasGRP1-mediated mTORC1 activity is independent of ERK signaling. HEK293 cells were grown as in (A) and replaced with AA+ or AA– media with DMSO (0.01%) or U0126 (10 μM) for 2 hours. Cell lysates were prepared and probed using Western blotting for indicated proteins. (D) Rapamycin abrogates RasGRP1-mediated mTORC1 signaling. Cells were transfected as in (A) followed by changing the medium to AA+ or AA– as in (C) with DMSO or rapamycin (500 nM) and probed for indicated proteins by Western blotting. (E) Wortmannin abrogates RasGRP1-mediated mTORC1 activity. Cells were transfected as in (A), and the AA+ and AA– media was treated with DMSO or wortmannin (100 nM) for 2 hours, followed by detection of indicated protein through Western blotting. (F) Relative inhibitory potency of different inhibitors on the RasGRP1-mediated mTORC1 activity. Error bars represent means ± SEM (n = 3 independent experiments). ***P ≤ 0.001 by unpaired Student’s t test. n.s., not significant. (G) Western blot showing Rheb and RasGRP1 binding in the striatum, in vivo. Blot is representative of three independent experiments. (H) Western blot showing recombinant Rheb and RasGRP1 protein interaction in vitro. Blot is representative of three independent experiments. (I) Western blot showing GST-RasGRP1 GEF domain and GST-RasGRP1-FL interaction with Rheb in vitro. Blot is representative of three independent experiments. (J) The Coomassie gel with recombinant GST-RasGRP1 purified from E. coli and PreScission Protease–cleaved RasGRP1 (closed arrow). Open arrow indicates GST tag. (K) Western blot to detect cleaved RasGRP1. (L) Concentration-dependent GEF activity (fluorescent assay, loading of mant-GTP) of RasGRP1 toward Rheb. (M and N) GEF assay for positive control (Dbs + Cdc42) and negative control (Dbs + Rac), respectively. Data are representative of three independent experiments.
Next, we tested the effects of ERK inhibition on RasGRP1-mediated mTORC1 signaling. While U0126, a potent inhibitor of mitogen-activated protein kinase (MEK) abrogated ERK signaling, it had negligible effects on RasGRP1-induced pS6K (T389) and p4EBP1 (S65; Fig. 4C). However, rapamycin, a mTORC1 inhibitor, which did not alter RasGRP1-induced ERK signaling, markedly attenuated RasGRP1-induced amino acid–mTORC1 signaling (Fig. 4D). Like rapamycin, the PI3K inhibitor wortmannin abolished RasGRP1-induced mTORC1 signaling (Fig. 4E). Collectively, rapamycin and wortmannin, but not U0126, blocked RasGRP1-induced mTORC1 activation (Fig. 4F). The depletion of endogenous RasGRP1 using short hairpin RNA (shRNA) in HEK293 cells also diminished mTORC1 signaling (fig. S5). Together, these data suggested that RasGRP1 physiologically activates mTORC1 signaling by regulating catalytically important residues on pS6K and p4EBP1 via a PI3K-sensitive pathway. In addition, using a pharmacological approach, we found no significant cross-talk between ERK and mTORC1 signaling induced by RasGRP1.
Interaction of RasGRP1 with Rheb in striatum and in vitro
According to the above results, we predicted that RasGRP1 would activate ERK and mTORC1 signaling in two independent and parallel pathways. Previous studies showed that RasGRP1 can activate ERK via GTPase H-Ras (8, 9), but we wondered how RasGRP1 activates amino acid–induced mTORC1. As Rheb, which directly binds and activates mTOR, mediates amino acid–induced mTORC1 activity (17), we hypothesized that RasGRP1 may promote mTORC1 activity by interacting with Rheb in the brain. To test this hypothesis, we coimmunoprecipitated Rheb and RasGRP1 from the brain’s striatum using Rheb antibody. As predicted, we observed that Rheb effectively coimmunoprecipitated with RasGRP1 and mTOR, a known Rheb interactor (Fig 4G). Rheb also coprecipitated with huntingtin (Htt), which was consistent with our previous report (18). However, the existing RasGRP1 antibody appeared inefficient for immunoprecipitation, as it only moderately enriched RasGRP1 (Fig. 4G). Next, we investigated whether RasGRP1 interacts directly with Rheb in vitro. Coincubation of bacterially purified glutathione S-transferase (GST)–FL-RasGRP1 and Rheb proteins revealed their robust interaction (Fig. 4H). As shown in Fig. 4I, the interaction appeared strong with FL-RasGRP1 compared to the RasGRP1 GEF domain (1 to 450 amino acids) (19). Next, we tested whether RasGRP1 can act as a GEF for Rheb by using a widely used in vitro fluorescent-based mant–guanosine 5′-triphosphate (GTP) assay (20). Figure 4 (J and K) shows the purified GST-RasGRP1 in Escherichia coli and was cleaved with PreScission Protease to produce untagged FL-RasGRP1 that is confirmed by Western blot. In mant-GTP loading assay, we found that RasGRP1 showed a robust and concentration-dependent GEF activity toward Rheb (Fig. 4L). At both (0.4 and 2 μM) concentration of RasGRP1, the steady-state exchange activity was reached quite rapidly, and there was a rapid exchange at the 2 μM compared to 0.4 μM RasGRP1 concentration (Fig. 4L). Therefore, we conclude that the RasGRP1 binds immediately and strongly to the Rheb and does not dissociate. Note that the positive control, Dbs [diffuse B-cell lymphoma’s (Dbl’s) big sister], which acts as a GEF toward cdc42, showed steady increase in exchange activity (Fig. 4M), whereas Dbs that is not a GEF for rac (21) showed no activity (Fig. 4N), as expected. Thus, we showed that RasGRP1 could directly interact with Rheb in the striatum and could act as its GEF in vitro. Collectively, these data suggested that RasGRP1 may promote mTORC1 activity in the brain via Rheb GTPase.
Quantitative striatal proteomic analysis of WT and RasGRP1−/− mice
To understand the mechanisms by which RasGRP1 might elicit LID, we undertook quantitative and comparative proteomics profiling of WT and RasGRP1 mice KO mice striatum with high-resolution mass spectrometry (MS) coupled to liquid chromatography–tandem MS (LC-MS/MS) based on tandem mass tags (TMTs) designed for phosphoprotein enrichment. We isolated the striatum (intact and 6-OHDA lesion side) 20 min after l-DOPA administration from three WT mice that showed severe dyskinesia and three RasGRP1 KO mice that showed no dyskinesia in response to l-DOPA in a PD model (Fig. 5A). We labeled each striatal lysate with TMT labeled six-plex reagents (Thermo Fisher Scientific) as indicated in Table 1.
Fig. 5. Quantitative proteomics of the striatum of WT and RasGRP1 KO dyskinesia animals.
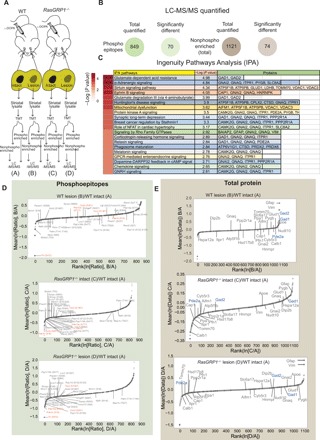
(A) Scheme of isolation of striatal tissue from the 6-OHDA–lesioned WT and RasGRP1 KO (RasGRP1−/−) after l-DOPA treatment, followed by LC-MS/MS. (B) Total number of quantifiable proteins that are enriched for phosphorylated epitopes and nonphosphorylated total protein. (C) Ingenuity Pathway Analysis (IPA) analysis for significantly altered nonphosphorylated proteins. Relative quantitation of phosphopeptide (D) and non-phosphopeptide [total protein; (E)] abundance between WT lesion/ WT intact, RasGRP1 KO intact/ WT intact and l RasGRP1 KO lesion/WT intact groups. Significant targets and nonsignificant targets were indicated in dark and light gray circles, respectively (n = 3 mice per group).
