

Abstract
Our understanding and utilization of Fecal Microbiota Transplantation (FMT) has jump-started over the past two decades. Recent technological advancements in sequencing and metabolomics have allowed for better characterization of our intestinal microbial counterparts, triggering a surge of excitement in the fields of mucosal immunology and microbiology. This excitement is well-founded, as evidenced by 90% relapse-free cure rates in FMT treatment for recurrent C. difficile infections. Growing evidence suggests that in addition to bacterial factors, the host immune response during C. difficile infection greatly influences disease severity. In this review, we discuss recent advancements in understanding the interplay between immune cells and the microbiota and how they may relate to recovery from C. difficile through FMT therapy.
Keywords: Gut Microbiota, Fecal Microbiota Transplantation, Immunity, Infection
Commensal dysbiosis is a risk factor for C. difficile infection.
Clostridioides (formerly Clostridium) difficile is a gram-positive, anaerobic, spore forming opportunistic pathogen and is the leading cause of hospital-acquired, antibiotic-associated diarrhea. It is also one of Center for Disease Control’s three most urgent infectious disease threats in the US. C. difficile colonizes the colon, causing inflammation and diarrhea via the expression of its epithelial-damaging toxins[1,2]. Over the past 15 years, C. difficile incidence has tripled and is attributed to the emergence of hypervirulent epidemic strains of C. difficile (referred to as NAP1/027 strains)(see Glossary) [3]. In a single year, C. difficile caused approximately 500,000 infections, $4.8 billion worth of excess health-care costs, and 29,000 deaths occurring in in the US alone[4]. Although a recent healthcare survey demonstrated an overall decline in the prevalence of health-care associated infections from 2011 to 2015, there was no reduction in the high prevalence of C. difficile infections (CDI) during that time[5]. Thus, reducing CDI is a priority, as emphasized in the NIAID’s Antimicrobial Resistance Program in 2014[6].
C. difficile disease is initiated by the depletion of protective commensal microbes commonly through antibiotic exposure[7,8]. Risk of hospital-acquired and community-acquired CDI is highest in individuals with prior exposure to cephalosporins, fluoroquinolones and clindamycin antibiotics[7,8]. Antibiotics can cause long-lasting alterations to the microbial community that normally would provide colonization resistance against C. difficile[9]. A single treatment with clindamycin confers murine susceptibility to infection and induces long-lasting alterations in the composition of the gut microbiome, eliminating ca. 90% of bacteria taxa for up to 28 days[10]. Similarly, in human subjects, a 7 day clindamycin regimen caused long-lasting deficiencies in the microbiota, especially a significant decline in the diversity of Bacteroides that over a span of two years, did not return to its original composition[9]. Furthermore, using next-generation sequencing, additional studies have demonstrated decreases in the diversity and species richness of CDI patients relative to controls[11–13]. Thus, it is clear that commensal dysbiosis is a risk factor for the development of C. difficile colitis.
During this susceptible state of intestinal dysbiosis, C. difficile flourishes in the colon whereby it releases epithelial-damaging toxins. Toxin damage initiates C. difficile diarrheal disease through the killing of colonic host-epithelial cells and breakdown of the gut barrier[14]. The clinical manifestations of C. difficile range from mild to severe diarrhea to life-threatening pseudomembranous colitis. The spectrum of C. difficile disease manifestations is dictated by the virulence factors of the infecting strain, the composition of the microbiome that provides colonization resistance, and the quality of the host inflammatory response (see Clinician’s Corner).
Box 1: Clinician’s Corner.
Diarrhea deaths have increased 4-fold in the last 35 years, despite an overall decrease in deaths due to infection.
The virulent epidemic strain of C. difficile is the leading hospital-acquired infection in North America and the likely reason for this increase.
One in five C. difficile infections relapse.
Fecal microbiota transplantation (FMT) is the most effective treatment for relapsing C. difficile infection.
Understanding the impact of the microbiota on the gut mucosal immune system will advance and ultimately allow for the refinement of FMT for C. difficile and potentially other intestinal diseases.
The importance of FMT therapy to treat C. difficile.
