

Abstract
The protein-tyrosine phosphatase SHP2 is an allosteric enzyme critical for cellular events downstream of growth factor receptors. Mutations in the SHP2 gene have been linked to many different types of human diseases, including developmental disorders, leukemia, and solid tumors. Unlike most SHP2-activating mutations, the T507K substitution in SHP2 is unique in that it exhibits oncogenic Ras-like transforming activity. However, the biochemical basis of how the SHP2/T507K variant elicits transformation remains unclear. By combining kinetic and biophysical methods, X-ray crystallography, and molecular modeling, as well as using cell biology approaches, here we uncovered that the T507K substitution alters both SHP2 substrate specificity and its allosteric regulatory mechanism. We found that although SHP2/T507K exists in the closed, autoinhibited conformation similar to the WT enzyme, the interactions between its N-SH2 and protein-tyrosine phosphatase domains are weakened such that SHP2/T507K possesses a higher affinity for the scaffolding protein Grb2-associated binding protein 1 (Gab1). We also discovered that the T507K substitution alters the structure of the SHP2 active site, resulting in a change in SHP2 substrate preference for Sprouty1, a known negative regulator of Ras signaling and a potential tumor suppressor. Our results suggest that SHP2/T507K's shift in substrate specificity coupled with its preferential association of SHP2/T507K with Gab1 enable the mutant SHP2 to more efficiently dephosphorylate Sprouty1 at pTyr-53. This dephosphorylation hyperactivates Ras signaling, which is likely responsible for SHP2/T507K's Ras-like transforming activity.
Keywords: phosphatase, cancer, transformation, structure–function, kinetics, signaling, mutation, SHP2
Introduction
Protein-tyrosine phosphatases (PTPs)3 are signaling enzymes that, in partnership with protein-tyrosine kinases (PTKs), modulate the homeostasis of protein tyrosine phosphorylation within the cell. Excessive tyrosine phosphorylation is frequently associated with oncogenic transformation, often thought of as a result of abnormal activation of PTKs. Hence, PTPs have traditionally been perceived as products of tumor suppressor genes. Interestingly, growing evidence has shown that PTPs can also act as oncoproteins. To this end, the Src homology 2 (SH2) domain containing protein-tyrosine phosphatase 2 (SHP2), encoded by PTPN11, mediates signal transduction in response to growth factors for full activation of Ras, a molecular switch that controls vital cellular processes such as proliferation, survival, and migration (1, 2). Unsurprisingly, abnormal activation or mutations in Ras are among the most common causative alterations in human cancers. Unfortunately, drugging oncogenic Ras proteins directly has historically been quite a challenge. Thus, there is heightened interest in therapeutic targeting of key regulatory molecules, such as SHP2, that contribute to the oncogenic potential of Ras.
As an allosteric enzyme, SHP2 is composed of two N-terminal SH2 domains (N-SH2 and C-SH2), a PTP catalytic domain in the middle, and a C-terminal tail. In the basal state, SHP2 exists primarily in an autoinhibited closed conformation, as a result of intramolecular interactions between the N-SH2 domain and the PTP active site (3). Upon growth factor stimulation, engagement of the SH2 domains by specific phosphotyrosine (pTyr) motifs in receptor-like PTKs (e.g. EGF receptor) and/or scaffold proteins (e.g. Gab1) localizes SHP2 to its intended substrates and concurrently lessens the binding between the N-SH2 and PTP domain, leading to an open and active conformation that is competent for SHP2 substrate turnover. The importance of SHP2 in human health is underscored by the large number of SHP2 mutations that are associated with a range of diseases, including developmental disorders such as Noonan syndrome and Noonan syndrome with multiple lentigines (NSML), various forms of leukemia, and several types of solid tumors (4). Nonetheless, despite our knowledge of various SHP2 mutations that are connected to different pathological conditions, the molecular basis for many disease-causing SHP2 variants remains poorly understood. A prime case in point is the Thr-507 to Lys (T507K) substitution in SHP2.
The SHP2/T507K mutation is found in several solid tumors, including neuroblastoma (5), glioblastoma (6), and hepatocellular carcinoma (7). Unlike Noonan syndrome or leukemia-specific SHP2 mutants, SHP2/T507K displays oncogenic Ras-like transforming activity (7). However, the underlying biochemical mechanism for T507K's transforming potential has not been fully delineated. Intriguingly, residue Thr-507 is highly conserved among the PTPs (8) and is located within the catalytic Q-loop in the SHP2 phosphatase-active site (Fig. S1), facing the N-SH2 domain in the basal autoinhibited state (Fig. S2). This raises the possibility that structural alteration of Thr-507 may affect SHP2 phosphatase activity and/or the regulatory mechanism. Here, we combined enzyme kinetics, X-ray crystallography, molecular modeling, hydrogen/deuterium exchange MS, and cell biology to elucidate the structural and mechanistic features of the SHP2/T507K variant that may be responsible for its transforming activity. We found that although SHP2/T507K still adopts the closed conformation like the native enzyme, its N-SH2/PTP inter-domain interaction is weakened, triggering an increased propensity for the mutant SHP2 to favor the open and active state. As a consequence, the SH2 domains of SHP2/T507K also possess a higher affinity for the scaffolding protein Gab1. We revealed that the Thr-507 to Lys mutation also alters the SHP2 active–site structure, resulting in a change in SHP2 substrate preference for Sprouty1, a known negative regulator of Ras signaling and a potential tumor suppressor. Our data showed that preferential association of SHP2/T507K with Gab1 enables the mutant SHP2 to more efficiently dephosphorylate Sprouty1 at Tyr-53, which likely accounts for SHP2/T507K's Ras-like transforming activity.
Results
Effects of T507K mutation on SHP2 phosphatase activity, substrate specificity, and regulatory mechanism
PTP catalysis cycles through a thiophosphoryl enzyme intermediate involving the active-site cysteine (Cys-459 in SHP2) in the P-loop (Fig. S3) (9). Formation of the phosphoenzyme intermediate is facilitated by a general acid (Asp-425 in SHP2) in the WPD loop, which neutralizes the build up of a negative charge on the leaving group. The breakdown of the phosphoenzyme intermediate occurs by the attack of a nucleophilic water aided by Asp-425, which now functions as a general base. Unique to the classical PTPs is an invariant Gln residue (Gln-506 in SHP2) located within the Q-loop, which holds both the general base Asp-425 and the nucleophilic water in place for efficient phosphoenzyme hydrolysis (10, 11). Given its proximity to Gln-506 and N-SH2 domain in the basal state, we speculated that substitutions at Thr-507 might alter SHP2 phosphatase activity and/or the allosteric regulatory mechanism.
To assess the impact of the T507K mutation on SHP2 phosphatase activity, we determined the kinetic parameters for recombinant WT as well as the T507K mutant SHP2-catalyzed reaction, using para-nitrophenyl phosphate (pNPP) as a substrate at pH 7.0 and 25 °C. Both the full-length SHP2 lacking the C-terminal tail (residues 1–528, designated as FL) and the PTP catalytic domain (residues 224–528, designated as CD) were utilized for the kinetic experiments. Because pNPP mimics pTyr, its hydrolysis by SHP2 is sensitive to structural perturbations in the active site (i.e. pTyr-binding site). Therefore, a comparison of the kinetic parameters for the WT and mutant SHP2 with pNPP as a substrate will reveal the effect of the T507K mutation on the intrinsic phosphatase activity of SHP2. Additionally, assessment of the kinetic properties of the FL and CD constructs of the WT and T507K mutant enzymes will also provide insight into the involvement of Thr-507 in SHP2 allosteric regulation. As shown in Table 1 and similar to previous findings (12, 13), FL SHP2 exhibits a kcat that is 50-fold lower than that of the CD (likely due to inhibition by the N-SH2 domain in the closed state), which is consistent with the closed and autoinhibited conformation observed for the FL SHP2 in the crystal structure (3). The kcat and Km values for the mutant CD-catalyzed pNPP hydrolysis are 20 and 49% lower than those of the WT counterpart, indicating that the T507K mutation has minimal impact on SHP2's intrinsic phosphatase activity. Interestingly, the kcat for FL T507K is still 23-fold lower than that of the corresponding CD, suggesting that the FL T507K mutant may also exist in a mostly closed state, albeit with a higher propensity for the open conformation as a result of decreased autoinhibition compared with the native enzyme (23-fold versus 50-fold) (Table 1).
Table 1.
Kinetic parameters of wild-type SHP2 and T507K mutant with pNPP as a substrate
| Enzyme | CD |
FL |
Extent of autoinhibition |
|||
|---|---|---|---|---|---|---|
| kcat | Km | kcat | Km | (CD/FL) kcat | (CD/FL) kcat/Km | |
| s−1 | mm | s−1 | mm | |||
| WT | 6.5 ± 0.3 | 4.3 ± 0.5 | 0.13 ± 0.02 | 3.4 ± 0.5 | 50 | 40 |
| T507K | 5.2 ± 0.1 | 2.2 ± 0.2 | 0.23 ± 0.01 | 1.7 ± 0.0 | 23 | 17 |
To further assess the effect of T507K mutation on SHP2 activity, we also studied the hydrolysis of physiologically relevant substrates, namely pTyr-containing peptides derived from previously described SHP2 substrate proteins, including Sprouty1/pY53 (GSNEpY53TEGPS) (14), paxillin/pY118 (EEHVpY118SFPN) (15), Gab1/pY589 (DSEENpY589VPMNPNL) (16), and EGFR/pY992 (DADEpY992LIPQQG) (17), by the native and mutant SHP2. To avoid complications due to potential binding between the N-SH2 domain and these pTyr-containing peptides, we determined the kcat/Km, a measure of catalytic efficiency as well as substrate specificity, under conditions where the substrate concentration was ≪Km. As shown in Table 2, the kcat/Km values for the WT CD-catalyzed dephosphorylation of the pTyr peptides are 12–24-fold higher than those of the FL SHP2, again in line with the latter being in the autoinhibited state. In support of the notion that SHP2/T507K is in a less autoinhibited state as compared with the WT protein, the kcat/Km values for the mutant CD-catalyzed reactions are 5–18-fold higher than those of the FL T507K. Unlike the situation with pNPP, the kcat/Km values for the mutant CD-catalyzed hydrolysis of paxillin/pY118, Gab1/pY589, and EGFR/pY992 peptides are 6–16-fold lower than those by the WT CD. Interestingly, the kcat/Km values associated with the Sprouty1/pY53 peptide hydrolysis by both the CD and FL SHP2/T507K constructs are similar to those of the WT counterparts. Taken together, the results suggest that although the T507K mutation has minimal effect on the intrinsic phosphatase activity (i.e. similar kcat values of pNPP for WT and T507K CDs), it may alter SHP2 substrate specificity and disrupt the autoinhibitory interaction between the N-SH2 and PTP domain.