Table 1. Sample groups and TMT labels.
|
WT-intact (A) |
WT-lesion (B) |
RasGRP1 KO intact (C) |
RasGRP1 KO lesion (D) |
|
| Plex 1 label | 126 (Kit 1) | 127 (Kit 1) | 128 (Kit 1) | 129 (Kit 1) |
| Plex 2 label | 128 (Kit 2) | 129 (Kit 2) | 130 (Kit 2) | 131 (Kit 2) |
| Plex 3 label | 126 (Kit 2) | 127(Kit 2) | 130 (Kit 1) | 131 (Kit 1) |
Loading bias was minimal and was removed by normalizing it with the total peptide amount. We quantified 849 phosphorylated epitopes in all groups. Analysis of variance (ANOVA) results indicated 70 phosphopeptides were significantly regulated between the comparison groups [B/A, C/A, and D/A; false discovery rate (FDR) = 0.10], and then Tukey’s honest significant difference (HSD) was used as post hoc test to find out which pairs were different from each other (α = 0.05; Fig. 5B and data files S1 and S2). Similarly, ANOVA results indicated that the levels of 74 proteins of 1121 identified nonphospho-enriched proteins (total) were significantly regulated between the comparison groups (B/A, C/A, and D/A; FDR = 0.10), and then Tukey’s HSD was used as post hoc test to find out which pairs were different from each other (α = 0.05; Fig. 5B and data files S3 to S5). Ingenuity Pathway Analysis revealed that signaling pathways related to glutamate-dependent acid resistance, α-adrenergic signaling, sirtuin signaling, ephrin signaling, glutamate degradation, Huntington disease pathways, mitochondrial dysfunction targets, protein kinase A signaling, and others were highly significantly regulated in WT but not in RasGRP1 KO mice striatum (Fig. 5C and data file S6).
We looked at some of the examples of phosphorylated epitopes (Fig. 5D and data files S1 and S2). Overall, the significantly (dark circles) affected phosphopeptides in the WT 6-OHDA–lesioned side show a diminished trend in RasGRP1 KO (Fig. 5D), indicating that RasGRP1 may regulate the phosphorylation status of these proteins in the striatum. For example, as expected, the phosphorylation status of TH at S472 is similar in the intact side but is down-regulated in the lesioned side of both RasGRP1 KO and WT (Fig. 5D, indicated in red). However, the phosphorylation at S261 of catechol-O-methyl transferase (Comt), an enzyme that catalyzes the degradation of catecholamines (including the neurotransmitters dopamine, epinephrine, and norepinephrine) is diminished more in RasGRP1 KO compared to WT mice (Fig. 5D) (22). Inhibitors of Comt can diminish dyskinesia in normal monkeys, and overexpression of Comt in mice can potentiate LID (23, 24). Similarly, phosphodiesterase (Pde), such as Pde10a, which is implicated in LID (25, 26), showed a diminished phosphorylation at S777 in RasGRP1 KO (Fig. 5D). Pde1b showed phosphorylation at three difference sites—such as S508, S465, and S18—but show significantly diminished phosphorylation only at S465 and S18 (Fig. 5D) in RasGRP1 KO striatum compared to WT (27). Notably, Tsc2, a GTPase activating protein for Rheb (28), showed a consistent reduction in the phosphorylation status at S1397 and S1367 in RasGRP1 KO striatum compared to WT (Fig. 5D). These data indicate that RasGRP1 physiologically regulates the phosphorylation of multiple targets in the striatum. However, the role or implications of these various phosphorylated targets in LID remains to be elucidated. A full list of phosphorylated targets can be found in data files S1 and S2.
Similarly, among total proteins, for example, Pde2a was higher in WT than RasGRP1 KO mice striatum (Fig. 5E, indicated in blue). Gfap up-regulation is found in 6-OHDA rodent model of PD (29). Similarly, we found a marked up-regulation of Gfap in the lesion side of both WT and RasGRP1 KO (Fig. 5E), although it was slightly lower in the RasGRP1 KO striatum (Fig. 5E). Gad1 and Gad2, also known as Gad67 and Gad65, respectively, which catalyzes the production of γ-aminobutyric acid, are up-regulated in WT but not in RasGRP1 KO striatum (Fig. 5E). A previous work showed that the loss of Gad67 has diminished LID in mouse models of PD (30), indicating that Gad67 might be a target of RasGRP1 in the striatum. Next, we validated some of the altered target proteins in RasGRP1 KO striatum. We used Western blotting with validated antibodies to confirm the up-regulation of Gad1, Gad2, Gfap, and Pde2a observed in quantitative LC-MS/MS in the lesioned area of WT mice but not in RasGRP1 KO striatum (Fig. 6, A to C). However, not all proteins were down-regulated in RasGRP1 KO mice in the proteomic analysis. For example, complexin-1, visinin-like protein, glutamate dehydrogenase 1, guanine nucleotide–binding protein G(q) subunit α, and lactate dehydrogenase were up-regulated in RasGRP1 KO mice but not in WT mice (data file S5). Collectively, LC-MS/MS data indicated that RasGRP1 acts upstream in response to l-DOPA and regulates a specific but diverse set of proteins to promote LID. This notion is strengthened by the fact that some of these proteins have been implicated in LID in independent studies (23, 25, 26).
Fig. 6. Validation of RasGRP1 and its targets and their up-regulation in monkey model of PD.
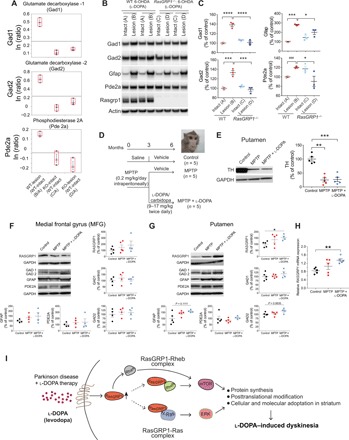
(A) Quantification (ratio) of selected examples from LC-MS/MS of proteins in indicated groups in comparison to intact WT striatum. (B and C) Western blotting and quantification of indicated proteins from the intact and lesioned striatum of WT and RasGRP1 KO mice after l-DOPA treatment. Error bars represent means ± SEM (n = 3). *P < 0.05, ***P < 0.001, and ****P < 0.0001 using one-way ANOVA followed by Tukey’s multiple comparison test. ###P < 0.001 by unpaired Student’s t test. Protein expression levels of RASGRP1 and its selected targets in the brain of parkinsonian and dyskinetic monkeys. (D) Experimental design. (E to G) Representative blot and Western blot analysis of striatal TH (E), RASGRP1, GAD1/2, GFAP, and PDE2A protein levels in the MFG (F) and putamen (G) of untreated (control), MPTP−, and MPTP + l-DOPA–treated monkeys. Error bars represent means ± SEM. [TH: control, MPTP, and MPTP + l-DOPA (n = 5); RASGRP1: control (n = 5), MPTP (n = 5), and MPTP + l-DOPA (n = 4); GAD1/2, GFAP, and PDE2A: MFG: control, MPTP, and MPTP + l-DOPA (n = 4); and putamen: control, MPTP (n = 5), and MPTP + l-DOPA (n = 4)]. Representative blots of each marker immunodensity comparing the experimental groups are shown. GAPDH was used to normalize for variations in loading and transfer. Dots represent the single values. *P < 0.05 by Mann-Whitney test compared to control group. (H) RASGRP1 transcript in MPTP-treated monkeys with or without l-DOPA administration. Error bars illustrate the means ± SEM [control, MPTP, and MPTP + l-DOPA (n = 5)]. **P < 0.01 by Mann-Whitney test compared to control group. (I) Mechanisms of RasGRP1-induced dyskinesia. Model depicts RasGRP1 is up-regulated during LID activates Rhes and forms complexes with H-Ras to signal ERK and with Rheb to signal mTOR. These dual complexes, in parallel, activates ERK and mTOR signaling, exerting a profound cellular and molecular changes in the striatum via protein synthesis and/or posttranslational modifications, which will influence the onset and progression of LID. Drugs or gene depletion strategies that block RasGRP1 may improve the therapeutic efficacy of l-DOPA by diminishing LID, the debilitating side effects observed in patients with PD. Photo credit: Erwan Bezard, Université de Bordeaux.
