

Abstract
Type 2 diabetes (T2D) is characterized by persistent hyperglycemia despite hyperinsulinemia, affects more than 400 million people worldwide, and is a major cause of morbidity and mortality. Insulin resistance, of which ectopic lipid accumulation in the liver [nonalcoholic fatty liver disease (NAFLD)] and skeletal muscle is the root cause, plays a major role in the development of T2D. Although lifestyle interventions and weight loss are highly effective at reversing NAFLD and T2D, weight loss is difficult to sustain, and newer approaches aimed at treating the root cause of T2D are urgently needed. In this review, we highlight emerging pharmacological strategies aimed at improving insulin sensitivity and T2D by altering hepatic energy balance or inhibiting key enzymes involved in hepatic lipid synthesis. We also summarize recent research suggesting that liver-targeted mitochondrial uncoupling may be an attractive therapeutic approach to treat NAFLD, nonalcoholic steatohepatitis, and T2D.
Keywords: type 2 diabetes, insulin resistance, ectopic lipids, liver-targeted mitochondrial uncoupling
INTRODUCTION
Diabetes mellitus is a major health issue that has reached epidemic proportions worldwide due to rapid urbanization, unhealthy eating, and a sedentary lifestyle (1). The International Federation Atlas (2018) estimates that over 415 million people have been diagnosed with diabetes, a figure that, by 2040, is projected to rise to more than 625 million (1). Of the three major types of diabetes, type 2 diabetes (T2D) is far more common than either type 1 diabetes or gestational diabetes, accounting for almost 90% of cases (2). The increasing prevalence of diabetes follows the spread of an epidemic of obesity, the single most important contributor to the pathogenesis of T2D. Indeed, by 2050, almost 90% of people in the United States are projected to be overweight or obese (3), and one in three are expected to have T2D (4). The tremendous costs of these related epidemics with regard to morbidity and mortality, as well as the financial costs of approximately $150–190 billion in medical spending (5, 6) and more than $4 billion in lost productivity (7) annually, demand the development of new approaches to prevent and treat T2D.
While progressive loss of pancreatic islet β-cell function is ultimately responsible for the progression from normoglycemia to hyperglycemia, insulin resistance predates β-cell dysfunction and plays a major role in the pathogenesis of T2D (8). After a carbohydrate-rich meal, glucose is primarily stored as glycogen in the muscle and liver (9, 10). Decreased insulin action in these organs leads to fasting and postprandial hyperglycemia (9, 10), the hallmarks of T2D and major risk factors for long-term microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular (cardiovascular disease) complications (11). As such, current therapy for T2D relies mainly on the following approaches intended to reduce hyperglycemia(12): lifestyle modifications, which include the adoption of a healthy diet, increased physical activity, and healthy body weight maintenance (13); metformin, which acts to reduce hepatic gluconeogenesis and hepatic glucose production (HGP) (14, 15); sulfonylureas (and related insulin secretagogues), which increase insulin secretion from pancreatic islets (16); sodium– glucose cotransporter (SGLT2) inhibitors, which block glucose reabsorption in the proximal renal tubule (17); incretin mimetics, which stimulate insulin secretion, delay gastric emptying, and suppress appetite (18); peroxisome proliferator-activated receptor-γ (PPARγ) agonists (thiazolidinediones), which enhance adipocyte lipid storage (19), decrease ectopic lipid accumulation in liver and skeletal muscle (19), and improve liver and muscle insulin sensitivity (19–21); α-glucosidase inhibitors, which interfere with gut glucose absorption; and insulin itself (22), which suppresses glucose production and increases glucose utilization (23). Unfortunately, these agents have met with limited success due to reduced efficacy, limited tolerability, and significant mechanism-based side effects (Table 1). Thus, new approaches to treat the root cause of T2D are needed.
Table 1.
Current pharmacological agents for T2D
| Class of medication | Representative agents | Mechanism of action | Glycemic efficacy (% HbAlc reduction) | CVD risks and benefits | Side effects |
|---|---|---|---|---|---|
| Biguanide | Metformin | Insulin sensitizer; ↓. HGP and gluconeogenesis | ↓ 1–2% | ↓ CVD risk factors; ↓ MI and coronary deaths | GI and lactic acidosis |
| Sulfonylureas | Glimepiride, glipizide, glyburide | ↑ Insulin secretion | ↓ 1–2% | ↑ CVD risk | Hypoglycemia |
| SGLT2 inhibitors | Canagliflozin, Dapagliflozin, Empagliflozin | ↓ Glucosuria; ↓ glucotoxicity | ↓ 0.5–0.7% | ↓ Blood pressure; improved lipid profile | Ketoacidosis, genital mycosis, bone fractures |
| Incretin mimetics | GIP, GLP-1 (Liraglutide, Exenatide, Dulaglutide) | ↑ Insulin secretion; ↓ glucagon secretion; delayed gastric emptying; ↑ satiety | ↓ 0.5–1.5% | ↓ CVD risk | Nausea, vomiting, pancreatitis |
| TZDs | Rosiglitazone, pioglitazone | Insulin sensitizer; ↑ adipocyte function; ↓ ectopic lipid accumulation; ↑ β-cell function | ↓ 0.5–1.4% | ↑ CVD risk | Weight gain, bladder cancer, bone fractures |
| α-Glucosidase inhibitors | Acarbose, voglibose, miglitol | ↓ Carbohydrate absorption | ↓ 0.8% | ↓ CVD risk | Diarrhea, abdominal pain, nausea, vomiting |
| Insulin | Short acting: humulin R, novolin R Intermediate acting: isophane Long acting: Lantus, Levemir, Tresiba Rapid acting: Lispro, Aspart, Apidra | ↓ HGP; ↑ glucose uptake | ↓ 1–2.5% | Neutral | Hypoglycemia, weight gain |
Abbreviations: CVD, cardiovascular disease; GI, gastrointestinal; GIP, gastric inhibitory polypeptide; GLP-1, glucagon-like peptide 1; HbA1c, hemoglobin A1c; HGP, hepatic glucose production; MI, myocardial infarction; SGLT2, sodium–glucose cotransporter; T2D, type 2 diabetes; TZD, thiazolidinedione.
Developing better treatment strategies requires a comprehensive understanding of the pathogenesis of T2D, in which insulin resistance plays an important role. In this review, we provide a brief overview of the pathogenesis of insulin resistance and how it is related to the development of new treatments for T2D. Additionally, we highlight several therapeutic strategies that have been developed to enhance insulin sensitivity by altering energy balance or inhibiting lipid synthesis. While several diabetes medications have also been studied as potential treatments for nonalcoholic fatty liver disease (NAFLD), the hepatic manifestation of insulin resistance, we refer readers to two recent reviews that discuss this in greater detail [by Samuel & Shulman (24) and Alkhouri et al. (25)]. Lastly, we examine the potential therapeutic utility of liver-targeted mitochondrial uncoupling, which would represent a new class of agents for the treatment of NAFLD, nonalcoholic steatohepatitis (NASH), and T2D.
PATHOPHYSIOLOGY OF TYPE 2 DIABETES
Although the pathophysiological mechanisms of T2D are not fully understood, there is compelling evidence that insulin resistance plays a major role in its development. Indeed, cross-sectional and longitudinal studies demonstrate that insulin resistance occurs 10–20 years before the onset of T2D and that it is the best predictor of whether an individual will become diabetic (26, 27). Insulin resistance arises when nutrient availability and demands can no longer be balanced and is present in multiple tissues, including skeletal muscle, liver, adipose tissue, kidney, gastrointestinal tract, vasculature, and brain (8). In the muscle, insulin resistance is manifested by impaired glucose uptake following ingestion of a carbohydrate-rich meal and results in postprandial hyperglycemia (28), which in turn can be attributed to decreased insulin-stimulated muscle glycogen synthesis (29) due to decreased insulin-stimulated glucose transport (30). Hepatic insulin resistance is characterized by an inability of insulin to stimulate hepatic glycogen synthesis and suppress HGP under postprandial conditions. In T2D patients, increased rates of HGP, due to increased rates of hepatic gluconeogenesis, contribute to both fasting and postprandial hyperglycemia (31). Recent studies have implicated increased rates of white adipose tissue lipolysis, due to macrophage-induced white adipocyte inflammation, as a major factor responsible for the increased rates of hepatic gluconeogenesis in T2D via increased hepatic acetyl-CoA content (derived from increased fatty acid delivery), allosteric activation of pyruvate carboxylase, and increased conversion of glycerol to glucose by a substrate push mechanism (32). Collectively, alterations in insulin responsiveness in these tissues place a major stress on the pancreatic β-cells to increase insulin secretion in an attempt to offset the defect in insulin action (33). As the disease progresses, however, β-cells begin to fail, and a vicious cycle ensues involving gluco- and lipotoxicity and, possibly, factors other than the β-cells, causing a further decline in insulin secretion, worsening of hyperglycemia, and overt T2D (Figure 1).
Figure 1.