Table 2.
kcat/Km values (×10−3m−1s−1) of wildtype SHP2 and T507K mutant with pTyr-peptides as substrates
| Enzyme | Substrate |
|||
|---|---|---|---|---|
| Sprouty1/pY53 | Paxillin/pY118 | Gab1/pY589 | EGFR/pY992 | |
| WT(CD) | 110 ± 0.9 | 123 ± 1.0 | 60 ± 0.7 | 1167 ± 23 |
| T507K(CD) | 50.0 ± 0.8 | 10.5 ± 0.2 | 11.0 ± 0.3 | 72.5 ± 1.5 |
| WT(FL) | 5.1 ± 0.1 | 5.2 ± 0.2 | 3.0 ± 0.07 | 95 ± 1.4 |
| T507K(FL) | 8.5 ± 0.1 | 0.8 ± 0.01 | 0.6 ± 0.01 | 15.0 ± 0.5 |
| Change in kcat/Km | ||||
| WT(CD)/T507K(CD) | 2.2 | 11.7 | 5.5 | 16.1 |
| WT(FL)/T507K(FL) | 0.6 | 6.5 | 5.0 | 6.3 |
| Extent of autoinhibition | ||||
| WT (CD/FL)kcat/Km | 21.6 | 23.7 | 20.0 | 12.3 |
| T507K (CD/FL)kcat/Km | 5.9 | 13.1 | 18.3 | 4.8 |
Structural perturbations in SHP2 introduced by the T507K mutation
To determine the structural impact of T507K mutation, we prepared crystals of the FL SHP2/T507K protein and solved its three-dimensional structure by X-ray crystallography at 2.25 Å (Table 3). The structure belongs to the P21212 space group with one SHP2/T507K molecule per asymmetric unit. Similar to the WT enzyme, SHP2/T507K crystallized in the closed autoinhibited conformation, in which the D′E-loop within the N-SH2 domain directly occludes access to the phosphatase active-site pocket formed by five loop fragments in the PTP domain, namely the P-loop, pY-loop, WPD-loop, Q-loop, and E-loop (Fig. 1A). The overall SHP2/T507K structure is similar to the previously reported WT SHP2 structure (PDB code 4DGP) (12) with a root-mean-square deviation (RMSD) value of 1.423 Å for 486 Cα atoms between the two structures. However, the conformations of two surface loops in SHP2/T507K deviate significantly from those in the native enzyme (Fig. 1A). One surface loop is the BG-loop in the N-SH2 domain, which creates an open groove with the adjacent EF-loop to enable binding interactions between the N-SH2 domain and its pTyr peptide ligands (3, 18). As shown in Fig. 1B, the groove between the EF- and BG-loop in WT SHP2 is ∼8 Å wide, which is incompetent for pTyr peptide binding (3). In contrast, the groove width widens to ∼11 Å in SHP2/T507K, which would likely better accommodate a phosphopeptide. Thus, it appears that the N-SH2 domain in SHP2/T507K has a more favorable conformation for engagement with pTyr-phosphorylated receptor-like PTKs and/or scaffold proteins in the basal state. The other surface loop is E-loop in the PTP domain, which constitutes one of five loops lining the active-site pocket and is important for substrate recognition and binding. As shown in Fig. 1C, the conformational change in E-loop significantly alters the shape and electrostatic properties of the active-site pocket, which may account for the observed change in T507K substrate specificity (Table 2 and below).
Table 3.
Data collection and structure refinement statistics
The data were collected from a single crystal. Values in parentheses are for the highest-resolution shell.
| SHP2/T507K | |
|---|---|
| Data collection | |
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 54.699, 203.176, 44.366 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 2.25 |
| Total observations | 105,858 |
| Unique observations | 23,674 |
| Completeness (%) | 95.7 (93.8) |
| Redundancy | 4.5 (4.0) |
| 〈I〉/σ | 12.7 (1.5) |
| Rmerge (%) | 14.0 (81.1) |
| Structure refinement | |
| Resolution (Å) | 2.25 |
| Rwork/Rfree | 0.198/0.262 |
| RMSD bond lengths (Å) | 0.008 |
| RMSD bond angles (°) | 1.005 |
| Ramachandran plot (%) | |
| Most favored | 87.4 |
| Additionally allowed | 11.0 |
| Generously allowed | 1.1 |
| Disallowed | 0.5 |
Figure 1.

Structural perturbations in SHP2 introduced by the T507K mutation. A, SHP2/T507K (purple) shows similar overall structure compared with WT SHP2 (gray, PDB code 4DGP) in the closed conformation, but with structural alterations around the BG-loop and E-loop, and its N-terminal SH2 domains shifting slightly away from the PTP domain. B, conformational change of the BG-loop leads to a more open BG/EF gate in SHP2/T507K, which potentially benefits the binding of N-SH2 domain with adapter proteins for the T507K mutant. To illustrate this effect, the structure of N-SH2·phosphopeptide (PDB code 1AYA) was superimposed, respectively, onto the N-SH2 domain in WT or T507K mutant SHP2, and the phosphopeptide (yellow ribbon with side chain) was shown to represent a bound conformation. C, conformational change for the E-loop in SHP2/T507K alters the catalytic active site pocket. D, potential driving force for SH2 domain shifting in SHP2/T507K. The T507K structure (purple) was superimposed onto the WT SHP2 structure (gray). The steric repulsions from the T507K mutation are presented by a red dashed line, and the newly-added hydrogen bond is indicated by a green dashed line.
In addition to the above-noted conformational changes in the BG- and E-loop, a slight shift in position of the two SH2 domains is also observed in the SHP2/T507K structure (Fig. 1A). A comparative analysis around the site of mutation reveals that replacement of Thr-507 by a Lys produces strong steric repulsion with residues Thr-59 and Gly-39 in the N-SH2 domain and also introduces a new H-bond between Lys-507 and the backbone oxygen of Asn-58, which draws Asn-58 closer to the Q-loop, causing a steric clash with the backbone of Gly-39 (Fig. 1D). Driven by these repulsive forces, the BC-loop (residues 34–41) shifts away from the PTP domain, which then pushes out the adjacent αA (residues 13–23), the N-terminal segment (residues 2–12), the linker between the two SH2 domains (residues 104–111), and most of the C-SH2 domain, which likely weakens the interactions between the N-SH2 domain and the PTP domain. The collective effect of these sequential movements is that in the closed conformation of SHP2/T507K, the N-SH2 and C-SH2 domains move away from the PTP domain by 1.3 Å, causing a destabilization of the autoinhibited conformation. To corroborate this conclusion, molecular dynamics (MD) simulation-based molecular mechanics–generalized born surface area (MM–GBSA) approach was employed to calculate the N-SH2/PTP domain interaction energy for the WT and T507K mutant SHP2. From the trajectory of a 30-ns MD simulation, the free energy of WT SHP2 or the T507K mutant was calculated and decomposed into pairwise residues, and the inter-domain interaction energy was determined by adding up the energy of all N-SH2/PTP interface residue pairs. As expected, the RMSD value of all backbone atoms during the MD simulation is significantly larger for SHP2/T507K, and the N-SH2/PTP interaction energy is ∼24 kcal/mol higher for T507K (−154.2 ± 9.0 kcal/mol) than that of the WT protein (−177.5 ± 8.7 kcal/mol) (Fig. S4), consistent with a more dynamic and weakened N-SH2/PTP interaction in SHP2/T507K as predicted from the structural analyses. Collectively, our structural and computational studies indicate that the SHP2/T507K mutant exists predominantly in the closed conformation, but with decreased N-SH2/PTP interdomain autoinhibitory interaction.