Up-regulation of RASGRP1 mRNA and protein levels in putamen of dyskinetic MPTP-lesioned nonhuman primate model of PD
The data described above established that RasGRP1 up-regulation is causally linked to the generation of LID in a mouse model of PD. Here, we investigated whether RASGRP1 and its downstream targets’ [glutamic acid decarboxylase 1/2 (GAD1/2), glial fibrillary acidic protein (GFAP), and PDE2A] protein levels are altered in the medial frontal gyrus (MFG) and putamen of a dyskinetic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–lesioned nonhuman primate model of PD (Fig. 6D). As shown in Fig. 6E, we found a marked loss of TH protein levels in the putamen of monkeys treated with MPTP and MPTP + l-DOPA, indicative of severe nigrostriatal degeneration in these animals. Western blotting experiments revealed no significant alterations of RASGRP1 levels within MFG in both parkinsonian and dyskinetic macaques compared to control group (Fig. 6F). Similarly, expression levels of GAD1/2, GFAP, and PDE2A were comparable in the MFG among experimental groups (Fig. 6F). Likewise, Western blot data showed that MPTP treatment alone did not significantly perturb the expression of RASGRP1 in the putamen of nonhuman primates (Fig. 6G). In contrast, we observed a significant increase in striatal RASGRP1 protein and mRNA content selectively in monkeys treated with MPTP + l-DOPA compared to control group (Fig. 6, G and H). Moreover, although statistical analysis failed to find significant difference, we found a clear trend of increase in both GAD2 and GFAP protein expression in the putamen of dyskinetic macaques (Mann-Whitney test, GAD2: control versus MPTP + l-DOPA, P = 0.0635; GFAP: control versus MPTP + l-DOPA, P = 0.1111; Fig. 6G) but not in parkinsonian monkeys (Fig. 6G). Yet, we found comparable levels of both GAD1 and PDE2A proteins in the putamen of MPTP-treated macaques with or without chronic l-DOPA treatment compared to the control group (Fig. 6G). Thus, consistent with mice observations (reported above) and previous studies in rats treated with l-DOPA (14, 15), we showed a significant RASGRP1 up-regulation under LID conditions also in nonhuman primate model of PD. Since monkey model for PD can mimic more signs and symptoms of human PD, our finding strengthens the translational relevance of RasGRP1 in PD treatment.
DISCUSSION
After about a decade of l-DOPA treatment, more than 95% of patients with PD develop dyskinesia. Only the N-methyl-d-aspartate receptor weak antagonist amantadine provides some clinical benefit but with limitations and nonmotor side effects (31); therefore, there is an immediate need to identify novel therapeutic targets for LID. In the present study, we demonstrate that RasGRP1 is a potential therapeutic target for LID, as our data indicated that (i) RasGRP1 is induced upon l-DOPA administration in parkinsonian mice and also nonhuman primates, (ii) RasGRP1 is causally linked to LID in mice, and (iii) RasGRP1 mediates l-DOPA–induced activation of ERK and mTOR pathways, known modulators of LID in striatum. RasGRP1 may represent a better target than mTOR and ERK, although mTOR inhibitors, such as rapamycin, or ERK inhibitors, such as U0126, have been shown to prevent LID in mouse models (2, 5, 32), these drugs are strong inhibitors of protein synthesis and have associated toxicity. In addition, they broadly inhibit targets in unwanted regions. Increasing number of data, including ours, indicates that RasGRP1 is up-regulated in the striatum of rat (14, 15), mouse, and monkey models of LID (Figs. 2, 3, and 6). However, the mechanism underpinning increased RasGRP1 expression under LID conditions remains unclear. Although dysfunctional D1R activation is well known to contribute to the generation of LID (33), its specific role in RasGRP1 modulation is unknown. We predict that RasGRP1 will be up-regulated in the dorsal striatum within D1R+ MSN in response to dopaminergic drugs under receptor super sensitivity, consistent with present IHC data (Fig. 3). The exact type of receptors and selective signal transduction pathway that may participate in inducing striatal RasGRP1 levels during LID is critical to further target the molecule for therapeutic purpose.
What are the mechanisms by which the up-regulated RasGRP1 induces ERK and mTOR signaling in the striatum during LID? Cell culture data demonstrated that there was no cross-talk between RasGRP1-induced ERK and mTORC1 signaling and suggest a role for PI3K in these interactions. We propose a model to show how RasGRP1 might activate both ERK and mTOR pathways during LID (Fig. 6I). We predicted that RasGRP1 would form complexes with more than one small GTPase in the dorsal striatum during LID. For example, up-regulated RasGRP1 can form two kinds of GEF complex, a RasGRP1-Ras complex and a RasGRP1-Rheb complex, to activate ERK and mTOR signaling, respectively, which in turn alter global protein translation and posttranslational modifications in the striatum (Fig. 6I). This notion is consistent with the actions of other GEFs that activate multiple small GTPases (34, 35). We predict that up-regulated RasGRP1 would act postsynaptically within MSNs like a “master GEF” for H-Ras, Rhes, and Rheb, and consistent with this notion, we found that RasGRP1 acts as GEF for Rheb. These multiple “GEF-GTPasome” complexes may afford the flexibility such as protein modifications needed to develop the AIMs observed in LID. Consistent with this, proteomics study revealed that RasGRP1 affect phosphorylation status and total striatal protein compositions that may elicit strong cellular, molecular, and anatomical alterations via regulating protein synthesis and/or posttranslational modifications. These biological effects may promote sustained alterations in striatal signaling that could trigger debilitating LID.
In addition to diminished ERK and mTOR signaling in RasGRP1 KO mice, we also found that pGluR1 (S845), a target of PKA, was significantly diminished. PKA pathway is reported to be critically involved in the development of LID (36). Thus, RasGRP1 can also affect D1/cAMP/PKA-dependent changes in pGluR1 (S845) levels under LID conditions presumably via Ras-cAMP signaling described in yeast (37). Although our work indicates a main influence of RasGRP1 in modulating striatal postsynaptic events occurring in MSNs under PD conditions, we cannot rule out that this protein could also affect indirectly via presynaptic regulation of dopamine homeostasis. Future studies are warranted to investigate this critical issue.
