

Abstract
Background
Recurrence after radiation therapy is nearly universal for glioblastomas, the most common form of adult brain cancer. The study aims to define clinically pertinent mechanisms underlying this recurrence.
Methods
microRNA (miRNA) profiling was performed using matched pre- and post-radiation treatment glioblastoma specimens from the same patients. All specimens harbored unmethylated O6-methylguanine-DNA methyltransferase promoters (umMGMT) and wild-type isocitrate dehydrogenase (wtIDH). The most altered miRNA, miR-603, was characterized.
Findings
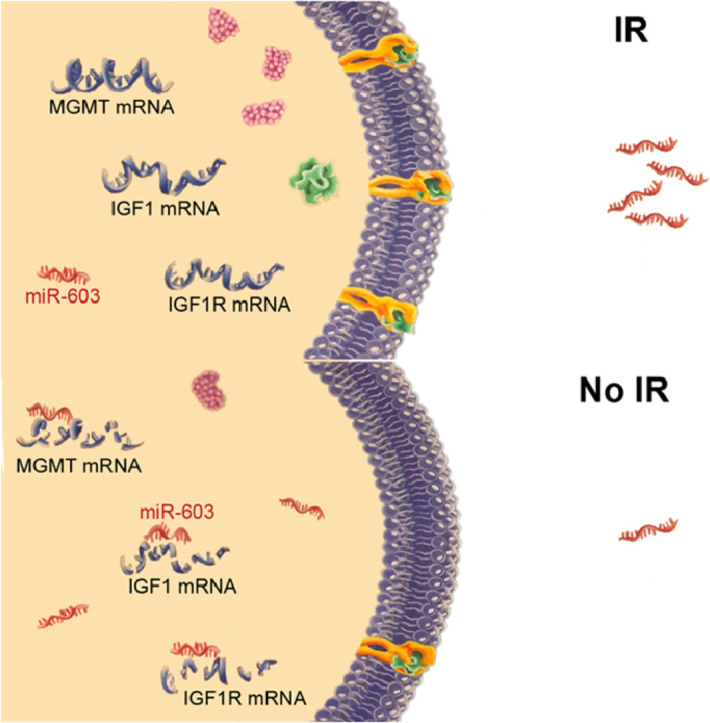
While nearly all miRNAs remained unchanged after treatment, decreased levels of few, select miRNAs in the post-treatment specimens were observed, the most notable of which involved miR-603. Unbiased profiling of miR-603 targets revealed insulin-like growth factor 1 (IGF1) and IGF1 receptor (IGF1R). Ionizing radiation (IR) induced cellular export of miR-603 through extracellular vesicle (EV) release, thereby de-repressing IGF1 and IGF1R. This de-repression, in turn, promoted cancer stem-cell (CSC) state and acquired radiation resistance in glioblastomas. Export of miR-603 additionally de-repressed MGMT, a DNA repair protein responsible for detoxifying DNA alkylating agents, to promote cross-resistance to these agents. Ectopic miR-603 expression overwhelmed cellular capacity for miR-603 export and synergized with the tumoricidal effects of IR and DNA alkylating agents.
Interpretation
Profiling of matched pre- and post-treatment glioblastoma specimens revealed altered homeostasis of select miRNAs in response to radiation. Radiation-induced EV export of miR-603 simultaneously promoted the CSC state and up-regulated DNA repair to promote acquired resistance. These effects were abolished by exogenous miR-603 expression, suggesting potential for clinical translation.
Funding
NIH 1R01NS097649-01, 9R44GM128223-02, 1R01CA240953-01, the Doris Duke Charitable Foundation Clinical Scientist Development Award, The Sontag Foundation Distinguished Scientist Award, the Kimmel Scholar Award, and BWF 1006774.01 (C.C.C).
Keywords: miR-603, IGF1, MGMT, Glioblastoma stem-cell state, Extracellular vesicles, Acquired radiation resistance
Graphical abstract
Research in context.
Evidence before this study
While studies of microRNAs (miRNAs) that are over- or under-expressed in clinical glioblastoma specimens have yielded insights into pathogenic mechanisms, there is little information on how miRNA expression changes as a function of ionizing radiation (IR) and contributes to acquired resistance. This question is of fundamental importance since altered miRNA expression is essential for cellular adaptation to environmental stress, including radiation exposure.
Added value of this study
We performed miRNA profiling of matched pre- and post-IR glioblastoma specimens. While the levels of nearly all miRNAs remained unchanged after treatment, we found decreased levels of select miRNAs in the post-treatment specimens, including miR-603. miR-603 suppressed the expression of insulin-like growth factor 1 (IGF1) and IGF1 receptor (IGF1R) to facilitate cellular exit from the glioblastoma cancer stem-cell state and confer radiation sensitivity. IR reduced cellular miR-603 by enhancing extracellular vesicle (EV)-mediated export of miR-603. Because miR-603 additionally suppressed expression of O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair protein responsible for detoxifying DNA alkylating agents, EV-mediated export of miR-603 conferred cross-resistance to those agents. These resistance mechanisms could be overcome by exogenous production of miR-603, which synergized with the anti-tumor activity of temozolomide (TMZ) and IR.
Implications of all the available evidence
Our findings suggest that EV-mediated secretion of miR-603 coordinates the regulation of DNA repair and the cancer stem-cell state to mediate acquired radiation resistance and cross-resistance to DNA alkylating agents. These findings provide a novel framework for glioblastoma therapeutic development.
Alt-text: Unlabelled box
1. Introduction
Glioblastoma is the most common form of primary adult brain cancer and one of the deadliest human cancers [1]. Radiation therapy is an essential element of the standard-of-care regimen, with established efficacy against glioblastomas [2]. However, glioblastoma recurrence after radiation is nearly universal [3]. The capacity of a glioblastoma cell to acquire or remain in a cancer stem-cell (CSC) like state is a key contributor to acquired resistance to ionizing radiation (IR) [4]. The concept of the glioblastoma CSC originated from flow-cytometric studies demonstrating that a subset of tumor cells isolated from clinical glioblastoma specimens expressed high-levels of neural stem cell markers, including CD133, Sox2, Musashi, and Olig2 [5,6]. These cells (1) possessed high capacity for tumorigenicity [7], (2) exhibited high levels of chemo-/radiation-resistance [4,5], and (3) altered gene expression in response to differentiation cues to suggest multi-potency [4,5]. While these CSC features were initially considered as “static” properties of select cancer cells [5], subsequent studies revealed that these properties can be dynamically lost or gained during glioblastoma cells transition between different cell states [8].
MicroRNAs (miRNAs) are a class of small (19–24 bp), non-coding RNAs that dampen gene expression by binding to the 3′ untranslated region (3′UTR) of the target mRNA [9]. In eukaryotes, master-regulatory miRNAs [10] coordinate the expression of genes regulated by distinct transcriptional programs toward a single phenotype. Major efforts have been invested in the study of miRNAs that are over- or under-expressed in clinical glioblastoma specimens relative to normal brain. The stability of miRNA during histologic processing, such as formalin fixation, renders it an attractive platform for clinical specimen analysis [11]. While these studies yielded insights into mechanisms of glioblastoma pathogenesis, how miRNA expression changes in response to treatment has not been carefully investigated. This question is of fundamental importance since altered miRNA expression is essential for cellular adaptation to stress, including radiation exposure [12].
A key aspect of miRNA homeostasis involves transport by extracellular vesicles (EVs). EVs are phospholipid membrane-bound organelles ranging 30–2000 nm that are secreted by all cell types, including glioblastoma [13]. EV-mediated export plays critical roles in a diverse spectrum of cellular physiologies including removal of intracellular metabolites and inter-cellular RNA transport [14]. Of note, the small RNA fraction contained in EVs is enriched for miRNAs [15]. Moreover, emerging literature suggests that select miRNAs are sorted into EVs [16], though the specific mechanisms responsible for this enrichment remain poorly understood.
Molecular characterization of glioblastomas revealed two determinants that profoundly influenced response to standard-of-care therapy (consisting of ionizing radiation (IR) and a DNA alkylating agent, temozolomide (TMZ)): mutation status in the isocitrate dehydrogenase (IDH) gene [17] and promoter methylation status of O6-methylguanine-DNA methyltransferase (MGMT) [18]. Mutant IDH produces high levels of the oncometabolite, D-2-hydroxyglutarate, which alters the epigenetic landscape [19] and DNA repair [20]. Glioblastomas with unmethylated MGMT promoter produce high levels of MGMT, a DNA repair enzyme that detoxifies the tumoricidal effects of TMZ [21]. Survival after standard-of-care treatment is poorest in patients afflicted with glioblastomas with wild-type IDH (wtIDH) and unmethylated MGMT promoter (umMGMT) [22]. >50% of all glioblastomas bear these features, and there is currently no effective therapeutic option for patients afflicted with these tumors.
Here, we performed miRNA profiling of wtIDH/umMGMT glioblastomas using matched pre- and post-standard-of-care treatment glioblastoma specimens. While the levels of most miRNAs remained unchanged after treatment, we found lower expression of few, select miRNAs in the post-treatment specimens, including miR-603. miR-603 suppressed post-transcriptional expression of insulin-like growth factor 1 (IGF1) and IGF1 receptor (IGF1R). IR induced EV-mediated export of miR-603 to de-repress IGF1 and IGF1R expression to promote glioblastoma CSC state and confer acquired radiation sensitivity. Because miR-603 additionally suppressed the expression of MGMT, EV-mediated export of miR-603 also conferred acquired cross-resistance to DNA alkylating agents.
2. Materials and methods
2.1. Cell lines, cell culture, and plasmids
Human glioblastoma cell lines LN340, A1207, LN18 (provided by Dr. Lynda Chin at University of Texas), T98G (ATCC), and U87MG (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-Glutamine and 1% Penicillin/Streptomycin in a humidified atmosphere at 37 °C with 5% CO2.
BT-147, BT-99 (provided by Dr. Keith Ligon at Dana-Farber Cancer Institute), CMK3 [23] and BT-83 cells [24] were grown as neurosphere in NeuroCult media supplemented with heparin, human epidermal growth factor (EGF), human fibroblast growth factor (FGF) in ultra-low attachment flasks and kept at 37 °C incubator with 5% CO2. Except for U87MG and BT-147 (both harbored wtIDH and methylated MGMT promoter), all glioblastoma lines used in this study were wtIDH/umMGMT .
