

Abstract
Although checkpoint immunotherapies have revolutionized the treatment of cancer, not all tumor types have seen substantial benefit. Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy in which very limited responses to immunotherapy have been observed. Extensive immunosuppressive myeloid cell infiltration in PDAC tissues has been postulated as a major mechanism of resistance to immunotherapy. Strategies concomitantly targeting monocyte or granulocyte trafficking or macrophage survival, in combination with checkpoint immunotherapies, have shown promise in preclinical studies and these studies have transitioned into ongoing clinical trials for the treatment of pancreatic and other cancer types. However, compensatory actions by untargeted monocytes, granulocytes, and/or tissue resident macrophages may limit the therapeutic efficacy of such strategies. CD11b/CD18 is an integrin molecule that is highly expressed on the cell surface of these myeloid cell subsets and plays an important role in their trafficking and cellular functions in inflamed tissues. Here, we demonstrate that the partial activation of CD11b by a small molecule agonist (ADH-503) leads to the repolarization of tumor-associated macrophages, reduction in the number of tumor-infiltrating immunosuppressive myeloid cells, and enhanced dendritic cell responses. These actions, in turn, improve anti-tumor T cell immunity and render checkpoint inhibitors effective in previously unresponsive PDAC models. These data demonstrate molecular agonism of CD11b reprograms immunosuppressive myeloid cell responses and potentially bypasses the limitations of current clinical strategies to overcome resistance to immunotherapy.
One Sentence Summary:
Agonism of CD11b overcomes myeloid cell-induced immunosuppression to render pancreatic cancer models responsive to checkpoint immunotherapy.
Introduction
The potential use of checkpoint immunotherapy to combat cancer has now been established in several tumor types. However, not all cancers respond. For example, despite a number of patients with adequate T cell infiltrates, immunotherapy has not led to clinical benefits in pancreatic ductal adenocarcinoma (PDAC) (1). Although there are multiple factors that could contribute to such therapeutic resistance to checkpoint immunotherapy, one major factor is the immunosuppressive myeloid cell populations present within the tumor tissues that can drive T cell exclusion and dysfunction (2–4). Therefore, one potential strategy is the targeting of these myeloid cell populations to improve T cell-mediated immunity. These preclinical strategies have included blocking the mobilization and trafficking of inflammatory monocytes [via C-C chemokine receptor-2 (CCR2)] or granulocytes [via C-X-C motif chemokine receptor (CXCR)-1 and −2 signaling]. Alternatively macrophage survival and/or pro-tumor polarization can be blocked through colony stimulating factor-1 receptor (CSF1R) inhibition. These strategies have all shown promise, in combination with checkpoint immunotherapies, in preclinical studies that have transitioned into ongoing clinical trials for the treatment of pancreatic and other cancers. However, compensatory actions by untargeted monocytes, granulocytes, and/or tissue resident macrophages may limit the therapeutic efficacy of such strategies. For example, targeting granulocytes can lead to the subsequent compensatory expansion of monocytes and macrophages (5, 6), suggesting that the nonselective targeting of all tumor-infiltrating myeloid cells may represent an optimal therapeutic strategy to promote anti-tumor immunity.
PDAC is characterized by abundant myeloid cell infiltrates that predominantly include monocytes, granulocytes, and macrophages (7–9). These infiltrates are associated with immunosuppression, fibrosis, and T cell dysfunction and poor prognosis in patients with PDAC (6, 8–10). The cells in these infiltrates rely on cell adhesion molecules for both their trafficking into tumors and their biological activity (11). Integrin αMβ2 (CD11b/CD18) is a multifunctional integrin expressed on myeloid cells that plays a well-established role in leukocyte adhesion to the vasculature, transendothelial migration, and tissue recruitment under inflammatory conditions. CD11b is the ligand binding subunit of the dimeric integrin CD11b/CD18 and a receptor for both fibrinogen and endothelial ICAM-1 and is expressed on most myeloid cells, including macrophages, monocytes, neutrophils and some dendritic cell (DC) subsets. CD11b also plays a key role in the phagocytosis of opsonized particles, including apoptotic cells, which can limiting inflammatory immune responses (12, 13). Once within tissues, CD11b can negatively regulate pro-inflammatory pathways, such as those involving TLR and FcRγ (14–16). Among its various functions, CD11b mediates cell adhesion, chemotaxis, migration, phagocytosis, and survival (17–20). Together these data suggesting that CD11b plays an important role in myeloid cell migration into and function within sites of inflammation.
Based on its involvement in pathologic inflammatory cell recruitment, small molecule and antibody antagonists of CD11b signaling were developed to prevent excessive myeloid cell infiltration (21–23). However, these approaches failed clinically, because such blockade strategies require the saturation CD11b, which is not achievable at tolerable doses in humans (23–26). To overcome this limitation, we developed a small molecule allosteric agonist, ADH-503, that result in a partially active CD11b conformation upon binding (24, 26). In inflammatory models, we demonstrated that this approach can by very effective at lower concentrations than those required for antagonist strategies (16, 24, 27–29). Mechanistically, ADH-503 suppresses myeloid cell infiltration into inflamed or infected sites by increasing CD11b-dependent cell adhesion to ICAM-1 on the endothelium, preventing subsequent extravasation (16, 24, 26–29). Pharmacologic activation of CD11b has also been shown to suppresses the TLR-dependent FOXO3/IRF7 pathway both in vitro and in vivo, suggesting that CD11b activation may directly affect macrophage functions beyond trafficking (16). Here in, we examined the impact of targeting multiple myeloid cells by ADH-503 on tumor immunity and response to immunotherapy.
Results
CD11b+ cells are abundant in human PDAC tissues
To determine the functional consequences that alterations in CD11b signaling might have on PDAC tumor immunity, we first assessed the abundance and identity of CD11b+ cells in the PDAC tumor microenvironment (TME). To determine abundance, we quantified CD11b+ cells in human pancreatic tissue samples containing paired tumor and adjacent normal tissues by immunohistochemistry (IHC). As expected, PDAC tissues had higher numbers and densities of CD11b+ leukocytes compared to adjacent normal tissues (Fig. 1A and fig. S1A). To determine the composition of these CD11b+ leukocytes, we profiled fresh PDAC tissues by mass cytometry (CyTOF). We observed that CD11b+ cells compose 25–42% of all tumor-infiltrating leukocytes (fig. S1B). As expected, CD11b was robustly expressed by tumor-infiltrating macrophages and granulocytes (Fig. 1C). Although CD11b has been reported to be expressed on activated B, T, and natural killer (NK) cells, we found that very few (< 3%) lymphocytes expressed CD11b (Fig. 1C). Additionally, analyses of the composition of CD11b+ cells revealed that greater than 95% of the cells were macrophages, monocytes, granulocytes, or conventional dendritic cells (cDCs) (Fig. 1D). To validate the results for the abundance of macrophages and granulocytes in PDAC tissues, we stained human tumors for CD14 and CD15, and found higher cell numbers and densities in PDAC tissues compared to adjacent normal tissues (Fig. 1E).
Fig. 1: Pancreatic ductal adenocarcinoma has a dense CD11b+ myeloid infiltrate.
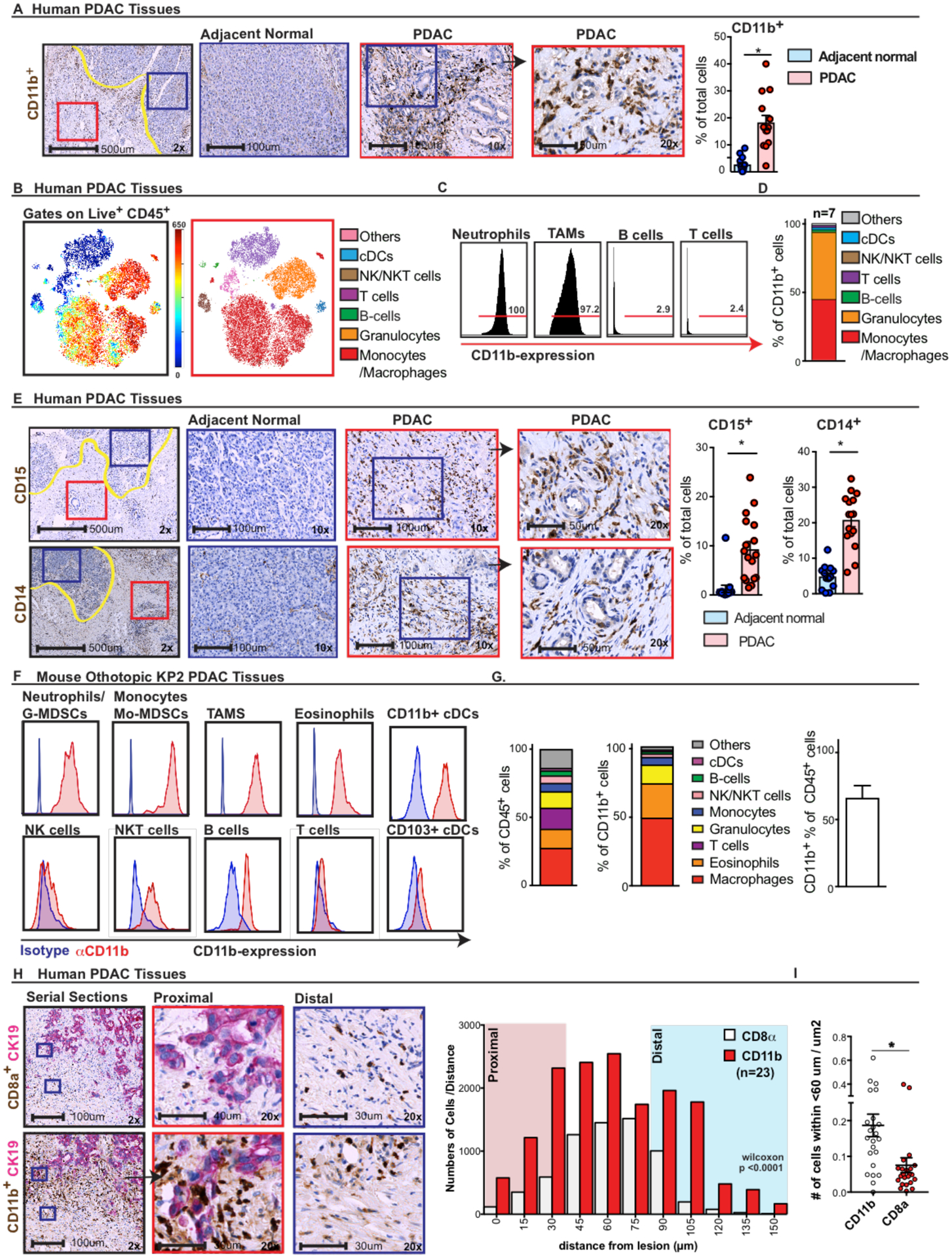
A) Representative images of human PDAC and adjacent normal tissues assessed for CD11b+ myeloid cells at 2.5×, 10×, and 20× (inset). Graphs show the frequency of the subsets in human PDAC and adjacent normal tissues from the same surgical sample (n = 13 paired samples). B–D) CD11b expression determined by CyTOF analysis of human PDAC tissue samples. B) Representative CyTOF tSNE plot showing monocytes, granulocytes, macrophages, T cells, and B cells. C) Expression analysis of CD11b in leukocyte populations. D) Relative frequencies of the composition of CD11b+ cells (n = 7). E) Representative images of CD15+ and CD14+ monocytes and macrophages by IHC. Graphs show the frequency of positive cells in human PDAC and adjacent normal tissues from the same surgical sample (n = 12–15 per paired samples). F–G) CD11b expression analysis of murine PDAC tissues. F) Representative flow cytometry plots showing expression of CD11b on pregated tumor-infiltrating immune cell populations in an orthotopic KP2 PDAC model. G) Graphs depicting the cell composition of total leukocytes (left) and CD11b+ cells, as well as the number of CD11b+ cells per total number of leukocytes (right) (n = 8 mice/graph). H and I) Leukocyte proximity analysis. H) Representative images of CD8a and CD11b (brown) co-staining with CK19 (pink). Histogram of relative CD8a+ or CD11b+ cell numbers binned by cellular distances from CK19+ cells. I) Mean number of positive cells per area within 60 μm of the CK19+ tumor cells (n = 23 PDAC samples).
Graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t test.
To better understand the relative localization of T cells and CD11b+ cells, we co-stained human PDAC tissues for either CD8a or CD11b and CK19 (a marker of PDAC cells). We observed that compared to CD8a+ T cells, CD11b+ cells were in closer proximity to CK19+ PDAC cells (Figs. 1D and S1F); more impressively CD11b+ cells had higher numbers at proximal distances to CK19+ PDAC cells (Fig. 1E, S1G). Taken together these data suggest that CD11b+ myeloid cells are numerically and physically positioned to regulate T cell exclusion in PDAC tissues.
We next examined genetically engineered mouse models (GEMMs) of PDAC (p48-CRE/LSL-KrasG12D/p53flox/flox, “KPC” mice) or cell line-derived PDAC GEMMs that were orthotopically implanted into syngeneic hosts (KP2 or KI). Similar to results obtained with human tumors, we observed that CD11b+ is highly expressed on PDAC-infiltrating monocytes, tumor-associated macrophages (TAMs), granulocytes, and CD11b+ cDCs, but more sparingly expressed on T, B, NK, and NK T cells (Figs. 1F and S1C and D). Overall, CD11b+ cells composed 50–60% of total leukocytes, and fewer than 15% of CD11b+ cells were B, T, and NK cells (Fig. 1F). Taken together, these data suggest that in both human and murine PDAC tumor tissue, CD11b+ is predominantly expressed on myeloid cells, which are highly abundant in the PDAC TME.
ADH-503 binds CD11b and reduces myeloid cell recruitment to PDAC tissues
To overcome the dosing limitation of CD11b blockade in previous studies, we developed a small molecule agonist (24, 26), ADH-503, whose binding achieves a partially active CD11b conformation (Fig. 2A and B).
Fig. 2: Changes in myeloid infiltrates after ADH-503 treatment.
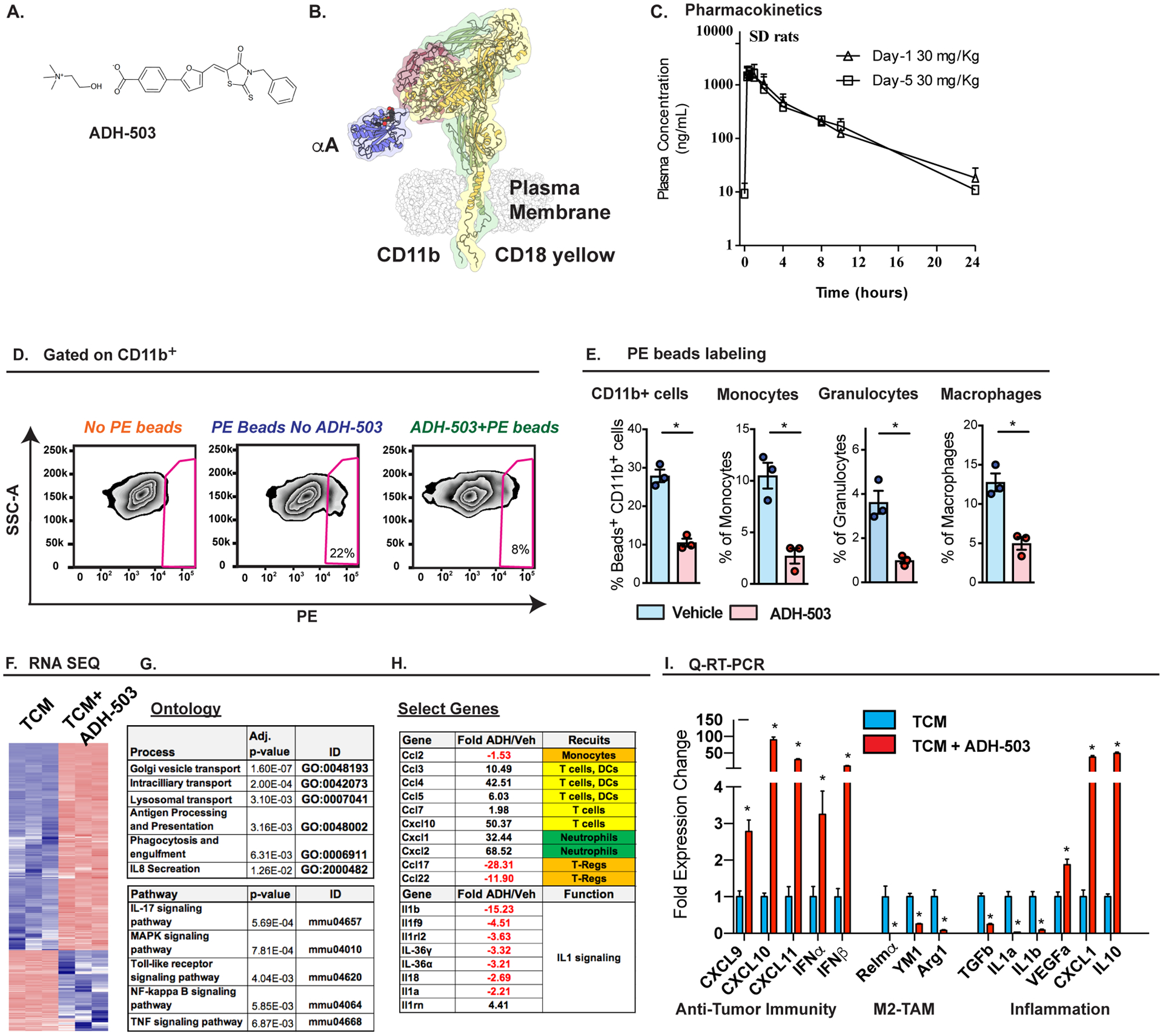
A) Chemical structure of ADH-503. B) A computational model of ADH-503 bound integrin CD11b/CD18, based on the published structure of αXβ2 (54). The integrin chains CD11b (yellow), CD18 (green and red) and the αA-domain of CD11b (blue) are labeled. The model also displays ADH-503 (space-filling model) docked in the activation-sensitive allosteric pocket of the CD11b αA-domain. C) Plasma concentration-time data following oral gavage administration of ADH503 at 30 mg/kg in male rats on Day 1 and Day 5. n=? D) Representative flow cytometry plots of PE+ beads taken up by CD11b+ cells in PDAC tissue from the KP2 orthotopic PDAC model. E) Quantification of PE+ beads taken up by tumor-infiltrating CD11b+ cells, monocytes, granulocytes, or macrophages with and without ADH-503 treatment (n = 3/group). F–H) RNA-seq expression analysis of bone marrow-derived macrophages treated with PDAC conditioned media ± ADH-503 for 7 hours. E) Heat map of differentially expressed genes, F) gene ontology table, and G) select gene changes are depicted. H) Q-PCR mRNA expression analysis of bone marrow-derived macrophages treated with PDAC conditioned media ± ADH-503 or vehicle for 7 hours. Changes in gene expression are depicted as the fold change from the vehicle baseline.
Graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t test.
To develop ADH-503 molecule for future clinical use we first tested its pharmacokinetics (PK) following oral administration and safety profile in several vertebrate species. We first determined that ADH-503 preforms equally well as an agonist using both human and mouse CD11b+ monocytes (Fig. S2A–B). Next, we performed pharmacokinetic studies in rats and C57/B6 mice (Fig. 2C, S2C–F). In rats the mean half-life of ADH503 at 30 and 100mg/kg dosing was 4.68 and 3.95 hours with a Cmax and AUC0-t in the plasma of 1716 and 2594 ng/mL and 6950 and 13962 ng.h/mL, respectively. Repeat dosing in rats was similar for these parameters as doses progressed (Fig. 2C, S2C–E). Dosing in C57/B6 mice has similar PK properties (Fig. S2D).
To determine the safety profile of ADH503, we performed pre-clinical toxicological studies in Sprague-Dawley rats and in Beagle dogs. Studies showed that ADH503 is well tolerated and displayed no adverse effects or toxicity after single dose or after repeated-dose for 28 days at doses up to 1500 mg/kg/d in rats and up to 1359 mg/kg/d in dogs. There was no mortality, clinical signs or body weight changes associated with ADH503 administrations and the compound was well-tolerated.
To test the impact of ADH-503 on myeloid cell trafficking in tumor-bearing mice, we used fluorescently labeled latex beads to monitor the trafficking of blood cells. Twenty-four hours after labeling, we found bead-positive inflammatory monocytes, neutrophils, and macrophages in PDAC tissues, suggesting the active recruitment of these inflammatory cells (Fig. 2D). However, in mice treated with ADH-503, we found a significant reduction in the number of beads positive cells in all three of these populations (Fig. 2E). These data suggest that ADH-503 can partially prevent the recruitment of myeloid cells from the circulation into PDAC tissues.
ADH-503 directly alters the cytokine profile of PDAC-activated macrophages
To examine the direct effects of ADH-503 on the responses of macrophages to tumor-derived factors, we treated bone marrow-derived macrophages with conditioned medium from KPC-derived PDAC cells in the presence or absence of ADH-503, and then examined the gene expression levels of key cytokines by Q-PCR and RNA sequencing (RNA-seq). RNA-seq analysis showed that greater than 8000 RNAs were changes by 2 fold within 6 hours of ADH-503 exposure (Fig. 2F). Ontological analyses of these gene sets showed key changes in genes involved in antigen presentation and processing, lysosomal trafficking, phagocytosis and interleukin (IL)-8 signaling (Fig. 2G). Further analyses of these data also show that ADH-503 rapidly decreased the genes involved in IL-1 signaling, increased the expression of cytokines involved in T cell and DC trafficking, and reduced the regulatory T cell recruitment of cytokines CCL17 and 22 (Fig. 2H). Results from a parallel Q-PCR analysis in a second experiment revealed that ADH-503 down-regulated TGFβ1, IL1α, and IL1β, and reduced the levels of alternative activation markers Arg1, YM1, and Retnlα, while upregulating type I interferons (IFNα1 and β) and T cell recruitment factors (CXCL9, 10, and 11, Fig. 2I). Taken together, these data suggest that ADH-503 results in the repolarization of macrophages towards a phenotype that could support anti-tumor T cell responses.