Fecal Microbiota Transplantation (FMT) therapy has had great success in the treatment of recurrent and severe CDI. Standard-of-care treatments for CDI include treatment with antibiotics such as vancomycin and fidaxomicin. However, high rates of recurrence are problematic and occur in up to 30% of patients due to sustained antibiotic-associated dysbiosis[3,15]. While C. difficile is susceptible to antibiotic therapy, the antibiotics used to treat the infection further disrupt the microbiota that provides colonization resistance and immune-mediated protection. Thus, while killing C. difficile, antibiotics also cause greater host susceptibility to reinfection and recurrent disease[16]. FMT combats this cyclical diseased state, by restoring a diverse community of microbes in the colon and restoring intestinal homeostasis (Figure 1).
Figure 1. FMT therapy restores microbial homeostasis and colonization resistance for long-term protection against C. difficile.
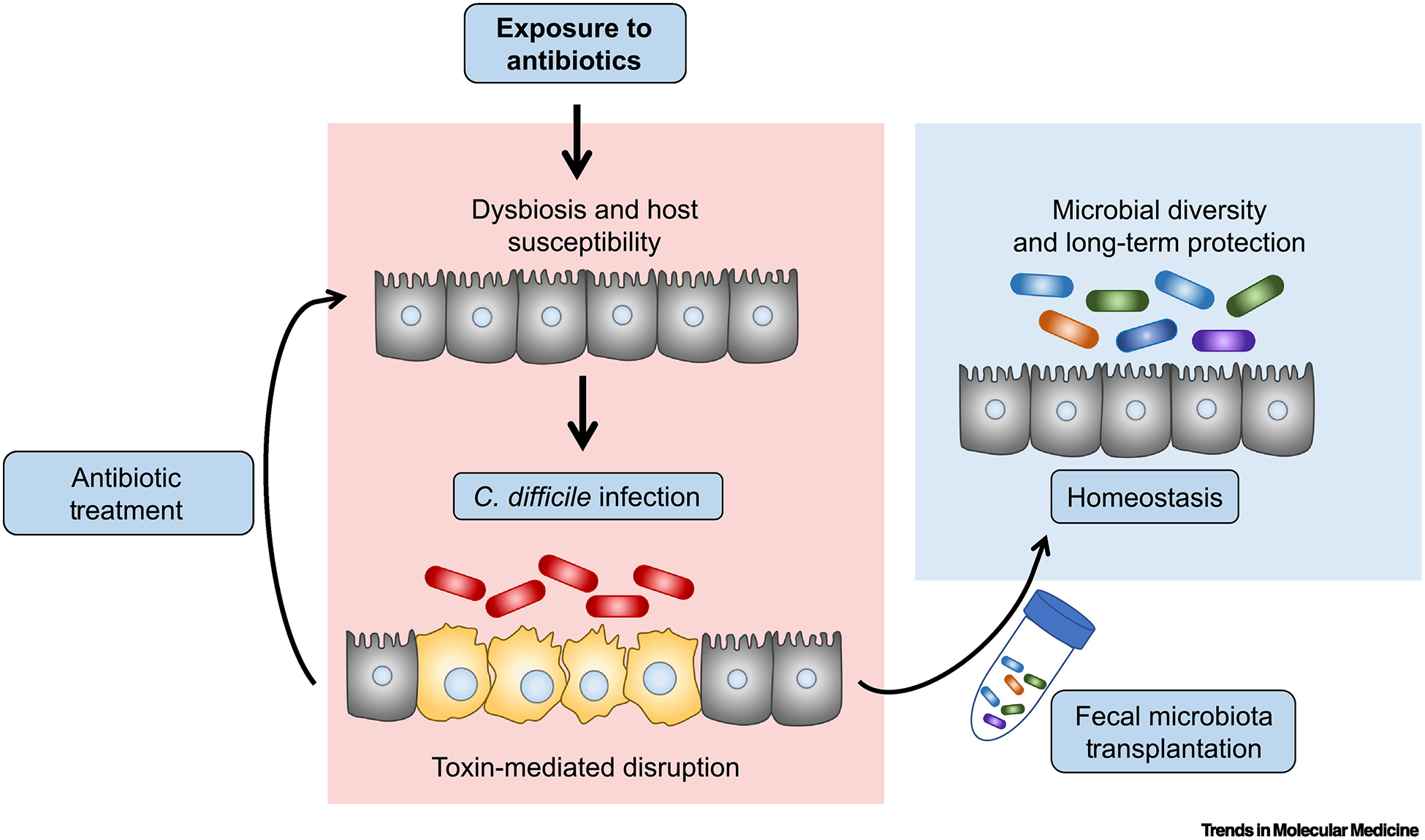
Commensal dysbiosis confers host susceptibility to C. difficile infection (red bacterial cells) and subsequent toxin expression within the gut lumen (left panels). C. difficile toxins initiate disease through killing of colonic host-epithelial cells (yellow cells) and breakdown of the gut barrier[14,15]. Antibiotic treatment of C. difficile maintains host susceptibility to reinfection. Fecal Microbiota Transplantation (FMT) therapy re-establishes commensal diversity and providing long-term protection from relapse (right panel).