Stable cell lines constitutively expressing miR-603 were established by transfecting pCMV-miR-603 miRNA expression vector (OriGene) into LN340, BT-83, or BT-99 cells and selected with neomycin as described previously [25]. Single clones were isolated and named as LN340(miR603)-clone number, BT-83(miR-603), BT-99(miR-603), respectively. The corresponding negative control cell lines were generated by transfecting empty control vector (OriGene) into LN340, BT-83, or BT-99 cells and selected with neomycin. A stable cell line constitutively expressing IGF1 was obtained by selection with neomycin after transfecting BT-83 cells with a pCMV-AC-GFP vector (OriGene) encoding human IGF1 cDNA. A negative control cell line was generated by transfecting corresponding empty expression vector. LN340-derived cell lines were cultured in DMEM medium supplemented with 10% FBS and 1% Penicillin/Streptomycin. Cell lines derived from BT-83 or BT-99 cells were grown as neurosphere in NeuroCult media supplemented with heparin, human EGF, human FGF in ultra-low attachment flasks and kept at 37 °C incubator with 5% CO2.
2.2. Immunofluorescence staining
CMK3 cells were plated onto laminin-coated glass coverslips and allowed to attach overnight. Cells were fixed with 10% formalin for 10 min at room temperature and permeabilized in 0.25% Triton X-100 for 8 min at room temperature. Non-specific binding was blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. Samples were incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: rabbit anti-Sox2 (Millipore, AB5603, 1:1000), rabbit anti-GFAP (Abcam, ab7260, 1:2500). The samples were then incubated with appropriate isotype-specific secondary fluorescent-labeled antibodies (Jackson ImmunoResearch) for 1 h at room temperature. Nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI). Fluorescent images were taken on Zeiss Axioplan 2 upright microscope.
2.3. miRNA transfection, miRNA target analysis, siRNA transfection
A1207, LN340, or BT-83 cells were transfected with human miR-603 mimic (Qiagen, MSY0003271) or the non-targeting control (Qiagen, AllStars Negative Control siRNA, 1027280) using HiPerfect transfection reagent (Qiagen) following the manufacturer's instructions. LN18 cells were transfected with anti-miR-603 (Qiagen, MIN0003271) or anti-miR-negative control (Qiagen, miScript Inhibitor Neg. Control, 1027271) using HiPerfect transfection reagent.
For profiling of miRNA targets, RNA was extracted 48 h after transfection of miR-603 mimic or biotinylated miR-603 as previously described [25], and profiled using Affymetrix HG-U133+PM microarray by the Beth Israel Deaconess Medical Center (BIDMC) Bioinformatics and Systems Biology Core. siRNA transfection were carried out as previously described [25]. The following siRNAs targeting human genes were used: MGMT siRNA (Dharmacon, M-008856-00-0005), and non-targeting control (Qiagen, AllStars Negative Control siRNA, 1027280).
2.4. RNA isolation and qPCR
Total RNA was isolated from cells using miRNeasy Kit (Qiagen) following the manufacturer's protocol. cDNA was synthesized using miScript II RT Kit (Qiagen). miRNA and mRNA transcripts were quantified using SYBR Green (Bio-Rad) on the Bio-Rad Chromo 4 DNA Engine Thermal Cycler. The following primers were used:
Hs_miR-603_3 miScript Primer (Qiagen, MS00037933),
IGF1: 5′-GCAGCACTCATCCACGATGC-3′ (forward primer),
5′- TGTGGAGACAGGGGCTTTTATTTC-3′ (reverse primer).
IGF1R: 5′- AAGTTCTGGTTGTCGAGGA-3′ (forward primer),
5′- GAGCAGCTAGAAGGGAATTAC −3′ (reverse primer).
MGMT: 5′-CCTGGCTGAATGCCTATTTC-3′ (forward primer),
5′-GATGAGGATGGGGACAGGATT-3′ (reverse primer).
18s: 5′-TTGCCCTCCAATGGATCCT-3′ (forward primer),
5′-GGGAGGTAGTGACGAAAAATAACAAT-3′ (reverse primer).
GAPDH: 5′-ACCCAGAAGACTGTGGATGG-3′ (forward primer),
5′-TTCTAGACGGCAGGTCAGGT-3′ (reverse primer).
SOX2: 5′-GAGCTTTGCAGGAAGTTTGC-3′ (forward primer),
5′-GCAAGAAGCCTCTCCTTGAA-3′ (reverse primer).
OLIG2: 5′- CAGAAGCGCTGATGGTCAT −3′ (forward primer),
5′- CGGCAGTTTTGGGTTATTC-3′ (reverse primer).
MUSASHI: 5′-GAGACTGACGCGCCCCAGCC-3′ (forward primer),
5′-CGCCTGGTCCATGAAAGTGACG-3′ (reverse primer).
NANOG: 5′-ACCTTGGCTGCCGTCTCTGG-3′ (forward primer),
5′-AGCAAAGCCTCCCAATCCCAAACA-3′ (reverse primer).
OCT4: 5′-TTTTGGTACCCCAGGCTATG-3′ (forward primer),
5′-GCAGGCACCTCAGTTTGAAT-3′ (reverse primer).
CD133: 5′-ACCAGGTAAGAACCCGGATCAA-3′ (forward primer),
5′-CAAGAATTCCGCCTCCTAGCACT-3′ (reverse primer).
2.5. Cell viability assay, clonogenic assay, and limiting dilution assay
Glioblastoma cell viability was assessed using CellTiter-Blu cell viability assay kit (Promega). For clonogenic assays, cells irradiated with X-rays were seeded in 6-well plates at the density of 500 cells per well. Cells were cultured until visible cell clones appeared, which took 2 weeks post irradiation. Cells were then fixed and stained with 0.1% crystal violet and the number of colonies was counted (50 or more cells were counted as one colony). For limiting dilution assays, single-cell suspensions were prepared, serially diluted and inoculated into 96-well ultra-low attachment plates at specific densities. Cells were allowed to grow for 14 days. Each well was then examined for the absence or presence of tumor sphere (at least one aggregate of ≥50 cells). The frequency of sphere-forming cells was then calculated using Extreme Limiting Dilution Analysis (ELDA, http://bioinf.wehi.edu.au/software/elda/).
2.6. γ-H2AX foci staining and comet assay
Cells were irradiated, fixed with 4% paraformaldehyde for 20 min at room temperature, and blocked with 2% BSA for 30 min. Cells were then stained with anti-γ-H2AX antibody (Millipore, 05–635) overnight at 4 °C, followed by incubation with Alexa Fluor 488-labelled secondary antibody. Nuclei were stained with DAPI. γ-H2AX foci were counted.
The comet assay was performed on 0 or 6 Gy-irradiated cells using neutral conditions as previously described [26]. DNA was stained with SYBR green I dye (Trevigen, 1:10,000). Comets were visualized by fluorescence microscope.
2.7. Immunohistochemistry (IHC)
GBM43 cells (provided by Dr. Jann Sarkaria at Mayo Clinic) were subcutaneously injected into the flanks of athymic male nude mice at 6 weeks of age following the procedure as described previously [27]. Once the average tumor volume reached 50 mm3, 250 pmol of miR-603 mimic or non-targeting miRNA mimic was intratumorally injected twice at 24-h interval. 48 h post the last treatment, mice were sacrificed, and tumors were collected, fixed in 10% formalin and embedded in paraffin sections.
Paraffin-embedded slides were placed in an oven at 60 °C for 1 h to remove paraffin layers. Antigen retrieval was accomplished by bringing slides to a boil in sodium citrate buffer (10 mM, pH 6.0) and incubating at sub-boiling temperature for 10 min. The following primary antibodies were used: rabbit anti-IGF1 (Abcam, ab9572), rabbit anti-IGF1 receptor (Abcam, ab39675), and mouse anti-PCNA (proliferating cell nuclear antigen) (Cell Signaling, 2586s). The sections were incubated with the respective primary antibodies at 4 °C overnight. The staining was visualized by species-specific-HRP/DAB staining. The sections were counterstained with hematoxylin. The images were captured using a light microscope.
2.8. Luciferase reporter assay
Luciferase reporters bearing the 3′UTR of IGF1 or IGF1R were constructed by subcloning the 3′UTRs into pSiCheck-2 dual luciferase vector (Promega), including full-length 3′UTR, truncated sections of 3′UTR containing predicted miR-603 miRNA Response Elements (MREs), or truncated sections of 3′UTR containing mutated miR-603 MREs. A1207 cells were co-transfected with miR-603 mimic and 3′UTR-luciferase reporter using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Luciferase activity was measured 48 h post transfection.
2.9. Western blot analysis and ELISA
Cells were lysed in RIPA buffer containing a mixture of protease inhibitors. Protein lysates were resolved by SDS/PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked using 5% BSA for 1 h at room temperature, followed by primary antibody incubation overnight at 4 °C. The primary antibodies used were the following: IGF1R (Abcam, ab16817 and Novus, 100-91823), MGMT (Abcam, ab39253), and α-tubulin (Sigma, T9026). After washing with 1x TBST buffer, membranes were incubated with species-specific secondary antibodies for 1 h at room temperature. Protein band intensity was quantified using ImageJ software.
IGF1 concentration in conditioned media was assessed using Human IGF1 ELISA kit (Abcam) based on manufacturer's instruction.
2.10. Extracellular vesicle isolation
Human glioblastoma cell line LN340 was cultured in DMEM medium supplemented with 10% exosome-depleted FBS (ThermoFisher Scientific, A2720803) for at least 24 h. Neurosphere line BT-83 was cultured in NeuroCult medium supplemented with growth factors as described in Cell lines, cell culture, and plasmids section. Cell culture medium was replenished prior to radiation treatment. Cell-produced EVs in the medium were isolated using Total Exosome Isolation Reagent (ThermoFisher Scientific, 4478359) or ExoQuick-TC ULTRA EV isolation kit for tissue culture media according to the manufacturers’ protocol. EV pellets were resuspended in 1x phosphate buffer saline (PBS) and stored at −20 °C for downstream nanoparticle tracking analysis and RNA extraction.