SHP2/T507K exhibits increased propensity to transition from the closed to open conformation
To provide further evidence that the T507K mutation diminishes the inter-domain interaction between the N-SH2 and PTP domain, we determined the affinity of the isolated N-SH2 domain for both the WT and SHP2/T507K catalytic domains. Consistent with the observation that the D′E-loop of N-SH2 domain occupies the PTP active site, the recombinant N-SH2 domain competitively inhibited the SHP2 catalytic domain-catalyzed pNPP hydrolysis with a Ki of 1.3 ± 0.1 μm. In line with the weaker interaction between the N-SH2 and PTP domain in SHP2/T507K, the Ki value (46.3 ± 1.9 μm) for the N-SH2 domain–mediated inhibition of the SHP2/T507K catalytic domain–catalyzed reaction is 36-fold higher than that for the WT PTP domain (Fig. S5). Moreover, to complement the crystallographic and computational results, we also carried out hydrogen/deuterium exchange MS (H/DX-MS) experiments to measure the dynamic and conformational changes in SHP2 as a result of the T507K mutation. H/DX-MS combined with peptidic mapping enables segment-specific detection of solvent-accessible exchange sites, which directly characterizes the dynamic/conformational changes within a protein (19, 20). To that end, we compared the overall solution dynamic properties of WT SHP2, SHP2/T507K, as well as SHP2/E76K, the most common leukemia-specific SHP2 mutant known to be in a fully open and activated conformation (13, 21, 22). We identified 38 peptides for each protein, covering ∼92% of the SHP2 sequence (residues 1–528). The backbone amide H/D exchange for each protein was determined; the difference in deuterium uptake between the mutant and WT for each peptide was calculated at various time points; the two-dimensional “heat map” (Fig. S6) and the three-dimensional “heat map” (Fig. 2) for the mutant displaying differential H/D exchange relative to the WT were generated. In Fig. 2, the differential H/D exchange for the SHP2/E76K mutant at 1 min is shown as a reference for the magnitude and location of H/D exchange experienced by SHP2 in the open conformation. Compared with WT SHP2, several peptides in SHP2/E76K, located at the interface of the N-SH2 and PTP domains, exhibit significant build up in deuterium incorporation. These peptides reside in the D′E loop and adjacent B, C, D, D′, E, and F β-strands, which are known to be involved in binding with the PTP domain. In the PTP domain, increased H/D exchange is also observed in peptides located within the loop fragments forming the active-site pocket, including the P-loop, pY-loop, E-loop, and Q-loop. The observed increase in the exchange of backbone amide hydrogen located between the N-SH2 and PTP domain interface with deuterium in bulk solvent is consistent with the N-SH2- and PTP-binding interface in SHP2/E76K being solvent-exposed and SHP2/E76K existing in an open, active conformation. As expected, the SHP2/T507K mutant also experiences increased deuterium uptake at the interface between the N-SH2 and PTP domain, although the extent of H/D exchange is attenuated compared with the SHP2/E76K mutant (Fig. 2). This finding suggests that residues within the SHP2/T507K N-SH2/PTP binding interface show increased solvent exposure/conformational flexibility and further supports our conclusion that the SHP2/T507K mutant has a higher tendency to transition from a closed autoinhibitory conformation to an open activated state.
Figure 2.
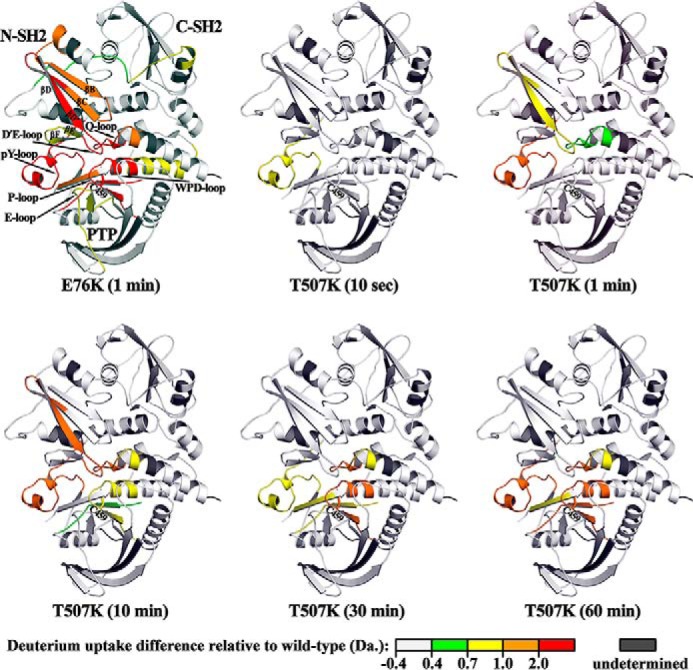
SHP2/T507K has higher conformational dynamics and propensity for the open conformation. The differential H/D exchange (mutant versus WT) at each time point is mapped on the WT SHP2 crystal structure to generate a 3D “heat map for conformational dynamics.” The heat map for the E76K mutant at 1 min is shown as a reference for differential H/D exchange magnitude and location in an open conformation. Starting from 10 s, SHP2/T507K shows increased deuterium uptake for several peptides at the N-SH2/PTP interface, indicating that this interface is more dynamic and solvent-exposed in the mutant compared with the WT enzyme.
Molecular basis for the altered substrate specificity of SHP2/T507K
To understand why SHP2/T507K and its WT counterpart have the same pNPP activity but exhibit different kcat/Km values toward pTyr-containing peptides, we compared the active-site structures between the two enzymes. As shown in Fig. 3A, the positioning of the catalytic residues in the P-loop (e.g. Cys-459 and Arg-465), WPD-loop (e.g. Asp-425), pY-loop (e.g. Tyr-279), and Q-loop (e.g. Gln-506 and Gln-510) is nearly identical within the active site of both the native and mutant enzymes. In addition, residue 507 is situated ∼11 Å away from pNPP and is not directly involved in substrate binding. Thus, it is understandable that the catalytic domains of SHP2/T507K and WT SHP2 display very similar phosphatase activity toward pNPP.
Figure 3.

Molecular basis for SHP2/T507K substrate specificity. A, comparison of active site residues in the WT SHP2 (green) and T507K mutant (cyan) with a modeled pNPP (yellow) binding at the active site. B, likely binding model for Sprouty1/pY53 peptide with SHP2 WT and T507K mutant catalytic domain. The electrostatic potential was mapped on the protein surface, and residues Thr-507 and Lys-507 are shown as sticks below the slightly transparent surface. C, sequence comparison among four pTyr-containing peptide substrates. D, atom fluctuation heat map for Sprouty1/pY53 peptide during a 5-ns MD simulation of two models in B. E, binding free energies for four indicated systems were calculated by MM-GBSA method.
In contrast to the lack of influence on SHP2's intrinsic phosphatase activity, replacement of Thr-507 by a Lys significantly reduces the catalytic efficiencies of SHP2/T507K toward peptide substrates paxillin/pY118, Gab1/pY589, and EGFR/pY992 (Table 2). Strikingly, the kcat/Km value for SHP2/T507K-catalyzed hydrolysis of Sprouty1/pY53 is comparable with that of the WT SHP2. To gain molecular insight into the change in SHP2/T507K substrate specificity, we modeled binding interactions of the Sprouty1/pY53 peptide with the catalytic domain of WT and the T507K mutant SHP2 (Fig. 3B). The peptide shows a similar overall binding mode in both models: the N-terminal residues (minus positions in Fig. 3C) are situated in a groove between the pY- and E-loop; the pTyr-53 inserts into the active-site pocket; and the C-terminal residues (plus positions in Fig. 3C) cross a saddle between pY-loop and Q-loop and extend to the previously reported second aryl phosphate-binding site in PTP1B (23). Because of the displacement of the E-loop in SHP2/T507K, the well-defined N-terminal peptide-binding groove in WT SHP2 is disrupted, leading to an impaired interaction with N-terminal residues of peptide substrates. On the other side, the T507K mutation also introduces a longer and positive side chain to make additional H-bonds with the glutamic acid at the +2 position of Sprouty1/pY53, which likely boosts the interaction of the C-terminal residues with SHP2. Consequently, it is plausible that the enhanced interactions between SHP2/T507K and the C-terminal residues of Sprouty1/pY53 may compensate for its reduced binding with the N-terminal residues such that the mutant enzyme displays similar or even better catalytic efficacy toward Sprouty1/pY53 than does WT SHP2.
Sequence comparison of all four peptide substrates (Fig. 3C) revealed that the +2 Glu is unique to Sprouty1/pY53. The hydrophobic residues (i.e. Phe, Pro, and Ile) at the +2 position in the other peptides cannot form H-bonds with Lys-507 to compensate for the reduced interaction at the N terminus, which may explain SHP2/T507K's reduced activity toward these peptides (Table 2). To provide further supporting evidence, we carried out molecular dynamics (MD) simulation–based MM-GBSA energy calculation. The atom fluctuation heat maps for Sprouty1/pY53 with the WT SHP2 and SHP2/T507K are shown in Fig. 3D. The pTyr residue in both systems exhibits practically the same mobility, a reflection of its stable engagement with the active site. The SHP2/T507K-bound Sprouty1/pY53 peptide shows larger fluctuations for the N-terminal residues but smaller ones for the C-terminal residues, which is a dynamic consequence of reduced and enhanced interactions of SHP2/T507K with the N and C termini, respectively. Based on the MD trajectory, we calculated that the binding free energy (BFE) for Sprouty1/pY53 with WT and SHP2/T507K was comparable (Fig. 3E), in accordance with the similar kcat/Km values for Sprouty1/pY53 dephosphorylation catalyzed by the WT SHP2 and SHP2/T507K. As a reference, we found that the BFE for EGFR/pY992 and SHP2/T507K is 7.02 kcal/mol higher than that for EGFR/pY992 and WT SHP2 (Fig. 3E), consistent with the greatly decreased kcat/Km value for the SHP2/T507K-catalyzed hydrolysis of EGFR/pY992.
Mechanism for SHP2/T507K's transforming activity
Unlike activating the SHP2 mutations associated with Noonan syndrome or leukemia, SHP2/T507K was reported to exhibit oncogenic Ras-like transforming activity in NIH3T3 mouse fibroblast cells (7). To further establish SHP2/T507K's transforming activity, we carried out soft agar colony formation assays using HEK293 cells. As shown in Fig. 4A, no appreciable colony formation was observed in vector control or WT SHP2-expressed cells. In contrast, SHP2/T507K cells formed numerous large colonies in soft agar, indicating that SHP2/T507K also exhibits transforming activity in HEK293 cells. Because uncontrolled cell growth and invasion are common features for cancer, we examined whether the T507K mutation alters SHP2's ability to promote cell proliferation and migration. Compared with the WT enzyme, SHP2/T507K increases HEK293 cell proliferation by 1.5-fold as measured by BrdU incorporation (Fig. 4B). Similarly, overexpression of SHP2/T507K in HEK293 cells led to a 1.6-fold higher rate of cell migration than that of the WT SHP2 cells (Fig. 4C). Collectively, we confirmed the transforming activity of SHP2/T507K in HEK293 cells and found that this SHP2 mutant can further promote cell proliferation and migration as compared with WT SHP2.