Note that RasGRP1 KO mice are fertile and have no significant changes in the basal motor activity (figs. S1 to S3) (10). RasGRP1 KO mice does show mild defects (20%) in thymocyte development (38). Notably, RASGRP1 up-regulation in the putamen of dyskinetic MPTP-lesioned primate model of PD further strengthens its therapeutic relevance in humans. Collectively, our study demonstrates that RasGRP1 has a causal role in LID in animal model of PD. Thus, drugs blocking RasGRP1, shRNA, or CRISPR-Cas9 approaches to reduce RasGRP1 in adults may produce beneficial effects against LID without inducing serious adverse effects.
MATERIALS AND METHODS
Reagents, plasmids, and antibodies
Chemicals and reagents were mainly purchased from Sigma-Aldrich. His-tagged rat CalDAG-GEFII was a gift from A. Graybiel [Massachusetts Institute of Technology (MIT)]. The myc-tagged RasGRP1 (pCMV-myc) and GST-tagged (pGEX-6P2) constructs were produced as described (10). Rheb GTPase was from Prospec (95% purity). The scrambled shRNA lentiviral control vector was from Addgene, and RasGRP1 shRNA was from Sigma-Aldrich (TRCN0000048268-72). Antibodies for RasGRP1 (1:1000; no. sc-8430), β-actin (1:20,000; no. sc-47778), GFAP (1:1000; no. sc-33673), glyceraldehyde-3-phosphate dehydrogenase (Gapdh) (1:1000; no. sc-32233), GST–horseradish peroxidase (HRP) (1:5000; no. sc-138), and Myc (1:3000; no. sc-40) were obtained from Santa Cruz Biotechnology. Antibodies against mTOR (1:4000; no. 2983), pmTOR S2481 (1:2000; no. 2974), pS6K T389 (1:1000; no. 9234), pS6 S235/236 (1:3000; no. 4858), p4EBP1 S65 (1:1000; no. 9451), p4EBP1 T37/46 (1:2000; no. 2855), pAkt S473 (1:5000; no. 4060), pAkt T308 (1:3000; no. 13038), p44/42 ERK1/2 (1:10,000; no. 9101), S6K (1:3000; no. 9202), S6 (1:15,000; no. 2217), 4EBP1 (1:15,000; no. 9644), Akt (1:15,000; no. 4691), ERK1/2 (1:30,000; no. 4695), Rheb (1:10,000; no. 13879), pGluR1 S845 (1:1000; no. 8084), total GluR1 (1:2000; no. 13185), and DARPP-32 (1:30,000; no. 2306) were from Cell Signaling Technology Inc. Htt (1:3000; no. MAB2166), TH (1:20,000; no. MAB318), and Gad65/67 (1:1000; no. ABN904) antibodies were from MilliporeSigma. Antibodies for GAD1 (1:1000; no. 10408), GAD2 (1:1000; no. 21760), PDE1C (1:1000; no. 13785), and PDE2A (1:1000; no. 55306) were from Proteintech. Rhes antibody (1:1000; no. RHES-101AP) was from FabGennix. RasGRP1 antibody for monkey tissue was from Millipore (1:1000; no. MABS146). HRP-conjugated secondary antibodies [1:10,000; no. 115-035-146 (goat anti-mouse) or 1:10,000; no. 111-035-144 (goat anti-rabbit)] were from Jackson ImmunoResearch Inc. Glutathione beads were from Amersham Biosciences, and protein G/protein A agarose beads were obtained from Santa Cruz Biotechnology. Rapamycin, U0126, and wortmannin were from Selleck Chemicals.
Animals
B6.129P3-Rasgrp1tm1Jstn/TbwnJ mice (stock no. 022353) and C57BL/6J mice (stock no. 000664) were obtained from the Jackson laboratory and maintained in our Scripps animal facility according to Institutional Animal Care and Use Committee (IACUC) instructions.
6-OHDA lesioning
Surgical procedures for unilateral 6-OHDA lesioning were performed as previously described (4). Briefly, 3- to 4-month-old male mice were used for the study, because male mice provided more robust and reliable results compared with female. Mice were deeply anesthetized by administration of isoflurane. The anesthetized animal was then mounted on a stereotaxic frame (David Kopf Instruments, Tujanga, CA) equipped with a mouse adaptor. 6-OHDA–HCl (Sigma-Aldrich, St Louis, MO) was dissolved in 0.02% ascorbic acid in saline at a final concentration of 2 μg of free-base 6-OHDA per μl. Each mouse received four injections of 6-OHDA (1 μl per injection) into the right striatum, according to the following coordinates (millimeter): anteroposterior (AP), +1.1; mediolateral (ML), −2; dorsoventral (DV), −3 and −4 and AP, +0.1; ML, −2.3; DV, −3.3 and −4.3. Animals were allowed to recover for 3 weeks before behavioral evaluations, and l-DOPA treatments were carried out. The efficacy of the lesion was assessed by the loss of TH in the lesioned mouse striatum (i.e., right) comparing to nonlesioned side (left). Only animals displaying ≥60% decrease in striatal TH immunoreactivity were included in statistical analyses of the turning test, drag test, open field, rotarod, and AIMs.
AIMs rating
AIMs were scored using the rating system described before (4). Briefly, 6-OHDA–lesioned (or sham) mice were treated for 17 consecutive days with one injection per day of l-DOPA (5 mg/kg) and benserazide (14 mg/kg) (4, 12). AIMs were assessed at days 1, 4, 7, 11, 14, and 17 by two observers who were fully blinded to the mice genotypes. On the day of each experiment, mice were habituated in single cages for 30 min and then received l-DOPA or vehicle injection. Twenty minutes after l-DOPA administration, three dyskinetic behaviors were assessed for 1-min monitoring period, which was repeated every 20 min for 120 min. Involuntary movements, distinguished from natural stereotyped behaviors (such as grooming, sniffing, rearing, and gnawing), were classified into three subtypes: locomotive AIMs (tight contralateral turns), axial AIMs (contralateral dystonic posture of the neck and upper body toward the side contralateral to the lesion), and limb AIMs (jerky and fluttering movements of the limb contralateral to the side of the lesion). Each subtype was scored on a severity scale from 0 to 4 (0, absent; 1, occasional; 2, frequent; 3, continuous; 4, continuous and not interruptible by outer stimuli). Statistical significance over days was determined by two-way ANOVA (genotype × days of treatment) with repeated measures. Statistical significance for the 120-min test session of each day was determined by two-way ANOVA (genotype × observation periods) with repeated measures, while total AIMs were analyzed by post hoc comparison.
Drag test
Mice were habituated in single cages 30 min before the test. Each mouse was held 1 cm from the base of the tail and lifted, forming an angle of 45° from the ground, allowing the support of the front limbs only. The animal was dragged backward and along a surface of 100-cm length at a constant speed of 20 cm/s for five consecutive times (each time alternating the direction of dragging). The animal was video recorded during the whole time. The number of each front limb stepping was counted later by two independent observers watching the videos at a slow pace. Drag test was evaluated 5 days before l-DOPA administration and days 3 and 16 after l-DOPA treatment (1 and 2 hours after injection).
Open field
Open-field test was used to measure the total activity. Briefly, animals were placed in the center of each square (50 cm × 50 cm) open top box under bright light and recorded via ceiling-mounted video camera for 40 min. Locomotor activity was assessed using the EthoVision XT 11.5 animal tracking software (Noldus) and showed as the total distance traveled in 40 min. Open-field test was made 4 days before starting the l-DOPA injections and at fifth day of l-DOPA treatment. To quantify the locomotor effect of l-DOPA injection, recording on fifth day of l-DOPA treatment was started after 60 min of the injection.