To determine the impact of CD11b agonists on myeloid cells in vivo, we examined immune infiltration in two distinct syngeneic orthotopic models (KI and KP2) and the genetic KPC model. In orthotopic tumors, we found that 8 days of treatment with ADH-503 reduced the numbers of total tumor-infiltrating CD11b+ cells as well as subsets of CD11b+ monocytes, granulocytes, eosinophils, and macrophages (Fig. 3A–C). Notably, at treatment durations shorter than 8 days we did not observe reduced macrophages or granulocytes numbers (Fig. S3B), suggesting 8 days is needed for the recruitment blockade to result in changed tumor myeloid numbers. Similar to orthotopic models, we observed reduced numbers of CD68+ and GR1+ cells in PDAC tissues from KPC mice treated with ADH-503 for 14 days (Fig. 3E). These reductions in the subsets of CD11b+ cells persisted for greater than 14 days orthotopic tumors and for more than 30 days in KPC GEMM PDAC tissues (Figs. 3E and S3A), suggesting that a limited compensatory mechanism exists. Other cell types, which express lower levels of CD11b (Fig. 1), including NK cells and B cells, remained unchanged after ADH-503 treatment (Fig. 3B–D). To determine whether the changes in the tumor-infiltrating myeloid cell subsets were due to systemic changes in circulating myeloid cells or their progenitors, we analyzed the blood, bone marrow, and spleens of mice treated with vehicle or ADH-503. We observed no differences in the numbers of myeloid cells in these peripheral tissues (Fig. S3B). These data suggest the possibility that high rate of transmigration of myeloid cells from circulation via CD11b likely requires high local density of CD11b ligands (e.g.; ICAM-1), which might be elevated near tumor sites v/s other normal tissues. Taken together, these data suggest that a CD11b agonist can functionally reduce the numbers of multiple myeloid cell subsets.
Fig. 3: ADH-503 alters innate responses in PDAC tissues.
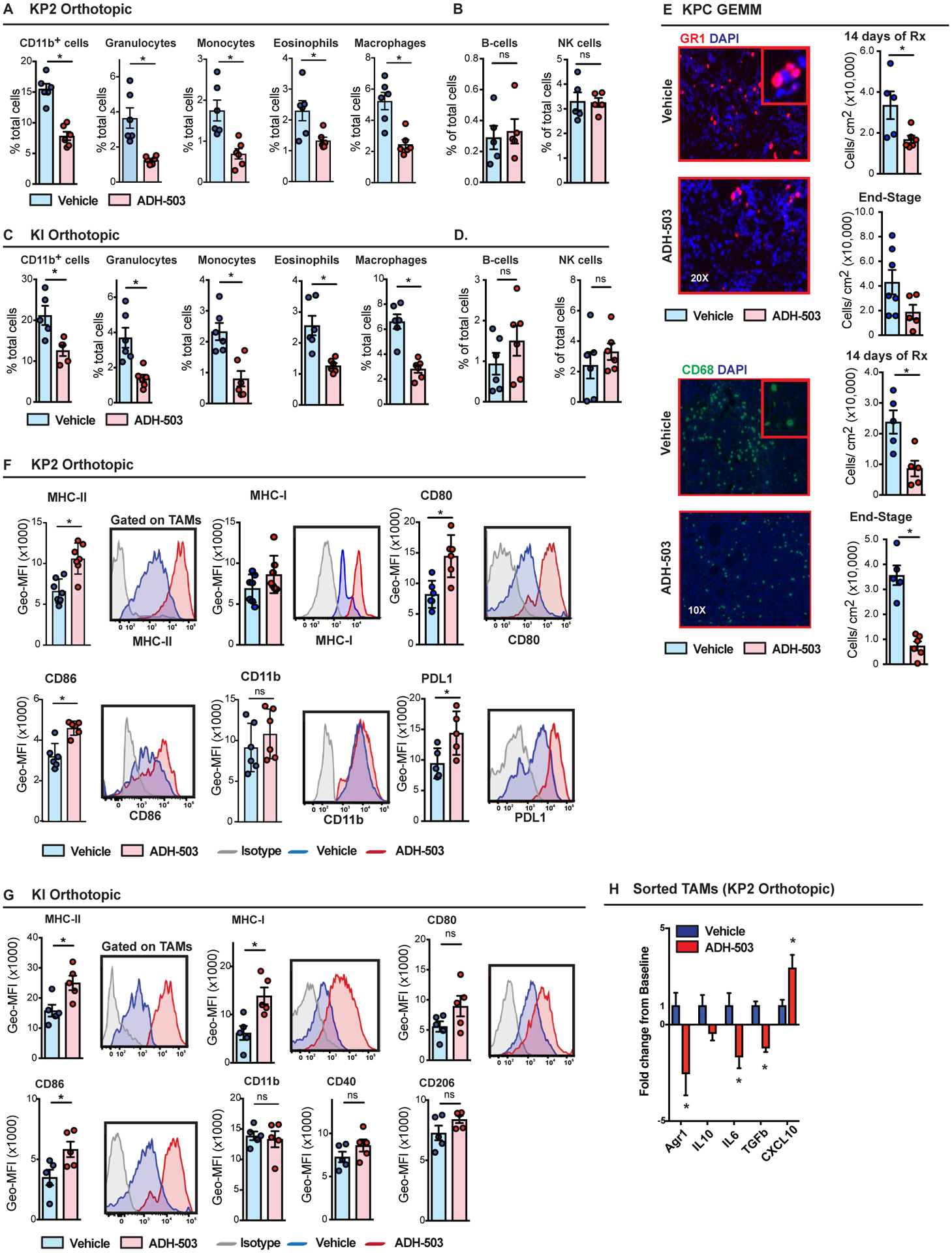
A–D) Relative frequencies of tumor-infiltrating granulocytes, monocytes, eosinophils, B cells, NK cells, and macrophages in orthotopic KP2 or KI PDAC models 10 days after treatment with ADH-503 or vehicle (n = 6/group). E) Representative immunofluorescent images of Gr-1 and CD68 in PDAC tissues from KPC mice treated with vehicle or ADH-503 for 14 days. Quantification is shown as the cell number per area in PDAC tissues from KPC mice treated for 14 days or until end-stage tumors developed (n = 5–7 mice/group). F and G) Flow cytometry analysis of antigen presentation markers on TAMs. Data are shown as histograms of geometric mean fluorescent intensity (GEO-MFI) data on TAMs in orthotopic KP2 and KI PDAC models treated with ADH-503 or vehicle for 10 or 12 days. n=? H) Q-PCR analysis on TAMs isolated by FACS from orthotopic KP PDAC tumors 10 days after treatment with ADH-503. Data are shown as the fold change from the vehicle baseline (n = 4 samples/group).
Graphs are shown as the mean ± standard error; * denotes P < 0.05 by two-sided t test. All flow cytometry data are representative of 2–3 independent in vivo experiments using both tumor models.
To assess the impact of ADH-503 on the tumor macrophage phenotype in vivo, we compared the expresison of macrophage markers by flow cytometry 10 days after treatment with ADH-503 in two syngeneic orthotopic models. We found that macrophages had higher expression levels of MHCI, MHCII, CD80, and CD86 after treatment with ADH-503, indicating improved antigen-presenting properties of the remaining macrophages (Figs. 3F and G, and S3C). The expression levels of CD11b, CD40, and CD206 were unchanged in the macrophages. These data suggest that ADH-503 not only decreases the number of total myeloid cells in the tumor but also reprograms the remaining macrophages and improves their antigen-presenting properties. Consistent with the alterations in the macrophage phenotype observed in vitro, gene expression profiling of TAMs sorted from tumors of ADH-503-treated mice showed reduced expression levels of immunosuppressive genes (IL6, TGFβ, Arginase-1, and IL10) and increased expression levels of the T cell chemokine CXCL10 (Fig. 3H). These data support the role of ADH-503 in enhancing a T cell-supportive TAM phenotype in vivo.
CD11b agonism improves T cell responses in vivo
To determine whether CD11b agonism can improve anti-tumor immunity, we explored changes in tumor T cell infiltration and phenotype in two orthotopic PDAC models and in KPC mice. We analyzed tumor-infiltrating T cells and found that ADH-503 treatment resulted in increased numbers of total CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ T effectors (Figs. 4A–D and S4A). In addition to increasing T cell frequency, ADH-503 also increased CD8+ CTL and CD4+ effector cell proliferation (Ki67+ CD8+ CTLs) and activation (CD44Hi CD62Lneg) (Fig. 4A–D). Notably, we also observed increases in PD1+, EomesHigh/PD1High, and Tim3High/PD1High CTLs, suggesting that activated CD8+ CTLs may have a more checkpoint-engaged phenotype (Fig. 4B). Similar increases in total CD8a+ T cell numbers were observed by IHC (Fig. S4B). In contrast, we found reduced numbers of FOXP3+ regulatory T cells (Treg) and a better CD8+ CTL to Treg ratio in PDAC tissues from ADH-503-treated mice (Figs. 4B and S4A–B). To determine whether ADH-503-induced immune changes could change the proximity of T cells relative to PDAC cells, we co-stained for CK19 and CD8a, and analyzed cell proximities (Fig. 4E). We found that although the distribution of CD8+ T cells was not markedly changed over distances of 30–150 μm, the number and frequency of CD8+ T cells in close proximity to PDAC cells were changed by ADH-503 treatment (Fig. 4E). In vehicle-treated mice, CD8+ T cells were in very limited numbers at distances of less than 25 μm, with nearly no CTLs at distances <10 μm. In contrast, ADH-503 treatment significantly increased the number of CD8+ CTLs in direct contact (< 5 μm) and in close proximity (< 25 μm) to CK19+ PDAC cells. Corresponding to this increase in T cell number, we also observed a marked increase in PD-L1 expression on the PDAC cells (Fig. 4C).
Fig. 4: CD11b-agonism stimulates T cell infiltration and function through augmentation of cDC1s.