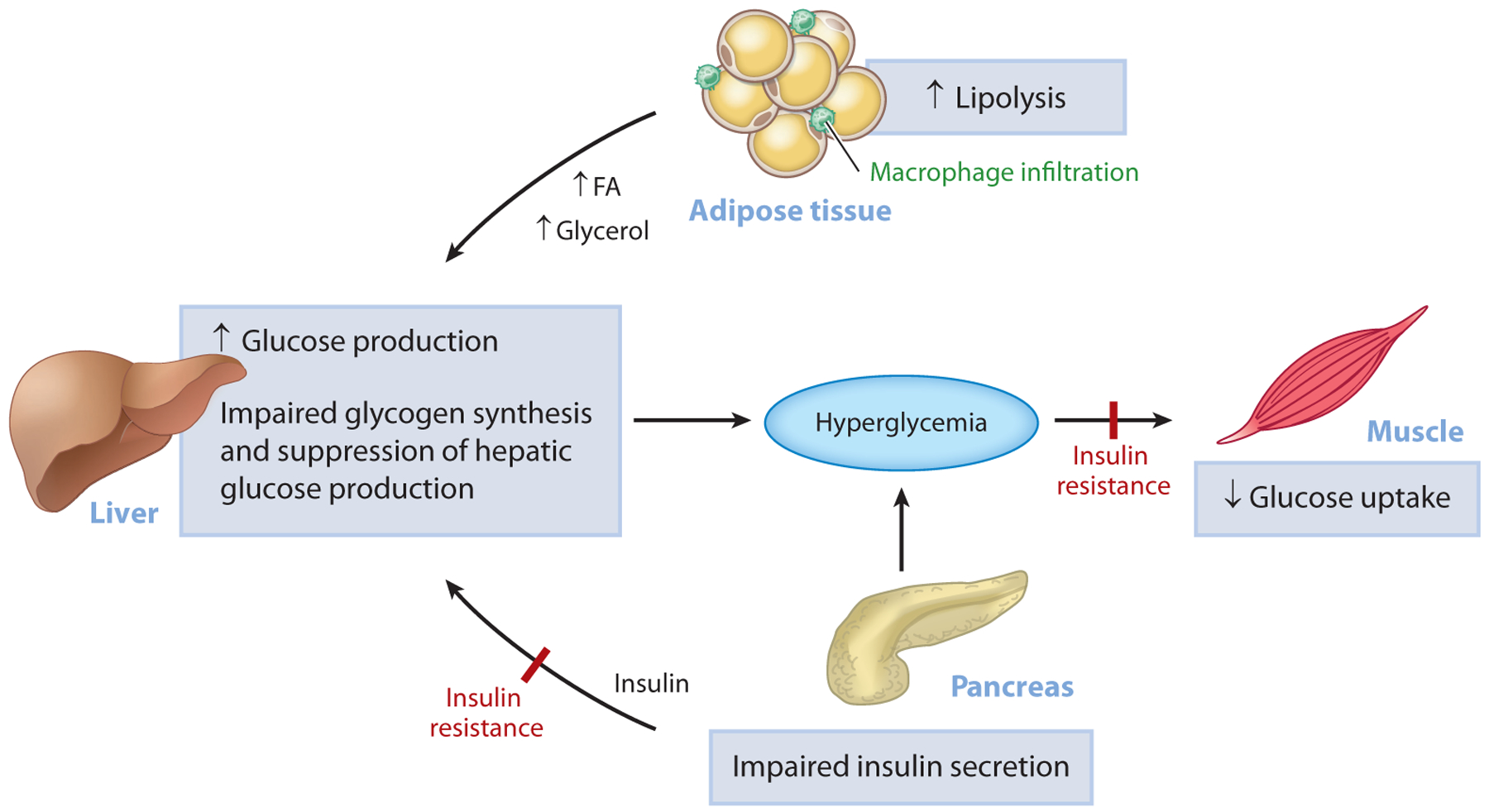
Pathogenesis of hyperglycemia in T2D. Uncontrolled hyperglycemia is a hallmark of T2D and is a major risk factor for long-term microvascular and macrovascular complications. While the progressive loss of pancreatic islet β-cell function is ultimately responsible for the progression from normoglycemia to hyperglycemia, insulin resistance predates β-cell dysfunction and plays a major role in the pathogenesis of T2D. In the muscle, insulin resistance is manifested as impaired glucose uptake following ingestion of a carbohydrate-rich meal and results in postprandial hyperglycemia. In the liver, insulin resistance is characterized by the inability of insulin to stimulate hepatic glycogen synthesis and suppress hepatic glucose production under postprandial conditions. In parallel, inappropriate increases in adipose tissue lipolysis (due to increases in inflammation) can drive hepatic gluconeogenesis through increases in hepatic acetyl-CoA content, allosteric activation of PC, and increased conversion of glycerol to glucose. Initially, the β-cell compensates for alterations in tissue insulin responsiveness by increasing insulin secretion; however, over time, this compensatory mechanism fails and β-cell mass declines, causing a further reduction in insulin secretion, worsening of hyperglycemia, and overt T2D. Abbreviations: FA, fatty acid; PC, pyruvate carboxylase; T2D, type 2 diabetes.
Ectopic lipid accumulation in the liver (NAFLD) and muscle (intramyocellular lipid) has been identified as a root cause of insulin resistance in these tissues (29, 34–39). Under conditions of overnutrition, bioactive lipid metabolites accumulate and cause cellular dysfunction and insulin resistance. While several lipid metabolites have been proposed to play a causal role in insulin resistance (40), a substantial body of work supports the role of diacylglycerol (DAG) accumulation and novel protein kinase C (PKC) activation in impairing insulin action in liver and skeletal muscle (Figure 2). Specifically, intrahepatic and intramyocellular DAG promote increased PKCε translocation in liver and PKCθ translocation in skeletal muscle, increasing the phosphorylation of insulin receptor threonine 1160 in liver (41) and serine 1101 in skeletal muscle (42, 43) and impairing insulin signaling and action in liver (41, 44–88) and muscle (42, 43, 50, 51, 60, 68, 69, 78, 80, 88–109).
Figure 2.
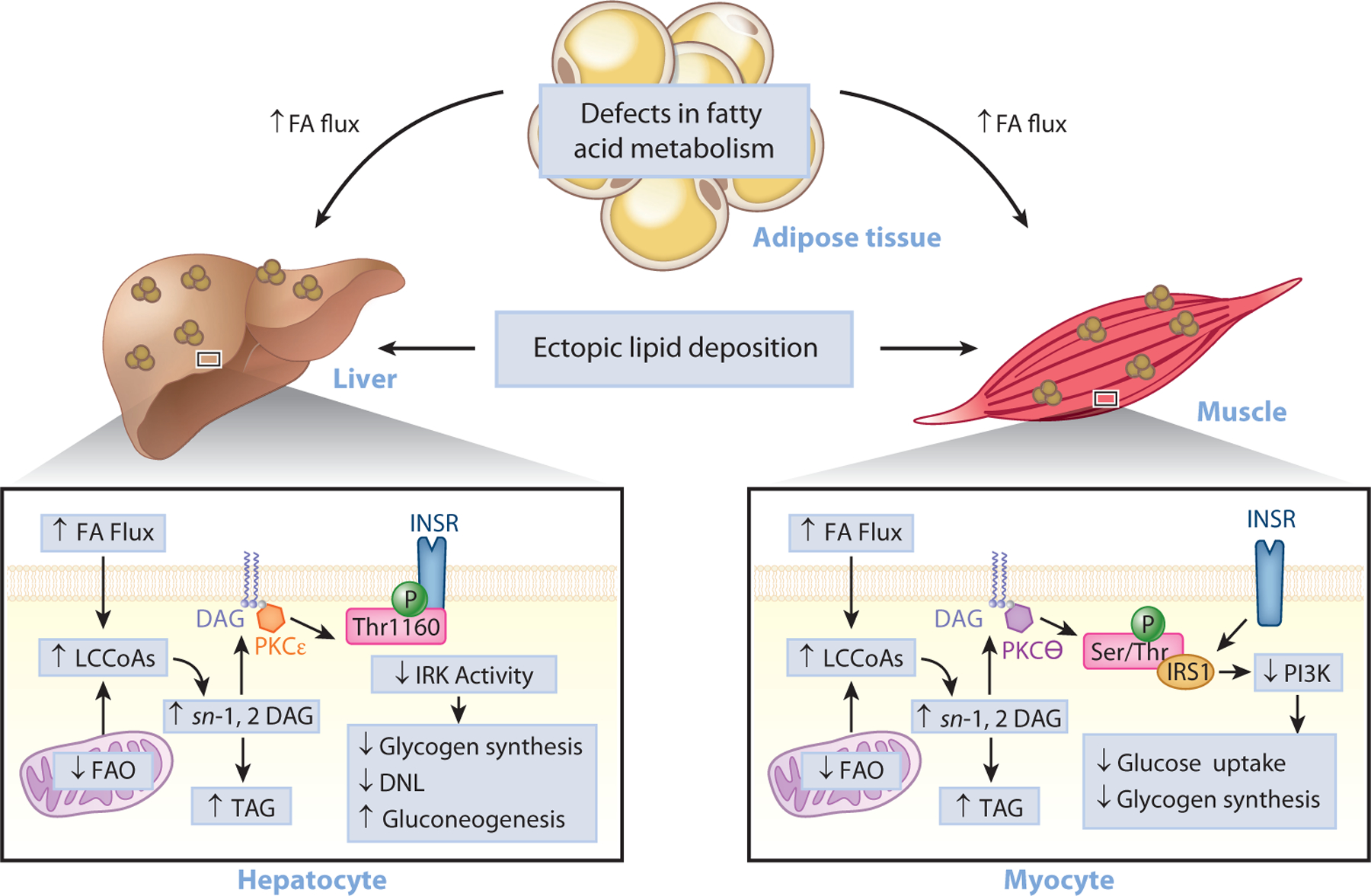
The role of ectopic lipids in insulin resistance. Under conditions of overnutrition or defective adipocyte fatty acid metabolism, lipids can be redistributed from eutopic sites (adipose tissue) to ectopic storage sites (liver and muscle) and lead to impaired insulin signaling, insulin resistance, and T2D. Lipid-induced hepatic insulin resistance may result from activation of the DAG–PKCε axis and the consequent inhibition of INSR signaling through inhibitory phosphorylation of INSR at Thr1160. This leads to impaired insulin stimulation of hepatic glycogen synthesis, impaired transcriptional upregulation of de novo lipogenic genes, and impaired transcriptional downregulation of gluconeogenic genes. Skeletal muscle insulin resistance, caused by increases in intramyocellular ectopic lipid, impairs insulin-stimulated glucose transport and glycogen synthesis through the activation of the DAG–PKCθ axis and the consequent inhibition of the PI3K pathway through inhibitory phosphorylation of IRS1. Abbreviations: DAG, diacylglycerol; FA, fatty acid; FAO, fatty acid oxidation; INSR, insulin receptor; IRK, insulin receptor kinase; IRS1, insulin receptor substrate 1; LCCoA, long-chain CoA; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; Ser/Thr, serine/threonine; T2D, type 2 diabetes; TAG, triglyceride; Thr1160, threonine 1160.
Evidence demonstrating a causal role for ectopic lipids in driving insulin resistance and T2D comes from studies showing that short-term caloric restriction, yielding minimal changes in body weight, reverses NAFLD and normalizes hepatic insulin sensitivity in obese humans (110, 111) and rodents (112). Although modest weight loss is highly effective at reversing T2D, with diabetes remission rates comparable to those of bariatric surgery (110, 113–119), weight loss is difficult to sustain, with approximately 70–95% of those who lose significant weight subsequently regaining it (120–123).