For GW4869 treatment, both cell lines were treated with GW4869 (10 μM) in EV-free medium (as described above) for 24 h before EV isolation. For siRab27a transfection, cells were transfected with Rab27a siRNA (Qiagen, SI02662744) or non-targeting control (Qiagen, AllStars Negative Control siRNA, 1027280) with HiPerfect transfection reagent (Qiagen) according to the manufacturer's instructions. Cells were treated for 24 h with siRNA, replated at equal concentration in EV-free medium, and cultured for another 24 h before EV isolation.
2.11. Nanoparticle tracking analysis (NTA)
Extracellular vesicle suspensions were measured using a Nanosight LM-10 instrument and NTA v3.2 software (NanoSight Ltd., Amesbury, UK) to determine the particle size and concentration. The resuspended EVs were diluted with 1x PBS to achieve measured particle concentration between 1 × 107/ml and 1 × 109/ml.
2.12. Absolute quantification of miR-603 using synthetic miRNA standards
Synthetic human miR-603 mimic (Qiagen, MSY0003271) were serially diluted to final concentration of 4 × 10−8 M, 4 × 10−9 M, 4 × 10−10 M, 4 × 10−11 M, 4 × 10−12 M, 4 × 10−13 M, 4 × 10−14 M, and 4 × 10−15 M. miR-603 mimic serial dilution and extracted EV-RNAs were reverse-transcribed to cDNA using miScript II RT Kit (Qiagen) in parallel. Those cDNAs generated from miR-603 mimic serial dilution were used for creating the standard curve for absolute quantification of miRNA copies. Standard curves for miR-603 were included in each plate of miR-603 qPCR assay to convert the cycle threshold (Ct) values of each sample into the corresponding number of miRNA copies.
2.13. Extracellular vesicle labeling
For PKH67 labeling, EVs (isolated from culture medium of IR-treated BT-83 cells) were stained with PKH67 Fluorescent Cell Linker Kits (Sigma-Aldrich). 50 μl of EV solution was resuspended in 250 μl of the diluent C plus 1.5 μl of the dye. After 10 min of incubation at room temperature, excessive dye was removed by using Exosome Spin Columns MW 3000 (Invitrogen) according to the manufacturer's protocol. For SYTO RNASelect staining, EVs were labeled with SYTO RNASelect Green Fluorescent Cell Stain (Invitrogen, USA) following the manufacturer's protocol. EVs were resuspended in 100 μl of PBS per labeling reaction. 1 µL of SYTO RNASelect dye stock solution was added to the EV sample to obtain a final dye concentration of 10 μM. The mixture of EVs and the dye were gently vortexed to get a homogenous distribution of the dye within the sample and incubated at 37 °C for 20 min. Excessive dye was then removed using Exosome Spin Columns MW 3000 following the manufacturer's protocol.
2.14. Xenograft studies
Animal studies were performed in accordance with the Animal Care and Use Rules at the University of California San Diego (UCSD). The animal study protocol was approved by the Institutional Animal Care and Use Committee of UCSD.
For subcutaneous xenograft experiments, patient-derived glioblastoma cells were subcutaneously injected into the flanks of 6-week-old nude mice according to the protocol previously described [27]. Tumor volume was measured daily with a caliper. Once the tumor volume reached 50 mm3, the mice were randomly assigned to treatment groups. At the end of a 7-week observation period, the mice were sacrificed, and the tumor tissues were removed for formalin fixation.
For intracranial xenograft experiments, patient-derived glioblastoma cells were dissociated into single-cell suspensions and stereotactically injected into the brains of nude mice at 6 weeks of age using murine stereotaxic system (Stoelting Co). The coordinates used were: 1.8 mm to the right of the bregma and 3 mm deep from the dura. Following tumor implantation, mice were maintained until the onset of overt neurological symptoms, including weight loss, lethargy, and hunched posture.
IR was administered at 3 Gy/day for two consecutive days starting 7 days after tumor implantation for the subcutaneous xenograft studies. IR was administered at 2 Gy/day for five consecutive days starting 7 days after tumor implant for the intracranial xenograft studies. For studies involving concurrent IR/TMZ, the mice underwent 5 days of 2 Gy/day radiation and the intraperitoneal administration of temozolomide at 50 mg/kg/day starting 7 days after tumor implantation as described in [28].
Kaplan-Meier survival curve was calculated. p values were determined using the log-rank test.
2.15. Clinical glioblastoma specimen
The research protocol (120345X) was approved by the Institutional Review Board of UCSD. All the patients gave informed consent before the study commenced. Only specimens that harbored wtIDH and umMGMT were collected for this study.
For the miRNA profiling of pre- and post-standard-of-care treatment glioblastomas, formalin-fixed, paraffin-embedded (FFPE) blocks of surgical specimens obtained at the time of the initial diagnosis and at the time of recurrence from the same patients were used. Slides were prepared and examined by a board-certified neuro-pathologist to identify those with >80% tumor component in specimens. 10-micron rolls were prepared from these blocks. Total RNAs were extracted from FFPE blocks, reverse-transcribed, and subjected to profiling using Qiagen Human Brain Cancer miScript miRNA PCR array in accordance with the manufacturer's instructions. This array was customized to consist of 86 miRNAs implicated in central nervous system-related carcinogenesis. A set of controls was included in this array to enable data analysis using the ΔΔCT method of relative quantification [29].
For the survival association study, glioblastoma specimens used in this study were obtained from patients who had undergone brain tumor removal. Those patient samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C. Patients were divided into progressive disease (n = 14) or stable disease (n = 12) group based on 6-month response according to Response Assessment in Neuro-Oncology (RANO) criteria by a board-certified neuro-radiologist.
All specimens studied harbored wtIDH and umMGMT based on CLIA-certified assays. IDH status was determined using IHC and confirmed by direct DNA sequencing. MGMT promoter methylation status was determined using methylation-specific PCR.
2.16. Statistical analysis
Data are presented as mean ± SE. Statistical analyses were conducted using GraphPad Prism software. The statistical significance was evaluated using Student's t-test or one-way ANOVA. p < 0.05 was considered statistically significant.
3. Results
3.1. Profiling of pre- and post-standard-of-care treatment glioblastoma specimens
Radiation resistance can be an intrinsic property of the tumor or acquired subsequent to radiation treatment. miRNAs mediating the former would be expected to be present in both the initial and recurrent glioblastoma specimens. In contrast, miRNAs mediating acquired resistance would exhibit altered expression in response to the treatment. Since we are interested in studying acquired resistance, we elected to characterize the latter.
To characterize altered miRNA homeostasis in response to standard-of-care therapy (IR/TMZ treatment), RNA was extracted from 10 matched pairs of pre- and post-treatment glioblastoma samples (all wtIDH/umMGMT) and analyzed using the Qiagen Human Brain Cancer miScript miRNA PCR Array (Fig. 1(a)). Consistent with the stability of miRNAs, we did not detect significant changes in the levels of most miRNAs between the pre- and post-treatment specimens. The three exceptions were: miR-603, miR-181d, and miR-124–3-p, the levels of which were lowered in the post-treatment specimens. We focused on miR-603 (shown as red dot in (Fig. 1(a)) since the magnitude of post-treatment decrease was greatest for this miRNA.
Fig. 1.

Ionizing radiation induces lower level of miR-603 in glioblastoma. (a) Scatter plot of the human miScript miRNome miRNA profiling depicted the relative expression of miRNA in the matched pre- (plotted on the x-axis) and post-treatment (plotted on the y-axis) clinical samples. All samples harbor wtIDH and umMGMT. Significant deviation from the solid 45° line indicates significant change in relative abundance after treatment. Each data point represents the average of triplicate results. Points lying outside of the dotted line indicate significant change in relative abundance. miR-603 is indicated in red. (b) Glioblastoma that recurred after IR/TMZ treatment exhibited decreased levels of miR-603. RNAs were extracted from FFPE samples obtained from the same patients (labelled as newly diagnosed and recurrent). Four matched pairs were analyzed. miR-603 levels were determined by qPCR. *p < 0.05 between newly diagnosed group and recurrent group (Student's t-test). (c) IR reduced cellular level of miR-603 in LN340 glioblastoma line. qPCR was performed to measure miR-603 levels in LN340 cells treated with 6 Gy IR at the indicated times. (d) IR reduced miR-603 level in freshly resected glioblastoma samples. miR-603 level was measured using qPCR in freshly resected human glioblastoma specimens or adjacent normal tissue after 2 Gy IR treatment. ***p < 0.001 compared to unirradiated group (Student's t-test).
First, we confirmed the decrease in the miR-603 levels in four additional matched pairs of pre- and post-IR/TMZ treatment wtIDH/umMGMT glioblastoma specimens. We observed consistent reduction in miR-603 levels in the post-treatment specimens (Fig. 1(b)), confirming the results of our initial profiling.
Since the standard-of-care treatment for glioblastoma consists of IR and TMZ, we next determined whether the reduction in miR-603 was related to IR or TMZ treatment. TMZ treatment of the glioblastoma line LN340 did not change the intracellular levels of miR-603 (Fig. S1(a)). In contrast, LN340 cells showed a time-dependent decrease in miR-603 level after 6 Gy IR, suggesting that IR contributed to the reduced miR-603 levels observed in the post-treatment specimens (Fig. 1(c)). Of note, such reduction was not observed for miR-21, a miRNA that plays key roles in glioblastoma pathogenesis [30] (Fig. S1(b)); these findings suggest that IR reduced cellular levels of select miRNAs rather than perturbing general miRNA hemostasis.
To confirm the clinical relevance of this observation, we used qPCR to assess miR-603 levels in freshly resected glioblastoma specimens. We found that 2 Gy IR treatment in these specimens was associated with a ∼2-fold decrease in miR-603 level relative to the unirradiated tumor specimen (Fig. 1(d)). Such decrease was not observed in brain tissue superficial to the tumor that was removed during surgical resection. Consistent with our cell line results, we did not observe a reduction in miR-21 (Fig. S1(c)) after IR treatment of the freshly resected tumor specimen.