Crude FMT therapy was first introduced into modern medicine in 1958 by Eisenmen and colleagues to treat pseudomembranous colitis with fecal enema[17]. Since then, multiple studies have demonstrated the effectiveness of FMT to treat recurrent CDI. However, the first open-label, randomized controlled trial was not completed until 2009 where 41 patients completed treatment with either a donor-feces infusion, standard vancomycin regimen, or standard vancomycin plus bowel lavage. Remarkably, 81.3 % relapse-free C. difficile cure rates were reported 10 weeks after the donor-feces infusion treatment. Furthermore, 2 additional patients were cured after receiving a second donor-feces infusion, resulting in a 94% overall donor infusion cure rate, surpassing conventional vancomycin treatment by 3-fold (NL1135)I [18]. The subsequent idea that a defined mixture of rationally designed, purified commensals, within a healthy stool are sufficient to transfer FMT-mediated protection was initially demonstrated by Tvede & Rask-Madsen in 1989. This initial human study demonstrated that the administration of 10 bacterial strains into 5 patients caused a loss of C. difficile and toxin levels within the infected bowel while replenishing Bacteroides species [19]. After this study, purified bacteriotherapy for CDI was limited and primarily focused on probiotics in combination with standard antibiotic care that had some success [20,21]. A resurgence of interest for rationally designed bacteriotherapy to treat C. difficile occurred in the mid-2000s. A seminal study by Lawley et al, demonstrated that a defined consortium of six bacteria strains (Staphylococcus, Enterococcus, Lactobacillus, Anaerostipes, Bacteroidetes, and Enterorhabdus) could suppress hypervirulent C. difficile 027/BI and promote recovery from antibiotic associated dysbiosis[22]. Another group importantly identified the Lachnospiraceae family of bacteria as a potential therapeutic target for bacteriotherapy[23]. A follow up study demonstrated that pre-colonization of germ-free mice with Lachnospiraceae leads to a dramatic reduction in subsequent C. difficile colonization and mortality after infection[24]. Additionally, administration of Clostridium scindens, an intestinal bacteria whose depletion is associated with more severe disease in humans and mice, was able to restore colonization resistance against CDI in mice and protection is associated with secondary bile acid synthesis[25]. Thus, great advancements have been made to improve our understanding of how defined species of bacteria can combat CDI.
Microbiota-elicited immunity and its importance in combating C. difficile.
While C. difficile toxins drive epithelial disruption and disease during CDI, recently many groups have demonstrated that the type of host immune response generated against C. difficile also dictates the severity of symptoms during infection, independent of bacterial factors[26–30]. The host immune system has a multifaceted role during CDI with some level of inflammation required to clear the infection. However, this inflammation must be tightly regulated to prevent overly robust damage to the host (Figure 2). Protective host defenses during CDI include: neutrophil-mediated bacterial killing; type-1 innate lymphoid cells (ILC1) and granulocyte-mediated bacterial clearance; and tissue-repair via type-2 innate lymphoid cells (ILC2) and eosinophil signaling[31–34]. Pathogenic immune responses include: Toll-like receptor (TLR) 2 activation; Interleukin (IL)-23 and IL-6 signaling; and adaptive type-17 T-cells (TH-17 cells) that further damage host intestinal tissue during infection[35–38]. Thus, the type of host immune response elicited during CDI plays an important role in dictating disease severity. Given the rise of FMT therapy to treat severe CDI and efforts to define specific, purified, commensals that elicit protection from disease, it is increasingly important to understand how members of the microbiota or their microbial-derived products influence the type of host-immune response generated during CDI[18,39]. In the subsequent sections of this review, mechanisms of host immunity that influence CDI recovery and their regulation by commensals are discussed.