Turning test
Recording of turning test was made using the open-field boxes. Before beginning the test, one bowl (diameter of 20 cm) was positioned in the center of the box, and then the recording was started after one mouse was placed inside the bowl. The number of contralateral or ipsilateral turns to the 6-OHDA lesion was automatically detected using the EthoVision XT 11.5 animal tracking software (Noldus). Software parameters were adjusted to consider only 360° spin as a quantified turn. Contralateral or ipsilateral turns were quantified during 40 min and expressed as ratio of total turns (right and left turns) as hundred. Turning test was made at 2 days before l-DOPA administration and days 12 of l-DOPA treatment (39).
Rotarod
The accelerating rotarod test was used to quantify the motor alterations in WT and RasGRP1 KO mice. After placing the mice on the rotating rod (diameter of 5 cm), they were tested using the speed of the rotarod accelerated from 4 to 40 rpm for 5-min period, and the total time spent on the rod was recorded. To identify that the hemi-parkinsonian phenotype, mice were tested on the rotarod after 3 weeks of 6-OHDA lesion, the average of three trials was used for analysis. Rotarod evaluation during l-DOPA treatment were made at day 2 and 15; in each day, mice were tested using one single trial at 0, 60, and 120 min after l-DOPA injection. In all the cases, animals were not trained before the test.
Immunohistochemistry
Twenty minutes after the last intraperitoneal l-DOPA injection, animals were anesthetized with 2% Avertin and transcardially perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Mouse brains were postfixed in 4% PFA, incubated in sucrose/PBS solution (10 to 30%) at 4°C for 3 days, and embedded in Tissue-Tek optimum cutting temperature (OCT) compound (Sakura). Coronal sections (25 μm) were collected on Superfrost/Plus slides and immunostained with the antibodies mentioned in Table 2.
Table 2. Antibodies used for immunohistochemistry.
| Primary antibody | Source | Company | Catalog number | Dilution |
Secondary antibody |
Source secondary antibody |
Dilution secondary antibody |
| RasGRP1 | Rabbit | Santa Cruz Biotechnology |
sc-8430 | 1:200 | Alexa Fluor 488 | Donkey anti-rabbit |
1:500 |
| D1R | Rat | MilliporeSigma | D2944 | 1:200 | Alexa Fluor 594 | Donkey anti-rat | 1:500 |
| TH | Mouse | MilliporeSigma | MAB318 | 1:200 | Alexa Fluor 488 | Donkey anti-mouse |
1:500 |
The secondary antibodies were from Thermo Fisher Scientific. Immunofluorescent brain sections were counterstained stained with DAPI (Sigma-Aldrich) and mounted with Fluoromount-G mounting medium (Thermo Fisher Scientific). Images were acquired by using the Zeiss LSM 880 confocal microscope system with 10×/63× objective.
Cell culture, transfections, amino acid treatments, and RasGRP1 shRNA experiments
HEK293 cells were cultured in growth medium containing Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) with 10% fetal bovine serum, 1% penicillin-streptomycin, and 5 mM glutamine. Cells seeded in 3.5- or 6-cm plates, after 1 day, were transfected with cDNA constructs using Polyfect (Qiagen) as per the manufacturer’s instructions. For the amino acid deprivation protocol, after 48 hours, the growth medium was replaced with either serum-free F12+ (D2906, Sigma-Aldrich) medium or F12− (D9785, Sigma-Aldrich; without l-Leucine) medium for 2 hours, and, wherever indicated, F12− containing cells were stimulated with leucine (3 mM) for 15 min, and cells were lysed to proceed with Western blotting. HEK293 cells were seeded in 12-well plates for 1 day and then were transfected with RasGRP1 shRNA along with control shRNA constructs using Polyfect (Qiagen), according to the manufacturer’s instructions. The growth medium was removed after 24 hours, and the cells were lysed to prepare for Western blotting. Jurkat cells were also cultured similarly.
Western blotting, coimmunoprecipitation, and recombinant protein purification
Protocol for Western blotting and coimmunoprecipitation experiments in striatum was described in our previous studies (10). Briefly, cells were lysed in lysis buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1.0% NP-40, and 10% glycerol] with 1× protease inhibitor cocktail (Roche, Sigma-Aldrich) and 1× phosphatase inhibitor (PhosSTOP, Roche, Sigma-Aldrich), sonicated three times for 5 s at 20% amplitude, and cleared by centrifugation for 10 min at 11,000g at 4°C. Protein concentration was determined with a bicinchoninic acid (BCA) protein assay reagent (Pierce). Equal amounts of protein (30 to 40 μg) were loaded and were separated by electrophoresis in 4 to 12% bis-tris Gel (Thermo Fisher Scientific), transferred to polyvinylidene difluoride membranes, and probed with the indicated antibodies. HRP-conjugated secondary antibodies (Jackson ImmunoResearch Inc.) were probed to detect bound primary immunoglobulin G [(IgG)] with a chemiluminescence imager (Alpha Innotech) using enhanced chemiluminescence from WesternBright Quantum (Advansta).
Twenty minutes after the last l-DOPA injection, mice were euthanized by decapitation, and brains were rapidly dissected. Striatum tissue was snap-frozen in liquid nitrogen. Tissues were homogenized in radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS] with 1× complete protease inhibitor cocktail and 1× phosphatase inhibitor (PhosSTOP), followed by a brief sonication two times for 5 s at 20% amplitude and cleared by centrifugation for 10 min at 11,000g at 4°C. Protein estimation was done using a BCA method and proceeded to Western Blotting as mentioned above. For coimmunoprecipitation experiment, striatum was homogenized in immunoprecipitation (IP) buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, and 1.0% CHAPS (3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate)] with protease and phosphatase inhibitors, followed by a brief sonication for 5 s at 20% amplitude, and cleared by centrifugation for 10 min at 11,000g at 4°C. Protein estimation was done using a BCA method, a concentration (1 mg/ml) of protein lysates was precleared with 35 μl of protein A/G beads for 1 hour, supernatant was incubated for 1 hour at 4°C in Rheb IgG or RasGRP1 IgG or control IgG, and then 60 μl of protein A/G beads was added and incubated overnight at 4°C. The beads were washed five times with IP buffer (without protease/phosphatase inhibitor), and the protein samples were eluted with 30 μl of 2× LDS (Lithium dodecyl sulfate) containing 1.5% β-mercaptoethanol and proceeded for Western blotting as mentioned above. Quantitative densitometric measurement of immunoblots was performed using ImageJ program [National Institutes of Health (NIH)] using actin as a loading control. Phosphorylated proteins were then normalized against the total protein levels.
GST-RasGRP1-FL or GST-RasGRP1 GEF domain (1 to 450 amino acids; in pGEX-6P2 vector) proteins were expressed as described earlier (10). Briefly, an E. coli BL21DE3 strain expressing pGEX-6P2 constructs was grown in 15-ml culture overnight, was transferred to 500-ml terrific broth culture, and grown until the log phase was reached (~3.5 hours). The cells were then treated with isopropyl-β-d-thiogalactopyranoside (500 μM) for another 3 hours at 37°C. Cells were lysed by sonication in a lysis buffer containing 50 mM tris-HCl (pH 8.0), 1 mM dithiothreitol (DTT), 100 mM NaCl, and 1% NP-40 with protease inhibitor cocktail containing EDTA. The supernatant was incubated with glutathione beads for 12 hours at 4°C, and the beads were washed three times with a lysis buffer for 30 min. The recombinant proteins were eluted after treating the GST-bound beads with PreScission protease [10 U] with overnight incubation in 50 mM tris-HCl (pH 8.0), 2 mM EDTA, 1 mM DTT, and 1% NP-40. The proteins were dialyzed against 20 mM tris-HCl (pH 7.4), 50 mM NaCl, 1 mM DTT, and 10% glycerol to remove EDTA and detergent. The purified proteins were used immediately or were stored in aliquots at −80°C until further use.