3.2. miR-603 suppressed the expression of IGF1 and IGF1R
We next profiled mRNAs regulated by miR-603. To this end, we transfected miR-603 mimic or non-targeting control miRNA into two glioblastoma cell lines with low miR-603 level, LN340 and T98G [25]. Both lines harbored wtIDH and umMGMT. mRNAs were extracted and profiled on the Affymetrix HG-U133+PM microarray to identify those suppressed by miR-603 transfection. In parallel, biotinylated miR-603 or biotinylated control miRNA were transfected into the same cell lines. Streptavidin-captured mRNAs were profiled to identify those preferentially bound to biotinylated miR-603. Because MGMT is a known miR-603 target [25], we considered mRNAs that scored higher than MGMT in both assays as potential candidates. Cross-referencing LN340 and T98G profiles yielded 849 shared candidate genes (Table S1). Pathways implicated by these candidate genes are listed in Fig. 2(a). Of these pathways, the insulin-like growth factor 1 (IGF1) pathway [31] stood out in terms of relevance to radiation resistance. Top scoring candidates in this pathway included IGF1 and IGF1R, two proteins critical for IGF1 signaling.
Fig. 2.

miR-603 suppresses the expression of IGF1 and IGF1R. (a) Identifying miR-603-regulated mRNAs. Venn diagram of mRNA silenced by miR-603 transfection and mRNAs bound to biotinylated miR-603 in human glioblastoma cell LN340 and T98G. (b) miR-603 suppressed mRNA expression of IGF1 and IGF1R. IGF1 and IGF1R mRNA level was measured in miR-603-transfected or non-targeting miRNA (miR-NT)-transfected BT-147 cells using qPCR. *p < 0.05, **p < 0.01 compared to miR-NT control (Student's t-test). (c) miR-603 suppressed IGF1R protein expression. Representative western blot of IGF1R (left panel) and densitometric quantification (right panel) in A1207 cells transfected with miR-603 or miR-NT. α-Tubulin was used as protein loading control. (d) miR-603 suppressed IGF1 production. IGF1 concentrations in conditioned media from A1207 cells were measured by ELISA after transfection with miR-603 or miR-NT. *p < 0.05 compared to miR-NT control (Student's t-test). (e) Anti-miR-603 de-repressed IGF1R expression. Representative western blot of IGF1R (left panel) and densitometric quantification (right panel) in human glioblastoma cell line LN18 (harboring high endogenous level of miR-603) transfected with anti-miR-603 or anti-miR non-targeting control (anti-miR-NT). α-Tubulin served as protein loading control. (f) Anti-miR-603 de-repressed IGF1 production. IGF1 concentrations were quantified by ELISA in culture medium of LN18 cells transfected with anti-miR-603 or anti-miR-NT. ***p < 0.001 compared to anti-miR-NT control (Student's t-test). (g) miR-603 suppressed IGF1R and IGF1 expression in vivo. Representative IHC staining images of IGF1R, IGF1, and PCNA in human GBM43 tumor xenografts received intratumoral injection of miR-603 or miR-NT. scale bar: 50 μm. (h) IR increased IGF1 mRNA level in freshly resected glioblastoma samples. IGF1 level was measured using qPCR in freshly resected human glioblastoma specimens or adjacent normal tissue after receiving 2 Gy IR. ***p < 0.001 versus unirradiated group (Student's t-test). (i) IGF1 and IGF1R mRNA co-precipitated with biotinylated miR-603 mimic. 48 h after biotinylated miR-603 or biotinylated miR-NT (30 nM) was transfected into A1207 cells, cells were lysed and incubated with streptavidin-coated magnetic beads. qPCRs were performed to determine the relative abundance of IGF1 and IGF1R mRNA bound to the magnetic beads. *p < 0.05 and **p < 0.01 versus miR-NT control (Student's t-test). (j) and k) miR-603 miRNA Response Elements (MREs) in IGF1R and IGF1 3′UTR were required for miR-603-mediated gene suppression. Predicted miR-603 MREs and mutated miR-603 MREs within 3′UTRs of IGF1R and IGF1 are shown. Empty vector or constructs bearing the various 3′UTR fragments of IGF1R (j) or IGF1 (k) were co-transfected with miR-603 mimic into A1207 cells. Luciferase activities were measured 48 h post transfection. *p < 0.05 and **p < 0.01 compared to corresponding luciferase reporter bearing unmutated miR-603 MRE (Student's t-test).
Transfection of miR-603 into the BT-147 glioblastoma line suppressed IGF1 and IGF1R mRNA expression by ∼70% and 90%, respectively (Fig. 2(b)). In terms of protein expression, miR-603 transfection reduced the expression of IGF1R by ∼90% (Fig. 2(c)) and reduced IGF1 concentration in the conditioned media by ∼50% (Fig. 2(d)). Importantly, transfection of anti-miR-603 into a glioblastoma cell line that expressed high levels of miR-603 [25], LN18, increased IGF1R protein expression by ∼2-fold (Fig. 2(e)) and increased IGF1 concentration in the conditioned media by ∼2-fold (Fig. 2(f)). These effects were not observed after transfection with a non-targeting anti-miR control.
To confirm that miR-603 suppressed the expression of IGF1 and IGF1R in vivo, xenografts generated from a patient-derived wtIDH/umMGMT glioblastoma line, GBM43 [25], were injected with miR-603 mimic or non-targeting control miRNA after the tumor volume reached 50 mm3. 48 h post the miRNA injection, tumor xenografts were extracted and processed for IHC staining. Glioblastoma xenografts that received miR-603 injection showed decreased staining for IGF1R and IGF1 relative to those injected with non-targeting control miRNA (Fig. 2(g)). Additionally, we observed that IR treatment (2 Gy) of freshly resected glioblastoma specimens induced a 2-fold increase in IGF1 mRNA level as compared to unirradiated tumor specimens (Fig. 2(h)). These results provide support for in vivo regulation of IGF1 by miR-603.
3.3. Interaction between miR-603 and the 3′UTRs of IGF1 and IGF1R
We wished to determine whether miR-603 interacts with IGF1 and IGF1R mRNA. Biotinylated miRNA pull-down assays were performed after transfecting biotinylated miR-603 mimic or biotinylated non-targeting miRNA into the A1207 cell line, a line that we previously used to optimize miRNA pull-down and 3′UTR luciferase assays. qPCR analyses were performed on the biotinylated miRNA pull-down to determine the relative abundance of IGF1 and IGF1R mRNA (Fig. 2(i)). We detected a ∼2-fold enrichment of IGF1 mRNA and a ∼4-fold enrichment of IGF1R mRNA in the biotinylated miR-603 pull-down relative to the biotinylated non-targeting miRNA pull-down.
To further examine the interaction between miR-603 and the 3′UTRs of IGF1R, we cloned its 3′UTRs into the pSiCheck-2 dual luciferase vector in ∼2 kbs fragments (Fig. 2(j)). Luciferase reporter constructs were transfected in A1207 cells along with miR-603 mimic. The luciferase activity of the reporter constructs bearing the first two putative miR-603 miRNA Response Elements (MREs) of the IGF1R 3′UTR (S1 and S2) was reduced by ∼50% when co-transfected with miR-603 in A1207 cells (Fig. 2(j)). Mutating the MREs in IGF1R 3′UTR fragments S1 and S2 abolished this suppression, suggesting that these MREs are required for miR-603 suppression of IGF1R.
For IGF1, each of its ∼2 kb 3′UTR fragment harboring two putative miR-603 MREs (S1, S2 or S3) were cloned into the pSiCheck-2 dual luciferase vector (Fig. 2(k)). The luciferase activity of each 3′UTR construct was suppressed by 30% to 50% when co-transfected with miR-603 in A1207 cells (Fig. 2(k)). Mutating the MREs in the IGF1 S1, S2, or S3 3′UTR fragment abolished this suppression, suggesting that MREs in all three 3′UTR fragments participated in miR-603-mediated IGF1 suppression.
3.4. miR-603 enhanced glioblastoma radiation sensitivity by suppressing IGF1 expression in vitro and in vivo
IGF1 signaling plays critical roles in mediating glioblastoma radiation resistance [32]. To test whether miR-603 contributes to radiation resistance through regulation of IGF1 signaling, we established independent clones of LN340 glioblastoma line that stably expressed miR-603 (LN340(miR-603)) or contained the parent empty-vector (LN340(miR-Empty)). We found that ectopic miR-603 expression was associated with increased radiation sensitivity at all radiation doses tested. The radiation sensitizing effect of miR-603 was most notable after 8 Gy radiation, with an 8- to 10-fold increase in sensitivity (Fig. 3(a)). The increased radiation sensitivity in these LN340(miR-603) clones was accompanied by increased comet tail moment and accumulation of γ-H2AX (Fig. S2(a) and S2(b)), two indicators of increased DNA damage accumulation. Exposure to 6 Gy IR in LN340 (miR-Empty) cells increased γ-H2AX accumulation and comet tail moment by ∼2-fold. These effects were amplified by an additional 2-fold increase in the presence of ectopic miR-603 expression. miR-603-induced radiation sensitization was reversed upon exogenous addition of IGF1 (Fig. 3(b)). IGF1 addition also abolished miR-603-associated increase in comet tail moment (Fig. 3(c)) and γ-H2AX accumulation (Fig. 3(d)) in response to IR. The radiation-sensitizing effects of miR-603 were comparable and epistatic to that associated with IGF1R silencing (Fig. 3(e) and (f)), further supporting the importance of the IGF1 signaling axis in mediating miR-603-associated radiosensitization.
Fig. 3.