Figure 2: Activation of the immune system during C. difficile infection.
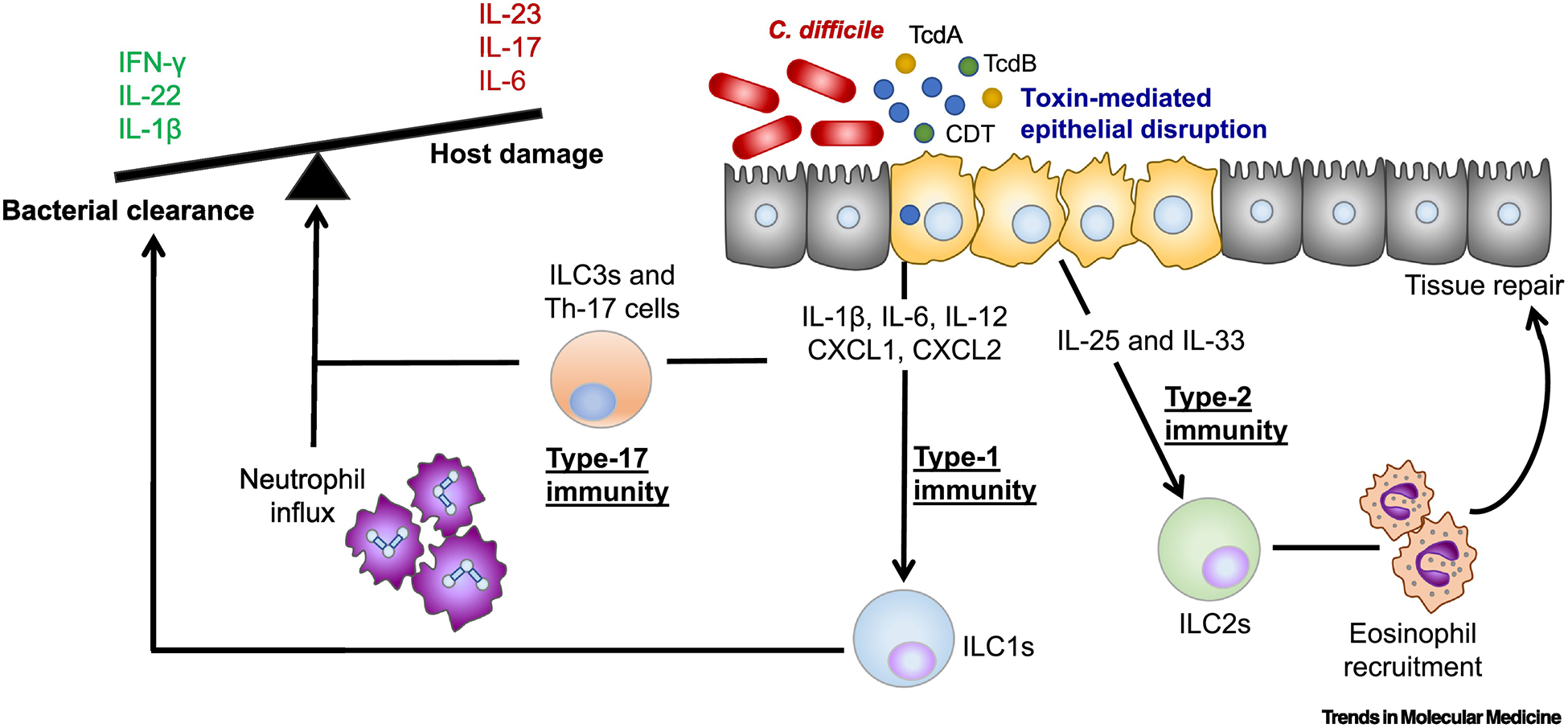
A schematic of the host immune defenses that influence disease severity during C. difficile infection. C. difficile drives epithelial disruption in the colon through toxin release. IFN-γ+ innate lymphoid cells (ILCs) promote recovery likely via downstream phagocytic mechanisms. Type-17 T cells (TH-17) cells elicited after previous gut barrier insult drive more severe inflammation and mortality in murine models of infection via IL-17 release. Neutrophils are required for bacterial clearance and survival from infection. However, an overly robust neutrophilic response is associated with more severe disease. Type-2 cytokines, IL-25 and IL-33 drive tissue repair mechanisms to protect from overly robust damage to the epithelium activating eosinophils during infection. Abbreviations, TcdA, Toxin A; TcdB, Toxin B; CDT, binary toxin.