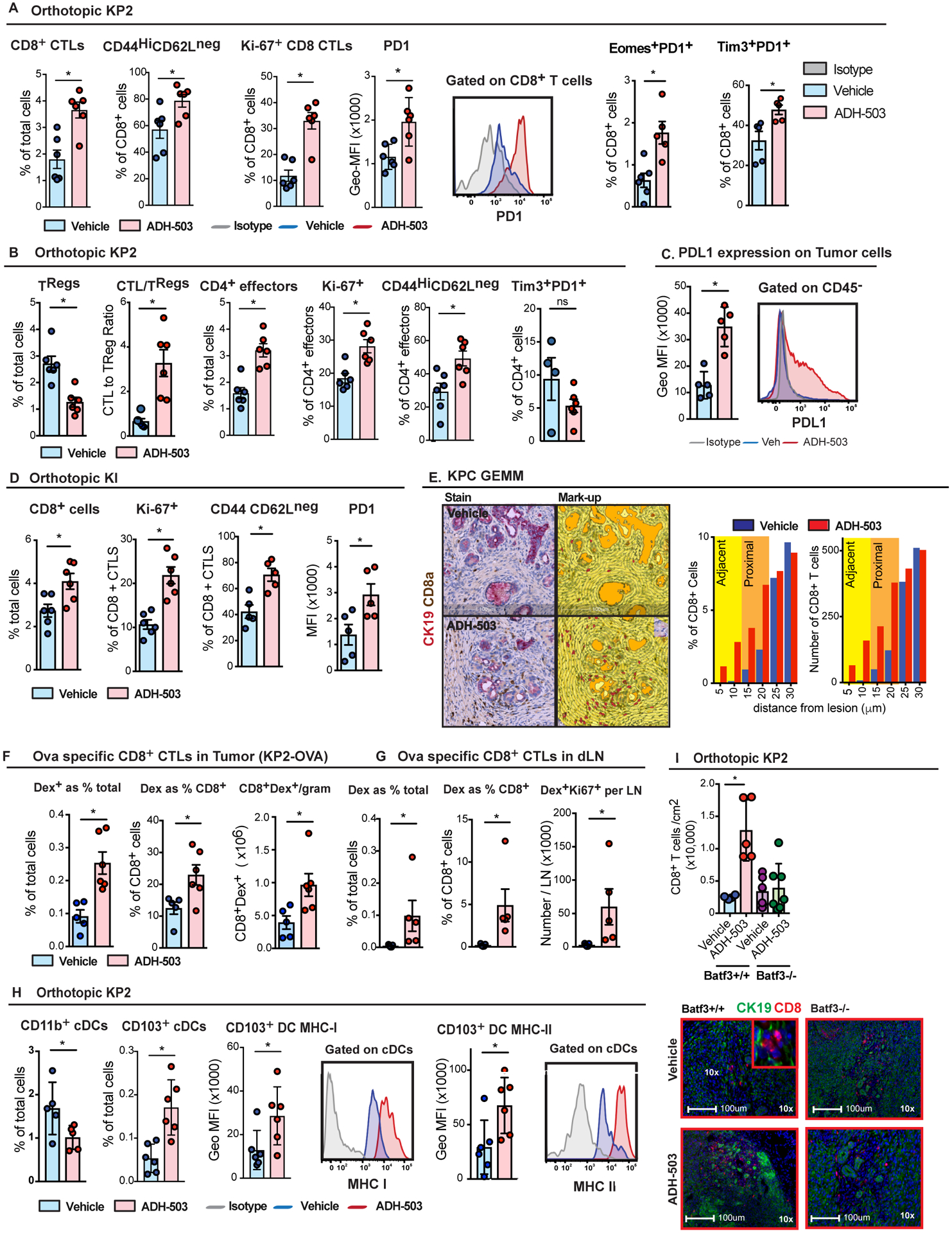
A and B) Frequencies of tumor-infiltrating CD8a+ CTLs, FOXP3+ regulatory T cells, and CD4+ effectors in orthotopic KP2 PDAC tissues from mice treated 10–12 days with ADH-503 or vehicle. Graphs show the mean CTL number and subsets of CD8+ CTLs marked by CD44HiCD62LLow, Ki-67, PD1High, Tim3+PD1+, or Eomes+PD1+ (n = 5–6/group). C) Measurement of PD-L1 expression by flow cytometry of the CD45− population in orthotopic KP2 PDAC tissues 10 days after treatment with ADH-503 or vehicle. Representative histograms and Geo-MFI are shown. D) Frequencies and phenotypes of tumor-infiltrating CD8a+ CTLs in orthotopic KI PDAC tissues from mice treated 12 days with ADH-503 or vehicle. Graphs show the mean CTL number and subsets of CD8a+ CTLs marked by CD44HiCD62LLow, Ki-67, or PD1High (n = 5–6/group). E) Representative IHC images of CD8a (brown) and CK19 (pink) in PDAC tissues derived from KPC mice treated with vehicle or ADH-503 for 14 days. The histogram shows the relative CD8a+ cell number or frequency binned by cellular distances from CK19+ cells. I) Mean number of positive cells per area within 60 μm of CK19+ tumor cells (n = 6/group). F and G) Frequencies and quantification of OVA-specific dextramer+ cells in PDAC tissues (F) and dLNs (G) using the orthotopic KP2-OVA PDAC model in mice treated with vehicle or ADH-503 for 10 days. H) Frequencies of CD11b+ and CD103+ DCs and MHC-I and MHC-II expression in CD103+ cDCs in KP2 PDAC tissues from mice treated with vehicle or ADH-503 for 12 days. Mean cell percentages and GEO-MFI data are shown. I) Quantification of CD8+ T cells in KP2-OVA PDAC tissues from wild-type and BATF3-deficient mice treated with vehicle or ADH-503 for 10 days. Mean cell number/area and representative IHC images are shown.
Graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t test (A-H) or Mann-Whitney test (G), depending on the data distribution or Kolmogorov-Smirnov test for immune cell proximity (E). All flow cytometry data are representative of 2–3 independent in vivo experiments using at least two PDAC models.
To determine whether ADH-503 improved tumor antigen-specific T cell responses, we analyzed OVA-specific dextramer+ CD8+ T cells in orthotopic KP2-OVA tumors. We observed that ADH-503 increased both the total number of dextramer+ CTLs as well as the relative frequency of dextramer+ cells among CD8+ CTLs, suggesting the effective expansion of tumor-specific T cells (Fig. 4C). We next examined changes in tumor-specific T cells in pancreas-draining lymph nodes (dLNs). We observed an increased frequency and proliferation of dextramer+ CD8+ CTLs in PDAC dLNs of ADH-503-treated mice compared to vehicle-treated mice (Fig. 4D). These data suggest that CD11b-agonism leads to the expansion of the tumor antigen-specific T cell population in tumor tissues and priming in the periphery.
ADH-503 treatment induces the accumulation of CD103+ cDCs in the tumor
Due to the increase in intratumoral T cell numbers and proliferation, we explored whether these effects are driven by changes in DCs. As expected, based on their CD11b expression, we observed reduced numbers of tumor-infiltrating CD11b+ cDC2s and monocyte-derived DCs after 12 days of ADH-503 treatment (Figs. 4H and S4D). In contrast, in ADH-503-treated mice, tumor-infiltrating CD103+ cDC1s (which express CD11b at extremely low levels) were markedly increased in both number and MCH-I and MHC-II expression (Fig. 4H). These data suggest that ADH-503 reduces the numbers of potentially tolerogenic and/or CD4+ T cell-priming DCs, while enhancing cross-presenting by CD103+ cDC1s. The identity of these cDC populations was confirmed using Zbtb46gfp/+ reporter mice (30) (Fig. S1D). To determine whether the changes in cDC1s were necessary for the increased CTL response observed in ADH-503-treated mice, we used BATF3−/− mice, which lack functional cDC1s (31). In contrast to wild-type controls, treatment with ADH-503 had no effect on T cell infiltration in BATF3-deficient mice (Fig. 4I). Taken together, these findings suggest that myeloid cell reprograming by ADH-503 drives cDC1 infiltration and function, leading to a reinvigorated anti-tumor T cell response.
ADH-503 impairs tumor growth and improves survival in orthotopic models and KPC GEMMs
To determine the impact of a CD11b agonist on tumor progression, we evaluated three syngeneic orthotopic PDAC models and KPC GEMMs (Fig. 5A–D). In all models, ADH-503 delayed tumor progression, leading to a significantly decreased tumor burden in time point analysis and improved overall survival (Fig. 5C and D). Importantly, ADH-503 had no direct effects on PDAC cell growth in vitro (Fig. S5A). To further confirm the specificity of ADH-503 for CD11b, we utilized CD11b-null (ITGAM-null) mice and found that unlike in wild-type mice, CD11b-null mice had similar tumor growth, regardless of treatment (Fig. 5E).
Fig. 5: CD11B agonism delays tumor progression.