miR-603-induced IGF1 suppression enhances glioblastoma sensitivity to ionizing radiation in vitro and in vivo. (a) Stable miR-603 expressing LN340 glioblastoma line exhibited increased sensitivity to IR. Clonogenic survival for two independent clones of LN340 with or without stable ectopic miR-603 expression (labeled as LN340(miR-603) or LN340(miR-Empty), respectively) were determined post IR treatment. *p < 0.05 and **p < 0.01 between LN340(miR-603)−1 and LN340(miR-Empty)−1 or −2 at indicated IR dose. #p < 0.05, ##p < 0.01, ###p < 0.001 between LN340(miR-603)−2 and LN340(miR-Empty)−1 or −2 at indicated IR dose (Student's t-test). (b) Exogenous IGF1 addition suppressed the radiation sensitizing effect of miR-603. Cell viability was determined 5 days after IR and normalized to that of LN340(miR-Empty)−1 cells without IR or IGF1 treatment. *p < 0.05 and **p < 0.01 between indicated groups (Student's t-test). (c) miR-603 expression increased comet tail moment in response to IR. Comet assay was performed in LN340(miR-Empty)−1 and LN340(miR-603)−1 cells (unirradiated or received 6 Gy IR) with or without IGF1 treatment. Representative images of olive moment and the quantification were shown. Results were normalized to comet tail moment in unirradiated LN340(miR-Empty)−1 cells without IGF1 treatment. ***p < 0.001 between indicated groups (Student's t-test). Scale bar is 50 μm. (d) miR-603 expression enhanced γ-H2AX foci accumulation in response to IR. Representative immunofluorescence images of γ-H2AX foci in LN340(miR-Empty)−1 and LN340(miR-603)−1 cells (unirradiated or received 6 Gy IR) with or without IGF1 treatment were shown (left panel). Quantification of γ-H2AX foci was provided (right panel). Results were normalized to γ-H2AX foci in unirradiated LN340(miR-Empty)−1 cells without IGF1 treatment. *p < 0.05 and ***p < 0.001 between indicated groups (Student's t-test). Scale bar is 5 μm. (e) and (f) Radiation sensitizing effects of miR-603 were epistatic to that of IGF1R silencing. The effects of radiation alone or combination with IGF1R silencing on colony formation in LN340(miR-Empty)−1 or −2 (e) and LN340(miR-603)−1 or −2 cells (f) were shown. * p < 0.05 between LN340(miR-Empty)−1+siIGF1R and LN340(miR-Empty)−1 or −2+control siRNA at indicated IR dose. # p < 0.05 between LN340(miR-Empty)−2+siIGF1R and LN340(miR-Empty)−1 or −2+control siRNA at indicated IR dose (Student's t-test). (g) and (h) Ectopic IGF1 expression suppressed radiation sensitizing effect of miR-603 in vivo. Subcutaneous xenograft tumor growth curves were plotted after tumor cell inoculation. BT-83(vector) or BT-83(IGF1) xenografts were injected with human miR-603 mimic or miR-NT twice at 24-hour interval after the tumor volume reached 50mm3. IR (3 Gy/day) was administered for 2 consecutive days after the last dose of miRNA injection. Panel g and panel h are derived from the same experiment but plotted separately for better visualization of the data. *p < 0.05, ***p < 0.001 between indicated groups (one-way ANOVA). (i) Kaplan–Meier survival curves of mice bearing various intracranial BT-83 implants with or without radiation treatment (2 Gy/day for 5 consecutive days). Implanted glioblastoma neursophere lines included: BT-83(vector), BT-83(IGF1)(with stable IGF1 expression), BT-83(miR-603)(with stable miR-603 expression), and BT-83(IGF1+miR-603)(with stable IGF1 and miR-603 expression). (j) miR-603 level in clinical glioblastoma specimens correlated with radiation response. Scatter plot of miR-603 expression levels in clinical glioblastoma specimen obtained from glioblastoma patients who underwent repeat radiation therapy due to tumor recurrence. Disease stability or progression were assessed approximately one year post re-radiation. 12 patients achieved stable disease, and 14 patients suffered disease progression. p = 0.0126 compared to progression group (Student's t-test). (k) miR-603 sensitized patient-derived glioblastoma cells to IR. Clonogenic potential of CMK3 cells (propagated as neurospheres) was determined using limiting dilution assays. CMK3 cells were transfected with miR-603 or miR-NT before IR treatment. ***p < 0.001 between indicated groups (Chi-square test).
We had demonstrated that miR-603 downregulated IGF1/IGF1R in vivo using the GBM43 line (Fig. 2(g)). We wished to validate this observation using a short-term passaged patient-derived glioblastoma cell line and selected the wtIDH/umMGMT BT-83 cell line for this purpose [24]. We directly injected miR-603 into established tumor xenografts derived from BT-83 cells with or without ectopic IGF1 expression (annotated as BT-83(IGF1) and BT-83(vector), respectively). miR-603 injection retarded the growth of BT-83(vector) xenografts. By day 36, the average tumor volume for the miR-603-treated xenograft was 29% of the tumor volume of xenografts injected with negative control miRNA (Fig. 3(g)). Treatment with IR similarly suppressed the xenograft growth. By day 36, the average volume for the IR treated tumor was 72% of that of the untreated xenograft. Combining miR-603 administration with IR produced profound suppression of xenograft growth. By day 36, the average volume for the miR-603+IR treated tumor was 9% of the untreated BT-83(vector) xenograft. Consistent with our in vitro studies, ectopic IGF1 expression abolished miR-603-induced radio-sensitizing effects in vivo (Fig. 3(h), dotted curves).
Next, BT-83 lines stably expressing IGF1 and/or miR-603 were intracranially implanted and subsequently treated with IR. The median survival for mice implanted with BT-83(vector; carrying parental vectors for the IGF1- and miR-603-expressing constructs) was 16 days (Fig. 3(i)). Ectopic expression of miR-603 or treatment with IR prolonged this median survival to 26.5 and 28 days, respectively (p < 0.0001). Combining IR with ectopic miR-603 expression further extended median survival to 58 days (p = 0.003 compared to IR treated mice). In mice harboring BT-83 ectopically expressing both IGF1 and miR-603 (BT-83(IGF1+miR-603)), IR treatment produced median survival comparable to those observed in IR treated mice harboring BT-83(IGF1) (13 and 16.5 days, respectively). These results suggest miR-603 modulates glioblastoma radiation response through regulation of IGF1 signaling.
If the radiation sensitizing effect of miR-603 bears clinical pertinence, we would anticipate that higher levels of miR-603 to be associated with improved clinical outcome after radiation therapy. To test this hypothesis, we correlated the levels of miR-603 in clinical glioblastoma specimens derived from 26 wtIDH/umMGMT glioblastoma patients who suffered recurrence and underwent repeat radiation therapy >1 year after initial radiation treatment. Disease control in response to radiation was classified using RANO criteria one year after the repeat radiation. Supporting our hypothesis, we found that specimens derived from patients with disease control (6 months after re-radiation) harbored higher levels of miR-603 relative to those who suffered disease progression (p = 0.0126, Fig. 3(j)).
MGMT is a published target of miR-603 [25]. We wished to determine whether MGMT expression influenced the effects of miR-603 on IR response. miR-603 transfection-induced radiation sensitivity in glioblastoma cell lines that harbored no detectable MGMT expression (including two short-term passaged patient-derived glioblastoma lines with stem cell properties: CMK3 and BT-147, as well as the U87MG cell line) (Figs. 3(k), S2(c)–S2(e)), suggesting that the radiation sensitizing effects of miR-603 did not require MGMT expression.
3.5. miR-603 suppressed glioblastoma stem-cell state through down-regulation of IGF1
In many tumors, IGF1 signaling contributed to radiation resistance by promoting CSC states [33,34]. We hypothesized that miR-603-mediated IGF1 downregulation in glioblastomas would suppress CSC states to promote radiation sensitivity. Supporting our hypothesis, miR-603 transfection into CMK3 cells (a short-term passaged patient-derived glioblastoma line with stem cell properties and low miR-603 expression) [23] suppressed expression of neural stem cell markers, including Sox2 (Fig. 4(a) and (b)), Olig2 (Figs. 4(c), (d), and S3(a)), Musashi (Figs. S3(b) and S3(d)), Nanog (Fig. S3(e)), Oct4 (Fig. S3(f)), and CD133 (Fig. S3(g)). Moreover, miR-603 transfection induced the expression of differentiation markers, including GFAP (Fig. 4(e)) and β-III-Tubulin (Fig. S3(c)). Importantly, these effects were reversed by exogenous addition of IGF1 (Figs. 4(a), (b), (d), (e), and S3(a)–S3(h)), suggesting IGF1 as the key mediator of miR-603′s effect on the CSC state. Similar results were also recapitulated in LN340 cells, another glioblastoma cell line with low miR-603 expression (Fig. S4).
Fig. 4.

miR-603 suppresses glioblastoma stem-cell state through down-regulation of IGF1. (a) miR-603 suppressed expression of the stem cell marker, Sox2. Representative immunofluorescence staining images of Sox2 (green) in the patient-derived glioblastoma neurosphere line CMK3 transfected with miR-603 or miR-NT. Studies were performed in the presence or absence of IGF1 treatment. DAPI (blue) staining was used to visualize nuclei. Quantifications of Sox2-positive cells were provided. ***p < 0.001 between indicated groups (one-way ANOVA). Scale bar represents 100 μm. (b) and (d) The mRNA levels of stem cell markers Sox2 (b) and Olig2 (d) were suppressed by miR-603 transfection in CMK3 cells, which was reversed by exogenous addition of IGF1. *p < 0.05 and **p < 0.01 between indicated groups (one-way ANOVA). (c) Olig2 expression in CMK3 cells transfected with miR-NT or miR-603 was assessed using western blotting. (e) miR-603 transfection induced expression of the differentiation marker, GFAP. Representative immunofluorescence staining images of GFAP (green) in CMK3 cells transfected with miR-603 or miR-NT in the presence or absence of IGF1 treatment. Quantifications of GFAP-positive cells were provided. ***p < 0.001 between indicated groups (one-way ANOVA). Scale bar represents 100 μm. (f) and (g) In vitro limiting dilution assay was performed in CMK3 cells transfected with miR-603 or miR-NT in the presence or absence of IGF1 treatment. Frequencies of clonogenic cells in each group were determined by ELDA analysis (g). **p < 0.01 between indicated groups (one-way ANOVA). Representative images of neurospheres formed in indicated experimental conditions were shown (f). Scale bar: 10 µm. (h) miR-603 transfection suppressed tumorigenicity of patient-derived glioblastoma neurosphere line BT-83. BT-83(vector) or BT-83(IGF1) cells transfected with miR-NT or miR-603 were injected into the subcutaneous compartment of nude mice (10,000 cells per injection). Twenty injections were performed for each experimental condition. *p < 0.05, ***p < 0.001 between indicated groups (Chi-square test).