Type-1 immunity drives CDI recovery and is regulated by the microbiota.
Type-1 immunity is typically characterized by expression of the cytokines Interferon gamma (IFN-γ), IL-12, and IL-18 which canonically have anti-viral functions. Type-1 associated cytokines, IFN-γ and IL-12, are upregulated in response to CDI in humans and mice and are critical for driving disease recovery[40–42]. Mice lacking ILCs have increased mortality during CDI and lack the ability to upregulate IFN-γ and IL-22[41,43]. Further investigation revealed that IFN-γ+ ILC1s were central to survival during CDI with a very limited role for IL-22+ ILC3s[41]. This important role for IFN-γ+ ILC1s was corroborated in humans, demonstrating that high levels of IFN-γ in the serum of CDI-patients is associated with less severe infection[29]. How IFN-γ protects during CDI requires further investigation but it may act by increasing phagocytic functions during infection.
Interestingly, type-1 immunity is regulated by commensal-derived signals[41]. For example, the commensal Bacteroides fragilis (B.fragilis) contributes to the maintenance of CD4+ T cell development, TH-1 differentiation, and lymphoid organogenesis through the production and subsequent immunological presentation of the zwitterionic polysaccharide PSA[44]. Within the TH-2/TH-1 imbalanced germ-free setting, colonization with PSA producing B. fragilis restores splenic IFN-y+ CD4+ T cells via antigenic presentation in a IL-12/STAT4 specific manner. In addition to adaptive TH-1 cells, innate NK cell function is also impaired in the absence of the microbiota. NK cells are cytotoxic type-1 innate lymphoid cells that require priming by mononuclear phagocytes in order to function[45]. Germ-free mice have defective type-1 interferon production from mononuclear phagocytes and thus lack primed IFNy+ NK cells[46]. As type-1 immunity is critical for recovery from C. difficile, further investigations are required to understand how FMT-derived commensals may contribute to C. difficile pathogenesis via modulation of type-1 interferon and IFNy+ signaling.
The microbiota maintains protective type-2 polarizing cytokines in the intestine.
Recent studies demonstrate that type-2 immune responses play an important role in recovery and survival during CDI[30,33,35]. Eosinophils, a type-2 associated myeloid subset, have protective roles in both human and murine studies of C. difficile colitis[30,33,35]. CDT toxin expressed by epidemic NAP1/027 isolates can kill protective eosinophils during infection, contributing to their heightened virulence[35]. IL-25, IL-33 and IL-5 are type 2 cytokines that contribute to eosinophil accumulation in the intestine, and are associated with survival and less severe disease during CDI[29,33,34,47]. IL-33 promotes ILC2 activation and subsequent eosinophilia during CDI and is essential for tissue repair and survival[34]. The maintenance of protective IL-33 protein in the colon was critically dependent upon the microbiota and could be rescued with FMT therapy[34]. Although microbiota-dependent IL-25 and IL-33 are important activators of ILC2s, ILC2 seeding of both the lung and intestine occurs independently of the microbiota, as germ-free mice have comparable ILC2s numbers[32,48]. While the microbiota does not dictate the seeding of intestinal ILC2s, it may have an important role in promoting ILC2 function. In line with this idea, human ILC2s express TLRs and can directly sense microbial-associated molecular patterns (MAMPs) resulting in increased expression of IL-5 and IL-13[49]. Furthermore, during a model of intestinal colitis, germ-free mice had reduced expression of type-2 cytokines, IL-4, IL-5, IL-13, and IL-33 in addition to lack of eosinophils which could be rescued by FMT therapy[50]. Given recent findings that IL-25 elicited eosinophils and IL-33 elicited ILC2s are protective during C. difficile, further studies are required to understand the connection between FMT regulation and possible restoration of protective type-2 immune function.