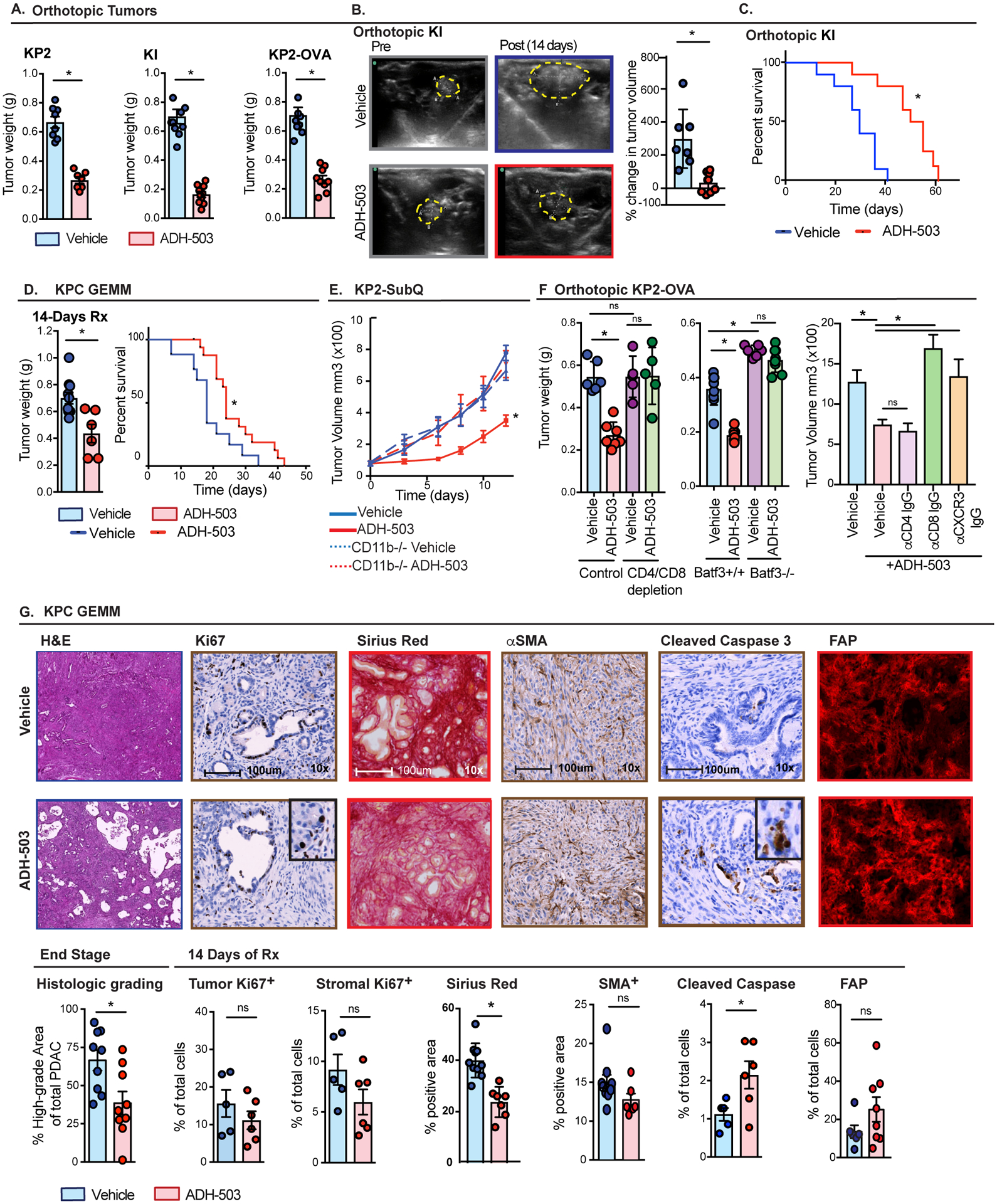
A) Tumor growth in syngeneic orthotopic models of PDAC KP2, KI, and KP2-OVA shown by tumor weights 10 days after treatment with vehicle or ADH-503 (n = 7–10/group). B) Changes in tumor volume as measured by ultrasound imaging. Representative ultrasound images of KI PDAC tumors and mean percent changes in tumor volume are depicted 14 days after treatment. Yellow line depicts tumor area. C) Kaplan-Meier survival analysis of orthotopic tumors KI tumors treated with vehicle or ADH-503 (n = 7–8/group). D) Genetic KPC mice were treated with vehicle or ADH-503. Tumor weight of mice treated for 14 days and Kaplan-Meier survival analysis are shown (n = 6–7 or 10–12/group, respectively). E) Subcutaneous KP2 tumor growth in wild-type or CD11b-deficient animals treated with vehicle or ADH-503 once the tumor reached 75–100 cm3. Tumor volume was measured by calipers. (n=?). F) Orthotopic KP-OVA tumor burden measured 10 days after treatment with vehicle or ADH-503 in wild-type or BATF3-deficient mice or in wild-type mice treated with CD4 and/or CD8-depleting IgGs or neutralizing IgGs against CXCR3 (n = 5–10/group). G) Analysis of PDAC pathology. Shown are representative H&E images with histological grading, IHC results of tumor and stromal Ki67 staining, tumor cleaved caspase 3 staining, Sirius red-stained collagen density, and SMA+ or FAP+ fibroblasts in PDAC tissues from KPC tumors treated with vehicle or ADH-503 for 14 days or at end stage (n = 5–10 mice/group).
Bar graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t -test, log-rank test or ANOVA as appropriate. Tumor burden data are representative of 2–3 independent in vivo experiments.
To understand the importance of the observed alterations in T cells and cDC1s on the efficacy of ADH-503, we depleted CD4+ and CD8+ T cells and implanted BATF3-deficient mice (30–32). We found that neither T cell depletion nor BATF3 deficiency markedly changed KP2-OVA tumor growth in vehicle-treated mice, suggesting that tumor immunity is dysfunctional, even when a neo-antigen (OVA) is engineered into these PDAC tumors (Fig. 5F). However, whereas ADH-503 significantly restrained the growth of KP2-OVA tumors, this effect was completely dependent on both T cells and BATF3dependent cDCs (Fig. 5F). More specific depletion of CD4 or CD8 T cells found that CD4+ T cells were dispensable for ADH-503 tumor growth suppression but CD8a+ T cells were necessary (Fig. 5F). Based on the observation that CXCL-9, −10, and −11 were highly unregulated in TAMs treated with ADH-503 (Fig. 2I) and are drivers of CD8 T cell recruitment, we neutralized CXCR3 and found this also abolished ADH-503’s anti-tumor activity (Fig. 5F). Taken together, these data suggest that ADH-503-induced improvements in cDC1 responses are critical for the orchestration of anti-tumor CD8+ T cell immunity and tumor restraining treatment efficacy.
To determine the impact that ADH-503 has on the pathology of PDAC tumors, we analyzed tissues from either KPC GEMMs treated for 14 days or at the time of terminal disease progression. Consistent with the lack of tumor cell intrinsic activity, we saw no change in PDAC cell proliferation in cells from ADH-503-treated mice. Instead, we observed increased cleaved caspase 3, a marker of apoptosis, in PDAC cells (Figs. 5G and S5B), which is consistent with an immune-mediated effect. In accord with the known invasion-promoting activities of myeloid cells (33), we found that ADH-503 decreased tumor grade (Figs. 5G and S5B).
Analysis of tumor-associated fibrosis revealed that ADH-503-treated mice had reduced collagen density, but no changes in α-smooth muscle actin or FAP+ fibroblasts (Fig. 5G). Consistent with these results, we saw no changes in stromal proliferation or apoptosis (Figs. 5G and S5B). These data suggest that ADH-503 can alter collagen density but not by decreasing fibroblast number.
ADH-503 enhances the efficacy of standard of care therapies to eliminate metastasis
To determine whether ADH-503 could augment the efficacy of chemotherapy, we treated mice bearing orthotopic KI tumors with gemcitabine (GEM) plus paclitaxel (PTX) alone or in combination with ADH-503. Ultrasound tumor volume measurements revealed that whereas GEM/PTX resulted in a 35% reduction in tumor progression, the combination of ADH-503 plus GEM/PTX resulted in significant disease control (> 90%, Fig. 6A), which translates into a marked increase in survival (Fig. 6B). Analysis of end-stage metastasis found that 80% vehicle-treated and 55% of and chemotherapy-treated animals developed gross hepatic metastases. However, in spite of living longer, we only observed metastasis in only 12.5% of animals treated with by ADH-503 and none of the animals treated with ADH-503 plus PTX/GEM (Fig. 6C). Intriguingly, immune profiling of tumors after 14 days of treatment showed that GEM/PTX therapy neither impaired nor synergized ADH-503 in stimulating tumor immunity (Fig. S6A). Together, these data suggest that the combination of ADH-503 with chemotherapy has a significant impact on disease progression and metastasis, but does not lead to a synergistic impact on tumor immunity. To determine whether ADH-503 could improve the efficacy of radiation therapy (RT), we treated mice bearing orthotopic KI tumors with 4Gy×5 RT (4Gy ×5) using a CT-image guided small animal radiation research platform (SARRP). We found that although both RT and ADH-503 lead to impaired disease progression, only the combination yielded substantial tumor regression (Fig. 6D). Taken together these data suggest CD11b-agonism might improve multiple types of conventional therapies.
Fig. 6: CD11B agonism improves the efficacy of chemotherapy.
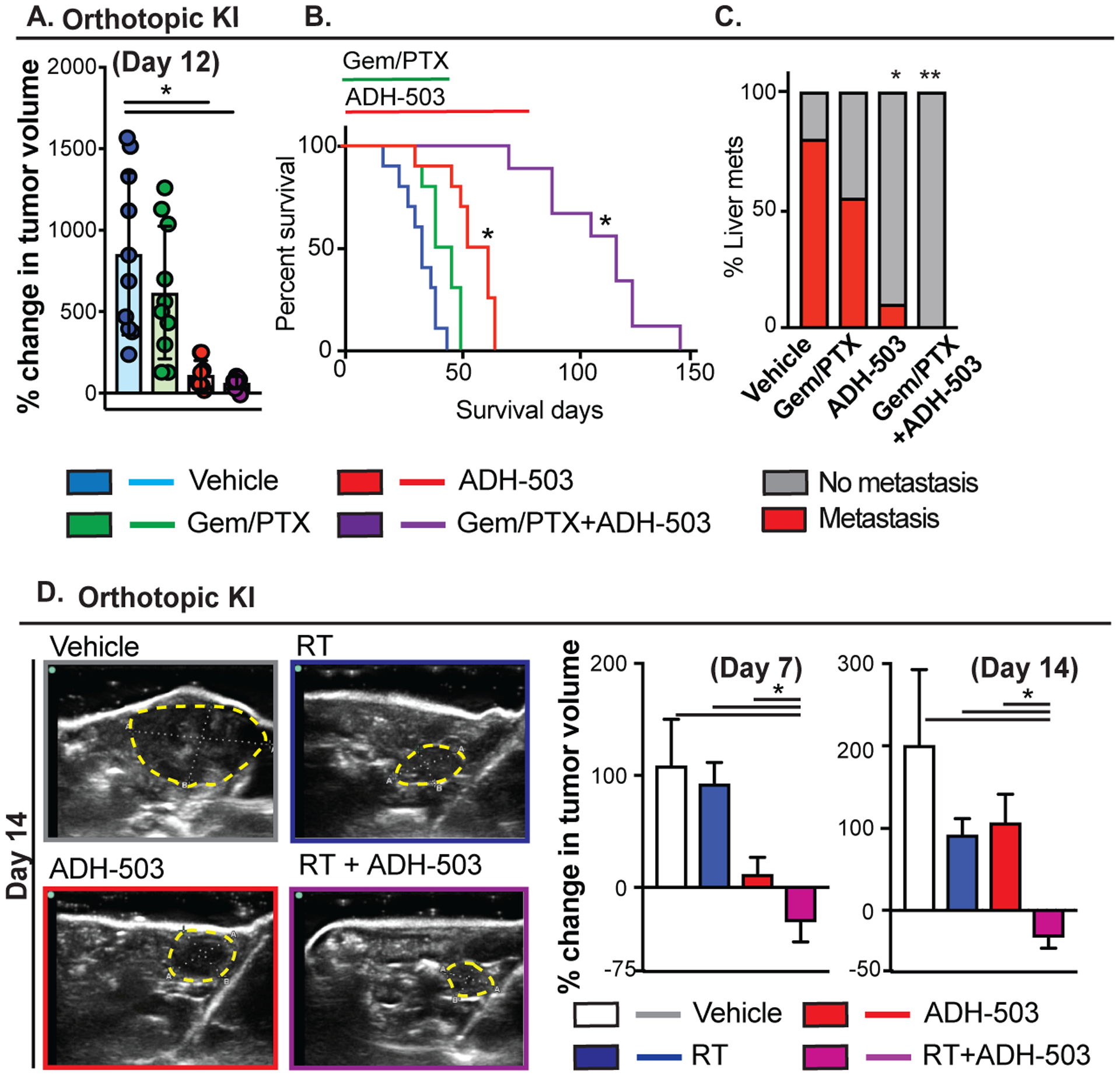
A) Changes in tumor volume as measured by ultrasound imaging. Animals were enrolled when the orthotopic KI tumor was greater than 0.4 cm in diameter and subsequently treated with vehicle or ADH-503 ± GEM/PTX. Representative ultrasound images and mean percent change in tumor volume are shown 12 days after treatment (n = 8–10/group). B) Kaplan-Meier survival analysis of mice from (A) (n = 8–10/group). C) Quantification of the percentage of mice bearing overt liver metastases on gross examination (n = 7–10/group). D) Changes in tumor volume as measured by ultrasound imaging. Animals were enrolled when the orthotopic KI tumor was greater than 0.4 cm in diameter and subsequently treated with vehicle or ADH-503 ± radiation therapy (4Gy × 5). Representative ultrasound images and mean percent change in tumor volume are shown 12 days after treatment (n = 8–10/group).