We next characterized the impact of miR-603 on CSC using an in vitro self-renewal assay. In this assay, sphere formation was associated with increased expression of “stemness” markers and suppressed expression of differentiation markers (Figure S3i). miR-603 transfection significantly decreased the sphere-forming cell frequency (Figs. 4(f), (g), and S3(j)), suggesting that miR-603 suppressed glioblastoma capacity for self-renewal. Importantly, the frequency of clonogenic miR-603-transfected CMK3 cells was restored by the exogenous addition of IGF1 (Figs. 4(f), (g), and S3(j)), indicating IGF1 as the mediator of miR-603′s effects on CSC.
Finally, we tested the effects of miR-603 on tumorigenicity, an in vivo phenotype closely associated with the glioblastoma CSC state. 13/20 injections of BT-83 cells transfected with negative control miRNA formed xenografts. In contrast, only 5/20 injections of BT-83 cells transfected with miR-603 formed xenografts (p = 0.011 relative to negative control miRNA-transfected cells), supporting our hypothesis that miR-603 suppressed the CSC state. Ectopic expression of IGF1 in BT-83 prior to miR-603 transfection restored tumorigenicity, with 17/20 of the injections forming xenografts (Fig. 4(h), p = 0.0001 relative to miR-603 transfected, non-IGF1 expressing cells).
Taken together, these results indicate that miR-603 suppressed glioblastoma stem-cell state by down-regulation of IGF1 expression.
3.6. Radiation-induced cross-resistance to DNA alkylating agents
MGMT is a down-stream target of miR-603 [25] and is the primary cellular determinant of cellular sensitivity to TMZ and nitrosoureas, a related class of DNA damaging agents frequently used in the treatment of recurrent glioblastomas [35]. As such, IR-induced reduction in glioblastoma miR-603 level should de-repress MGMT expression in addition to IGF1 and IGF1R. De-repression of MGMT would be expected to enhance glioblastoma resistance to DNA alkylating agents. Supporting this hypothesis, independent clones of IR-resistant LN340 (isolated after 6 Gy IR, annotated as LN340R) showed reduced miR-603 expression (Fig. 5(a)) and increased MGMT expression (Fig. 5(b)). The increase in MGMT expression was associated with increased TMZ resistance. ∼50% of the parental LN340 cells survived after 250 µM TMZ treatment (Fig. 5(c)). In contrast, IR-resistant LN340 clones (LN340R-1, 2, and 3 clones) were essentially insensitive to TMZ treatment. This radiation-induced resistance to TMZ was abolished by treatment with an siRNA against MGMT (siMGMT) or by miR-603 (Figs. 5(c), S5). The effects of miR-603 were epistatic to that of siMGMT in this regard, suggesting MGMT up-regulation as a key modulator of radiation-induced TMZ cross-resistance.
Fig. 5.

Ionizing radiation decreases miR-603 level to induce cross-resistance to DNA alkylating agents. (a and b) IR-resistant clones of LN340 (LN340R-1, -2, -3) showed reduced level of miR-603 (a) and increased MGMT level (b) compared to parental LN340 cells. *p < 0.01 and ***p < 0.001 compared to parental LN340 group (one-way ANOVA). (c) IR induced cross-resistance to TMZ through up-regulation of MGMT. Parental LN340 and LN340R-3 cells were transfected with control, siMGMT, or miR-603 24 h prior to TMZ treatment. Cell viability was measured 7 days after TMZ (250 µM) treatment and normalized to untreated LN340 cells without TMZ treatment. **p < 0.01, ***p < 0.001, n.s.p > 0.05 between indicated groups (one-way ANOVA). (d) IR induced cross-resistance to BCNU through up-regulation of MGMT. Parental LN340 and LN340R cells were transfected with control, siMGMT, or miR-603 24 h prior to BCNU (50 µM) treatment. Cell viability was measured 7 days after BCNU treatment and normalized to untreated LN340 without BCNU treatment. ***p < 0.001, n.s.p > 0.05 between indicated groups (Student's t-test). (e) Glioblastoma that recurred after IR/TMZ treatment exhibited increased levels of MGMT. RNAs were extracted from FFPE samples obtained from the same patients (newly diagnosed and recurrent). Four matched pairs were analyzed. MGMT levels were determined by qPCR. *p < 0.05 between pre- and post-treatment group (Student's t-test).
Bis-chloroethylnitrosourea (BCNU) belong to a class of bi-functional DNA alkylating agent frequently used as treatment for glioblastoma that recurred after stand-of-care treatment [35]. MGMT is a major determinant of BCNU sensitivity in glioblastomas [36]. We therefore explore whether radiation-induced decrease in miR-603 influenced BCNU sensitivity in glioblastomas. IR-resistant LN340R cells showed an approximate 3-fold increased resistance to BCNU compared to parental LN340 cells, which was abolished with miR-603 or siMGMT transfection (Fig. 5(d)). The effects of miR-603 were epistatic to that of siMGMT in this regard. Supporting the clinical relevance of these observations, we observed that the reductions of miR-603 in the post-treatment FFPE specimens were associated with increase in MGMT expression (Fig. 5(e)). These results suggest that IR-induced reduction in miR-603 de-repressed MGMT expression to confer BCNU cross-resistance.
3.7. Radiation induced miR-603 export through EV release
To investigate the mechanism underlying radiation-induced decrease in cellular miR-603 level, we first examined whether miR-603 synthesis or degradation were affected by IR. IR treatment did not decrease the level of pre-miR-603 in LN340 cells (Fig. S6(a)). Additionally, silencing of RNAses implicated in miRNA degradation [37] did not significantly impact the effect of IR on miR-603 level in the same cell line (Figs. S6(b) and S6(c)).
In this context, we explored other mechanisms that affect miRNA homeostasis. Since miRNA export through EV release can contribute to decreased cellular miRNA levels [38], we tested whether IR induced EV-mediated miR-603 export. EVs isolated from culture media of two independent glioblastoma lines (BT-83 and LN340) were characterized based on MISEV guideline 2018 [39] (Fig. 6(a)). IR treatment reduced the number of EVs by approximately 50% (Fig. 6(b)). This decrease, however, was accompanied with a 6- to 9-fold increase in the absolute copy number of miR-603 contained in each released EV (Figs. 6(c), S6(d)–S6(f)). Accounting for the EV number, there is a 3- to 4-fold increase in total miR-603 released after radiation treatment (Fig. 6(d)). We excluded the possibility that the EV-miR-603 detected in our study resides in miRNA aggregates located outside of the EVs by the following experiments. Quantitative assessment of EV-associated miR-603 was performed after combinations of RNase, Triton X-100, or proteinase K treatment. Without Triton X-100 treatment of EVs, RNase +/- proteinase K treatment did not decrease the level of EV-miR-603. In contrast, miR-603 was undetectable after RNase +/- proteinase K treatment in the Triton X-100-treated EV samples. These results suggest that EV-miR-603 we detected is located in the EVs and the EV membrane protected EV-miR-603 from the RNase +/- proteinase K treatment (Fig. 6(e), Fig. S6(g)).
Fig. 6.

Ionizing radiation stimulates extracellular vesicle miR-603 export. (a) Western blotting detection of EV markers in EVs secreted by BT-83 cells with or without IR treatment (6 Gy). The EV markers CD9, CD81, TSG101, and HSC70 were used to identify the presence of EVs, ApoA1 was served as an EV negative marker, and α-tubulin was used as an intracellular marker not incorporated into EVs. EV pellets were run with equivalent total numbers of EV particles, as determined by NTA. Cell pellets were run with equivalent total protein input amounts, as determined by α-tubulin levels. (b) IR lowered the number of released EVs in BT-83 and LN340 cells. Culture medium of BT-83 cells (top panel) and LN340 cells (bottom panel) with or without IR treatment was collected and subjected to EV isolation and EV quantification. *p < 0.05 versus unirradiated control group (Student's t-test). (c) and (d) IR increased extracellular secretion of EV-miR-603. miR-603 copy number per EV (c) and total miR-603 copy number (d) were compared between unirradiated control group and irradiated group. *p < 0.05, **p < 0.01 versus unirradiated control group (Student's t-test). (e) EVs secreted by irradiated or unirradiated BT-83 cells were isolated using ExoQuick-TC ULTRA EV isolation kit for tissue culture media and incubated with the indicated treatments before isolating RNA and measuring the levels of miR-603. The EV pellets were incubated with RNase A (0.5 µg/µl) alone or were incubated with proteinase K (0.4 µg/µl) prior to RNase treatment in the presence or absence of 2% Triton X-100. ***p < 0.001, n.s.p > 0.05 between indicated groups (one-way ANOVA). (f) Representative fluorescence microscopy images for PKH67-labelled EVs uptake by human microglia HMC3 cells after 24 h incubation. Green: PKH67; red: Alexa Fluor 594 phalloidin; blue: DAPI. Scale bar: 25 µm. (g) Pharmaceutical blocking of EV secretion by GW4869 repressed IR-induced miR-603 reduction in GBM cells. BT-83 cells (left panel) and LN340 cells (right panel) were pretreated with GW4860 (10 µM, 24 h) or DMSO before receiving IR treatment. Cells were collected at indicated time points and subjected to miR-603 quantification using qPCR. *p < 0.05, **p < 0.01 and ***p < 0.001 compared to DMSO group (Student's t-test). (h) Repression of EV secretion through Rab27a knockdown rescued IR-induced cellular miR-603 decrease. BT-83 cells (left panel) and LN340 cells (right panel) were transfected with siRNA targeting Rab27a (siRab27a) or negative control siRNA (siNT) before receiving IR treatment. Cells were collected at indicated time points and the level of miR-603 was measured by qPCR. *p < 0.05 versus siNT-transfected group (Student's t-test).
Notably, total miR-21 released by EVs of BT-83 or LN340 cells was not altered by IR treatment (Fig. S6(h)). Similar to previous reports of tumor secreted EVs [40], EVs released by glioblastoma cells can be taken up by other types of cells in the tumor microenvironment, including microglia [41]. Our results showed EVs isolated from BT-83 cells post IR treatment were taken up by the human microglia cell line HMC3 (Figs. 6(f), S6(i)).