Figure 4.

SHP2/T507K mutant has transforming activity in HEK293 cells. A, only SHP2/T507K but not the vector or WT SHP2 overexpression leads to colony formation. B, SHP2/T507K-expressed cells have higher BrdU incorporation than that of the WT SHP2 cells. In all scatter plots hereafter, the bold black line and thin gray line show the mean and standard deviation of three or four independent experiments, and the star means statistical significance with p < 0.05 from Student's t test. C, SHP2/T507K cells display a higher rate of cell migration than that of WT as measured by the transwell migration assay.
Given that SHP2 is a positive mediator of receptor tyrosine kinase-driven signaling, we investigated the effect of SHP2/T507K on ERK1/2 and AKT activation upon stimulation by several growth factors. We found that the T507K mutation further promoted (versus WT SHP2) hepatocyte growth factor- (HGF) or epidermal growth factor (EGF)-stimulated ERK1/2 and AKT activation (Fig. S7A), with HGF being more potent than EGF. Thus, we focused our mechanistic study under HGF-stimulated conditions. As shown in Fig. 5A, SHP2/T507K is more efficient than WT SHP2 in activating both ERK1/2 and AKT. The data also indicate that although SHP2/T507K does not shift the HGF-stimulated ERK1/2 and AKT activation kinetics, it produces a heightened and sustained ERK1/2 and AKT activation at later time points (Fig. S8). Consistent with the weakened intramolecular interaction between the N-SH2 and PTP domains, we also found that the SHP2/T507K-mediated ERK1/2 and AKT activation is more sensitive to HGF stimulation than the WT SHP2 (Fig. S7B). We also examined the total and phospho-Met upon HGF stimulation in both WT SHP2 and SHP2/T507K-expressing cells. There is no obvious difference in Met expression or Met phosphorylation at Tyr-1234/1235, which is crucial for its kinase activity, at all time points between WT SHP2 and SHP2/T507K overexpressed cells upon HGF stimulation (Fig. S7C). This observation excludes the possibility that SHP2/T507K overactivates ERK1/2/AKT through up-regulating Met.
Figure 5.

SHP2/T507K preferentially binds with Gab1 and overactivates Ras, ERK1/2, and AKT. A, compared with WT SHP2, the T507K mutant can further activate ERK1/2 and AKT upon HGF stimulation at later time points (i.e. 30 and 60 min). The overactivation of ERK1/2 by the SHP2/T507K mutant was defined as SHP2/T507K-induced ERK1/2 activation (i.e. pERK1/2/ERK1/2 in SHP2/T507K-expressing cells) over WT SHP2-induced ERK1/2 activation (i.e. pERK1/2/ERK1/2 in WT SHP2-expressing cells). The overactivation of AKT was calculated the same way but using the ratio pAKT/AKT. B, SHP2/T507K mutant promotes Ras activation at later time points versus WT SHP2. C, Ras–SOS binding inhibitor BAY-293 blocks ERK1/2 and AKT activation in WT SHP2 and the T507K mutant cells. D, SHP2/T507K preferentially binds and stays longer with Gab1 upon HGF stimulation. E, mutation of Arg-32 or Arg-138 to Met in SHP2/T507K abolishes its binding with Gab1 and significantly suppresses HGF-induced ERK1/2 and AKT activation. F, Gab1 knockdown impairs the ERK1/2 or AKT activation in both WT SHP2- and SHP2/T507K-overexpressed cells and eliminates the excessive ERK1/2 or AKT activation for SHP2/T507K. The results of Western blottings are representative of three independent experiments, and the star in the figure indicates statistical significance from Student's t test (p < 0.05). IP, immunoprecipitation; TCL means total-cell lysate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
SHP2 phosphatase activity is required for full activation of Ras, thus playing a pivotal role in regulating several cancer-associated cellular processes, including cell proliferation, migration, and survival (1, 2). Ras cycles between the inactive GDP-bound state and the active GTP-bound state. The GTP-bound Ras (Ras–GTP) recruits its downstream effector molecules such as the Raf and PI3K kinases to commence the ERK1/2 and AKT pathways, which in turn elicit post-translational and transcriptional programs that shape appropriate cellular responses (24). To assess the effect of the SHP2/T507K mutation on Ras activation, a purified GST fusion protein of the Ras-binding domain of Raf (GST–Raf–RBD) was used to pull down the activated Ras from the cell. As shown in Fig. 5B, expression of SHP2/T507K caused more than a 2-fold increase in Ras–GTP after 30 min of HGF stimulation, and the elevated Ras activity persisted well up to 60 min. These observations indicate that compared with WT SHP2, the SHP2/T507K mutant is endowed with an abnormally higher capacity to promote and sustain Ras activation. As a consequence, both ERK1/2 and AKT were also correspondingly up-regulated in SHP2/T507K cells (Fig. 5A). To further demonstrate that SHP2/T507K up-regulates ERK1/2 and AKT via Ras, we blocked Ras activation by a Ras–SOS-binding inhibitor BAY-293, which dramatically inhibited the activation of both ERK1/2 and AKT in either WT or SHP2/T507K-overexpressing cells (Fig. 5C). This result suggests that the overactivation of ERK1/2 and AKT by SHP2/T507K is mediated by Ras.
To investigate the mechanism underlying the observed aberrant Ras activation in SHP2/T507K cells, we determined the ability of SHP2/T507K to associate with Gab1, a molecular event known to be essential for SHP2-mediated Ras activation (15, 25, 26). Given the weakened interaction between the N-SH2 and PTP domain within SHP2/T507K, we predicted that the SH2 domains of the mutant SHP2 should exhibit enhanced affinity for the tyrosine-phosphorylated Gab1. This prediction is borne out by the observed negative cooperativity between the two nonoverlapping ligand-binding sites in the SHP2 N-SH2 domain: an intermolecular interaction with pTyr-peptides from docking proteins and an intramolecular interaction with the PTP domain in SHP2 (3, 12). Indeed, compared with WT SHP2, the SHP2/T507K mutant displayed a stronger and more sustained binding with Gab1 upon HGF stimulation (Fig. 5D). To ascertain whether Gab1 engagement is required for the SHP2/T507K-mediated Ras activation, we mutated either Arg-32 in the N-SH2 domain or Arg-138 in the C-SH2 domain, both of which are known to be critical for SHP2 binding of pTyr motifs from adapter proteins (27, 28). As expected, mutation of either Arg-32 or Arg-138 abolished SHP2/T507K binding to Gab1 and completely suppressed ERK1/2 and AKT phosphorylation (Fig. 5E), supporting the importance of SHP2/T507K-Gab1 association for Ras activation. We further showed that Gab1 knockdown also blocked ERK1/2 and AKT activation in both WT SHP2 and the T507K mutant–expressed cells (Fig. 5F). Importantly, the ERK1/2 and AKT activation status was comparable in WT and SHP2/T507K cells upon Gab1 knockdown, indicating that a decrease in Gab1 levels removes the excessive ERK1/2 and AKT activation by SHP2/T507K. Collectively, the results support that enhanced Gab1 engagement by SHP2/T507K is required for the mutant SHP2–mediated Ras pathway overactivation.
Our kinetic studies revealed that although the Thr-507 to Lys mutation significantly impaired SHP2's ability to dephosphorylate paxillin/pY118, Gab1/pY589, and EGFR/pY992, SHP2/T507K exhibits WT-like catalytic efficacy toward the Sprouty1/pY53 peptide (Table 2). The Sprouty proteins are endogenous inhibitors of the Ras pathways and have been implicated as tumor suppressors in several types of cancer (29–36). Biochemically, Tyr-53 phosphorylated Sprouty1 binds to and sequesters Grb2 to prevent the recruitment of the Grb2–SOS complex for Ras activation by its guanine nucleotide exchange factor SOS (14, 37). In response to growth factor stimulation, SHP2 directly dephosphorylates the pTyr-53 negative regulation site in Sprouty1 to attenuate its inhibitory action on Ras activation (14, 38, 39). Given the comparable catalytic activity exhibited by WT SHP2 and the SHP2/T507K mutant toward the Sprouty1/pY53 peptide, as well as the preferential and sustained binding of SHP2/T507K with the adapter protein Gab1, which may help prolong SHP2 substrate turnover (12, 13), we speculated that SHP2/T507K could dephosphorylate Sprouty1 at Tyr-53 much more efficiently and therefore hyperactivate Ras signaling. To test this hypothesis, we first examined the overall tyrosine phosphorylation of Sprouty1 in WT SHP2- and SHP2/T507K-expressing HEK293 cells. Consistent with the prediction, the level of total Sprouty1 tyrosine phosphorylation in SHP2/T507K cells is 40–50% that in WT SHP2 cells (Fig. 6A). We then determined the ability of WT and T507K mutant SHP2 to dephosphorylate pTyr-53 in Sprouty1 inside the cell. As shown in Fig. 6B, the level of Sprouty1/pY53 in SHP2/T507K cells is ∼40% that in WT SHP2 cells. As a consequence, the Sprouty1-bound Grb2 in SHP2/T507K cells is only half that in WT SHP2 cells (Fig. 6C), which is in line with the 2-fold higher Ras–GTP concentration in the mutant SHP2 cells (Fig. 5B). To further confirm the inhibitory role of Sprouty1 in SHP2/T507K-induced Ras pathway activation, we knocked down Sprouty1 in both WT- and SHP2/T507K-overexpressed cells. Compared with the scrambled siRNA control, Sprouty1 siRNA effectively reduced Sprouty1 expression and increased pERK1/2 and pAKT levels (Fig. 6D). More importantly, we note that SHP2/T507K can further promote ERK1/2 and AKT activation over WT SHP2 in both scrambled siRNA control cells and Sprouty1-knockdown cells, but the magnitude of SHP2/T507K-induced ERK1/2 and AKT overactivation is reduced in Sprouty1-knockdown cells as compared with that in the scrambled siRNA control cells (Fig. 6D). The results are in line with the notion that the preferential dephosphorylation of Sprouty1 by SHP2/T507K may be responsible for the overactivated ERK1/2 and AKT activation in SHP2/T507K cells. Collectively, the results suggest that SHP2/T507K promotes cell transformation by aberrantly activating Ras and Ras-mediated ERK1/2 and AKT pathways through more efficient dephosphorylation of pTyr-53 in Sprouty1, which abrogates the sequestration of Grb2–SOS complex by Sprouty1.