In vitro binding
For the in vitro binding assay, an equimolar concentration of recombinant purified GST or GST-tagged RasGRP1 (FL or GEF domain) were incubated with Rheb for 16 hours at 4°C with glutathione beads in binding buffer containing 50 mM tris-HCl (pH 8.2), 1 mM DTT, 100 mM NaCl, and 1% NP-40, and the Rheb was detected using Western blotting with a Rheb antibody, as described earlier (4, 10).
GEF exchange assay
GEF assay was carried out using mant-GTP–based assay (BK100, Cytoskeleton) according to the manufacturer’s instructions. Briefly, 2 μl of 15 μM Rheb GTPase/Cdc42/Rac was added to 7.5 μl of 2× exchange reaction buffer [40 mM tris (pH 7.5), 100 mM NaCl, 20 mM MgCl2, and 1.5 mM mant-GTP] in 384-well plates. Plates were then read immediately (excitation, 360 nm; emission, 440 nm) in a plate reader (Molecular Devices). After six readings (for a total of 180 s), 5.5 μl of 1.2 μM or 5.5 μM RasGRP1-FL or 3 μl of 2.5 μM Dbs or dH2O was added to the wells, with an immediate resumption of the reading (70 readings for a total of 35 min). Three independent assays were performed.
TMT labeling, phosphopeptide enrichment, and MS experimental methods
Striatum from WT (n = 3) and RasGRP1 KO (n = 3; see scheme in Fig. 5A) mice isolated after l-DOPA treatment was lysed in RIPA buffer precipitated with ice-cold acetone overnight, and protein was resolubilized with 100 μl of 6 M urea/50 mM tris-HCl (pH 8.0). The protein was subsequently reduced for 45 min at 55°C using 3 μl of 0.5 M DTT and then alkylated in the dark for 30 min using 6 μl of 0.55 M iodoacetamide. Following reduction and alkylation, protein was once again precipitated with ice-cold acetone overnight. The protein pellet was resolubilized with 150 μl of TEAB (triethylammonium bicarbonate), and digestion was performed overnight at 37°C with 6 μg of trypsin (Promega). Following the digestion, the peptides were quantified using the Pierce quantitative colorimetric peptide assay (Thermo Fisher Scientific). One hundred micrograms of peptides per sample was subsequently labeled with varying TMT labels according to the manufacturer’s instructions (Thermo Fisher Scientific) and pooled. The pooled plexed samples (400 μg in total) were vacuum-dried, resolubilized in 1% trifluoroacetic acid, then desalted using Oasis HLB 1-cm3 solid phase extraction cartridges (Waters), and then dried once again using vacuum. TMT-labeled phosphopeptides for MS were enriched using the High-Select Fe–NTA (nitrilotriacetic acid) Phosphopeptide Enrichment Kit from the Thermo Fisher Scientific according to the manufacturer’s instructions. The TMT-labeled nonphoshopeptide complement was cleaned up for MS using a C18 ZipTip according to the manufacturer’s instructions (Millipore).
For MS, dried TMT-labeled peptides (phoshopeptides and nonphosphopeptides) were reconstituted in 5 μl of 0.1% formic acid and on line eluted into a Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) from an Acclaim PepMap RSLC nano Viper analytical column [75 μm (inner diameter) × 15 cm, Thermo Fisher Scientific] using a gradient of 5 to 25% solvent B (80:20 acetonitrile/water and 0.1% formic acid) in 180 min, followed by 25 to 44% solvent B in 60 min, 44 to 80% solvent B in 0.1 min, a 5-min hold of 80% solvent B, a return to 5% solvent B in 0.1 min, and, last, a 20-min hold of solvent B. All flow rates were 300 nl/min delivered using a nEasy-LC1000 nano liquid chromatography system (Thermo Fisher Scientific). Solvent A consisted of water and 0.1% formic acid. Ions were created at 2.0 kV using the Nanospray Flex ion source (Thermo Fisher Scientific). A synchronous precursor selection (SPS)–MS3 MS method was used by scanning between 380 to 2000 mass/charge ratio (m/z) at a resolution of 120,000 for MS1 in the Orbitrap mass analyzer and by performing collision-induced dissociation (CID) at top speed in the linear ion trap of peptide monoisotopic ions with charges 2 to 8 using a quadrupole isolation of 0.7 m/z and a CID energy of 35%. The top 10 MS2 ions in the ion trap between 400 and 1200 m/z were then chosen for higher-energy collisional dissociation (HCD) at 65% energy and detection in the Orbitrap at a resolution of 60,000 and an automatic gain control (AGC) target of 1 × 105 and an injection time of 120 ms (MS3). Data were analyzed as described below.
Proteomic data processing and statistical analysis
Quantitative analysis of the TMT experiments was performed simultaneously to protein identification using Proteome Discoverer 2.3 software. The precursor and fragment ion mass tolerances were set to 10 parts per million and 0.2 Da, respectively. Enzyme was Trypsin with a maximum of two missed cleavages, and Uniprot Mouse proteome FASTA file was used in SEQUEST searches. The impurity correction factors obtained from the Thermo Fisher Scientific for each kit was included in the search and quantification. The following settings were used to search the phospho enriched data and dynamic modifications: Oxidation, +15.995 Da (M); deamidated, +0.984 Da (N, Q); phospho, +79.966 Da (S, T, Y); static modifications of TMT 6plex, +229.163 Da (N terminus, K); and carbamidomethyl, +57.021 (C). Only unique+ Razor peptides were considered for quantification purposes. Percolator feature of Proteome Discoverer 2.3 was used to set an FDR of 0.01. The IMP–ptmRS node was used to calculate probability values for each putative phosphorylation site. Total peptide abundance normalization method was used to adjust for loading bias, and protein abundance–based method was used to calculate the protein level ratios. Coisolation threshold and SPS Mass Matches threshold were set to 50 and 65, respectively. After natural log (In) transformation of the raw ion counts, each channel was divided with reference sample (WT intact). The resulting list of proteins was further filtered by removing proteins that were not quantified in all plexes. Proteins passing this cut-off value were exported to JMP (SAS) 13.2.1 software for data cleaning and statistical analysis. The nonenriched (also known as total) dataset was analyzed in the same fashion except for omission phosphorylation in SEQUEST search and phosphoRS node in Proteome Discoverer workflow.
Proteins (including the ones identified by a single peptide) were only included in subsequent analyses if they met the following requirement: The peptides must be quantified in all samples of a given treatment group. Last, we used one-way ANOVA where category was fixed effect to identify proteins that are regulated across comparison groups (category). The multiple testing correction as per Benjamini Hochberg was applied to identify a “top tier” of significant proteins and limit identification of false positives with an FDR of 10%. P < 0.1 was considered statistically significant. The significantly regulated proteins were further interrogated to identify the different treatment group by using Tukey’s HCD.