NOVEL AND EMERGING TARGETS TO IMPROVE INSULIN SENSITIVITY
Novel therapeutics aimed at reducing ectopic lipids and improving insulin sensitivity may be of great benefit for the treatment of T2D. In particular, several strategies have been developed to influence ectopic lipid deposition by broadly altering energy balance or inhibiting key enzymes involved in lipid synthesis (24). In this section, we highlight one nonpharmacological approach (bariatric surgery) and two pharmacological targets currently under clinical development [fibro-blast growth factor 21 (FGF21) and acetyl-CoA carboxylase (ACC)] for the treatment of T2D. We also discuss the potential use of liver-targeted mitochondrial uncoupling agents to treat NAFLD, NASH, and T2D and present our view on what will be needed to generate liver-targeted mitochondrial uncouplers that are appropriate for clinical use.
Bariatric Surgery
Recently, bariatric surgery has emerged as an effective treatment option for obesity, NAFLD, and T2D (124, 125), resulting in long-term sustained weight loss with significant improvements in glycemic parameters (30% decrease in endogenous glucose production), histological features in NAFLD, insulin resistance, and cardiovascular disease risk factors [40% decrease in very low-density lipoprotein (VLDL) production] (126, 127). These beneficial effects likely relate, at least in part, to weight loss, although an improved incretin effect (115) and forced reduction in caloric intake [leading to a reduction in hepatic acetyl-CoA content, glycogenolysis, and DAG–PKCε activation (128)], may contribute to the improvements in glycemic control. Nonetheless, longer-term studies have demonstrated that the metabolic effects of bariatric surgery persist for an extended period; Schauer et al. showed that surgery improved diabetes control over a 5-year period (129). However, a study of a larger cohort of patients undergoing sleeve gastrectomy, the most common bariatric procedure in the United States (130), found weight regain at 3–5 years after surgery and relapse of diabetes in a significant number of patients (131). As such, while bariatric surgery can greatly reduce weight and resolve T2D, it may not be appropriate for all patients.
FGF21
FGF21, a distinctive member of the FGF family that functions as an endocrine hormone, is a key mediator of energy homeostasis and lipid and glucose metabolism (132). Circulating FGF21 is predominantly derived from the liver but is also found in the gut, brain, adipose tissue, muscle, and pancreas (133). FGF21 may coordinate the metabolic shift from the fed to fasted states and regulates hepatic ketogenesis, gluconeogenesis, and adipose lipolysis (133, 134). Liver-specific knockout of FGF21 causes glucose intolerance in high-fat diet (HFD)-fed mice (135), whereas mice overexpressing FGF21 are resistant to diet-induced obesity and have enhanced glucose tolerance (136). While FGF21 appears to exert its antidiabetic effects through multiple tissues (132), recent pharmacological studies in rodents have shown that exogenous FGF21 normalizes glucose and lipid homeostasis by increasing cellular energy expenditure independently of brown adipose tissue activation (69, 137). In particular, these improvements were associated with a reduction in liver and muscle triglycerides, DAGs, and novel PKC (nPKC) translocation in the liver and muscle (69). FGF21 has also been shown to improve β-cell function (possibly secondary to the reversal of systemic insulin resistance) and survival in T2D mice (138).
Although FGF21 levels are positively associated with obesity and insulin resistance (139), preclinical and clinical studies suggest that pharmacological supplementation may be beneficial. Analogs in clinical trials (LY2405319, PF-052313023, and BMS-986036) have yielded mixed results. LY2405319 was tested in obese patients with T2D in a 28-day proof-of-concept trial and reduced body weight, reduced plasma insulin concentrations, and decreased the low-density lipoprotein (LDL) to high-density lipoprotein cholesterol ratio compared to baseline (140). Despite this, its effect on fasting plasma glucose concentrations was not as robust as anticipated based on prior studies in rodent and monkey models of T2D (141, 142); only modest dose-dependent reduction in plasma glucose was observed (140). In addition to LY2405319, PF-052313023, a long-acting FGF21 analog, also showed therapeutic promise. Specifically, PF-052313023 treatment decreased body weight and improved the lipid profile in rodents, monkeys, and patients with T2D. Unfortunately, PF-052313023 treatment produced dose-dependent changes in bone turnover markers (143), raising concern over the long-term use of this FGF21 analog to treat T2D patients. A recent phase 2 study with a pegylated analog of FGF21 (BMS-986036) has shown the most promise. In patients with NAFLD, NASH, and T2D, once-weekly injections decreased liver fat, serum transaminases, plasma triglycerides, and LDL cholesterol without any significant adverse events, paving the way for longer phase 3 studies (144). However, BMS-986036 did not significantly alter plasma glucose concentrations or HbA1c after 12 weeks of treatment (144). Future studies are therefore warranted to determine the long-term beneficial and potential adverse effects of FGF21 administration in T2D patients.
Acetyl-CoA Carboxylase Inhibition
Another therapeutic option to reduce ectopic lipids and improve insulin sensitivity is through inhibition of ACC. ACC catalyzes carboxylation of acetyl-CoA into malonyl-CoA, which is the rate-limiting step in fatty acid synthesis and primary regulator of mitochondrial fatty acid oxidation (145). ACC1, a primarily cytosolic isoform of ACC, is highly expressed in liver and adipose tissue and catalyzes the first committed step in de novo lipogenesis (145). The mitochondria membrane– bound ACC2 is expressed in oxidative tissues, such as the muscle and heart, and produces localized malonyl-CoA, which inhibits carnitine palmitoyltransferase I and the transfer of long-chain CoAs into the mitochondria for fatty acid oxidation (146). Thus, ACC is an intriguing therapeutic target to reduce lipid storage by simultaneously modulating fatty acid synthesis and oxidation (147).
Several rodent and human studies have shown favorable impact of ACC inhibition on NAFLD, NASH, and T2D. In a diet-induced rat model of obesity, antisense oligonucleotide (ASO)-mediated reduction of hepatic ACC1 and ACC2 resulted in marked reductions in hypertriglyceridemia, hepatic triglyceride, and DAG content and the reversal of hepatic insulin resistance, which was associated with a reduction in PKCε activation (62). Additionally, a novel liver-specific allosteric inhibitor of ACC1 and ACC2 (ND-630) is currently being developed for the treatment of NASH and has recently been shown to reduce hepatic steatosis and improve dyslipidemia in rodent models of obesity with no adverse effects (148). Moreover, ND-630 improved glucose metabolism in high-sucrose diet–fed, HFD-fed and Zucker diabetic fatty rats (148). However, these results are not generalizable to all ACC inhibitors; Bourbeau & Bartberger demonstrated that long-term inhibition of ACC markedly increased fasting plasma glucose and worsened glucose intolerance in a diet-induced mouse model of obesity (149). In addition, we have recently observed that long-term allosteric inhibition of ACC significantly increased basal rates of glucose production, most likely due to increases in hepatic acetyl-CoA content and allosteric activation of pyruvate carboxylase (150). More concerning, allosteric inhibitors currently under development for the treatment of NAFLD and NASH (MK-4074 and GS-0976) were associated with a significant increase in fasting plasma triglyceride concentrations, despite a significant reduction in hepatic steatosis (151–153). Subsequent studies in mice and rats demonstrated that the hypertriglyceridemia was mediated by a reduction in hepatic polyunsaturated fatty acids and disequilibrium in nuclear hormone receptor signaling that increased hepatic VLDL secretion and reduced systemic triglyceride clearance (150, 151). Interestingly, cotreatment with a PPARα agonist reduced the hypertriglyceridemia associated with ACC inhibition, suggesting that ACC inhibitors may be a viable treatment option if given in conjunction with fibrates (150, 151). However, given that cardiovascular disease is the leading cause of death in T2D patients (154), additional studies are crucial to determine the therapeutic potential of ACC inhibition for the treatment of T2D.
Liver-Targeted Mitochondrial Uncouplers
Liver-targeted hepatic mitochondrial uncoupling, whereby the mitochondrial proton gradient is dissipated, thereby dissipating stored energy (fat) in the liver (155), has recently gained increasing attention as a potential therapeutic approach to burn fat and combat the life- and health-limiting consequences of T2D. The first mitochondrial uncoupler, 2,4-dinitrophenol (DNP), was originally used as a component of explosives during World War I (156, 157). After it was observed that many of the workers who handled this compound lost weight, researchers began to investigate the possibility of using DNP as a weight loss drug, and studies by multiple groups demonstrated the efficacy of this approach in obese humans (158–161). The drug was available as an over-the-counter medication and was widely taken for weight loss in the United States, but reports of toxic effects, including several deaths, led to its withdrawal from the market by the US Food and Drug Administration in 1938 (162). Despite the withdrawal from the US market, Russian soldiers continued to take DNP to stay warm on the Eastern Front during the frigid winters of World War II (163), and DNP is currently obtained illegally over the Internet and taken by body builders and individuals trying to lose weight (162, 164–175).
Rodent studies have provided ample evidence for the potential of tissue-targeted mitochondrial uncoupling to improve whole-body glucose and lipid metabolism in vivo. Overexpression of uncoupling proteins 1 or 3 in skeletal muscle has been shown to lower body weight (176–182) and improve insulin sensitivity in HFD-fed rodents (177, 178, 181). Pharmacologic mitochondrial uncoupling is similarly effective at reversing diet-induced obesity and insulin resistance: Five days of treatment with low doses of DNP reduced weight gain, reduced hepatic steatosis, reduced DAG–PKCε activation, and improved hepatic insulin sensitivity in HFD-fed rats (183).
Like DNP, thyroid hormone has long been considered a potential therapeutic agent for the treatment of obesity because of its ability to increase mitochondrial respiration (184–186). However, studies examining the impact of thyroid hormone on glucose metabolism have yielded mixed results, with some investigators suggesting that treatment of euthyroid subjects with exogenous thyroid hormone improves glucose metabolism (187–189), but others reporting no impact (190, 191) or a deleterious effect (192) on metabolism. More problematic, treatment with supraphysiologic concentrations of thyroid hormone has been documented to cause deleterious side effects such as tachycardia, cardiomyopathy, and sarcopenia (193–196).
However, new approaches leveraging thyroid hormone action specifically in the liver have proved beneficial. In particular, the liver-selective, cytochrome P450–activated prodrug MB07811 markedly reduced hepatic steatosis and plasma lipids in rats (197) and was well tolerated and efficacious at reducing LDL cholesterol and triglycerides in patients with mild hypertriglyceridemia (198). In 2016, Finan et al. used a novel glucagon–T3 hybrid molecule to target T3 to the liver and showed that it markedly increased energy expenditure, fat mass, and plasma lipids independent of food intake (199). Importantly, this compound also reduced hepatic lipids and improved glucose tolerance without causing cardiac or bone toxicity (199), thereby suggesting that liver-specific thyroid hormone may be a therapeutic option for the treatment of obesity-associated ectopic lipid and insulin resistance.