Figure 6.

Preferential dephosphorylation of Sprouty1 by SHP2/T507K is responsible for Ras pathway overactivation. A, SHP2/T507K is more efficient than WT SHP2 in dephosphorylating Sprouty1. TCL means total-cell lysate. Sprouty1 was immunoprecipitated by anti-Sprouty1 antibody, and the total phosphorylation of Sprouty1 (pTyr Sprouty1) was blotted with 4G10 anti-phosphotyrosine antibody. B, SHP2/T507K promotes dephosphorylation of Sprouty1/pY53. The phosphorylation of Sprouty1 at Tyr-53 in total-cell lysates was blotted with anti-pY53 Sprouty1 antibody. C, enhanced dephosphorylation of Sprouty1 decreases sequestration of Grb2. WT SHP2 and SHP2/T507K mutant were co-transfected with Flag-tagged Grb2; Sprouty1 was immunoprecipitated (IP) by anti-Sprouty1 antibody, and the co-immunoprecipitated Grb2 was blotted with anti-Flag antibody. D, Sprouty1 siRNA effectively knocks down Sprouty1, increases pERK1/2 and pAKT, and blunts the SHP2/T507K-induced overactivation effect. At each time point, the ratio of pERK/ERK (or pAKT/AKT) for WT in scrambled siRNA-expressed cells is taken as reference (labeled as ref.), then the ratios for other three lanes at the same time point were normalized to the reference. The overactivation is calculated based on the normalized values (TK/WT). The results of Western blots are representative of three independent experiments, and the star in the figure indicates a statistical significance from Student's t test (p < 0.05). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Finally, to further support the proposed mechanism and demonstrate that the profound transformation in HEK293 cells is unique to SHP2/T507K, we compared the colony-forming potential of SHP2/T507K with other SHP2 gain-of-function mutations frequently occurring in leukemia (e.g. E76K), Noonan syndrome (e.g. N308D), and NSML (e.g. Q506P). Consistent with SHP2/T507K's remarkable transforming potential, the number of colonies formed by SHP2/T507K cells are 13.8-fold higher than those by vector control, WT SHP2, and SHP2/N308D cells and are 5.5- and 6.0-fold higher than those by SHP2/E76K and SHP2/Q506P cells (Fig. 7, A and B). Similarly, SHP2/E76K and SHP2/N308D were also much less transforming than SHP2/T507K in NIH3T3 cells (7). We then investigated the ability of WT and SHP2 mutants to dephosphorylate Sprouty1 and activate Ras as well as its downstream molecules ERK1/2 and AKT (Fig. 7C). The results show that SHP2/E76K, SHP2/N308D, and SHP2/Q506P mutants do not dephosphorylate Sprouty1 and activate Ras to the extent as does SHP2/T507K and further strengthen the conclusion that SHP2/T507K's unique transforming activity stems from its ability to more efficiently dephosphorylate Sprouty1 and therefore hyperactivate Ras signaling.
Figure 7.

Profound Ras-like transformation is unique to SHP2/T507K. A, SHP2/T507K shows the most profound transforming activity in HEK293 cells over other representative SHP2 gain-of-function mutants observed in leukemia (e.g. E76K), Noonan syndrome (e.g. N308D), and NSML (e.g. Q506P). B, relative transforming activity of SHP2 WT and mutants. The quantification was done by setting the number of colonies induced by WT SHP2 as a reference, and the number of colonies induced by the vector or SHP2 mutants were normalized to the reference. C, compared with WT SHP2 and other gain-of-function SHP2 mutants, SHP2/T507K is the most efficient in dephosphorylating Sprouty1 and activating Ras as well as its downstream targets ERK1/2 and AKT. The results of Western blottings are representative of three independent experiments, and the star in the figure indicates a statistical significance from Student's t test (p < 0.05). IP, immunoprecipitation; TCL, total-cell lysate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
SHP2 plays a key role in regulating cellular events downstream of growth factor receptors. Most studies on SHP2 have so far focused on the WT enzyme in the context of receptor tyrosine kinase signaling. However, several dozens of mutations in SHP2 have been found in leukemia, cancer, as well as developmental disorders (4). Despite our growing understanding of the molecular and structural features that govern the regulation of SHP2 phosphatase activity, the exact mechanism by which a mutated SHP2 provokes illnesses is poorly understood. The majority of disease-causing SHP2 mutations (including those that are linked to Noonan syndrome and leukemia) occur at the N-SH2/PTP interface and disrupt the autoinhibitory allosteric regulatory mechanism, resulting in constitutive activation of SHP2 phosphatase activity. Interestingly, SHP2 mutations that are associated with Noonan syndrome with multiple lentigines (NSML) decrease its catalytic activity but engender gain-of-function disease phenotypes similar to those associated with Noonan syndrome (12, 13). Obviously, the “activity-centric” model cannot account for aberrant behaviors for all SHP2 variants. To that end, Thr-507 is highly conserved among the PTPs and is known to reside in close proximity to the two conserved and catalytic Gln residues in the Q-loop. A previous study found that the T507K mutation had little effect on the activity of the WT SHP2 and the fully-activated SHP2/E76K mutant using pNPP as a substrate (7). Enigmatically, SHP2/T507K differs from the Noonan syndrome and leukemia-specific activating SHP2 mutants in that it exhibits oncogenic Ras-like transforming activity (7). Importantly, the T507K mutation is associated with neuroblastoma (5), hepatocellular carcinoma (7), and glioblastoma (6). Unfortunately, the precise mechanism by which SHP2/T507K elicits transformation is not well-understood. In addition, the molecular underpinning of why different SHP2 mutations give rise to such diverse disease phenotypes remains to be illuminated. Improved knowledge of how mutations in SHP2 lead to diseases is essential for designing new therapeutic strategies for improved cancer treatment.
To begin to define the molecular basis by which SHP2/T507K elicits transformation, we characterized the structural and functional consequences of SHP2 T507K mutation. We confirmed that replacement of Thr-507 by Lys has little effect on the kinetic parameters for the SHP2-catalyzed hydrolysis of pNPP, an artificial small molecule substrate that mimics pTyr. However, kinetic analysis using more physiological pTyr-containing peptides derived from putative SHP2 substrate proteins revealed that the T507K mutation significantly compromised the catalytic efficiency of SHP2-mediated dephosphorylation of paxillin/pY118 (EEHVpY118SFPN), Gab1/pY589 (DSEENpY589VPMNPNL), and EGFR/pY992 (DADEpY992LIPQQG). Interestingly, the same mutation had little impact on SHP2's ability to dephosphorylate Sprouty1/pY53 (GSNEpY53TEGPS). A comparison of the WT and mutant SHP2 crystal structures reveals that the T507K mutation does not alter the conformation of any vital residues within the active site that are directly involved in pNPP binding and hydrolysis. In contrast, molecular modeling of the Sprouty1/pY53 peptide binding with the WT and mutant SHP2 suggests that whereas the T507K mutation lessens SHP2 interactions with the N-terminal residues of the peptide substrate, it also gains new H-bonds with the +2 Glu residue in Sprouty1/pY53, which makes up the decreased binding with the N-terminal region. The same compensatory interactions are not present between SHP2 and other peptides. Thus, it appears that the T507K mutation alters SHP2 substrate specificity such that the mutant enzyme exhibits a strong preference for Sprouty1/pY53.
In addition to changing SHP2 substrate specificity, our data show that the T507K mutation also perturbs SHP2's allosteric regulatory mechanism. Thus, although we found SHP2/T507K predominantly in the closed and autoinhibited conformation in both the crystalline state and in solution, results from our kinetic, H/DX-MS, X-ray crystallographic, and molecular modeling experiments indicate that the T507K mutation substantially weakens the interaction between the N-SH2 domain and the PTP domain. As a result, the T507K mutant SHP2 has acquired a higher propensity (compared with the WT enzyme) to adopt the open and active conformation. A direct consequence of this heightened tendency for the open state is the increased affinity of the N-SH2 domain for its pTyr ligands, thereby enabling SHP2/T507K to be more responsive to growth factor stimulation and to bind upstream adapter and scaffolding molecules more preferentially (12, 13). Indeed, we showed that the SHP2/T507K mutant displays a more sustained and stronger binding with Gab1, and the enhanced Gab1 engagement by SHP2/T507K is essential for the mutant SHP2-induced excessive Ras activation. Consistent with this observation, we found that less HGF is required to stimulate the SHP2/T507K-mediated ERK1/2 and AKT activation, which should confer growth advantage when growth factors are limiting. Moreover, it has been previously shown that prolonged association with the scaffolding protein Gab1 enables more efficient substrate turnover by SHP2 (12, 13). Thus, we hypothesized that SHP2/T507K may be more efficient than the WT SHP2 in dephosphorylating its target substrate Sprouty1, even though the kcat/Km values for the WT and T507K mutant SHP2-catalyzed hydrolysis of the Sprouty1/pY53 peptide are comparable.