Commensal regulated Type-17 immunity is a double-edged sword during CDI
Type-17 associated cytokines, IL-1β, IL-23, IL-17a, IL-22 and IL-6 are upregulated in the gut in response to C. difficile infection[29,37]. Both pathogenic and protective outcomes are associated with type-17 immunity during C. difficile colitis. IL-23 signaling is pathogenic during CDI, contributing to increased mortality and tissue pathology, whereas IL-22 contributes to survival and systemic pathobiont elimination through the complement system [37,38,51]. The type-17 cytokine network is a key driver of neutrophil production and accumulation during mucosal infections. In accordance, neutrophils are characteristically recruited at high levels into the colon during C. difficile colitis via MyD88 signaling[31,52]. Neutrophils have important anti-microbial functions during infection through their phagocytic activity, granule formation, and production of antimicrobial proteases, peptides, and reactive oxygen species[52]. However, dual roles for neutrophils have been reported during C. difficile colitis. Complete depletion of neutrophils with anti-GR1 antibody increases CDI-associated mortality; however inhibition of neutrophil recruitment through blockage of CD18 or MIP-2 or inhibition of IL-23 reduces tissue pathology and mortality during infection[31,37,38,53,54]. Similarly in human patients, high neutrophil counts are associated with more severe disease whereas neutropenia is a risk factor during infection[55,56]. Thus, type-17 associated immunity is essential for pathogen elimination, however if left unregulated, can contribute to profound tissue damage during infection.
The maintenance of a robust and effective type-17 immunity is critically dependent upon commensal-derived signals from the microbiota. For example, adaptive type-17 T-cell (TH-17) differentiation is impaired in the small intestine of germ-free mice and rescued with fecal transplant[57]. The commensal organism segmented filamentous bacteria (SFB) drives TH-17 differentiation in the gut and can aggravate colitis and other auto-immune diseases[58–60]. SFB makes direct contact with intestinal epithelial cells, activating a IL-22/IL-23 dependent circuit between ILC3s, epithelial cells, and TH-17 cells[61,62]. In addition to adaptive TH-17 cells, the microbiota can additionally shape the function of type-3 innate lymphoid cells (ILC3s). Microbiota-induced TNF-like ligand 1A (TL1A) released from CX3CR1+ mononuclear phagocytes can increase the production of IL-22 by ILC3s[63]. Thus, FMT-derived colonization of certain commensals may contribute to CDI-associated disease depending on their alterations of type-17 dynamics.
Certain microbes can elicit immunosuppression after colonization of the intestine.
In addition to mounting anti-microbial defenses, the microbiota is also essential for mounting immune suppression. One of the prime examples of this is the induction of regulatory Foxp3+ T cells (T-regs) by B. fragilis and Clostridia commensals. B. fragilis-derived PSA inhibits IL-17 production in the intestine by enhancing the production of anti-inflammatory IL-10 and T-regs to protect from experimental colitis via TLR2 signaling[64]. Thus, in addition to its role in maintaining a balanced TH2/TH1 ratio, B. fragilis-derived PSA also supports intestinal T-regs. Interestingly, in this specific pathogen-free experimental setting, B. fragilis did not increase inflammatory TH-1 (IFNy+) CD4+ T cells in gut tissue as had previously been demonstrated in spleens of germ-free mice[44]. This indicates that the same commensal-derived molecule may have opposing functions depending on compartmentalized tissue specific microenvironments and the endogenous microbiota. Commensal Clostridia also contribute to IL-10+ T-reg accumulation in the gut by increasing the expression of TGF-β from intestinal epithelial cells[65]. Additionally, a consortia of 17 Clostridia strains, falling within clusters IV, XIVa and XVIII, were rationally identified from a human fecal sample based on their T-reg inducing activity, and could attenuate colitis, quantified by a reduction in colon tissue pathology and diarrheal scores, upon administration to mice[66]. Therefore, sensing of the microbiota additionally maintains homeostasis through immune suppression, preventing an overly robust immune response to host-proximal commensals.