Next-generation mitochondrial uncouplers.
Several novel mitochondrial and tissue-targeted chemical uncouplers have recently been developed (Table 2). In 2010, Skulachev and colleagues discovered that synthesized plastoquione derivatives, SkQ1 and C12TPP, potentiate fatty acid–induced uncoupling of respiration and oxidative phosphorylation in mitochondria isolated from rat liver (200). While SkQ1 was further investigated as a potential treatment for Alzheimer’s disease (201), mitochondria-targeted C12TPP was shown to abolish diet-induced obesity in mice by reducing food intake and increasing resting metabolic rate and fatty acid oxidation (202). Similarly, Rhodamine 19 butyl ester (C4R1), a short-chain alkyl derivative of Rhodamine 19, dose-dependently reduced food intake, body weight, and fat mass in HFD-fed mice (203).
Table 2.
Next-generation mitochondrial uncouplers
| Compound | Target tissue | Model | Dose | Body weight | Food intake | Whole-body energy expenditure | FA oxidation | Liver TAGs | Muscle TAGs | Plasma glucose | Plasma insulin | Insulin sensitivity | Plasma lipids | Adverse side effects | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C12TPP | Systemic | HFD-fed C57BL/6 mice (6 weeks) | 50 μM/kg/day × 16 days | ↓ | ↓ | ↑ | ↑ | ND | ND | ND | ND | ND | ND | None | 202 |
| C4R1 | Systemic | HFD-fed C57BL/6 mice (8 weeks) | 12–14, 30 nM/kg/day × 30 days | ↓ | ↓ | ↑ | ↑ | ND | ND | ND | ND | ND | ND | Reduction in lean body mass | 203 |
| db/db mice | 50 mg/kg × 4 weeks | - | - | ND | ND | ND | ND | ↓ | - | ↑ | - | None | |||
| CZ5 | Systemic (in vitro uncoupling in muscle and adipose tissue) | Chow-fed C57BL/6 mice | 30 mg/kg/day × 30 days | - | ↑ | ↑ | ↑ | ND | ND | - | - | - | ND | None | 205 |
| HFD-fed C57BL/6 mice (8 weeks) | 10 mg/kg/day × 5 weeks | ↓ | ↓ | ↑ | ND | - | ↓ | ↓ | ↓ | ↑ | ↓ Cholesterol | None | |||
| db/db mice | 150 mg/kg/day × 60 days | - | - | ND | ND | ND | ND | ↓ | - | ND | ND | None | |||
| NPP | Liver | HFD-fed C57BL/6 mice (8 weeks) | 125 mg/kg/day × 8 weeks | ↓ | - | ND | ND | ↓ | ND | ↓ | ↓ | ↑ | ND | None | 207 |
| T2D rat model | 5 mg/kg/day × 14 days | - | - | ND | ND | ↓ | ND | ↓ | ↓ | ↑ | 4. TAGs | None | |||
| CRMP | Liver | HFD-fed SD rats (3 weeks) | 1 mg/kg/day × 5 days | - | - | - | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ | |TAGs | None | 50 |
| ZDF rats | 1 mg/kg/day × 14 days | - | ND | ND | ND | ↓ | ↓ | ↓ | ↓ | ↑ | |TAGs | None | |||
| MCD-fed rats (8 weeks) | 1 mg/kg/day x× 6 weeks | - | ND | ND | ND | ↓ | ND | ND | ND | ND | ND | None | |||
| A-ZIP/F-1 mice | 2 mg/kg/day × 4 weeks | - | - | - | ND | ↓ | ↓ | ↓ | ↓ | ↑ | |TAGs | None | 60 |
In addition to systemic mitochondrial uncouplers, novel tissue-specific uncoupling agents are also being developed, including the small molecule compounds C1 and CZ5. Acute administration of C1 increased AMPK activity and fat oxidation in chow-fed mice, while chronic C1 treatment reduced hyperglycemia and improved glucose tolerance in diabetic db/db mice (204). CZ5 treatment also reduced body weight and improved glucose and lipid metabolism in HFD-fed mice by increasing whole-body energy expenditure and reducing energy uptake (205). Lastly, niclosamide ethanolamine (NEN), an anthelmintic drug that uncouples the mitochondria, has recently emerged as a potential therapeutic agent for obesity-associated insulin resistance. By increasing energy expenditure, NEN reduced fasting plasma glucose and improved glucose and insulin tolerance in mice with diet-induced obesity (206). A related compound, niclosamide piper-azine, may also hold similar promise for treatment of obesity-associated insulin resistance (207), although the weight-lowering effects of these next-generation chemical uncouplers, despite being an on-target effect of mitochondrial uncoupling, may limit their utility in clinical practice.
Liver-targeted mitochondrial uncouplers.
Systemic mitochondrial uncoupling agents (e.g., DNP) have a narrow therapeutic window due to the on-target effects of these agents to promote hyperthermia. Our group examined whether the therapeutic index could be significantly increased by targeting a mitochondrial uncoupler to the liver. In this regard, we developed a liver-targeted mitochondrial uncoupling agent, DNP–methyl ether (DNPME), which both prevented and reversed diet-induced hepatic insulin resistance without affecting body weight (51). Surprisingly, despite its liver specificity, DNPME also decreased intramyocellular ectopic lipid content and reversed muscle insulin resistance in HFD-fed rats due to reduced hepatic VLDL export. Targeting DNP to the liver improved its toxic to effective dose ratio 50-fold, in association with marked reductions in peak plasma DNP concentrations relative to standard DNP administration. Based on these data, we hypothesized that the toxicity of DNP is related to its peak (Cmax) concentrations, whereas its efficacy is related to the area under the curve of DNP exposure throughout the day. Consistent with that hypothesis, adding an extended-release coating to DNP to generate a controlled-release mitochondrial protonophore (CRMP) increased the toxic to effective dose ratio even further, with a ratio of toxic to effective dose 200-fold higher than that of nontargeted DNP(50). We demonstrated that, akin to DNPME, CRMP (by virtue of its first pass uptake by the liver following ingestion) is a liver-targeted mitochondrial uncoupler (208) that is able to reverse insulin resistance, hepatic inflammation, and hepatic fibrosis in rodent models of T2D, NASH, and lipodystrophy (50, 51, 60). The reversal of hyperglycemia and hepatic insulin resistance by CRMP was attributed to increased fat oxidation exclusively in the liver, with reductions in hepatic triglycerides, DAGs, and PKCε translocation as well as reductions in hepatic acetyl-CoA content and pyruvate carboxylase activity (50). Moreover, CRMP treatment also lowered hepatic VLDL export, thereby reducing intramyocellular ectopic lipid (DAG) content, reducing PKCθ activity, and reversing muscle insulin resistance. Overall, these improvements in liver and muscle insulin resistance, caused by reductions in ectopic lipid in liver and skeletal muscle, as well as in hepatic acetyl-CoA leading to reductions in pyruvate carboxylase activity and gluconeogenesis, produced a reversal of liver inflammation, fibrosis, and diabetes in rodent models of NASH and T2D (50) (Figure 3).
Figure 3.
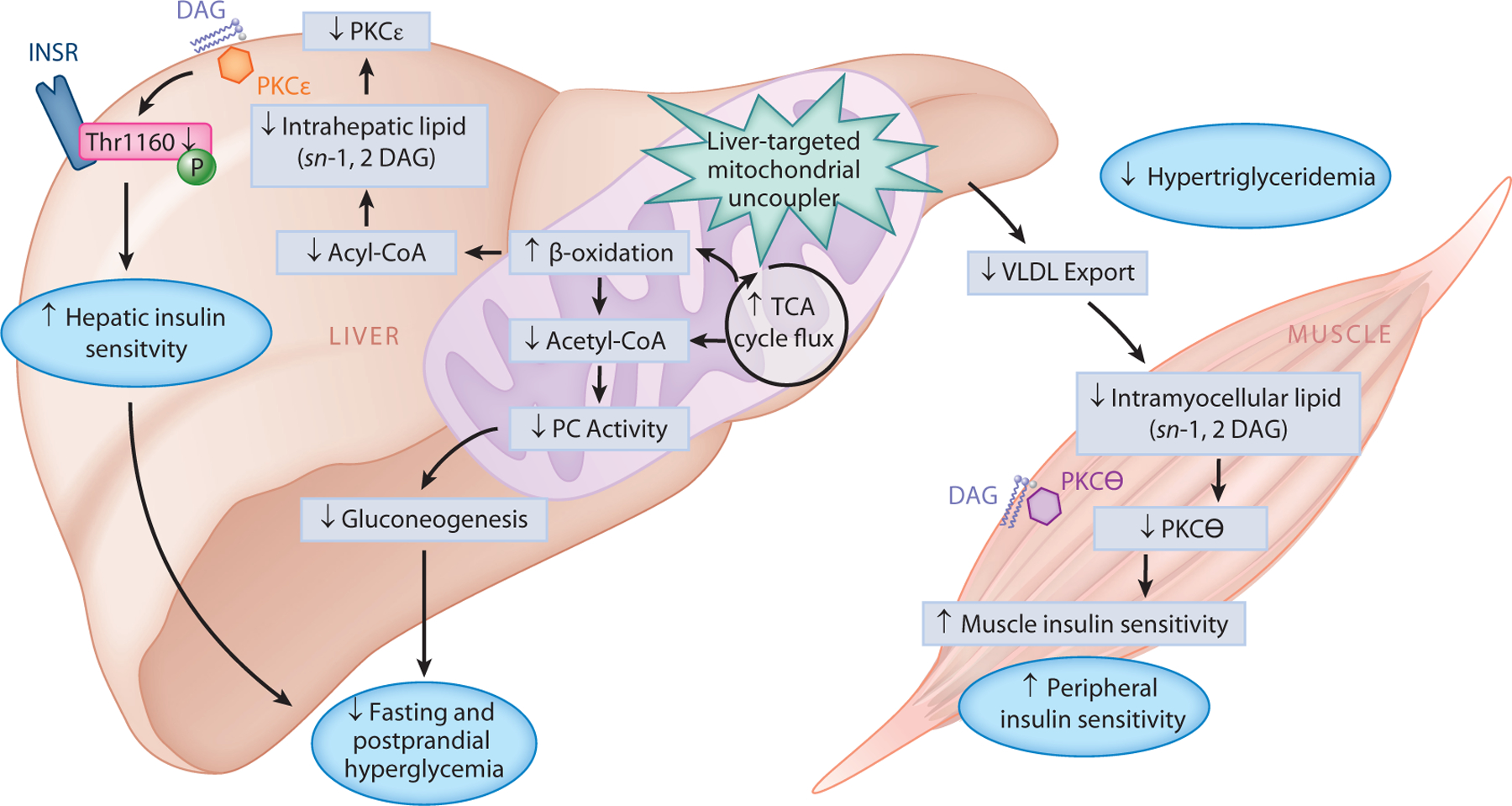
Therapeutic potential of liver-targeted mitochondrial uncouplers for the treatment of T2D. Promoting increased hepatic cellular energy expenditure through the use of liver-targeted mitochondrial uncoupling agents (such as DNP analogs, DNPME, and CRMP) holds therapeutic promise for the treatment of NAFLD, NASH, and T2D. By increasing fat oxidation exclusively in the liver, DNPME and CRMP lower hepatic triglycerides, DAGs, and PKCε translocation, which increases hepatic insulin sensitivity. Liver-targeted mitochondrial uncoupling also increases TCA cycle flux, which reduces hepatic acetyl-CoA content, PC activity, and gluconeogenesis; collectively, this leads to reduced fasting and postprandial hyperglycemia. Moreover, DNPME and CRMP also lower hepatic VLDL export, thereby reducing muscle DAG content, reducing PKCθ activity, and reversing muscle insulin resistance. Overall, these improvements in liver and muscle insulin resistance can reverse diabetes in rodent models of NASH and T2D and suggest that liver-targeted mitochondrial uncoupling agents may be a therapy for the treatment of T2D in humans. Abbreviations: CRMP, controlled-release mitochondrial protonophore; DAG, diacylglycerol; DNP, 2,4-dinitrophenol; DNPME, DNP–methyl ether; INSR, insulin receptor; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PC, pyruvate carboxylase; PKC, protein kinase C; T2D, type 2 diabetes; TCA, tricarboxylic acid; Thr1160, threonine 1160; VLDL, very low-density lipoprotein.