The Sprouty proteins are tumor suppressors that are found down-regulated in a number of malignancies (29–36). The Sprouty proteins function as negative regulators of Ras, through their ability to sequester Grb2 upon Tyr-53 (in Sprouty1) phosphorylation, which prevents the Grb2–SOS complex from catalyzing the production of the GTP-bound activated Ras (14, 37). One mechanism by which SHP2 promotes Ras signaling is by dephosphorylating Sprouty1 at pTyr-53, thereby attenuating its inhibitory effects on Ras activation (14, 38, 39). Consistent with this notion, expression of Sprouty2/Y55F, in which Tyr-55 was replaced with a nonphosphorylatable phenylalanine, led to enhanced cell proliferation and formed larger and greater numbers of colonies in soft-agar assays (34). Here, we confirmed the transforming activity of SHP2/T507K and found that expression of SHP2/T507K causes a persistent overactivation of Ras and concomitant activation of ERK1/2 and AKT pathways, two of the most frequently dysregulated signaling modules in human cancers. We further demonstrated that compared with the WT SHP2, the T507K mutant is a more efficient enzyme for Sprouty1/Tyr-53 dephosphorylation, likely assisted by its increased association with Gab1, thus releasing Grb2 for SOS-mediated Ras activation. To further strengthen this mechanism, we confirmed the profound transforming potential of SHP2/T507K over other SHP2 gain-of-function mutations frequently observed in leukemia (e.g. E76K), Noonan syndrome (e.g. N308D), and NSML (e.g. Q506P). Moreover, we showed that, among the SHP2 mutants, SHP2/T507K is the most efficient in dephosphorylating Sprouty1 and activating Ras as well as its downstream targets ERK1/2 and AKT. Together, the data support the conclusion that SHP2/T507K's unique Ras-like transforming activity stems from its ability to more efficiently dephosphorylate Sprouty1 and therefore hyperactivate Ras signaling.
In summary, this work provides a comprehensive characterization of a point mutation that convert the WT SHP2 protein to an oncoprotein, SHP2/T507K, with powerful Ras-like transforming properties. Our results reveal that the T507K mutation alters SHP2 substrate specificity in favor of Sprouty1 and perturbs the allosteric regulatory mechanism such that SHP2/T507K possesses a higher affinity for the scaffolding protein Gab1. We conclude that SHP2/T507K acquires its transforming potential through enhanced activity toward Sprouty1/Tyr-53, leading to aberrant Ras activation. Our findings offer the first example where a point mutation in SHP2 can perturb both SHP2 substrate specificity and regulatory mechanism. Thus, disease-causing SHP2 mutations could instigate distinct pathophysiology through their varied effects on phosphatase activity, targeting/activation mechanism, and/or substrate specificity. Detailed understanding of these mechanisms should help develop drugs that target mutant SHP2 specifically.
Experimental procedures
Enzyme kinetic analyses of WT SHP2 and the T507K mutant
Initial rate measurements for the enzyme-catalyzed hydrolysis of pNPP were conducted at 25 °C in a pH 7.0 assay buffer (50 mm 3,3-dimethylglutarate, 1 mm DTT, 1 mm EDTA, 150 mm NaCl). The assays were performed in 96-well plates. Substrate concentrations ranging from 0.2 to 5 Km were used to determine the kcat and Km values. Reactions were started by the addition of an appropriate amount of enzyme into 100 μl of pNPP solution. The reaction mixtures were quenched with 50 μl of 5 m sodium hydroxide, and the absorbance at 405 nm was detected using SpectraMax Plus 384 microplate spectrophotometer (Molecular Devices). The steady-state kinetic parameters were determined by fitting the data to the Michaelis-Menten equation in SigmaPlot.
To determine the kcat/Km values for pTyr-containing peptide substrates, the reaction (0.5 ml) was carried out in a 1-ml quartz cuvette at 25 °C in a pH 7.0 assay buffer (50 mm 3,3-dimethylglutarate, 1 mm DTT, 1 mm EDTA, 150 mm NaCl). The reaction rate was monitored by the increase in fluorescence of the dephosphorylated peptide product at 305 nm with excitation at 280 nm on a luminescence spectrometer LS50B (PerkinElmer Life Sciences) (40). The reaction was performed at a substrate concentration much lower than its Km, and the enzyme concentration was at least 10 times lower than the substrate concentration. Under these conditions, the Michaelis-Menten equation reduces to the following equation: v = (kcat/Km)·[E]·[S]; the reaction is first-order with respect to [S], and the observed apparent first-order rate constant is equal to (kcat/Km)·[E], which can be determined by fitting the reaction's time-course data (Fig. S9) into a first-order rate equation in SigmaPlot. The kcat/Km value was calculated from the first-order rate constant divided by a given enzyme concentration.
Inhibition assay of SHP2 PTP domain by the N-SH2 domain
PTP activity was assayed using pNPP as a substrate at 25 °C in a pH 7.0 assay buffer (50 mm 3,3-dimethylglutarate, 1 mm DTT, 1 mm EDTA, 150 mm NaCl). The assays were performed in 96-well plates with total reaction volume of 200 μl. The reaction was initiated by the addition of enzyme (WT SHP2 or the T507K mutant catalytic domain) to a reaction mixture containing pNPP and the isolated SHP2 N-SH2 domain. For determination of Ki, the pNPP concentration was varied at three different concentrations of the N-SH2 domain. The reaction rate was measured using a SpectraMax Plus 384 microplate spectrophotometer (Molecular Devices). The Ki values were determined by fitting the data using the EnzymeKinetics module in SigmaPlot.
Cloning, expression, and purification of SHP2 proteins
The WT SHP2 catalytic domain (residues 224–528), full-length lacking the C-terminal tail (residues 1–528), as well as the N-SH2 domain (residues 4–103) were cloned into the pET-21a+ vector using NdeI and XhoI restriction enzymes (New England Biolabs), which generated recombinant proteins with a C-terminal His6 tag. The SHP2 catalytic domains and full-length constructs lacking the C-terminal tail containing T507K mutations were generated using the QuikChange mutagenesis kit (Stratagene). All proteins were expressed in Escherichia coli BL21(DE3). Proteins used for kinetic or inhibition assays were purified using Ni-NTA resin (Qiagen) with >90% purity. Proteins used for crystallography studies were sequentially purified by Ni-NTA resin, chromatography of HiPrep 26 desalting column (GE Healthcare), cation-exchange column packed with SP-Sepharose (GE Healthcare), and Superdex 75 gel-filtration column (GE Healthcare). The purities were more than 95% as determined by SDS-PAGE and Coomassie staining.
Crystallization, data collection, and structure determination
The crystals of SHP2/T507K (residues 1–528) were grown at 20 °C in the hanging drops containing 1.5 μl of protein (10 mg/ml) and 1.5 μl of reservoir solution. The protein storage buffer contains 20 mm Tris-base, 50 mm NaCl, 2 mm DTT, and 1 mm EDTA, and the pH was adjusted to 7.8 by HCl. The reservoir solution contains 16% PEG3350, 200 mm LiCl. The crystals were transferred into the cryoprotectant buffer, containing the same components in reservoir solution except for increasing the concentration of PEG3350 to 30%, and were flash-frozen by liquid nitrogen. Data were collected at 19-ID at the Advanced Photon Source (APS) and were processed with HKL3000 (41). The structures were solved by molecular replacement with Molrep (42) using the coordinates of our previously reported SHP2 WT structure (PDB code 4DGP) (12) as search model. The structure refinements were carried out iteratively using phenix.refine program in the PHENIX software suite (43).
Hydrogen/deuterium exchange MS
Stock solutions of WT SHP2 and SHP2/T507K mutant (residues 1–528) were prepared in a (1H2O)-based buffer (pH 7.8). Deuterium exchange was initiated by dilution of each enzyme 20-fold in a similar deuterium (2H2O)-based buffer (pD 7.8). At set deuterium exchange time points (10 s and 1, 10, 30, and 60 min), the reaction was quenched by the addition of a cold (1H2O)-based 100 mm Na3PO4 buffer (pH 2.3) at equal volume. For peptide-based MS analysis, the quench solution contained pepsin endoproteinase (Sigma) that would make a pepsin/enzyme = 1.5:1 (w/w) ratio during digestion. Quenched sample was digested on ice for 4 min and then loaded by autosampler onto an XBridge C18 2.5-micron 2.1 × 50-mm (Waters) column that was submerged under ice. A Surveyor MS pump (Finnigan) was used to generate the chromatographic gradients. Peptides were separated in time using a steep gradient of acetonitrile (10–35% in 7.5 min) and electrosprayed into an LTQ mass spectrometer (Finnigan). Sequest (Thermo Fisher Scientific) was used to identify the SHP2 peptic peptides. Peptides were accepted for analysis based upon XCorr value significance and identification in multiple sample runs. All peptide-based samples were manually prepared and run in triplicate with a general standard deviation of <0.2 Da at each time point. Unbiased peptide precursor ion peak envelope centroiding was performed using HX-express software (44). Significant deuterium exchange differences in T507K mutant relative to WT were mapped onto the crystal structure in a heat map format.