We next tested whether inhibition of EV secretion suppressed IR-induced decrease in cellular miR-603 level. First, we inhibited EV release by treatment with the sphingomyelinase inhibitor, GW4869. Consistent with previous studies, treatment with GW4869 attenuated BT-83 and LN340 EV secretion by approximate 3-fold (Fig. S6(j)) without affecting cellular viability (Fig. S6(k)). Importantly, pre-treatment of BT-83 or LN340 with GW4869 for 24 h reversed IR-induced decrease in cellular miR-603 level (Fig. 6(g)). To further substantiate this result, we silenced Rab27a, a member of Rab family of GTPase that is required for EV secretion. >80% silencing of Rab27a (Fig. S6(l)) resulted in 2-fold decreased EV secretion in BT-83 and LN340 cells (Fig. S6(m)) without affecting cellular viability (Fig. S6(n)). Recapitulating results observed with GW4869, siRab27a-transfection eliminated IR-induced decrease in cellular miR-603 level (Fig. 6(h)). As a whole, these results suggest that IR induced select enrichment of miR-603 within EVs. Subsequent release of these miR-603 enriched EVs, in turn, decreased cellular level of miR-603.
3.8. miR-603 synergized with the tumoricidal effects of ionizing radiation and temozolomide in an unmethylated glioblastoma xenograft model
Our results indicate that radiation induced a decrease in cellular levels of miR-603, which in turn, de-repressed expression of IGF1/IGF1R (to induce radiation resistance) and MGMT (to induce TMZ resistance). In this context, we hypothesized that ectopic miR-603 expression would simultaneously suppress radiation-induced expression of IGF1/IGF1R and MGMT to synergize with the tumoricidal effects of the current standard of care for glioblastoma (IR+TMZ). We first tested this hypothesis in vitro using a short-term passaged patient-derived glioblastoma line, BT-99 (wtIDH/umMGMT), which harbored the highest level of MGMT expression in our collection of lines. Due to the high levels of endogenous MGMT expression (Fig. S2(c)), BT-99 cells were refractory to the tumoricidal effect of 100 µM TMZ. We observed a ∼2-fold decrease in the frequencies of clonogenic cells after 6 Gy IR or 6 Gy IR+TMZ treatment. Ectopic expression of miR-603 in BT-99 cells enhanced the tumoricidal effect of the IR+TMZ combination by an additional ∼5-fold (Fig. 7(a) and S7), supporting our hypothesis.
Fig. 7.

miR-603 synergizes with IR and TMZ treatment in BT-99, an unmethylated glioblastoma xenograft model. (a) miR-603 synergized with IR (6 Gy) and TMZ treatment (100 µM) in BT-99 in vitro. Limiting dilution assay was performed using BT-99, an MGMT promoter unmethylated glioblastoma line with high MGMT expression, with or without stable miR-603 expression. The frequency of sphere-forming cells in each group was calculated using ELDA analysis. **p < 0.01, ***p < 0.001, n.s.p > 0.05 between indicated groups (one-way ANOVA). (b) miR-603 synergized with IR and TMZ treatment in BT-99 in vivo. Kaplan–Meier survival curves of mice bearing intracranial BT-99 implants with or without ectopic miR-603 expression after various treatments. The mice underwent 5 days of 2 Gy/day radiation and the intraperitoneal administration of TMZ at 50 mg/kg/day starting 7 days after tumor implant.
Similar patterns of therapeutic response were observed when treatments were administered after BT-99 was intracranially implanted. The median survival of mouse bearing BT-99 with or without TMZ treatment were similar: at 25 and 24 days, respectively (p = 0.7985). Exposure to radiation prolonged the median survival to 36.5 days (p = 0.0016 relative to control). As previously observed with the BT-83 glioblastoma model (Fig. 3(i)), ectopic miR-603 expression suppressed BT-99 tumorigenicity and prolonged median survival to 34 days in the absence of additional treatment (p = 0.0007, Fig. 7(b)). In contrast to BT-99 with the empty vector, BT-99 with ectopic miR-603 expression was sensitive to TMZ, with median survival of 51.5 days after TMZ treatment (p < 0.0001 compared to TMZ-treated BT-99(miR-Empty)). This result confirmed the importance of miR-603 in MGMT regulation. Consistent with our previous observations with BT-83 (Fig. 3(i)), ectopic miR-603 expression increased sensitivity to IR, with median survival of 68.5 days after IR treatment (p < 0.0001 compared to IR-treated BT-99(miR-Empty)).
IR+TMZ combination improved median survival relative to TMZ or IR treatment alone in BT-99(miR-Empty)-implanted mice (median survival of 47, 24, 36.5 days for the IR+TMZ, TMZ, and IR groups, respectively; p < 0.0001 and =0.0118 compared to TMZ- and IR-treated mice, respectively). Ectopic miR-603 expression significantly prolonged median survival in mice treated with IR+TMZ, with 80% of the treated mice remaining viable after 120 days (p < 0.0001 compared to TMZ+IR-treated BT-99(miR-Empty) bearing mice).
4. Discussion
While studies in vitro and in vivo models have yielded mechanisms that potentially explain acquired radiation resistance in glioblastomas [42], the pertinence of these findings in the clinical setting requires interrogation of specimens secured pre- and post-treatment. There are several challenges in the securement of these specimens. Many patients do not undergo resection at the time of recurrence, and for those who undergo resection, discrepancy in the relative abundance of tumor and reactive cells between the original and the recurrent specimens render meaningful comparisons difficult [43]. These challenges are magnified when considering the inter-tumoral heterogeneity in IDH and MGMT status [44] in the context of the relative rarity of glioblastomas. The stability of miRNA in FFPE samples [11], which is available for all surgical patients, allows the maximal number of samples to be considered for the proposed study. Our strategy, therefore, was to identify miRNAs whose level changed after standard-of-care treatment and then to characterize their function as means to understand clinically pertinent biology. We studied miR-603 in this context.
Characterization of miR-603 revealed IGF1 and IGF1R as key downstream targets. Our study demonstrates that radiation induced EV-mediated export of miR-603, a master regulatory miRNA that governs multiple aspects of glioblastoma resistance. Decreased level of miR-603 in the cytoplasm de-represses IGF1 and IGF1R, to promote cellular transition into stem-cell like states. This transition, in turn, confers acquired resistance to IR. Additionally, EV export of miR-603 de-represses another key target, MGMT, which confers cross-resistance to DNA alkylating agents routinely used in glioblastoma treatment. These findings shed light on a previously unappreciated interaction between EV biology, DNA repair, and stem cell state regulation and define a novel framework for therapeutic development. In this context, we demonstrate that therapeutic resistance through EV-mediated miR-603 export can be overcome by exogenous miR-603 expression, suggesting plausibility of gene therapy in this context. Equally important to be noted, while there is increasing appreciation of the impact of EV-mediated genetic information transfer on the recipient cell, the consequences of this transfer on the EV secreting cell remain poorly characterized. Our results suggest that EV-mediated export of miR-603 has a fate-determining impact on the cell releasing the EVs.
The available literature suggests that CSC state and DNA repair independently contribute to glioblastoma resistance to DNA alkylating agents. For instance, the level of MGMT expression in glioblastoma cell lines with stem-cell properties remains a key determinant of TMZ sensitivity [21]. However, induction of the CSC state also increases glioblastoma resistance to DNA damaging agents through MGMT-independent mechanisms [45,46]. Interestingly, maintenance of the CSC state and increased MGMT expression appears to be co-regulated [47,48]. For instance, intra-tumoral hypoxia [47] or exposure to DNA damaging agents [48] simultaneously induces MGMT expression as well as properties associated with CSC states. Our results suggest that EV-mediated export of the miR-603 contributes to this coordination.
Of note, patients afflicted with wtIDH/umMGMT glioblastoma exhibit poor response to concurrent IR/TMZ therapy and essentially have no meaningful therapeutic option at the present time. The compelling efficacy of the exogenous miR-603 expression in synergizing with the standard-of-care treatment in unmethylated glioblastoma models is striking in this context and suggests opportunities for clinical translation, particularly in light of the recent approval of the first RNA-based therapy [49]. In addition to consideration of miR-603 as an up-front therapy in combination with IR/TMZ, the effectiveness of miR-603 in augmenting the tumoricidal effects of BCNU against recurrent glioblastomas suggests potential for clinical translation of miR-603-based therapy in this setting. It remains unclear whether the mechanisms of acquired resistance described in this study are applicable to glioblastomas with methylated MGMT promoter or mutant IDH (mIDH). It is likely that the absence of or low MGMT expression in glioblastoma with methylated MGMT promoters renders miR-603-mediated post-transcriptional regulation of MGMT inconsequential.
The critical importance of the IGF1 signaling axis in maintaining the glioblastoma stem-cell state and in radiation resistance has been reported by independent investigators [4,31]. Importantly, radiation delivery simultaneously up-regulates both IGF1 secretion and IGF1R expression [4]. Mechanism underlying this coordinated up-regulation remains unknown until this study. Our results indicate IR-induced EV export of miR-603 simultaneously up-regulates IGF1 and IGF1R. Supporting the clinical pertinence of this miR-603/IGF1 axis, we observed (1) significant correlation between miR-603 and IGF1 levels in clinical glioblastoma specimens, (2) statistically significant association between miR-603 level in clinical specimens and glioblastoma response to radiation. Additionally, single nucleotide polymorphisms in the IGF1 3′UTR that disrupted a miR-603 MRE is associated with cancer predisposition [50], potentially attributable to de-repression of IGF1.
In summary, our results suggest that concordant regulation of DNA repair and the CSC state in response to IR is mediated through EV-miR-603 export. Such export decreases cellular miR-603 level, resulting in de-repression of genes (IGF1, IGF1R, and MGMT) that mediate radiation resistance and cross-resistance to DNA alkylating agents. Importantly, the findings of this study were recapitulated using independent short-term and long-term passaged, patient-derived glioblastoma lines, clinical glioblastoma specimens, as well as independent in vitro and in vivo glioblastoma models. From a therapeutic perspective, our study suggests that miR-603-based therapeutic platforms hold translational potential in the treatment of wtIDH/umMGMT glioblastomas.
Declaration of Competing Interest
B.S.C. and C.C.C. received personal fees from Tocagen Co. Other authors declare no conflicts of interest.