Similarly, via the same mechanisms, CRMP administration in a mouse model of severe lipodystrophy (fatless A-ZIP/F1 mice) reversed hepatic insulin resistance, hepatic inflammation, and diabetes, lowering fasting plasma glucose by approximately 75 mg/dl and fasting plasma insulin by approximately 75% (60). Taken together, these data suggest that CRMP or other liver-targeted mitochondrial protonophores with a similarly high ratio of toxic to effective dose represent an attractive class of agents for potential use to resolve NAFLD and its downstream consequences, including NASH, liver fibrosis, insulin resistance, and T2D.
CONCLUSIONS
T2D and its downstream sequelae, including an increased risk of end-stage vascular dysfunction and cardiovascular disease, are well known to reduce the quality and duration of life. Unfortunately, current treatments have had limited success and do not address a major contributor to T2D: ectopic lipid-related insulin resistance. Over the past years, our understanding of the pathogenesis of insulin resistance and T2D has improved, and accordingly, new pharmacological targets have emerged. Unfortunately, the development and successful use of new treatments have proved to be challenging due to the complexity of insulin resistance and the presence of multiple feedback loops that make it difficult to predict the consequences of a particular intervention (209). For example, liver-specific inhibition of ACC increased plasma triglycerides via the derepression of nuclear receptor signaling as a means of compensating for reduced hepatic lipids (151). Moving forward, identifying therapeutic targets for blocking biochemical pathways involved in glucose and lipid metabolism may prove to be difficult in practice. Therefore, we propose that promoting increased hepatic cellular energy expenditure through the use of liver-targeted mitochondrial uncoupling agents may hold more promise. Indeed, animal studies suggest that liver-targeted mitochondrial uncoupling has a wide therapeutic index and can safely reverse NAFLD, NASH, liver fibrosis, and diabetes in rodent models of NASH, cirrhosis, and T2D (50, 51, 60, 206, 207). Liver-targeted mitochondrial agents are now being developed by several pharmaceutical companies, and studies in humans will be required to determine whether this approach can safely reverse the related epidemics of NAFLD, NASH, and T2D.
SUMMARY POINTS.
Ectopic lipid (DAG) accumulation in skeletal muscle promotes muscle insulin resistance by activation of PKCθ, leading to impaired insulin signaling.
Ectopic lipid (DAG) accumulation in liver promotes hepatic insulin resistance by activation of PKCε, leading to increased insulin receptor threonine 1160 phosphorylation, which leads to decreased insulin receptor tyrosine kinase activity.
Increased hepatic acetyl-CoA content promotes increased rates of hepatic gluconeogenesis and fasting hyperglycemia in T2D by allosteric activation of pyruvate carboxylase.
Several agents aimed at reducing ectopic lipid accumulation in the liver by promoting increased hepatic fat oxidation, inhibiting hepatic lipid synthesis, or increasing hepatic mitochondrial energy expenditure are currently under development for the treatment of NAFLD, NASH, and T2D.
Liver-targeted mitochondrial uncoupling has been shown to reverse liver and muscle insulin resistance and diabetes in rodent models of NAFLD and T2D by decreasing hepatic acetyl-CoA content and DAG–nPKC activation in liver and skeletal muscle.
Liver-targeted mitochondrial uncoupling has been shown to safely reverse NAFLD, NASH, liver fibrosis, and T2D in rodent models of these diseases.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R01 DK040936 and R01 DK113984 to G.I.S., F32 DK114954 to L.G., and K99/R00 CA215315 to R.J.P.) and from the Howard Hughes Medical Institute (to G.I.S.). The authors apologize to those colleagues whose work could not be cited due to space limitations.
Footnotes
DISCLOSURE STATEMENT
G.I.S. serves on the scientific advisory boards for Aegerion, AstraZeneca, Celgene, Merck, and Novo Nordisk and receives investigator-initiated support from Gilead Sciences. R.J.P. and G.I.S. are inventors on Yale patents for liver-targeted mitochondrial uncoupling agents and controlled-release mitochondrial uncoupling agents for the treatment of nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, type 2 diabetes, and related metabolic disorders. L.G. is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, et al. 2017. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract 128:40–50 [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, et al. 2015. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 1:15019. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. 2008. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity 16:2323–30 [DOI] [PubMed] [Google Scholar]
- 4.Boyle JP, Honeycutt AA, Narayan KM, Hoerger TJ, Geiss LS, et al. 2001. Projection of diabetes burden through 2050: impact of changing demography and disease prevalence in the U.S. Diabetes Care 24:1936–40 [DOI] [PubMed] [Google Scholar]
- 5.Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. 2009. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. 28:w822–31 [DOI] [PubMed] [Google Scholar]
- 6.Cawley J, Meyerhoefer C. 2012. The medical care costs of obesity: an instrumental variables approach.J. Health Econ 31:219–30 [DOI] [PubMed] [Google Scholar]
- 7.Cawley J, Rizzo JA, Haas K. 2007. Occupation-specific absenteeism costs associated with obesity and morbid obesity. J. Occup. Environ. Med 49:1317–24 [DOI] [PubMed] [Google Scholar]
- 8.Shulman GI. 2000. Cellular mechanisms of insulin resistance. J. Clin. Invest 106:171–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor R, Magnusson I, Rothman DL, Cline GW, Caumo A, et al. 1996. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Invest 97:126–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. 1990. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med 322:223–28 [DOI] [PubMed] [Google Scholar]
- 11.Nazimek-Siewniak B, Moczulski D, Grzeszczak W. 2002. Risk of macrovascular and microvascular complications in Type 2 diabetes: results of longitudinal study design. J. Diabetes Complic 16:271–76 [DOI] [PubMed] [Google Scholar]
- 12.Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, et al. 2012. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 35:1364–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuso P 2014. Prediabetes and lifestyle modification: time to prevent a preventable disease. Perm. J 18:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, et al. 2000. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49:2063–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, et al. 1998. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N. Engl. J. Med 338:867–72 [DOI] [PubMed] [Google Scholar]
- 16.Roumie CL, Hung AM, Greevy RA, Grijalva CG, Liu X, et al. 2012. Comparative effectiveness of sulfonylurea and metformin monotherapy on cardiovascular events in type 2 diabetes mellitus: a cohort study. Ann. Intern. Med 157:601–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdul-Ghani MA, Norton L, Defronzo RA. 2011. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr. Rev 32:515–31 [DOI] [PubMed] [Google Scholar]
- 18.Lovshin JA, Drucker DJ. 2009. Incretin-based therapies for type 2 diabetes mellitus. Nat. Rev. Endocrinol 5:262–69 [DOI] [PubMed] [Google Scholar]
- 19.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, et al. 2002. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51:797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petersen KF, Krssak M, Inzucchi S, Cline GW, Dufour S, Shulman GI. 2000. Mechanism of troglitazone action in type 2 diabetes. Diabetes 49:827–31 [DOI] [PubMed] [Google Scholar]
- 21.Tonelli J, Li W, Kishore P, Pajvani UB, Kwon E, et al. 2004. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes 53:1621–29 [DOI] [PubMed] [Google Scholar]
- 22.Van de Laar FA, Lucassen PL, Akkermans RP, Van de Lisdonk EH, Rutten GE, Van Weel C. 2005. Alpha-glucosidase inhibitors for type 2 diabetes mellitus. Cochrane Database Syst. Rev 8:CD003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weng J, Li Y, Xu W, Shi L, Zhang Q, et al. 2008. Effect of intensive insulin therapy on β-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet 371:1753–60 [DOI] [PubMed] [Google Scholar]
- 24.Samuel VT, Shulman GI. 2018. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27:22–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alkhouri N, Poordad F, Lawitz E. 2018. Management of nonalcoholic fatty liver disease: lessons learned from type 2 diabetes. Hepatol. Commun 2:778–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Jarvinen H, et al. 1988. Impaired glucose tolerance as a disorder of insulin action: longitudinal and cross-sectional studies in Pima Indians. N. Engl. J. Med 318:1217–25 [DOI] [PubMed] [Google Scholar]
- 27.Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. 1990. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann. Intern. Med 113:909–15 [DOI] [PubMed] [Google Scholar]
- 28.DeFronzo RA, Tripathy D. 2009. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl. 2):S157–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shulman GI. 2014. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl.J. Med 371:1131–41 [DOI] [PubMed] [Google Scholar]
- 30.Rothman DL, Shulman RG, Shulman GI. 1992. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate: evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J. Clin. Invest 89:1069–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Defronzo RA. 2009. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58:773–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, et al. 2015. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160:745–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeFronzo RA. 1988. Lilly lecture 1987. The triumvirate: β-cell, muscle, liver—a collusion responsible for NIDDM. Diabetes 37:667–87 [DOI] [PubMed] [Google Scholar]
- 34.Petersen MC, Shulman GI. 2017. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol. Sci 38:649–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuel VT, Shulman GI. 2016. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest 126:12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boren J, Taskinen MR, Olofsson SO, Levin M. 2013. Ectopic lipid storage and insulin resistance: a harmful relationship. J. Intern. Med 274:25–40 [DOI] [PubMed] [Google Scholar]
- 37.Lettner A, Roden M. 2008. Ectopic fat and insulin resistance. Curr. Diabetes Rep 8:185–91 [DOI] [PubMed] [Google Scholar]
- 38.Szendroedi J, Roden M. 2009. Ectopic lipids and organ function. Curr. Opin. Lipidol 20:50–56 [DOI] [PubMed] [Google Scholar]