Molecular modeling
The MD simulation–based interface interaction energy calculations were described previously (12). To build a likely binding model of the Sprouty1/pY53 peptide with a catalytic domain of WT SHP2 or the T507K mutant, the structure of the SHP1·pY–peptide complex (PDB code 1FPR) (45) was superimposed onto the structure of the catalytic domain of WT SHP2 (PDB code 4DGP) (12) or the T507K mutant by fitting all Cα atom pairs of conserved residues between SHP1 and SHP2 in UCSF Chimera (46). The coordinates of the pY-peptide were then transferred into a SHP2 catalytic domain, and mutations were made at specific residues according to the sequence of Sprouty1/pY53-peptide to create a rough binding model of the Sprouty1/pY53-peptide with the SHP2 catalytic domain. Each complex was then neutralized by Na+ counterion and solvated in a truncated octahedral box of TIP3P water with 10 Å buffer between the solute and box edge, and the amber ff03 force field was employed to generate the topology and parameter files. Each system was energy-minimized using the sander program in the Amber14 software package (47) in the following order: 1) minimizing added water only for 5000 steps; 2) minimizing water and side chain for another 5000 steps; 3) minimizing the whole system for 50,000 steps. Both energy-minimized complex structures were considered as likely binding models of Sprouty1/pY53-peptide with WT or T507K for comparative analysis, and they were also used as initial structures for next MD simulations and MM-GBSA–binding free-energy calculations. The calculation details were the same as what we described previously (12) except the MD simulation time was 5 ns in this study. The binding free energy of the EGFR/pY992-peptide with the catalytic domain of WT SHP2 or T507K mutant was obtained exactly the same way as described above; the only difference was that mutations of the peptide were made according to the sequence of the EGFR/pY992-peptide during an early step of building the likely binding model.
Cell culture, immunoblotting, and immunoprecipitation
HEK293 cells (American Tissue Culture Collection) were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (Corning) supplemented with 10% fetal bovine serum (HyClone). WT or SHP2 mutant (T507K, T507K/R32M, T507K/R138M, E76K, N308D, and Q506P) cloned in the mammalian expression vector pCN-HA, a modified version of pcDNA3.1 that generated proteins with N-terminal HA tag, were transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Twenty four hours post-transfection, cells were serum-starved overnight and then either left unstimulated or stimulated with 10 ng/ml HGF (Sigma) for the indicated time. For immunoblotting, the cell lysates were electrophoresed on a 10% polyacrylamide gel; the separated proteins were transferred to a nitrocellulose membrane and probed with the following antibodies: anti-phospho-ERK1/2 (Cell Signaling); anti-ERK1/2 (Cell Signaling); anti-phospho-AKT (Cell Signaling); anti-AKT (Cell Signaling); anti-Gab1 (Cell Signaling); anti-phospho-Met (1234/1235) (Cell Signaling); anti-Met (Cell Signaling); anti-Sprouty1 (Cell Signaling); anti-pY53 Sprouty1 (a generous gift from Professor Mark A. Krasnow (14)); anti-HA (Santa Cruz Biotechnology); anti-Ras (EMD Millipore); anti-phosphotyrosine antibody 4G10 (EMD Millipore); anti-β-actin (Santa Cruz Biotechnology); anti-glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology); and anti-Flag (Sigma) followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling). The blots were developed with the SuperSignalTM West Pico chemiluminescent substrate (Thermo Fisher Scientific). For immunoprecipitation, HEK293 cells transiently transfected with WT SHP2 or mutants were serum-starved overnight, stimulated with 10 ng/ml HGF for the indicated time, and then lysed on ice for 30 min in lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 10% glycerol) supplemented with a complete protease inhibitor tablet and PhosSTOP tablets (Roche Applied Science). Cell lysates were cleared by centrifugation at 14,000 rpm for 20 min. Anti-HA–agarose antibody (Sigma) or Sprouty1 (Cell Signaling) antibody with protein A/G–agarose beads (Santa Cruz Biotechnology) was added to the cell lysate and incubated at 4 °C for 3 h. After being washed with lysis buffer three times, the protein complex was boiled in Laemmli sample buffer and subjected to electrophoresis and immunoblotting. Ras–GTP pulldown experiments were carried out following the protocol previously published (48).
Soft agar assay
1000 HEK293 cells with stable overexpression of vector, WT SHP2, T507K, E76K, N308D, or Q506P mutants were respectively suspended in 0.35% agar-containing medium and plated on a layer of 0.6% agar in each 35-mm plate. The cells were incubated at 37 °C for 9 days. The colonies were stained with crystal violet and scanned with a scanner, and the number of colonies were counted using ImageJ software.
Cell proliferation assay
BrdU labeling and detection kit (Roche Applied Science) was used to determine cell proliferation according to the manufacturer's instructions. HEK293 cells were seeded in a 24-well plate and transfected with SHP2 WT or T507K mutant at ∼90% confluency. 6 h later, 6000 cells were reseeded in a 96-well plate. After recovery overnight, cells were starved in serum-free DMEM for 6 h followed by being maintained in DMEM with 50 ng/ml HGF for another 2 days. BrdU was added for the last 3 h of culture. The incorporated BrdU was assayed by immunostaining with anti-BrdU–POD antibody. Absorbance was read using an ELISA microplate reader at wavelengths of 450 and 690 nm.
Cell migration assay
The assay was performed in 6-well Transwell plates (Corning). Transiently-transfected HEK293 cells (1 × 106) were placed in the upper chamber in 1.5 ml of serum-free medium, and the lower chamber was loaded with 2 ml of medium containing 10% fetal bovine serum. After incubation at 37 °C and 5% CO2 for 22 h, the cells that migrated into the lower chamber were trypsinized and counted with a hemocytometer.
RNAi studies
Small interfering RNA (siRNA) for Gab1 and Sprouty1 were purchased from Sigma with SKU-Pack size of EHU077111 and EHU087521, respectively. The scrambled siRNA was purchased from Santa Cruz Biotechnology with catalog no. sc-37007. siRNAs were transfected into HEK293 cells using LipofectamineTM RNAiMAX transfection reagent (Invitrogen) according to the protocol recommended by the manufacturer.
Statistical analysis
Student's t test was performed to assess the statistical significance. Results are represented in “mean ± standard deviation” format. The results of Western blottings shown in Figs. 5–7 are representative of three independent experiments, and the star in figures indicates a statistical significance from Student's t test (p < 0.05).
Data availability
The atomic coordinates and crystallographic structure factors for the SHP2/T507K protein have been deposited in the Protein Data Bank under accession code 5BK8. All other data are contained within the manuscript and supporting information.
Author contributions
R.-Y. Z., Z.-H. Y., and Z.-Y. Z. conceptualization; R.-Y. Z., Z.-H. Y., L. C., C. D. W., S. Z., and L. W. data curation; R.-Y. Z., Z.-H. Y., L. C., C. D. W., S. Z., and L. W. formal analysis; R.-Y. Z., Z.-H. Y., and Z.-Y. Z. writing-original draft; Z.-Y. Z. supervision; Z.-Y. Z. funding acquisition; Z.-Y. Z. project administration.
Supplementary Material
Acknowledgment
We thank Dr. Mark A. Krasnow for providing the anti-pY53 Sprouty1.
This work was supported in part by National Institutes of Health Grants RO1 CA069202 and RO1 CA207288 (to Z.-Y. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
- PTP
- protein-tyrosine phosphatase
- PTK
- protein-tyrosine kinase
- SH2
- Src homology 2
- pNPP
- para-nitrophenyl phosphate
- MM-GBSA
- molecular mechanics–generalized born surface area
- MD
- molecular dynamics
- HGF
- hepatocyte growth factor
- EGFR
- epidermal growth factor receptor
- DMEM
- Dulbecco's modified Eagle's medium
- NSML
- Noonan syndrome with multiple lentigines
- RMSD
- root-mean-square deviation
- PDB
- Protein Data Bank
- BFE
- binding free energy
- Ni-NTA
- nickel-nitrilotriacetic acid
- H/DX-MS
- hydrogen/deuterium exchange MS.