Acknowledgments
Acknowledgments
We thank Drs. Subree Subramanian, David Largaespada, Tomoyuki Koga, and Ming Li for careful review of this manuscript.
Funding sources
The work is supported by NIH 1R01NS097649-01, NIH 9R44GM128223-02, 1R01CA240953-01, the Doris Duke Charitable Foundation Clinical Scientist Development Award, The Sontag Foundation Distinguished Scientist Award, the Kimmel Scholar Award, and BWF 1006774.01 (C.C.C). Funders did not have any role in study design, data collection, data analysis, interpretation, or writing of the report.
Author contributions
Conceptualization, V.R., B.X., and C.C.C.; Methodology, V.R., B.X., and C.C.C.; Investigation, V.R. and B.X.; Writing – Original Draft, B.X. and C.C.C.; Writing – Review & Editing, V.R., J.A., T.N., J.M., S.D., J.N., Y.M., W.H., E.K., F.F., B.S.C. and C.C.C.; Funding Acquisition, C.C.C.; Resources, Y.M., W.H., F.F., and B.S.C.; Supervision, C.C.C.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.ebiom.2020.102736.
Appendix. Supplementary materials
References
- 1.Bartek J., Jr., Ng K., Bartek J., Fischer W., Carter B., Chen C.C. Key concepts in glioblastoma therapy. J Neurol Neurosurg Psychiatry. 2012;83(7):753–760. doi: 10.1136/jnnp-2011-300709. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Henriksson R., Asklund T., Poulsen H.S. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: a review. J Neurooncol. 2011;104(3):639–646. doi: 10.1007/s11060-011-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osuka S., Van Meir E.G. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Investig. 2017;127(2):415–426. doi: 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Graham V., Khudyakov J., Ellis P., Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39(5):749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 7.Singh S.K., Clarke I.D., Terasaki M., Bonn V.E., Hawkins C., Squire J. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 8.Suva M.L., Rheinbay E., Gillespie S.M., Patel A.P., Wakimoto H., Rabkin S.D. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157(3):580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai E.C... Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30(4):363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 10.Garofalo M. Croce CM. microRNAs: master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol. 2011;51:25–43. doi: 10.1146/annurev-pharmtox-010510-100517. [DOI] [PubMed] [Google Scholar]
- 11.de Biase D., Visani M., Morandi L., Marucci G., Taccioli C., Cerasoli S. miRNAs expression analysis in paired fresh/frozen and dissected formalin fixed and paraffin embedded glioblastoma using real-time pCR. PLoS ONE. 2012;7(4):e35596. doi: 10.1371/journal.pone.0035596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhry M.A., Omaruddin R.A., Kreger B., de Toledo S.M., Azzam E.I. Micro RNA responses to chronic or acute exposures to low dose ionizing radiation. Mol Biol Rep. 2012;39(7):7549–7558. doi: 10.1007/s11033-012-1589-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akers J.C., Gonda D., Kim R., Carter B.S., Chen C.C. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113(1):1–11. doi: 10.1007/s11060-013-1084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raposo G., Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldie B.J., Dun M.D., Lin M., Smith N.D., Verrills N.M., Dayas C.V. Activity-associated miRNA are packaged in MAP1B-enriched exosomes released from depolarized neurons. Nucl Acids Res. 2014;42(14):9195–9208. doi: 10.1093/nar/gku594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shurtleff M.J., Temoche-Diaz M.M., Karfilis K.V., Ri S., Schekman R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife. 2016;5 doi: 10.7554/eLife.19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hegi M.E., Diserens A.C., Gorlia T., Hamou M.F., de Tribolet N., Weller M. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 19.Turcan S., Rohle D., Goenka A., Walsh L.A., Fang F., Yilmaz E. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nunez F.J., Mendez F.M., Kadiyala P., Alghamri M.S., Savelieff M.G., Garcia-Fabiani M.B. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med. 2019;11(479) doi: 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W., Zhang J., Hoadley K., Kushwaha D., Ramakrishnan V., Li S. miR-181d: a predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol. 2012;14(6):712–719. doi: 10.1093/neuonc/nos089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akers J.C., Ramakrishnan V., Kim R., Skog J., Nakano I., Pingle S. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): a platform for glioblastoma biomarker development. PLoS ONE. 2013;8(10):e78115. doi: 10.1371/journal.pone.0078115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozono D., Li J., Nitta M., Sampetrean O., Gonda D., Kushwaha D.S. Dynamic epigenetic regulation of glioblastoma tumorigenicity through LSD1 modulation of MYC expression. Proc Natl Acad Sci USA. 2015;112(30):E4055–E4064. doi: 10.1073/pnas.1501967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kushwaha D., Ramakrishnan V., Ng K., Steed T., Nguyen T., Futalan D. A genome-wide miRNA screen revealed miR-603 as a MGMT-regulating miRNA in glioblastomas. Oncotarget. 2014;5(12):4026–4039. doi: 10.18632/oncotarget.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y., Li J., Nitta M., Futalan D., Steed T., Treiber J.M. Orthogonal targeting of EGFRVIII expressing glioblastomas through simultaneous EGFR and PLK1 inhibition. Oncotarget. 2015;6(14):11751–11767. doi: 10.18632/oncotarget.3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlson B.L., Pokorny J.L., Schroeder M.A., Sarkaria J.N. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. 2011 doi: 10.1002/0471141755.ph1416s52. Chapter 14:Unit 14 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parrish K.E., Cen L., Murray J., Calligaris D., Kizilbash S., Mittapalli R.K. Efficacy of PARP inhibitor RUCAPARIB in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther. 2015;14(12):2735–2743. doi: 10.1158/1535-7163.MCT-15-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak K.J., Schmittgen T.D... Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Chan J.A., Krichevsky A.M., Kosik K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65(14):6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 31.Turner B.C., Haffty B.G., Narayanan L., Yuan J., Havre P.A., Gumbs A.A. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res. 1997;57(15):3079–3083. [PubMed] [Google Scholar]
- 32.Osuka S., Sampetrean O., Shimizu T., Saga I., Onishi N., Sugihara E. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31(4):627–640. doi: 10.1002/stem.1328. [DOI] [PubMed] [Google Scholar]
- 33.Hart L.S., Dolloff N.G., Dicker D.T., Koumenis C., Christensen J.G., Grimberg A. Human colon cancer stem cells are enriched by insulin-like growth factor-1 and are sensitive to figitumumab. Cell Cycle. 2011;10(14):2331–2338. doi: 10.4161/cc.10.14.16418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu C., Xie D., Yu S.C., Yang X.J., He L.R., Yang J. Beta-Catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res. 2013;73(10):3181–3189. doi: 10.1158/0008-5472.CAN-12-4403. [DOI] [PubMed] [Google Scholar]
- 35.Weller M., Cloughesy T., Perry J.R., Wick W. Standards of care for treatment of recurrent glioblastoma – are we there yet? Neuro Oncol. 2013;15(1):4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marathi U.K., Kroes R.A., Dolan M.E., Erickson L.C. Prolonged depletion of O6-methylguanine DNA methyltransferase activity following exposure to O6-benzylguanine with or without streptozotocin enhances 1,3-bis(2-chloroethyl)-1-nitrosourea sensitivity in vitro. Cancer Res. 1993;53(18):4281–4286. [PubMed] [Google Scholar]
- 37.Das S.K., Sokhi U.K., Bhutia S.K., Azab B., Su Z.Z., Sarkar D. Human polynucleotide phosphorylase selectively and preferentially degrades microRNA-221 in human melanoma cells. Proc Natl Acad Sci USA. 2010;107(26):11948–11953. doi: 10.1073/pnas.0914143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson M.B., Woodworth-Hobbs M.E., Zheng B., Rahnert J.A., Blount M.A., Gooch J.L. miR-23a is decreased during muscle atrophy by a mechanism that includes calcineurin signaling and exosome-mediated export. Am J Physiol Cell Physiol. 2014;306(6):C551–C558. doi: 10.1152/ajpcell.00266.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thery C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1) doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulcahy L.A., Pink R.C., Carter D.R. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3 doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abels E.R., Maas S.L.N., Nieland L., Wei Z., Cheah P.S., Tai E. Glioblastoma-Associated microglia reprogramming is mediated by functional transfer of extracellular miR-21. Cell Rep. 2019;28(12):3105. doi: 10.1016/j.celrep.2019.08.036. –19 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J., Li Y., Yu T.S., McKay R.M., Burns D.K., Kernie S.G. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan U.A., Rennert R.C., White N.S., Bartsch H., Farid N., Dale A.M. Diagnostic utility of restriction spectrum imaging (RSI) in glioblastoma patients after concurrent radiation-temozolomide treatment: a pilot study. J Clin Neurosci. 2018;58:136–141. doi: 10.1016/j.jocn.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Yang P., Zhang W., Wang Y., Peng X., Chen B., Qiu X. IDH mutation and MGMT promoter methylation in glioblastoma: results of a prospective registry. Oncotarget. 2015;6(38):40896–40906. doi: 10.18632/oncotarget.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shou J., Ali-Osman F., Multani A.S., Pathak S., Fedi P., Srivenugopal K.S. Human DKK-1, a gene encoding a WNT antagonist, responds to DNA damage and its overexpression sensitizes brain tumor cells to apoptosis following alkylation damage of DNA. Oncogene. 2002;21(6):878–889. doi: 10.1038/sj.onc.1205138. [DOI] [PubMed] [Google Scholar]
- 46.Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 47.Pistollato F., Abbadi S., Rampazzo E., Persano L., Della Puppa A., Frasson C. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells. 2010;28(5):851–862. doi: 10.1002/stem.415. [DOI] [PubMed] [Google Scholar]
- 48.Auffinger B., Tobias A.L., Han Y., Lee G., Guo D., Dey M. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Diff. 2014;21(7):1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mullard A. FDA approves landmark RNAi drug. Nat Rev Drug Discov. 2018;17(9):613. doi: 10.1038/nrd.2018.152. [DOI] [PubMed] [Google Scholar]
- 50.Jiang H., Wang H., Ge F., Wu L., Wang X., Chen S. The functional variant in the 3′UTR of IGF1 with the risk of gastric cancer in a Chinese population. Cell Physiol Biochem. 2015;36(3):884–892. doi: 10.1159/000430263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.