- 39.Perry RJ, Samuel VT, Petersen KF, Shulman GI. 2014. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510:84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel VT, Shulman GI. 2012. Mechanisms for insulin resistance: common threads and missing links. Cell 148:852–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, et al. 2016. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest 126:4361–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, et al. 2014. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. PNAS 111:9597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Soos TJ, Li X, Wu J, Degennaro M, et al. 2004. Protein kinase C θ inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J. Biol. Chem 279:45304–7 [DOI] [PubMed] [Google Scholar]
- 44.Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, et al. 2017. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 19:1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, et al. 2011. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. PNAS 108:16381–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magkos F, Su X, Bradley D, Fabbrini E, Conte C, et al. 2012. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142:1444–46.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, et al. 2016. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol 64:1167–75 [DOI] [PubMed] [Google Scholar]
- 48.Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, et al. 2005. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2:55–65 [DOI] [PubMed] [Google Scholar]
- 49.Nagle CA, An J, Shiota M, Torres TP, Cline GW, et al. 2007. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J. Biol. Chem 282:14807–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. 2015. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347:1253–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, et al. 2013. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 18:740–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, et al. 2007. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem 282:22678–88 [DOI] [PubMed] [Google Scholar]
- 53.Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, et al. 2007. Mitochondrial dysfunction due to long-chain acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. PNAS 104:17075–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jornayvaz FR, Birkenfeld AL, Jurczak MJ, Kanda S, Guigni BA, et al. 2011. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. PNAS 108:5748–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang D, Christianson J, Liu ZX, Tian L, Choi CS, et al. 2010. Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 11:402–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryu D, Seo WY, Yoon YS, Kim YN, Kim SS, et al. 2011. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes 60:1072–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryu D, Oh KJ, Jo HY, Hedrick S, Kim YN, et al. 2009. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab. 9:240–51 [DOI] [PubMed] [Google Scholar]
- 58.Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, et al. 2013. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 57:1763–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, et al. 2007. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest 117:739–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abulizi A, Perry RJ, Camporez JP, Jurczak MJ, Petersen KF, et al. 2017. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J. 31:2916–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, et al. 2011. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 54:1447–56 [DOI] [PubMed] [Google Scholar]
- 62.Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, et al. 2006. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Invest 116:817–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, et al. 2006. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes 55:2042–50 [DOI] [PubMed] [Google Scholar]
- 64.Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, et al. 2007. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. PNAS 104:16480–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaworski K, Ahmadian M, Duncan RE, Sarkadi-Nagy E, Varady KA, et al. 2009. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med 15:159–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erion DM, Ignatova ID, Yonemitsu S, Nagai Y, Chatterjee P, et al. 2009. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab. 10:499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galbo T, Perry RJ, Jurczak MJ, Camporez JP, Alves TC, et al. 2013. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. PNAS 110:12780–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Camporez JP, Jornayvaz FR, Lee HY, Kanda S, Guigni BA, et al. 2013. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology 154:1021–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, et al. 2013. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 154:3099–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jornayvaz FR, Jurczak MJ, Lee HY, Birkenfeld AL, Frederick DW, et al. 2010. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am. J. Physiol. Endocrinol. Metab 299:E808–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, et al. 2011. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14:184–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee HY, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, et al. 2011. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54:1650–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jornayvaz FR, Lee HY, Jurczak MJ, Alves TC, Guebre-Egziabher F, et al. 2012. Thyroid hormone receptor-αgene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 153:583–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, et al. 2012. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J. Biol. Chem 287:2558–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brown WH, Gillum MP, Lee HY, Camporez JP, Zhang XM, et al. 2012. Fatty acid amide hydrolase ablation promotes ectopic lipid storage and insulin resistance due to centrally mediated hypothyroidism. PNAS 109:14966–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun Z, Miller RA, Patel RT, Chen J, Dhir R, et al. 2012. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med 18:934–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montgomery MK, Brown SH, Lim XY, Fiveash CE, Osborne B, et al. 2016. Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: a beneficial role for very long-chain sphingolipid species. Biochim. Biophys. Acta 1861:1828–39 [DOI] [PubMed] [Google Scholar]
- 78.Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, et al. 2013. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56:1638–48 [DOI] [PubMed] [Google Scholar]
- 79.Zhu L, Martinez MN, Emfinger CH, Palmisano BT, Stafford JM. 2014. Estrogen signaling prevents diet-induced hepatic insulin resistance in male mice with obesity. Am. J. Physiol. Endocrinol. Metab 306:E1188–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Camporez JP, Kanda S, Petersen MC, Jornayvaz FR, Samuel VT, et al. 2015. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J. Lipid Res 56:526–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jordy AB, Kraakman MJ, Gardner T, Estevez E, Kammoun HL, et al. 2015. Analysis of the liver lipidome reveals insights into the protective effect of exercise on high-fat diet-induced hepatosteatosis in mice. Am. J. Physiol. Endocrinol. Metab 308:E778–91 [DOI] [PubMed] [Google Scholar]
- 82.Aroor AR, Habibi J, Ford DA, Nistala R, Lastra G, et al. 2015. Dipeptidyl peptidase-4 inhibition ameliorates Western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function. Diabetes 64:1988–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shang J, Castro-Perez JM, Shen X, Zhu Y, Liu H, et al. 2016. Duodenal-jejunal bypass surgery induces hepatic lipidomic alterations associated with ameliorated hepatic steatosis in mice. Obesity 24:1938–45 [DOI] [PubMed] [Google Scholar]
- 84.Popov VB, Jornayvaz FR, Akgul EO, Kanda S, Jurczak MJ, et al. 2015. Second-generation antisense oligonucleotides against β-catenin protect mice against diet-induced hepatic steatosis and hepatic and peripheral insulin resistance. FASEB J. 30:1207–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. 2016. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 157:570–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Montgomery MK, Fiveash CE, Braude JP, Osborne B, Brown SH, et al. 2015. Disparate metabolic response to fructose feeding between different mouse strains. Sci. Rep 5:18474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baumeier C, Kaiser D, Heeren J, Scheja L, John C, et al. 2015. Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim. Biophys. Acta 1851:566–76 [DOI] [PubMed] [Google Scholar]
- 88.Jelenik T, Sequaris G, Kaul K, Ouwens DM, Phielix E, et al. 2014. Tissue-specific differences in the development of insulin resistance in a mouse model for type 1 diabetes. Diabetes 63:3856–67 [DOI] [PubMed] [Google Scholar]
- 89.Lee HY, Lee JS, Alves T, Ladiges W, Rabinovitch PS, et al. 2017. Mitochondrial-targeted catalase protects against high-fat diet-induced muscle insulin resistance by decreasing intramuscular lipid accumulation. Diabetes 66:2072–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao N, Lou MD, Lu YT, Yang LL, Liu Q, et al. 2017. Ginsenoside Rg5 attenuates hepatic glucagon response via suppression of succinate-associated HIF-1αinduction in HFD-fed mice. Diabetologia 60:1084–93 [DOI] [PubMed] [Google Scholar]
- 91.Goossens GH, Moors CC, Jocken JW, van der Zijl NJ, Jans A, et al. 2016. Altered skeletal muscle fatty acid handling in subjects with impaired glucose tolerance as compared to impaired fasting glucose. Nutrients 8:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu C, Chen Y, Cline GW, Zhang D, Zong H, et al. 2002. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem 277:50230–36 [DOI] [PubMed] [Google Scholar]
- 93.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, et al. 1999. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C θ and alterations in the insulin signaling cascade. Diabetes 48:1270–74 [DOI] [PubMed] [Google Scholar]
- 94.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, et al. 1999. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest 103:253–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lam YY, Hatzinikolas G, Weir JM, Janovska A, McAinch AJ, et al. 2011. Insulin-stimulated glucose uptake and pathways regulating energy metabolism in skeletal muscle cells: the effects of subcutaneous and visceral fat, and long-chain saturated, n-3 and n-6 polyunsaturated fatty acids. Biochim. Biophys. Acta 1811:468–75 [DOI] [PubMed] [Google Scholar]
- 96.Newsom SA, Everett AC, Park S, Van Pelt DW, Hinko A, Horowitz JF. 2015. Lipid mixtures containing a very high proportion of saturated fatty acids only modestly impair insulin signaling in cultured muscle cells. PLOS ONE 10:e0120871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee JS, Pinnamaneni SK, Eo SJ, Cho IH, Pyo JH, et al. 2006. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: role of intramuscular accumulation of lipid metabolites. J. Appl. Physiol 100:1467–74 [DOI] [PubMed] [Google Scholar]
- 98.Coudray C, Fouret G, Lambert K, Ferreri C, Rieusset J, et al. 2016. A mitochondrial-targeted ubiquinone modulates muscle lipid profile and improves mitochondrial respiration in obesogenic diet-fed rats. Br.J. Nutr 115:1155–66 [DOI] [PubMed] [Google Scholar]