Bar graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t test or log-rank test.
ADH-503 renders PDAC tumors responsive to checkpoint immunotherapy
As described above, we observed that CD11b agonists improved T cell infiltration and function but also upregulate PD1/PDL1 checkpoint molecule expression on CD8 T cells, TAMs, and PDAC cells (Fig. 3 and 4). These data suggest that ADH-503 might synergize with anti-PD-1 or other checkpoint therapies. To examine this supposition, we combined ADH-503 with αPD-1 IgG in two orthotopic PDAC models. Although neither model showed benefit from treatment with αPD-1 antagonists (34–36), the combination of ADH-503 and αPD-1 IgG resulted in significant tumor regression over 14 days of treatment (Fig. 7A). This short-term tumor regression led to complete tumor regression by days 21–30 and survival past 120 days, even after cessation of treatment (Fig. 7B). Analysis of tissues from ADH-503 plus αPD-1-treated mice showed the restoration of normal pancreas morphology, minimal scar tissue, and no evidence of malignant cells, either by H&E or CK19 staining (Fig. 7B). This experiment was repeated in two independent cohorts with the same outcomes: 100% survival past 120 days and no residual disease. Re-challenge of mice with subcutaneous KI tumor cells resulted in 100% tumor growth in naïve control mice, but no detectable tumor in mice previously treated with ADH-503 plus αPD-1. These data demonstrate that ADH-503 strongly synergizes with αPD-1, converting completely unresponsive PDAC tumors into responsive tumors with marked tumor regression, long-term survival, and lasting immunologic memory.
Fig. 7: CD11B agonism renders PDAC tumors responsive to checkpoint immunotherapy.
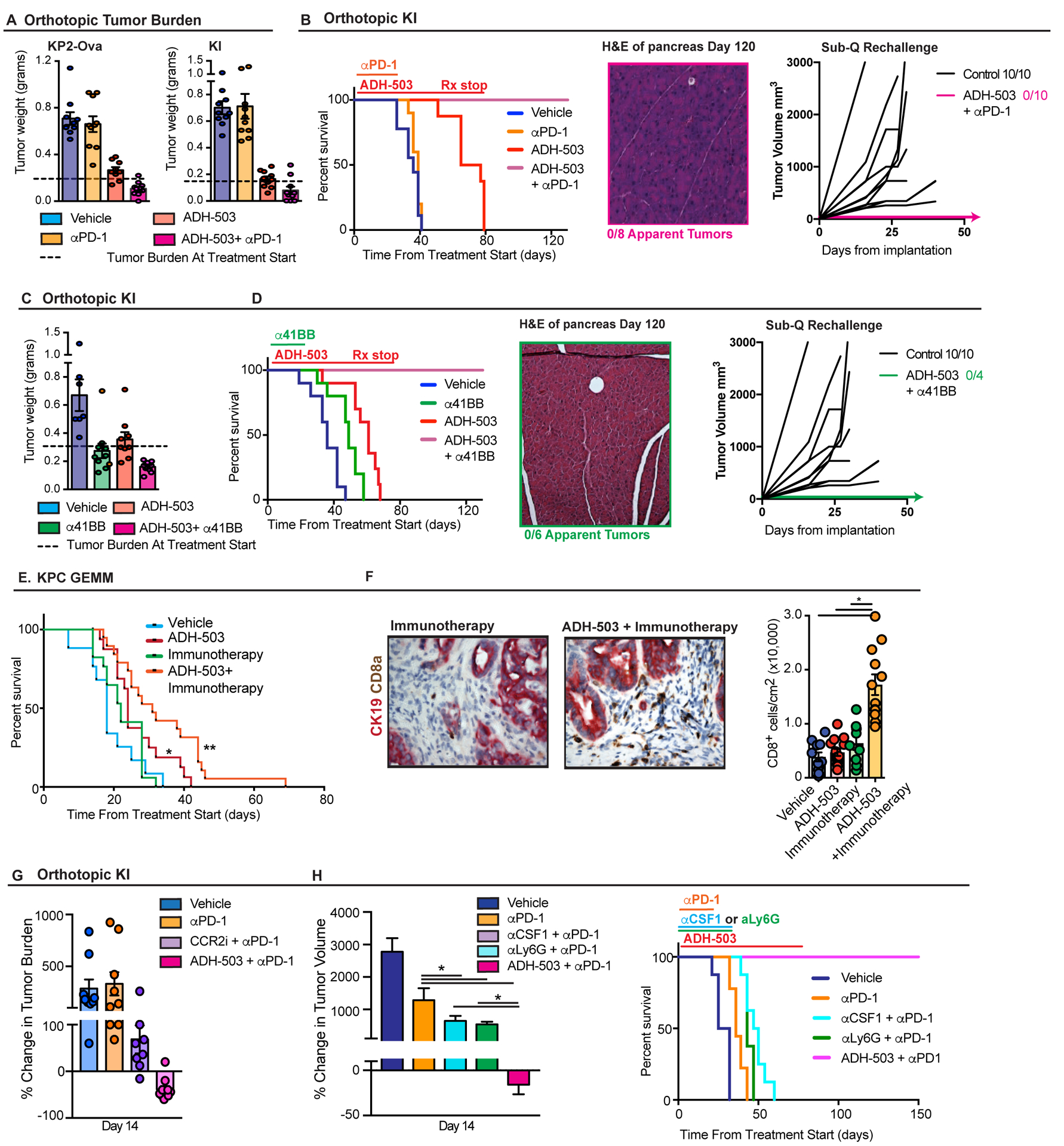
A) Tumor burden 12–14 days after treatment with vehicle or ADH-503 ± anti-PD1 in orthotopic KP2-OVA or KI models (n = 8–10/group). Dashed line depicts tumor burden from five parallel mice taken at start of treatment. B) Analyses of survival and rechallenge. KI tumor-bearing mice from (A) were assessed for Kaplan-Meier survival analysis (left), pathological analysis of the pancreas was performed in mice surviving more than 120 days (center), and mice treated with this regimen were re-challenged with KI tumor cells subcutaneously (right). “Control” mice were animals who had never been exposed to KI tumor cells prior to subcutaneous injection (n = 8–10/group). C) Tumor burden 12 days after treatment with vehicle or ADH-503 ± anti-41BB in mice bearing established (> 0.4 cm) orthotopic KI PDAC tumors (n = 7–9/group). D) Analyses of survival and rechallenge. KI tumor-bearing mice from (C) were assessed for Kaplan-Meier survival analysis (left), pathological analysis of the pancreas was performed in mice surviving more than 120 days (center), and mice were re-challenged with KI tumor cells subcutaneously (right). “Control” mice were animals who had never been exposed to KI tumor cells prior to subcutaneous injection (n = 4–10/group). E) Kaplan-Meier survival analysis of genetic KPC mice treated with vehicle or ADH-503 ± “Immunotherapy” (50 mg/kg gemcitabine + anti-PD1 + anti-CTLA4). Mice were enrolled in the study when tumors were greater than 0.4 cm in diameter (n = 10–15/group). F) Analysis of CD8a+ cells that infiltrated into PDAC tissues in end-stage KPC tumors from mice in (E). Representative images and quantitation results are shown. G) Comparison of ADH-503 and CCR2 inhibition (CCR2i, PF-04136309). Tumor burden 14 days after treatment with vehicle, ADH-503, or CCR2i ± anti-PD1 in mice bearing established (> 0.4 cm) orthotopic KI PDAC tumors (n = 7–9/group). Data are depicted as the change in tumor burden compared to five untreated animals sacrificed at the beginning of treatment. H) Comparison of ADH-503 and CSF1/CSF1R or granulocyte inhibition. The left panel shows the observed changes in tumor volume 14 days after treatment with vehicle, ADH-503, anti-CSF1 IgG (5A1), or Ly6G-depleting IgG (1A8) ± anti-PD1 in mice bearing established (> 0.4 cm) orthotopic KI PDAC tumors. The right panel shows the Kaplan-Meier survival analysis in the same animals.
Bar graphs show the mean ± standard error; * denotes P < 0.05 by two-sided t -test, log-rank test or ANOVA as appropriate.
To understand whether CD11b-agonism could improve additional checkpoints strategies, we combined ADH-503 with CTLA4-antagonistic and 41BB (CD137)-agonist IgGs. Unlike αPD1, α41BB IgG show significant tumor-restraining properties as single agents (Fig. 7C and D). More impressively, ADH-503, in combination with α41BB IgG, acted synergistically to induce marked tumor regression, long-term survival of treated animals past 120 days, and complete resistance in re-challenge with KI tumor cells (Fig. 7C and D). In contrast to 41BB, αCTLA4 IgGs had no impact on tumor burden as a single-agent and no synergy with ADH-503 (Fig. S7A), suggesting that either the PD1 checkpoint was still dominant in this context or that there is specificity in the checkpoint combinations that synergize with CD11b agonists.
To validate these results in an autologous model, we treated KPC GEMMs with a combination of αPD1, αCTLA4, and low-dose GEM (“PCG”). As we have previously shown this combination is not effective alone in KPC GEMMs (37), similarly we did not see any efficacy in this study (Fig. 7E). In contrast, the combination of ADH-503 plus PCG significantly increased the survival of KPC mice, with the majority of mice dying of pancreatic insufficiency-induced weight loss rather than tumor progression (Figs. 7E and S7B). Corresponding to these effects on increased survival, while PCG alone did not increase tumor infiltration by CD8+ T cells, ADH-503 plus PCG resulted in a marked increase in CD8+ T cell infiltration (Fig. 7F). These findings demonstrate that such combination therapy can overcome resistance to the penetration of CTLs into the TME, even in otherwise immunologically cold GEMM tumors.
CD11b-agonism is superior to macrophage or granulocyte depletion for enhancing immunotherapeutic responsiveness
Several published reports have shown that depletion of either macrophages or granulocytes with CSF1R, CXCR1/2, or CCR2 inhibitors, or depleting antibodies against Ly6G or CSF1, can synergize with checkpoint inhibitors in pancreatic and other cancer models (5–7, 38–41). To determine how CD11b-agonism compares to these approaches, we conducted head-to-head efficacy studies of ADH-503 against CCR2 inhibitors (CCR2i), CSF1 neutralization, and granulocyte depletion using αLy6G IgGs. As described above, single-agent αPD1 IgG treatment was ineffective. Also, as expected, αPD-1 agonists in combination with CCR2 inhibition, αCSF1 neutralizing IgG, or Ly6G-depleting IgGs all synergized to restrain tumor progression and augment animal survival (Fig. 7G and H).