References
- 1. Neel B. G., Gu H., and Pao L. (2003) The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 10.1016/S0968-0004(03)00091-4 [DOI] [PubMed] [Google Scholar]
- 2. Frankson R., Yu Z.-H., Bai Y., Li Q., Zhang R.-Y., and Zhang Z.-Y. (2017) Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 77, 5701–5705 10.1158/0008-5472.CAN-17-1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., and Shoelson S. E. (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 10.1016/S0092-8674(00)80938-1 [DOI] [PubMed] [Google Scholar]
- 4. Tajan M., de Rocca Serra A., Valet P., Edouard T., and Yart A. (2015) SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 58, 509–525 10.1016/j.ejmg.2015.08.005 [DOI] [PubMed] [Google Scholar]
- 5. Bentires-Alj M., Paez J. G., David F. S., Keilhack H., Halmos B., Naoki K., Maris J. M., Richardson A., Bardelli A., Sugarbaker D. J., Richards W. G., Du J., Girard L., Minna J. D., Loh M. L., et al. (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 64, 8816–8820 10.1158/0008-5472.CAN-04-1923 [DOI] [PubMed] [Google Scholar]
- 6. Brennan C. W., Verhaak R. G., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H., Beroukhim R., Bernard B., Wu C. J., Genovese G., Shmulevich I., et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miyamoto D., Miyamoto M., Takahashi A., Yomogita Y., Higashi H., Kondo S., and Hatakeyama M. (2008) Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 27, 3508–3515 10.1038/sj.onc.1211019 [DOI] [PubMed] [Google Scholar]
- 8. Andersen J. N., Mortensen O. H., Peters G. H., Drake P. G., Iversen L. F., Olsen O. H., Jansen P. G., Andersen H. S., Tonks N. K., and Møller N. P. (2001) Structural and evolutionary relationships among protein-tyrosine phosphatase domains. Mol. Cell. Biol. 21, 7117–7136 10.1128/MCB.21.21.7117-7136.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu Z.-H., and Zhang Z.-Y. (2018) Regulatory mechanisms and novel therapeutic targeting strategies for protein-tyrosine phosphatases. Chem. Rev. 118, 1069–1091 10.1021/acs.chemrev.7b00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y., Wu L., Noh S. J., Guan K.-L., and Zhang Z.-Y. (1998) Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction: probing the function of the invariant glutamine residues. J. Biol. Chem. 273, 5484–5492 10.1074/jbc.273.10.5484 [DOI] [PubMed] [Google Scholar]
- 11. Pannifer A. D., Flint A. J., Tonks N. K., and Barford D. (1998) Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 273, 10454–10462 10.1074/jbc.273.17.10454 [DOI] [PubMed] [Google Scholar]
- 12. Yu Z.-H., Xu J., Walls C. D., Chen L., Zhang S., Zhang R.-Y., Wu L., Wang L., Liu S., and Zhang Z.-Y. (2013) Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J. Biol. Chem. 288, 10472–10482 10.1074/jbc.M113.450023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu Z.-H., Zhang R.-Y., Walls C. D., Chen L., Zhang S., Wu L., Liu S., and Zhang Z.-Y. (2014) Molecular basis for gain-of-function LEOPARD syndrome associated SHP2 mutations. Biochemistry 53, 4136–4151 10.1021/bi5002695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jarvis L. A., Toering S. J., Simon M. A., Krasnow M. A., and Smith-Bolton R. K. (2006) Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development 133, 1133–1142 10.1242/dev.02255 [DOI] [PubMed] [Google Scholar]
- 15. Ren Y., Meng S., Mei L., Zhao Z. J., Jove R., and Wu J. (2004) Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J. Biol. Chem. 279, 8497–8505 10.1074/jbc.M312575200 [DOI] [PubMed] [Google Scholar]
- 16. Edouard T., Combier J. P., Nédélec A., Bel-Vialar S., Métrich M., Conte-Auriol F., Lyonnet S., Parfait B., Tauber M., Salles J. P., Lezoualc'h F., Yart A., and Raynal P. (2010) Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3β signaling. Mol. Cell. Biol. 30, 2498–2507 10.1128/MCB.00646-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agazie Y. M., and Hayman M. J. (2003) Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23, 7875–7886 10.1128/MCB.23.21.7875-7886.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee C. H., Kominos D., Jacques S., Margolis B., Schlessinger J., Shoelson S. E., and Kuriyan J. (1994) Crystal structures of peptide complexes of the amino-terminal SH2 domain of the Syp tyrosine phosphatase. Structure 2, 423–438 10.1016/S0969-2126(00)00044-7 [DOI] [PubMed] [Google Scholar]
- 19. Hoofnagle A. N., Resing K. A., and Ahn N. G. (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32, 1–25 10.1146/annurev.biophys.32.110601.142417 [DOI] [PubMed] [Google Scholar]
- 20. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 21. LaRochelle J. R., Fodor M., Vemulapalli V., Mohseni M., Wang P., Stams T., LaMarche M. J., Chopra R., Acker M. G., and Blacklow S. C. (2018) Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 9, 4508 10.1038/s41467-018-06823-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pádua R. A. P., Sun Y., Marko I., Pitsawong W., Stiller J. B., Otten R., and Kern D. (2018) Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun. 9, 4507 10.1038/s41467-018-06814-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puius Y. A., Zhao Y., Sullivan M., Lawrence D. S., Almo S. C., and Zhang Z.-Y. (1997) Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc. Natl. Acad. Sci. U.S.A. 94, 13420–13425 10.1073/pnas.94.25.13420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Young A., Lyons J., Miller A. L., Phan V. T., Alarcón I. R., and McCormick F. (2009) Ras signaling and therapies. Adv. Cancer Res. 102, 1–17 10.1016/S0065-230X(09)02001-6 [DOI] [PubMed] [Google Scholar]
- 25. Cunnick J. M., Dorsey J. F., Munoz-Antonia T., Mei L., and Wu J. (2000) Requirement of SHP2 binding to Grb2-associated binder-1 for mitogen-activated protein kinase activation in response to lysophosphatidic acid and epidermal growth factor. J. Biol. Chem. 275, 13842–13848 10.1074/jbc.275.18.13842 [DOI] [PubMed] [Google Scholar]
- 26. Cunnick J. M., Mei L., Doupnik C. A., and Wu J. (2001) Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 276, 24380–24387 10.1074/jbc.M010275200 [DOI] [PubMed] [Google Scholar]
- 27. Mayer B. J., Jackson P. K., Van Etten R. A., and Baltimore D. (1992) Point mutations in the abl SH2 domain coordinately impair phosphotyrosine binding in vitro and transforming activity in vivo. Mol. Cell. Biol. 12, 609–618 10.1128/MCB.12.2.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugimoto S., Wandless T. J., Shoelson S. E., Neel B. G., and Walsh C. T. (1994) Activation of the SH2-containing protein-tyrosine-phosphatase, SH-PTP2, by phosphotyrosine-containing peptides derived from insulin-receptor substrate-1. J. Biol. Chem. 269, 13614–13622 [PubMed] [Google Scholar]
- 29. Wakioka T., Sasaki A., Kato R., Shouda T., Matsumoto A., Miyoshi K., Tsuneoka M., Komiya S., Baron R., and Yoshimura A. (2001) Spred is a Sprouty-related suppressor of Ras signaling. Nature 412, 647–651 10.1038/35088082 [DOI] [PubMed] [Google Scholar]
- 30. Fong C. W., Chua M. S., McKie A. B., Ling S. H., Mason V., Li R., Yusoff P., Lo T. L., Leung H. Y., So S. K., and Guy G. R. (2006) Sprouty 2, an inhibitor of mitogen-activated protein kinase signaling, is down-regulated in hepatocellular carcinoma. Cancer Res. 66, 2048–2058 10.1158/0008-5472.CAN-05-1072 [DOI] [PubMed] [Google Scholar]
- 31. Frank M. J., Dawson D. W., Bensinger S. J., Hong J. S., Knosp W. M., Xu L., Balatoni C. E., Allen E. L., Shen R. R., Bar-Sagi D., Martin G. R., and Teitell M. A. (2009) Expression of sprouty2 inhibits B-cell proliferation and is epigenetically silenced in mouse and human B-cell lymphomas. Blood 113, 2478–2487 10.1182/blood-2008-05-156943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishida M., Ichihara M., Mii S., Jijiwa M., Asai N., Enomoto A., Kato T., Majima A., Ping J., Murakumo Y., and Takahashi M. (2007) Sprouty2 regulates growth and differentiation of human neuroblastoma cells through RET tyrosine kinase. Cancer Sci. 98, 815–821 10.1111/j.1349-7006.2007.00457.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kwabi-Addo B., Wang J., Erdem H., Vaid A., Castro P., Ayala G., and Ittmann M. (2004) The expression of sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Res. 64, 4728–4735 10.1158/0008-5472.CAN-03-3759 [DOI] [PubMed] [Google Scholar]
- 34. Lo T. L., Yusoff P., Fong C. W., Guo K., McCaw B. J., Phillips W. A., Yang H., Wong E. S., Leong H. F., Zeng Q., Putti T. C., and Guy G. R. (2004) The ras/mitogen-activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res. 64, 6127–6136 10.1158/0008-5472.CAN-04-1207 [DOI] [PubMed] [Google Scholar]
- 35. Macià A., Gallel P., Vaquero M., Gou-Fabregas M., Santacana M., Maliszewska A., Robledo M., Gardiner J. R., Basson M. A., Matias-Guiu X., and Encinas M. (2012) Sprouty1 is a candidate tumor-suppressor gene in medullary thyroid carcinoma. Oncogene 31, 3961–3972 10.1038/onc.2011.556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schutzman J. L., and Martin G. R. (2012) Sprouty genes function in suppression of prostate tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 20023–20028 10.1073/pnas.1217204109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanafusa H., Torii S., Yasunaga T., and Nishida E. (2002) Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 4, 850–858 10.1038/ncb867 [DOI] [PubMed] [Google Scholar]
- 38. Hanafusa H., Torii S., Yasunaga T., Matsumoto K., and Nishida E. (2004) Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor sprouty. J. Biol. Chem. 279, 22992–22995 10.1074/jbc.M312498200 [DOI] [PubMed] [Google Scholar]
- 39. Quintanar-Audelo M., Yusoff P., Sinniah S., Chandramouli S., and Guy G. R. (2011) Sprouty-related Ena/vasodilator-stimulated phosphoprotein homology 1-domain-containing protein (SPRED1), a tyrosine-protein phosphatase non-receptor type 11 (SHP2) substrate in the Ras/extracellular signal regulated kinase (ERK) pathway. J. Biol. Chem. 286, 23102–23112 10.1074/jbc.M110.212662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Z.-Y., Maclean D., Thieme-Sefler A. M., Roeske R. W., and Dixon J. E. (1993) A continuous spectrophotometric and fluorimetric assay for protein-tyrosine phosphatase using phosphotyrosine-containing peptides. Anal. Biochem. 211, 7–15 10.1006/abio.1993.1224 [DOI] [PubMed] [Google Scholar]
- 41. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 42. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 10.1107/S0021889897006766 [DOI] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weis D. D., Engen J. R., and Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 10.1016/j.jasms.2006.07.025 [DOI] [PubMed] [Google Scholar]
- 45. Yang J., Cheng Z., Niu T., Liang X., Zhao Z. J., and Zhou G. W. (2000) Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 275, 4066–4071 10.1074/jbc.275.6.4066 [DOI] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 47. Case D. A., Cheatham T. E. 3rd., Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Castro A. F., Rebhun J. F., and Quilliam L. A. (2005) Measuring Ras-family GTP levels in vivo–running hot and cold. Methods 37, 190–196 10.1016/j.ymeth.2005.05.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and crystallographic structure factors for the SHP2/T507K protein have been deposited in the Protein Data Bank under accession code 5BK8. All other data are contained within the manuscript and supporting information.