- 99.Rivas DA, McDonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. 2016. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol 310:R561–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Coen PM, Menshikova EV, Distefano G, Zheng D, Tanner CJ, et al. 2015. Exercise and weight loss improve muscle mitochondrial respiration, lipid partitioning, and insulin sensitivity after gastric bypass surgery. Diabetes 64:3737–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kase ET, Feng YZ, Badin PM, Bakke SS, Laurens C, et al. 2015. Primary defects in lipolysis and insulin action in skeletal muscle cells from type 2 diabetic individuals. Biochim. Biophys. Acta 1851:1194–201 [DOI] [PubMed] [Google Scholar]
- 102.Rahimi Y, Camporez JP, Petersen MC, Pesta D, Perry RJ, et al. 2014. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. PNAS 111:16508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zachariah Tom R, Garcia-Roves PM, Sjogren RJ, Jiang LQ, Holmstrom MH, et al. 2014. Effects of AMPK activation on insulin sensitivity and metabolism in leptin-deficient ob/ob mice. Diabetes 63:1560–71 [DOI] [PubMed] [Google Scholar]
- 104.Holloway GP, Han XX, Jain SS, Bonen A, Chabowski A. 2014. Chronic muscle stimulation improves insulin sensitivity while increasing subcellular lipid droplets and reducing selected diacylglycerol and ceramide species in obese Zucker rats. Diabetologia 57:832–40 [DOI] [PubMed] [Google Scholar]
- 105.Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, et al. 2014. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 63:1881–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mathew D, Zhou P, Pywell CM, van der Veen DR, Shao J, et al. 2013. Ablation of the ID2 gene results in altered circadian feeding behavior, and sex-specific enhancement of insulin sensitivity and elevated glucose uptake in skeletal muscle and brown adipose tissue. PLOS ONE 8:e73064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jocken JW, Goossens GH, Boon H, Mason RR, Essers Y, et al. 2013. Insulin-mediated suppression of lipolysis in adipose tissue and skeletal muscle of obese type 2 diabetic men and men with normal glucose tolerance. Diabetologia 56:2255–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Badin PM, Vila IK, Louche K, Mairal A, Marques MA, et al. 2013. High-fat diet-mediated lipotoxicity and insulin resistance is related to impaired lipase expression in mouse skeletal muscle. Endocrinology 154:1444–53 [DOI] [PubMed] [Google Scholar]
- 109.Dube JJ, Amati F, Toledo FG, Stefanovic-Racic M, Rossi A, et al. 2011. Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54:1147–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. 2005. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54:603–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Taylor R, Leslie WS, Barnes AC, Brosnahan N, Thom G, et al. 2018. Clinical and metabolic features of the randomised controlled Diabetes Remission Clinical Trial (DiRECT) cohort. Diabetologia 61:589–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, et al. 2018. Mechanisms by which a very low calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab. 27:210–17.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Isbell JM, Tamboli RA, Hansen EN, Saliba J, Dunn JP, et al. 2010. The importance of caloric restriction in the early improvements in insulin sensitivity after Roux-en-Y gastric bypass surgery. Diabetes Care 33:1438–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Campos GM, Rabl C, Peeva S, Ciovica R, Rao M, et al. 2010. Improvement in peripheral glucose uptake after gastric bypass surgery is observed only after substantial weight loss has occurred and correlates with the magnitude of weight lost. J. Gastrointest. Surg 14:15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laferrere B, Teixeira J, McGinty J, Tran H, Egger JR, et al. 2008. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. J. Clin. Endocrinol. Metab 93:2479–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Plum L, Ahmed L, Febres G, Bessler M, Inabnet W, et al. 2011. Comparison of glucostatic parameters after hypocaloric diet or bariatric surgery and equivalent weight loss. Obesity 19:2149–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Henry RR, Scheaffer L, Olefsky JM. 1985. Glycemic effects of intensive caloric restriction and isocaloric refeeding in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab 61:917–25 [DOI] [PubMed] [Google Scholar]
- 118.Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. 2011. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 54:2506–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jackness C, Karmally W, Febres G, Conwell IM, Ahmed L, et al. 2013. Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and β-cell function in type 2 diabetic patients. Diabetes 62:3027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stunkard A, McLaren-Hume M. 1959. The results of treatment for obesity: a review of the literature and report of a series. AMA Arch. Intern. Med 103:79–85 [DOI] [PubMed] [Google Scholar]
- 121.Wing RR, Phelan S. 2005. Long-term weight loss maintenance. Am. J. Clin. Nutr 82:222–25S [DOI] [PubMed] [Google Scholar]
- 122.McGuire MT, Wing RR, Hill JO. 1999. The prevalence of weight loss maintenance among American adults. Int. J. Obes. Relat. Metab. Disord 23:1314–19 [DOI] [PubMed] [Google Scholar]
- 123.Kraschnewski JL, Boan J, Esposito J, Sherwood NE, Lehman EB, et al. 2010. Long-term weight loss maintenance in the United States. Int. J. Obes 34:1644–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baskota A, Li S, Dhakal N, Liu G, Tian H. 2015. Bariatric surgery for type 2 diabetes mellitus in patients with BMI <30 kg/m2: a systematic review and meta-analysis. PLOS ONE 10:e0132335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, et al. 2009. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am. J. Med 122:248–56.e5 [DOI] [PubMed] [Google Scholar]
- 126.Klein S, Mittendorfer B, Eagon JC, Patterson B, Grant L, et al. 2006. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology 130:1564–72 [DOI] [PubMed] [Google Scholar]
- 127.Mattar SG, Velcu LM, Rabinovitz M, Demetris AJ, Krasinskas AM, et al. 2005. Surgically-induced weight loss significantly improves nonalcoholic fatty liver disease and the metabolic syndrome. Ann. Surg 242:610–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, et al. 2018. Mechanisms by which a very-low-calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab. 27:210–17.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Aminian A, et al. 2017. Bariatric surgery versus intensive medical therapy for diabetes: 5-year outcomes. N. Engl. J. Med 376:641–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Khorgami Z, Shoar S, Andalib A, Aminian A, Brethauer SA, Schauer PR. 2017. Trends in utilization of bariatric surgery, 2010–2014: sleeve gastrectomy dominates. Surg. Obes. Relat. Dis 13:774–78 [DOI] [PubMed] [Google Scholar]
- 131.Golomb I, Ben David M, Glass A, Kolitz T, Keidar A. 2015. Long-term metabolic effects of laparoscopic sleeve gastrectomy. JAMA Surg. 150:1051–57 [DOI] [PubMed] [Google Scholar]
- 132.So WY, Leung PS. 2016. Fibroblast growth factor 21 as an emerging therapeutic target for type 2 diabetes mellitus. Med. Res. Rev 36:672–704 [DOI] [PubMed] [Google Scholar]
- 133.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, et al. 2007. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5:415–25 [DOI] [PubMed] [Google Scholar]
- 134.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. 2007. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5:426–37 [DOI] [PubMed] [Google Scholar]
- 135.Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, et al. 2014. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63:4057–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, et al. 2012. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife 1:e00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xu J, Stanislaus S, Chinookoswong N, Lau YY, Hager T, et al. 2009. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models: association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab 297:E1105–14 [DOI] [PubMed] [Google Scholar]
- 138.Wente W, Efanov AM, Brenner M, Kharitonenkov A, Koster A, et al. 2006. Fibroblast growth factor-21 improves pancreatic β-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 55:2470–78 [DOI] [PubMed] [Google Scholar]
- 139.Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, et al. 2008. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57:1246–53 [DOI] [PubMed] [Google Scholar]
- 140.Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, et al. 2013. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 18:333–40 [DOI] [PubMed] [Google Scholar]
- 141.Adams AC, Halstead CA, Hansen BC, Irizarry AR, Martin JA, et al. 2013. LY2405319, an engineered FGF21 variant, improves the metabolic status of diabetic monkeys. PLOS ONE 8:e65763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, et al. 2013. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLOS ONE 8:e58575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wei W, Dutchak PA, Wang X, Ding X, Wang X, et al. 2012. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. PNAS 109:3143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanyal A, Charles ED, Neuschwander-Tetri B, Loomba R, Harrison S, et al. 2017. BMS-986036 (pegylated FGF21) in patients with non-alcoholic steatohepatitis: a phase 2 study. J. Hepatol 66:S89–90 [Google Scholar]
- 145.Brownsey RW, Zhande R, Boone AN. 1997. Isoforms of acetyl-CoA carboxylase: structures, regulatory properties and metabolic functions. Biochem. Soc. Trans 25:1232–38 [DOI] [PubMed] [Google Scholar]
- 146.McGarry JD, Mannaerts GP, Foster DW. 1977. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest 60:265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tong L, Harwood HJ Jr. 2006. Acetyl-coenzyme A carboxylases: versatile targets for drug discovery.J. Cell Biochem 99:1476–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harriman G, Greenwood J, Bhat S, Huang X, Wang R, et al. 2016. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. PNAS 113:E1796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bourbeau MP, Bartberger MD. 2015. Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J. Med. Chem 58:525–36 [DOI] [PubMed] [Google Scholar]
- 150.Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, et al. 2018. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. In press. 10.1002/hep.30097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, et al. 2017. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26:394–406.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lawitz EJ, Poordad F, Coste A, Loo N, Djedjos CS, et al. 2017. Acetyl-CoA carboxylase (ACC) inhibitor GS-0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRIPDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH. J. Hepatol 66:S34 [Google Scholar]