Discussion
Several lines of evidence suggest that targeting tumor-infiltrating myeloid cells might improve either the proportion of cancer patients who respond to immunotherapy or the extent of their responses. However, it is also clear that compensatory mechanisms exist within the myeloid cell lineage to render such approaches inert, in at least a subset of patients. Our data highlight a promising strategy to target multiple lineages of immunosuppressive myeloid cells with a single agent that activates CD11b signaling. We show that ADH-503 blunts both monocyte and granulocyte trafficking, while simultaneously directly stimulating the anti-tumor properties in tumor-residing macrophages. These immune changes overcome resistance to checkpoint immunotherapy in PDAC models, supporting the concept of using a CD11b agonists to overcome resistance to checkpoint immunotherapies.
Preclinical data for targeting the monocyte/macrophage axis in PDAC and other cancers to improve tumor immunity are strong (9, 42–45). Mechanisms of action include the expansion of tumor-infiltrating T cells by proliferation or recruitment, and/or the augmentation of DC responses (9, 38, 42–46). These preclinical data have led to a considerable number of clinical trials that examined a combination of cytotoxic and immunotherapeutic approaches (47). In PDAC, early clinical data suggested that blocking CCR2 improved responsiveness to chemotherapy with associated improvements in tumor immunity (8) and that CSF1R blockade improved PD1 checkpoint immunotherapy (48). However, both of these clinical outcomes, while positive, were only modest improvements over conventional treatments, suggesting that additional strategies might be more effective. In support of this premise, preclinical testing of combined CCR2 inhibition to target monocytes and CXCR2 inhibition to target granulocytes in PDAC models showed improved responses to chemotherapy compared to either agent alone (6). It remains to be seen what the toxicity profile of a combined strategy might be.
Consistent with previous reports in inflammatory models (26, 29), the use of ADH-503, a CD11b agonist, resulted in significantly reduced numbers of trafficked monocytes and granulocytes, and differentiated macrophages in tumors. Intriguingly, we also observed that macrophages remaining in the tumor expressed high amounts of MHC-I, MHC-II, CD80, and CD86, which support improved antigen presentation within the tumors. The mechanisms by which CD11b signaling can control a macrophage’s antigen-presenting activity are not known. Recently, work by Judith Varner’s group showed that loss of CD11b expression in TAMs results in tumor progression by driving macrophage polarization to support vascular maturation (49). Intriguingly, our data also suggest that the presence of different CD11b ligands in the TME, which are often extracellular matrix (ECM) molecules, might regulate the macrophage phenotype. Although the impact of ECM signaling on the TAM phenotype has been studied for some ECM molecules (50, 51), these interactions are still poorly understood, particularly in vivo. Nonetheless, ADH-503 appears to have a direct impact on the cytokine profiles of macrophages in vitro, which is consistent with changes observed in vivo. It remains to be seen whether this change in macrophage phenotype mimics normal biologic signaling or is a de novo activity of ADH-503 as a molecular agonist of CD11b. Both interpretations likely would have clinical impact.
Although ADH-503 did not target T cells directly, our data suggest that it can augment anti-tumor T cell responses. We found that ADH-503 bolstered both CD8+ and CD4+ effector T cell responses by increasing their numbers, activation, and proliferative status. Interestingly, we observed increased proximity of CD8+ T cells to PDAC cells following ADH-503 treatment, suggesting that myeloid cells actively exclude T cells from contact with target malignant cells in these PDAC models. We did not determine whether changes in T cell proximity are due to reduced numbers of infiltrating myeloid cells, corresponding increases in numbers of cDC1s, or both. Certainly, the responses of cDC1s, which express extremely low levels of CD11b, appear to be markedly improved, with increases in cDC numbers and MHC expression in PDAC tissues, and cross-priming of CTLs in dLNs. Taken together, these data suggest myeloid cell and cDC responses are intimately associated for the regulation of T cell responses in both tumor tissues and the periphery.
Our data demonstrate that the successful reprograming of the innate immune compartment by CD11b agonism can render tumors more sensitive to checkpoint blockade. We demonstrate this effect for PD1 and 41BB combination therapies, but intriguingly, not for CTLA4 treatment. ADH-503 is currently on track for phase I single agent clinical testing next year and the data presented here suggest several potential therapeutic combinations that may have activity in PDAC and other cancers.
Materials and Methods
Study design
All studies were designed with the help of the Siteman Cancer Center Biostatistics core. The core staff provided us sample size estimated and power calculations and post data analysis help. All experiments were replicated 2–4 times and all critical observation were made with different cell lines and/or genetic models. To blind the research team only animal number not treatment groups were used when investigators made measurements or conducted analysis, for example flowcytometry analysis, survival or ultrasound measurements. Primary data are reported in data file S1.
Murine PDAC Models
KPC mice (p48-CRE/Lox-stop-Lox(LSL)-KrasG12D/p53flox/flox) used in these studies have been previously described (52) and were backcrossed to C57BL/6 background and screened for C57BL/6 identity using congenic markers. KP2 cell line was derived from tumors of 6-month-old p48-CRE+/LSL-Lox KrasG12D/p53flox/+ mouse. Kras-INK (KI) cells were obtained from Dr. Hanahan’s laboratory at the EPFL institute. All cell lines were negative for MAP and mycoplasma. To establish orthotopic pancreas tumors, either 100,000 or 200,000 KP2, KP2ova or KI cells in 50 μL of Cultrex (Trevigen) were injected into the pancreas of 8–12-week-old C57BL/6 or FVB/NJ mice according to published protocol (53). All animals were randomized and assigned a treatment groups at time of tumor diagnosis >0.4cm in diameter. CD11b deficient (Itgam−/−), Zbtb46-GFP (B6.129S(C)-Zbtb46tm1.1Kmm/J) and Batf3−/− (B6.129S(C)-Batf3tm1Kmm/J) in the C57BL/6 background were obtained from Jackson Laboratories (30–32). Washington University School of Medicine Institutional Animal Studies Committee approved all animal studies (protocol #20160265).
Inhibitors, neutralizing antibodies, and in vivo IgGs
Cell-based assays have determined it has biochemical half-maximal effective concentrations (EC50) of 4 uM for ADH-503. For animal experiments, ADH-503 was given at 30, 60, or 120 mg/kg and is specified in the text when not at 60mg/kg. ADH-503 was formulated for treatment in 0.5% carboxymethyl cellulose and 0.1 % Tween-80 (Sigma-Aldrich) in sterile water and administered by oral gavage twice a day (BID). Immunotherapy IgGs were given i.p. and anti-PD-1 (200ug/dose clone RMP1–14) was given every 3–4 days, while anti-CTLA4 (250ug UC10–4F10–11) and anti-41BB/CD137 (200ug/dose clone 3H3) were given every 4–5 days. Anti-CTLA4 and anti-41BB were discontinued after four doses whereas anti-PD1 was continued until day 30. All were purchased form BioXCell. For T cell depletion, CD4- and CD8- neutralizing IgG antibodies (anti-mCD4 clone GK1.5, anti-mCD8 clone 2.43, BioXCell) were administered via i.p. injection every 4–5 days, with the 1st injection containing 500 μg before tumor implantation and subsequent injections containing 250 μg. Mice were treated with anti-Ly6G IgGs (clone 1A8, BioXCell), anti-CSF1 IgG (5A1 clone, BioXCell) or matched isotype control. For Ly6G the first dose 400 μg i.p. followed by 100μg every 3 days and for CSF1 IgG 1.0 mg followed by 500 ug every 5 days. CCR2i PF-04136309 (Tocris) was given at 100mg/kg BID.
Bead labeling blood cells
To selectively label blood cells, 250 uL of liposomes containing clodronate were injected intraveneously (i.v.), followed by i.v. injection of 250 uL of PE-conjugated plain microspheres 16–18 hours later (1.0 um, 2.5% solids [wt/vol]; Polysciences, diluted 1:4 in PBS). Tissues were processed for flow cytometry analyses at indicated time points after bead injection.
Mouse Tissue Isolation and Flow Cytometry
Mice were euthanized by intracardiac perfusion using 20 mL of PBS-heparin. Tumor tissues were manually minced and digested in 25 mL of Dulbecco’s Modified Eagle Medium (DMEM) containing 2 mg/mL of collagenase A (Roche) and DNase I (Sigma) for 30 minutes at 37°C. Digestion was quenched in 5 mL of fetal bovine serum and filtered through 40 μm Nylon mesh. Single cell suspensions were subsequently labeled with fluorophore-conjugated anti-mouse antibodies at recommended dilutions following manufacturers’ recommendations (Table S1). Data were acquired on LSR-II or X-20 or sorted on an Aria-II (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Human PDAC Tissues
Human PDAC tissues were obtained from surgically resected specimens from patients diagnosed in the Department of Pathology at Washington University. Patients underwent pancreaticoduodenectomy and did not receive neoadjuvant therapy. Preparation of tissues used for CyTOF are described below. Tissues for histology were embedded in paraffin blocks and processed into 6 μm-thick sections for immunofluorescence staining. All tissues were collected under informed consent from patients. Washington University Ethics committee approved the study under IRB protocol #201108117.
Histology
Tissues were fixed in 10% formalin overnight, incubated in graded ethanol, embedded in paraffin, and cut into 6 μm-thick sections. All immunohistochemistry (IHC) reagents are listed in Table S2. For Cleaved Caspase-3, Ki67 and αSMA, CD8a or CK19 analysis tissues were stained using Bond Rxm (Leica Biosystems) following citrate-based epitope retrieval (AR9661, Leica Biosytems). Immunostaining was visualized using the Bond Polymer Refined Detection and/or Bond Intense R Detection Systems (DS9263, Leica Biosytems). For CD68 and Ly6G and a subset of CD8a staining we used immunofluorescent staining on frozen slides. 5 μm-thick sections were air-dried and fixed in 4% PFA (Ted Pella, Inc.). Slides were incubated with the primary antibodies listed in Table S2 and detected with either appropriate fluorescent secondary or using Tyramide Signal Amplification (Perkin-Elmer) kit.
CyTOF:
Tumor samples were digested in HBSS (lonza) supplemented with 2mg/ml collagenase A(Roche), 2.5 unit/ml hyaluronidase (Sigma) and DNase (Sigma) at 37°C for 30 minutes with agitation to generate single-cell suspension. Cell suspensions were then counted and stained in 5uM cisplatin, incubated with FcR-blocking reagents plus the surface antibody cocktail (Table S3). Cells were then fixed with 4% PFA for 10 minutes on ice and permeabilized with permeabilization buffer (Invitrogen) for 40 minutes containing the intracellular stain cocktail (Table S3). Stained cells were then washed twice with PBS and stained with 200ul of DNA intercalator per 1×106 cells. Cells were acquired on a CyTOF 2 mass cytometer and data were uploaded to cytobank for further analysis.