Nonhuman primate studies
Captive bred female macaques (Macaca mulatta, Xierxin, Beijing, Peoples Republic of China; mean age = 5 ± 1 years; mean weight = 5.3 ± 0.8 kg) were housed in individual primate cages under controlled conditions of humidity (50 ± 5%), temperature (24° ± 1°C), and light (12-hour light/12-hour dark cycles; time lights on, 8:00 a.m.), allowing visual contacts and interaction with macaques housed in the adjacent cages. Food and water were available ad libitum, and animal care was supervised daily by veterinarians skilled in the healthcare and maintenance of nonhuman primates. Experiments were carried out in accordance with European Communities Council Directive (2010/63/EU) for care of laboratory animals in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care)–accredited facility following the acceptance of study design by the Institute of Lab Animal Science IACUC (Chinese Academy of Medical Sciences, Beijing, China). The tissues used in the present work have been obtained from an experimental brain bank used in several occasions whose experimental conditions are described elsewhere in great details (40). As shown in Fig. 6D, MPTP-treated nonhuman primate PD model macaques (n = 10) received daily MPTP hydrochloride injections [0.2 mg/kg, intravenous (i.v.)] until parkinsonian signs appeared. Once PD motor signs were stable, some of the animals (n = 5) were treated twice daily with an individually titrated dose of l-DOPA that provided maximum reversal of parkinsonian motor signs (Madopar, l-DOPA/carbidopa, 4:1 ratio; range, 9 to 17 mg/kg). This dose of l-DOPA, defined as 100% dose, was used for chronic l-DOPA treatment, which lasted for 4 to 5 months until dyskinesia stabilized. Animals then received l-DOPA twice a week to maintain a consistent level of dyskinesia before acute drug tests were carried out using a within subject experimental design. At the end of the experiment, all animals were killed by sodium pentobarbital overdose (150 mg/kg, i.v.) 1 hour after the last dose of vehicle or L-DOPA (i.e., at peak of antiparkinsonian effect), and the brains were removed quickly after death. Each brain was bisected along the midline, and the two hemispheres were immediately frozen by immersion in isopentane (−45°C) and then stored at −80°C. Punches of brain tissue were taken for the following regions: putamen and MFG. An average sample size of 6 ± 2 mg was obtained.
Western blotting analysis of monkey brain tissues
Preparation and immunoblotting were performed as previously described (40). Frozen, powdered samples from postmortem monkey brains were sonicated in 1% SDS and boiled for 10 min. Aliquots (1 μl) of the homogenate were used for the protein determination by a Bio-Rad Protein Assay kit (Bio-Rad). Equal amounts of total proteins (30 μg) for each sample were loaded on precast 4 to 20% gradient gel (Bio-Rad). Proteins were separated by SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes through Trans-Blot Turbo System. Membranes were then immunoblotted overnight using the following primary antibody: anti-RasGRP1 (1:1000; no. MABS146), anti-GFAP (1:1000; no. sc-33673), anti-PDE2A (1:1000; no. 55306-1-AP), anti-GAD1/2 (1:1000; no. ABN904), and anti-TH (1:2000; no. MAB318). Blots were then incubated in horseradish peroxidase–conjugated secondary antibodies. Immunoreactivity was detected by enhanced chemiluminescence (GE Healthcare) and quantified by Quantity One software (Bio-Rad). Optical density values were normalized to GAPDH (1:1000; no. SC-32233) for variation in loading and transfer. Normalized values were then averaged and used as dependent variable. Statistical analysis was performed by using Mann-Whitney test.
Quantitative real-time polymerase chain reaction analysis
Total RNAs were extracted using TRIzol reagent (Thermo Fisher Scientific), according to the manufacturer’s instructions. Total RNA was purified to eliminate potentially contaminating genomic DNA by a recombinant deoxyribonuclease. A 0.2 μg of total RNA per sample was used to synthesize cDNA by the QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time polymerase chain reaction (PCR) amplifications were performed with LightCycler 480 SYBR Green I Master (Roche Diagnostic) in a LightCycler 480 real-time thermocycler (Roche). The following protocol was used: 10 s for initial denaturation at 95°C followed by 40 cycles consisting of 10 s at 94°C for denaturation, 10 s at 65°C for annealing, and 6 s for elongation at 72°C temperature. RasGRP1 cDNA was amplified with following primers: RasGRP1 (forward, ACCCAGTTCCGAATGATGGT) and RasGRP1 (reverse, GACACAGGTTTCCATCCGCA). Transcripts quantities were normalized by the geometric mean of the two housekeeping genes, β-actin (Actin Beta, ACTB) and cyclophilin A (Peptidylprolyl isomerase A, PPIA), which were amplified using the following primers: ACTB (forward, CTGTGCTATGTCGCCCTAGA; reverse, GGAAGGTTGGAAGAGAGCCT) and PPIA (forward, TGCTGGACCCAACACAAATG; reverse, GTCCACAGTCAGCAATGGTG. All measurements from each participant were performed in duplicate. mRNA expression was calculated by using the relative quantification method (2−ΔΔCt). Statistical analyses were performed with Mann Whitney test.
Statistical analysis
Data were expressed as means ± SEM as indicated. All experiments were performed at least in biological triplicate and repeated at least twice. Statistical analysis was performed with a Student’s t test or repeated measure two-way ANOVA followed by post hoc Bonferroni multiple comparison test or one-way ANOVA followed by Tukey’s multiple comparison test (GraphPad Prism 7) as indicated in the figure legends.
Supplementary Material
Acknowledgments
We would like to thank M. Carta, M. Morelli, M.E. De Stefano, and F. Errico for the critical reading of the manuscript. We would like to thank M. Benilous for administrative help, and members of the labs Subramanian and Usiello for continuous support and collaborative atmosphere. We like to thank the members at the Scripps animal facility, behavior core, and proteomics core for the help and expertise. Funding: T.N., A.D.R., and A.U. were supported by a grant from Fondazione Cariplo and Ricerca Ateneo, Università Campania (L. Vanvitelli). This research was supported by grant awards from NIH/NINDS R01-NS087019-01A1 and NIH/NINDS R01-NS094577-01A1 and partly from the Cure for Huntington Disease Initiative (CHDI) Foundation to S.Su. Author contributions: S.Su. conceptualized the project and codesigned the project with A.U. M.E. and U.N.R.-J. carried out the lesion and related behaviors under A.U. supervision. U.N.R.-J. carried out IHC analysis. N.S. carried out biochemical (including GEF assay) and proteomics work. S.Sw. performed cell culture work. N.G. helped in Western blotting. O.R. purified RasGRP1 and carried out in vitro binding experiments. G.T. and C.S.-T. carried out LC-MS/MS. G.C. carried out quantification analysis and bioinformatics. T.N. and A.D.R. performed Western blotting and real-time PCR analysis on monkey tissues. Q.L., M.-L.T., and E.B. provided monkeys samples. S.Su. wrote the paper with input from A.U and the coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (41) partner repository with the dataset identifier PXD017434. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz7001/DC1
REFERENCES AND NOTES