Recently, experimental evidence has linked the effectiveness of FMT for colitis with immunosuppression. FMT therapeutically controlled gut inflammation and colitis via the induction of IL-10 and TGF-β, cytokines critical for T-reg accumulation in the intestine[67,68]. Whether FMT-directed immunosuppression aids in the recovery from C. difficile requires further investigation. This area of research is of utmost importance as patients with recurrent CDI have increased pro-inflammatory IL-17+ or IFNY+ CD4+ T cells circulating in their peripheral blood[69] and additionally, TH-17 cells are both necessary and sufficient to enhance the severity of CDI in settings of prior gut insult like IBD colitis[36]. Thus, FMT-induced IL-10+ T-regs could plausibly be involved in the inhibition of pathogenic TH-17 cells during recurrent CDI or in patients with prior exposure to gut commensals such as those with IBD or Crohns disease.
Commensal derived signals that orchestrate host immunity.
Commensal bacteria are dynamic- their nutrient acquisition, production of metabolites and MAMPSs, and expression of anti-microbial factors are all important mechanisms of colonization resistance against C. difficile. The microbial-microbial interactions by which commensals and FMT therapy limit C. difficile growth have been outlined beautifully in other review articles[16,70]. Here we focus on how signals from the microbiota interact with the host immune system to possibly confer additional protection during C. difficile colitis (Figure 3).
Figure 3. C. difficile relevant host immune responses are impacted by the presence of the microbiota.
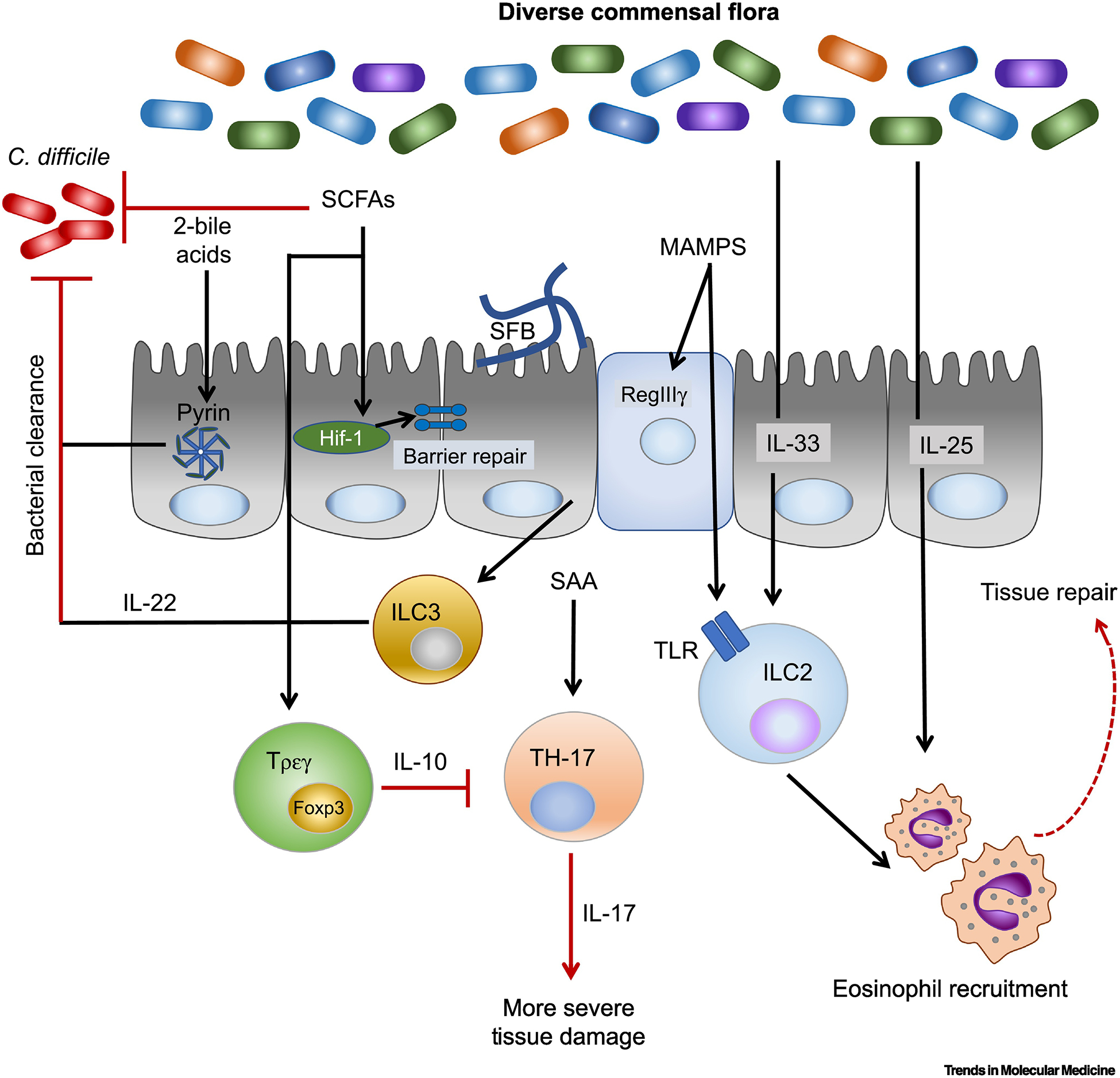
A diverse microflora protects from C. difficile infection. Commensals that produce secondary bile acids and short chain fatty acids (SCFA) can inhibit C. difficile growth through bacterial competition. Commensals also promote disease-limiting immune responses. Secondary bile acids analogs can activate the pyrin inflammasome which is important for anti-microbial immune defenses against toxins. SCFAs promote epithelial barrier tight-junction through butyrate stabilization of HIF-1. The SCFAs butyrate and propionate aid in regulatory T-cells (T-regs) generation via histone deacetylase inhibition. T-reg