- 153.Stiede K, Miao W, Blanchette HS, Beysen C, Harriman G, et al. 2017. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: a randomized, double-blind, crossover study. Hepatology 66:324–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yuan G, Al-Shali KZ, Hegele RA. 2007. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ 176:1113–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. 2010. Mitochondrial proton and electron leaks. Essays Biochem. 47:53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Perkins RG. 1919. A study of the munitions intoxications in France. Public Health Rep. 34:2335–7419314692 [Google Scholar]
- 157.Cutting WC, Mehrtens HG, Tainter ML. 1933. Actions and uses of dinitrophenol: promising metabolic applications. JAMA 101:193–95 [Google Scholar]
- 158.Tainter ML, Stockton AB, Cutting WC. 1933. Use of dinitrophenol in obesity and related conditions: a progress report. JAMA 101:1472–75 [Google Scholar]
- 159.Tainter ML, Stockton AB. 1935. Dinitrophenol in the treatment of obesity: final report. JAMA 101:322–36 [Google Scholar]
- 160.Cutting WC, Tainter ML. 1933. Metabolic actions of dinitrophenol with the use of balanced and unbalanced diets. JAMA 101:2099–102 [Google Scholar]
- 161.Simkins S 1937. Dinitrophenol and desiccated thyroid in the treatment of obesity: a comprehensive clinical and laboratory study. JAMA 108:2110–18 [Google Scholar]
- 162.Grundlingh J, Dargan PI, El-Zanfaly M, Wood DM. 2011. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J. Med. Toxicol 7:205–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kurt TL, Anderson R, Petty C, Bost R, Reed G, Holland J. 1986. Dinitrophenol in weight loss: the poison center and public health safety. Vet. Hum. Toxicol 28:574–75 [PubMed] [Google Scholar]
- 164.Swamy SA. 1953. Suicidal poisoning by dinitrophenol. J. Indian Med. Assoc 22:504–5 [PubMed] [Google Scholar]
- 165.Cann HM, Verhulst HL. 1960. Fatality from acute dinitrophenol derivative poisoning. Am. J. Dis. Child 100:947–48 [DOI] [PubMed] [Google Scholar]
- 166.Bartlett J, Brunner M, Gough K. 2010. Deliberate poisoning with dinitrophenol (DNP): an unlicensed weight loss pill. Emerg. Med. J 27:159–60 [DOI] [PubMed] [Google Scholar]
- 167.Tewari A, Ali T, O’Donnell J, Butt MS. 2009. Weight loss and 2,4-dinitrophenol poisoning. Br. J.Anaesth 102:566–67 [DOI] [PubMed] [Google Scholar]
- 168.Siegmueller C, Narasimhaiah R. 2010. “Fatal 2,4-dinitrophenol poisoning… coming to a hospital near you”. Emerg. Med. J 27:639–40 [DOI] [PubMed] [Google Scholar]
- 169.McFee RB, Caraccio TR, McGuigan MA, Reynolds SA, Bellanger P. 2004. Dying to be thin: a dinitrophenol related fatality. Vet. Hum. Toxicol 46:251–54 [PubMed] [Google Scholar]
- 170.Hsiao AL, Santucci KA, Seo-Mayer P, Mariappan MR, Hodsdon ME, et al. 2005. Pediatric fatality following ingestion of dinitrophenol: postmortem identification of a “dietary supplement.” Clin. Toxicol 43:281–85 [DOI] [PubMed] [Google Scholar]
- 171.Suozzi JC, Rancont CM, McFee RB. 2005. DNP 2,4-dinitrophenol: a deadly way to lose weight. JEMS 30:82–91 [PubMed] [Google Scholar]
- 172.Miranda EJ, McIntyre IM, Parker DR, Gary RD, Logan BK. 2006. Two deaths attributed to the use of 2,4-dinitrophenol. J. Anal. Toxicol 30:219–22 [DOI] [PubMed] [Google Scholar]
- 173.Politi L, Vignali C, Polettini A. 2007. LC-MS-MS analysis of 2,4-dinitrophenol and its phase I and II metabolites in a case of fatal poisoning. J. Anal. Toxicol 31:55–61 [DOI] [PubMed] [Google Scholar]
- 174.Holborow A, Purnell RM, Wong JF. 2016. Beware the yellow slimming pill: fatal 2,4-dinitrophenol overdose. BMJ Case Rep. 2016:214689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lu YQ, Jiang JK, Huang WD. 2011. Clinical features and treatment in patients with acute 2,4-dinitrophenol poisoning. J. Zhejiang Univ. Sci. B 12:189–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Clapham JC, Arch JR, Chapman H, Haynes A, Lister C, et al. 2000. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 406:415–18 [DOI] [PubMed] [Google Scholar]
- 177.Li B, Nolte LA, Ju JS, Han DH, Coleman T, et al. 2000. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med 6:1115–20 [DOI] [PubMed] [Google Scholar]
- 178.Poher AL, Veyrat-Durebex C, Altirriba J, Montet X, Colin DJ, et al. 2015. Ectopic UCP1 overexpression in white adipose tissue improves insulin sensitivity in Lou/C rats, a model of obesity resistance. Diabetes 64:3700–12 [DOI] [PubMed] [Google Scholar]
- 179.Kopecky J, Clarke G, Enerback S, Spiegelman B, Kozak LP. 1995. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Invest 96:2914–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kopecky J, Hodny Z, Rossmeisl M, Syrovy I, Kozak LP. 1996. Reduction of dietary obesity in aP2-Ucp transgenic mice: physiology and adipose tissue distribution. Am. J. Physiol 270:E768–75 [DOI] [PubMed] [Google Scholar]
- 181.Bernal-Mizrachi C, Weng S, Li B, Nolte LA, Feng C, et al. 2002. Respiratory uncoupling lowers blood pressure through a leptin-dependent mechanism in genetically obese mice. Arterioscler. Thromb. Vasc. Biol 22:961–68 [DOI] [PubMed] [Google Scholar]
- 182.Couplan E, Gelly C, Goubern M, Fleury C, Quesson B, et al. 2002. High level of uncoupling protein 1 expression in muscle of transgenic mice selectively affects muscles at rest and decreases their IIb fiber content. J. Biol. Chem 277:43079–88 [DOI] [PubMed] [Google Scholar]
- 183.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, et al. 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem 279:32345–53 [DOI] [PubMed] [Google Scholar]
- 184.Byrom FB. 1933. Nature of myxoedema. Clin. Sci 1:273–85 [Google Scholar]
- 185.Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, et al. 2001. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J. Clin. Invest 108:733–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mitchell CS, Savage DB, Dufour S, Schoenmakers N, Murgatroyd P, et al. 2010. Resistance to thyroid hormone is associated with raised energy expenditure, muscle mitochondrial uncoupling, and hyperphagia. J. Clin. Invest 120:1345–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vazquez-Anaya G, Martinez B, Sonanez-Organis JG, Nakano D, Nishiyama A, Ortiz RM. 2017. Exogenous thyroxine improves glucose intolerance in insulin-resistant rats. J. Endocrinol 232:501–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.da Silva Teixeira S, Filgueira C, Sieglaff DH, Benod C, Villagomez R, et al. 2017. 3,5-diiodothyronine (3,5-T2) reduces blood glucose independently of insulin sensitization in obese mice. Acta Physiol. 220:238–50 [DOI] [PubMed] [Google Scholar]
- 189.Iannucci LF, Cioffi F, Senese R, Goglia F, Lanni A, et al. 2017. Metabolomic analysis shows differential hepatic effects of T2 and T3 in rats after short-term feeding with high fat diet. Sci. Rep 7:2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.van der Valk F, Hassing C, Visser M, Thakkar P, Mohanan A, et al. 2014. The effect of a diiodothyronine mimetic on insulin sensitivity in male cardiometabolic patients: a double-blind randomized controlled trial. PLOS ONE 9:e86890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vatner DF, Snikeris J, Popov V, Perry RJ, Rahimi Y, Samuel VT. 2015. 3,5 Diiodo-L-thyronine (T2) does not prevent hepatic steatosis or insulin resistance in fat-fed Sprague Dawley rats. PLOS ONE 10:e0140837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Vatner DF, Weismann D, Beddow SA, Kumashiro N, Erion DM, et al. 2013. Thyroid hormone receptor-β agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am. J. Physiol. Endocrinol. Metab 305:E89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Klein I, Levey GS. 1984. New perspectives on thyroid hormone, catecholamines, and the heart. Am. J.Med 76:167–72 [DOI] [PubMed] [Google Scholar]
- 194.Burgi U, Burgi-Saville ME, Burgherr J, Clement M, Lauber K. 1990. T3 plus high doses of beta-blockers: effects on energy intake, body composition, BAT and heart in rats. Int. J. Obes 14:1023–38 [PubMed] [Google Scholar]
- 195.Kyle LH, Ball MF, Doolan PD. 1966. Effect of thyroid hormone on body composition in myxedema and obesity. N. Engl. J. Med 275:12–17 [DOI] [PubMed] [Google Scholar]
- 196.Abraham RR, Densem JW, Davies P, Davie MW, Wynn V. 1985. The effects of triiodothyronine on energy expenditure, nitrogen balance and rates of weight and fat loss in obese patients during prolonged caloric restriction. Int. J. Obes 9:433–42 [PubMed] [Google Scholar]
- 197.Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, et al. 2007. Targeting thyroid hormone receptor-β agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. PNAS 104:15490–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ligand Pharm. 2009. Study of MB07811 in subjects with hypercholesterolemia. Rep. NCT00879112, US Natl.Libr. Med., Natl. Inst. Health, Bethesda, MD: https://clinicaltrials.gov/ct2/show/NCT00879112 [Google Scholar]
- 199.Finan B, Clemmensen C, Zhu Z, Stemmer K, Gauthier K, et al. 2016. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell 167:843–57.e14 [DOI] [PubMed] [Google Scholar]
- 200.Severin FF, Severina II, Antonenko YN, Rokitskaya TI, Cherepanov DA, et al. 2010. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. PNAS 107:663–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stefanova NA, Kozhevnikova OS, Vitovtov AO, Maksimova KY, Logvinov SV, et al. 2014. Senescence-accelerated OXYS rats: a model of age-related cognitive decline with relevance to abnormalities in Alzheimer disease. Cell Cycle 13:898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kalinovich AV, Mattsson CL, Youssef MR, Petrovic N, Ost M, et al. 2016. Mitochondria-targeted dodecyltriphenylphosphonium (C12TPP) combats high-fat-diet-induced obesity in mice. Int. J. Obes 40:1864–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kalinovich AV, Shabalina IG. 2015. Novel mitochondrial cationic uncoupler C4R1 is an effective treatment for combating obesity in mice. Biochemistry 80:620–28 [DOI] [PubMed] [Google Scholar]
- 204.Qiu BY, Turner N, Li YY, Gu M, Huang MW, et al. 2010. High-throughput assay for modulators of mitochondrial membrane potential identifies a novel compound with beneficial effects on db/db mice. Diabetes 59:256–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Fu YY, Zhang M, Turner N, Zhang LN, Dong TC, et al. 2013. A novel chemical uncoupler ameliorates obesity and related phenotypes in mice with diet-induced obesity by modulating energy expenditure and food intake. Diabetologia 56:2297–307 [DOI] [PubMed] [Google Scholar]
- 206.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. 2014. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med 20:1263–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Guo J, Tao H, Alasadi A, Huang Q, Jin S. 2018. Niclosamide piperazine prevents high-fat diet-induced obesity and diabetic symptoms in mice. Eat Weight Disord. In press [DOI] [PubMed] [Google Scholar]
- 208.Perry RJ, Peng L, Cline GW, Butrico GM, Wang Y, et al. 2017. Non-invasive assessment of hepatic mitochondrial metabolism by positional isotopomer NMR tracer analysis (PINTA). Nat. Commun 8:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Altaf QA, Barnett AH, Tahrani AA. 2015. Novel therapeutics for type 2 diabetes: insulin resistance. Diabetes Obes. Metab 17:319–34 [DOI] [PubMed] [Google Scholar]