- 1.Bezard E., Gross C. E., Qin L., Gurevich V. V., Benovic J. L., Gurevich E. V., L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol. Dis. 18, 323–335 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Santini E., Heiman M., Greengard P., Valjent E., Fisone G., Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2, ra36 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Fasano S., Bezard E., D'Antoni A., Francardo V., Indrigo M., Qin L., Doveró S., Cerovic M., Cenci M. A., Brambilla R., Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 107, 21824–21829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramaniam S., Napolitano F., Mealer R. G., Kim S., Errico F., Barrow R., Shahani N., Tyagi R., Snyder S. H., Usiello A., Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat. Neurosci. 15, 191–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brugnoli A., Napolitano F., Usiello A., Morari M., Genetic deletion of Rhes or pharmacological blockade of mTORC1 prevent striato-nigral neurons activation in L-DOPA-induced dyskinesia. Neurobiol. Dis. 85, 155–163 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Hartzell C., Ksionda O., Lemmens E., Coakley K., Yang M., Dail M., Harvey R. C., Govern C., Bakker J., Lenstra T. L., Ammon K., Boeter A., Winter S. S., Loh M., Shannon K., Chakraborty A. K., Wabl M., Roose J. P., Dysregulated RasGRP1 responds to cytokine receptor input in T cell leukemogenesis. Sci. Signal. 6, ra21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapoport M. J., Bloch O., Amit-Vasina M., Yona E., Molad Y., Constitutive abnormal expression of RasGRP-1 isoforms and low expression of PARP-1 in patients with systemic lupus erythematosus. Lupus 20, 1501–1509 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Ebinu J. O., Bottorff D. A., Chan E. Y., Stang S. L., Dunn R. J., Stone J. C., RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science 280, 1082–1086 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Kawasaki H., Springett G. M., Toki S., Canales J. J., Harlan P., Blumenstiel J. P., Chen E. J., Bany I. A., Mochizuki N., Ashbacher A., Matsuda M., Housman D. E., Graybiel A. M., A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. U.S.A. 95, 13278–13283 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shahani N., Swarnkar S., Giovinazzo V., Morgenweck J., Bohn L. M., Scharager-Tapia C., Pascal B., Martinez-Acedo P., Khare K., Subramaniam S., RasGRP1 promotes amphetamine-induced motor behavior through a Rhes interaction network ("Rhesactome") in the striatum. Sci. Signal. 9, ra111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavón N., Martin A. B., Mendialdua A., Moratalla R., ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Ryan M. B., Bair-Marshall C., Nelson A. B., Aberrant Striatal Activity in Parkinsonism and L-DOPA-Induced Dyskinesia. Cell Rep. 23, 3438–3446.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morissette M., Samadi P., Hadj Tahar A., Bélanger N., Di Paolo T., Striatal Akt/GSK3 signaling pathway in the development of L-Dopa-induced dyskinesias in MPTP monkeys. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 446–454 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Konradi C., Westin J. E., Carta M., Eaton M. E., Kuter K., Dekundy A., Lundblad M., Cenci M. A., Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiol. Dis. 17, 219–236 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crittenden J. R., Cantuti-Castelvetri I., Saka E., Keller-McGandy C. E., Hernandez L. F., Kett L. R., Young A. B., Standaert D. G., Graybiel A. M., Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc. Natl. Acad. Sci. U.S.A. 106, 2892–2896 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara K., Yonezawa K., Weng Q. P., Kozlowski M. T., Belham C., Avruch J., Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273, 14484–14494 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Avruch J., Long X., Ortiz-Vega S., Rapley J., Papageorgiou A., Dai N., Amino acid regulation of TOR complex 1. Am. J. Physiol. Endocrinol. Metab. 296, E592–E602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pryor W. M., Biagioli M., Shahani N., Swarnkar S., Huang W.-C., Page D. T., MacDonald M. E., Subramaniam S., Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington's disease. Sci. Signal. 7, ra103 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Iwig J. S., Vercoulen Y., Das R., Barros T., Limnander A., Che Y., Pelton J. G., Wemmer D. E., Roose J. P., Kuriyan J., Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife 2, e00813 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen E., Tohme M., Hedayat M., Leick M., Kumari S., Ramesh N., Massaad M. J., Ullas S., Azcutia V., Goodnow C. C., Randall K. L., Qiao Q., Wu H., Al-Herz W., Cox D., Hartwig J., Irvine D. J., Luscinskas F. W., Geha R. S., A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J. Clin. Invest. 126, 3837–3851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitehead I. P., Lambert Q. T., Glaven J. A., Abe K., Rossman K. L., Mahon G. M., Trzaskos J. M., Kay R., Campbell S. L., der C. J., Dependence of Dbl and Dbs transformation on MEK and NF-kappaB activation. Mol. Cell. Biol. 19, 7759–7770 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J., Song J., Yuan P., Tian Q., Ji Y., Ren-Patterson R., Liu G., Sei Y., Weinberger D. R., Orientation and cellular distribution of membrane-bound catechol-O-methyltransferase in cortical neurons: Implications for drug development. J. Biol. Chem. 286, 34752–34760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearce R. K., Heikkilä M., Lindén I. B., Jenner P., L-dopa induces dyskinesia in normal monkeys: Behavioural and pharmacokinetic observations. Psychopharmacology 156, 402–409 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Solís O., García-Montes J.-R., Garcia-Sanz P., Herranz A. S., Asensio M.-J., Kang G., Hiroi N., Moratalla R., Human COMT over-expression confers a heightened susceptibility to dyskinesia in mice. Neurobiol. Dis. 102, 133–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck G., Maehara S., Chang P. L., Papa S. M., A selective phosphodiesterase 10a inhibitor reduces L-Dopa-induced dyskinesias in parkinsonian monkeys. Mov. Disord. 33, 805–814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picconi B., Bagetta V., Ghiglieri V., Paillè V., di Filippo M., Pendolino V., Tozzi A., Giampà C., Fusco F. R., Sgobio C., Calabresi P., Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain 134, 375–387 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Kuroiwa M., Snyder G. L., Shuto T., Fukuda A., Yanagawa Y., Benavides D. R., Nairn A. C., Bibb J. A., Greengard P., Nishi A., Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP-32 signaling cascade in frontal cortex. Psychopharmacology 219, 1065–1079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoki K., Li Y., Xu T., Guan K.-L., Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bortolanza M., Cavalcanti-Kiwiatkoski R., Padovan-Neto F. E., da-Silva C. A., Mitkovski M., Raisman-Vozari R., Del-Bel E., Glial activation is associated with l-DOPA induced dyskinesia and blocked by a nitric oxide synthase inhibitor in a rat model of Parkinson's disease. Neurobiol. Dis. 73, 377–387 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Zhang K., Chammas C., Soghomonian J.-J., Loss of glutamic acid decarboxylase (Gad67) in striatal neurons expressing the Drdr1a dopamine receptor prevents L-DOPA-induced dyskinesia in 6-hydroxydopamine-lesioned mice. Neuroscience 303, 586–594 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Pajo A. T., Espiritu A. I., Jamora R. D. G., Efficacy and safety of extended-release amantadine in L-DOPA-induced dyskinesias: A meta-analysis. Neurodegener Dis Manag 9, 205–215 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Decressac M., Björklund A., mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J. Parkinsons. Dis. 3, 13–17 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Darmopil S., Martín A. B., De Diego I. R., Ares S., Moratalla R., Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Schmidt A., Hall A., Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 16, 1587–1609 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Rossman K. L., Der C. J., Sondek J., GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Lebel M., Chagniel L., Bureau G., Cyr M., Striatal inhibition of PKA prevents L-DOPA-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol. Dis. 38, 59–67 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Toda T., Uno I., Ishikawa T., Powers S., Kataoka T., Broek D., Cameron S., Broach J., Matsumoto K., Wigler M., In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 40, 27–36 (1985). [DOI] [PubMed] [Google Scholar]
- 38.Kortum R. L., Rouquette-Jazdanian A. K., Samelson L. E., Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. 34, 259–268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Francardo V., Recchia A., Popovic N., Andersson D., Nissbrandt H., Cenci M. A., Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson's disease. Neurobiol. Dis. 42, 327–340 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Nuzzo T., Punzo D., Devoto P., Rosini E., Paciotti S., Sacchi S., Li Q., Thiolat M.-L., Véga C., Carella M., Carta M., Gardoni F., Calabresi P., Pollegioni L., Bezard E., Parnetti L., Errico F., Usiello A., The levels of the NMDA receptor co-agonist D-serine are reduced in the substantia nigra of MPTP-lesioned macaques and in the cerebrospinal fluid of Parkinson's disease patients. Sci. Rep. 9, 8898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz7001/DC1