Radiation Therapy (RT)
Ten days post tumor implantation, cohorts of mice were randomized into different treatment groups using either gross tumor diameter or tumor volume (length*(width2)/2) determined by ultrasound. Mice were given daily fractionated doses of RT (4Gy × 5) using the Small Animal Radiation Research Platform (SARRP) 200 (XStrahl Life Sciences). Mice were injected i.p. with an iodine contrast agent (4200 mg/kg) before being placed on the irradiation platform one at a time and fitted with a nose cone for isoflurane anesthesia. Cone beam computed tomography imaging was performed for each individual mouse to pinpoint the pancreas tumors and images were imported into Muriplan and used to select an isocenter. The tumor was then irradiated to 4Gy using anterior-posterior-opposed beams using the 5mm × 5mm collimator at a dose rate of 3.9 Gy/min. Tumor size was assessed by portable SonoSite m-Turbo ultrasound apparatus. Ultrasound images were taken at the beginning of RT and one and two weeks post the start of RT treatment to monitor tumor growth changes.
Statistical Analyses
Day to day statistical analysis was performed using Prism 7 and Unpaired Student’s t-test, Mann-Whitney U test, Wilcoxon matched-pairs signed rank test, Kolmogorov-Smirnov test or ANOVA analysis were used as appropriate for the data set.
Supplementary Material
Acknowledgements:
R.Z.P. was supported by the Surgical Oncology Basic Science, Translational Research Training program, Surgical oncology training grant (NIH5T32CA00962128), National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH; T32CA009621-28). D.G.D. was supported by the Department of Defense (W81XWH-15-1-0385) and the National Cancer Institute (NCI; P50CA196510, R01CA177670, and R01CA203890). JKS was supported by NCI R01CA181745 and the Small Animal Radiation Research Platform was purchased with the assistance of S10 OD020136. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analyses. The Center is partially supported by the NCI Cancer Center (P30CA91842; Siteman Cancer Center) and by ICTS/CTSA (UL1RR024992) from the NCRR and the NIH Roadmap for Medical Research.
D.G.D. managed the project and coordinated author activities. R.Z.P. designed and performed experiments including mouse modeling, flow cytometry analysis, and gene expression analysis. B.L.K and J.M.H. supported experimental design and execution. C.Z. designed and performed experiments, including human and mouse CyTOF staining and analysis. S.H. performed CD8 immunofluorescence staining and proximity analysis. W.G.H., and R.C.F. collected human patient samples and assisted in patient outcome analysis. R.Z.P. performed statistical analysis and support patient outcome analysis. J.K.S. and B.E.R. helped with the RT experiments. Adhaere Pharmaceuticals provided ADH-503 for the in vivo and vitro studies, and V.G. and XL provided intellectual input on the project. D.G.D. and R.Z.P. wrote the manuscript with input from all authors.
Footnotes
Competing interests: VG and DGD have competing financial interest related to ADH-503. VG has partial ownership of Adhaere Pharmaceuticals and DGD is a scientific advisory board member.
Data and materials availability: ADH-503 can be obtained through material transfer agreement with Adhaere Pharmaceuticals or synthesized commercially from the structure in Figure 2A. All data associated with this study are available in the paper or Supplementary Materials.
References and Notes
- 1.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA, Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33, 828–833 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollard JW, Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol 84, 623–630 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW, CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian BZ, Pollard JW, Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR, Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63, 1769–1781 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, Fields RC, DeNardo DG, Hawkins WG, Goedegebuure P, Linehan DC, Targeting both tumourassociated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG, CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74, 5057–5069 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, Wang-Gillam A, Eberlein TJ, Denardo DG, Goedegebuure SP, Linehan DC, Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 19, 3404–3415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, Goedegebuure P, Linehan DC, DeNardo DG, Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73, 1128–1141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirstrom K, West BL, Coussens LM, Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1, 54–67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid MC, Varner JA, Myeloid cells in tumor inflammation. Vasc Cell 4, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupuy AG, Caron E, Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci 121, 1773–1783 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P, Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203, 25692575 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han C, Jin J, Xu S, Liu H, Li N, Cao X, Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 11, 734–742 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Means TK, Luster AD, Integrins limit the Toll. Nat Immunol 11, 691–693 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, Kumar V, Armstrong AR, CarmonaRivera C, Dorschner JM, Schnaith AM, Li X, Ghodke-Puranik Y, Moore E, Purmalek M, Irizarry-Caro J, Zhang T, Day R, Stoub D, Hoffmann V, Khaliqdina SJ, Bhargava P, Santander AM, Torroella-Kouri M, Issac B, Cimbaluk DJ, Zloza A, Prabhakar R, Deep S, Jolly M, Koh KH, Reichner JS, Bradshaw EM, Chen J, Moita LF, Yuen PS, Li Tsai W, Singh B, Reiser J, Nath SK, Niewold TB, Vazquez-Padron RI, Kaplan MJ, Gupta V, CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest 127, 1271–1283 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sica A, Mantovani A, Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122, 787–795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid MC, Avraamides CJ, Foubert P, Shaked Y, Kang SW, Kerbel RS, Varner JA, Combined blockade of integrin-alpha4beta1 plus cytokines SDF-1alpha or IL-1beta potently inhibits tumor inflammation and growth. Cancer Res 71, 6965–6975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gahmberg CG, Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr Opin Cell Biol 9, 643–650 (1997). [DOI] [PubMed] [Google Scholar]
- 20.McFarland HI, Nahill SR, Maciaszek JW, Welsh RM, CD11b (Mac-1): a marker for CD8+ cytotoxic T cell activation and memory in virus infection. J Immunol 149, 1326–1333 (1992). [PubMed] [Google Scholar]
- 21.Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ, Smith CW, Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology 17, 915–923 (1993). [PubMed] [Google Scholar]
- 22.Rogers C, Edelman ER, Simon DI, A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A 95, 10134–10139 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dove A, CD18 trials disappoint again. Nat Biotechnol 18, 817–818 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, Barth CJ, Lugo G, Donnelly M, Nayer A, Moita LF, Schurer S, Traver D, Ruiz P, Vazquez-Padron RI, Ley K, Reiser J, Gupta V, Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal 4, ra57 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harlan JM, Winn RK, Leukocyte-endothelial interactions: clinical trials of anti-adhesion therapy. Crit Care Med 30, S214–219 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Celik E, Faridi MH, Kumar V, Deep S, Moy VT, Gupta V, Agonist leukadherin-1 increases CD11b/CD18-dependent adhesion via membrane tethers. Biophys J 105, 2517–2527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jagarapu J, Kelchtermans J, Rong M, Chen S, Hehre D, Hummler S, Faridi MH, Gupta V, Wu S, Efficacy of Leukadherin-1 in the Prevention of Hyperoxia-Induced Lung Injury in Neonatal Rats. Am J Respir Cell Mol Biol 53, 793–801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan SQ, Guo L, Cimbaluk DJ, Elshabrawy H, Faridi MH, Jolly M, George JF, Agarwal A, Gupta V, A Small Molecule beta2 Integrin Agonist Improves Chronic Kidney Allograft Survival by Reducing Leukocyte Recruitment and Accompanying Vasculopathy. Front Med (Lausanne) 1, 45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faridi MH, Altintas MM, Gomez C, Duque JC, Vazquez-Padron RI, Gupta V, Small molecule agonists of integrin CD11b/CD18 do not induce global conformational changes and are significantly better than activating antibodies in reducing vascular injury. Biochim Biophys Acta 1830, 3696–3710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM, Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209, 1135–1152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseno CG, Durai V, Bagadia P, Haldar M, Schonheit J, Rosenbauer F, Murphy TL, Murphy KM, Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol 16, 708–717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joyce JA, Pollard JW, Microenvironmental regulation of metastasis. Nat Rev Cancer 9, 239–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zippelius A, Schreiner J, Herzig P, Muller P, Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res 3, 236–244 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Luheshi NM, Coates-Ulrichsen J, Harper J, Mullins S, Sulikowski MG, Martin P, Brown L, Lewis A, Davies G, Morrow M, Wilkinson RW, Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget 7, 18508–18520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eissler N, Mao Y, Brodin D, Reutersward P, Andersson Svahn H, Johnsen JI, Kiessling R, Kogner P, Regulation of myeloid cells by activated T cells determines the efficacy of PD-1 blockade. Oncoimmunology 5, e1232222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG, Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, Tille L, Homicsko K, Damsky W, Maby-El Hajjami H, Klaman I, Danenberg E, Ioannidou K, Kandalaft L, Coukos G, Hoves S, Ries CH, Fuertes Marraco SA, Foukas PG, De Palma M, Speiser DE, T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Science translational medicine 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guerin M, Biton J, Ouakrim H, Regnier F, Lupo A, Alifano M, Damotte D, Donnadieu E, Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 115, E4041–E4050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A, Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, Eberlein C, Candido JB, Clarke M, Nixon C, Connelly J, Jamieson N, Carter CR, Balkwill F, Chang DK, Evans TRJ, Strathdee D, Biankin AV, Nibbs RJB, Barry ST, Sansom OJ, Morton JP, CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 29, 832–845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ, Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7, e50946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D, Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer cell 21, 836–847 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH, Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer cell 21, 822–835 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moran AE, Polesso F, Weinberg AD, Immunotherapy Expands and Maintains the Function of High-Affinity Tumor-Infiltrating CD8 T Cells In Situ. J Immunol 197, 2509–2521 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, C. H, Pryer N, Daniel D, Hwang ES, Rugo H, Coussens LM, Macrophage IL10 Blocks CD8+ T Cell-Dependent Responces to Chemotherapy by Supressing IL12 Expression in Intratumoral Dendritic Cells. Cancer Cell 26, 623–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ngambenjawong C, Gustafson HH, Pun SH, Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev 114, 206–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wainberg ZA, Piha-Paul SA, Luke J, Kim EJ, Thompson JA, Britten CD, Johnson JM, Pfanzelter N, Gordon M, Rasco DW, Hodi FS, Weise A, Inamdar S, Perna S, Ma C, Powers J, Lee Y, Ghoddusi M, Carleton M, Xiang H, Zhou L, Collins H, Lee J, J, First-in-Human Phase 1 Dose Escalation and Expansion of a Novel Combination, Anti–CSF-1 Receptor (cabiralizumab) Plus Anti–PD-1 (nivolumab), in Patients With Advanced Solid Tumors. SITC-Meeting Abstract, O42 (2017). [Google Scholar]
- 49.Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shepard R, Louis TL, Anand S, Woo G, Leem C, Faridi MH, Geraghty T, Rajagopalan A, Gupta S, Ahmed M, Vazquez-Padron RI, Cheresh DA, Gupta V, Varner JA, Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun 9, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McWhorter FY, Davis CT, Liu WF, Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci 72, 1303–1316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V, Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol 184, 1049–1061 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE, Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 4, 1670–1680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA, Structure of an integrin with an alphaI domain, complement receptor type 4. The EMBO journal 29, 666–679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.