mediated IL-10 expression can suppress pathogenic TH-17 cells. TH-17 cells increase C. difficile disease severity in cases of prior gut insult and are promoted by the gut commensal, segmented filamentous bacteria (SFB). ILC2s are protective from C. difficile infection. Human ILC2s express toll-like receptors (TLRs) that can sense microbiota-associated molecular patterns (MAMPS). ILC2 activating cytokines, IL-33 and IL-25, prevent mortality during murine models of C. difficile colitis and their expression are maintained by the presence of a diverse microbiota.
SCFAs affect the fitness of C. difficile and also promote host epithelial regeneration and immunoregulation.
Breakdown of carbohydrates provides an energy source for commensals, whereas antibiotic treatment alters the balance of commensal fermenters, leading to reduced short-chain fatty acid (SCFA) production[71,72]. Lawley et al. demonstrated that in the presence of hypervirulent C. difficile, the microbiota of infected mice was marked by low diversity and reductions in SCFAs, with most notable reductions in acetate and butyrate[22]. Similarly, an additional study characterized the metabolome of susceptible, antibiotic-treated mice and demonstrated reduced fermenting commensal activity with increases in sugar alcohols and carbohydrates and reciprocal reductions in SCFAs[73]. Aligning with these studies, 16S rRNA sequencing of the human flora demonstrated reductions in the families Ruminococcaceae, Lachnospiraceae, and butyrate-producing C2 to C4 anaerobic fermenters in human CDI patients[12]. Finally, a recent study demonstrated that microbiota-accessible carbohydrate (MAC) diets caused outgrowth of MAC-utilizing bacteria (eg. Bacteroides). This outgrowth was associated with increased fermentation end products acetate, propionate, and butyrate, as well as decreased C. difficile fitness in vivo[74,75].
While SCFAs have effects on C. difficile fitness, they are also important communicators with the intestinal immune system, being sensed by epithelial cells and immune cells via G-protein coupled receptors, GPR41 and GPR43; and by maintaining the transcription of many cytokine and chemokines within the intestine[76]. Recently it was demonstrated that butyrate protected from C. difficile colitis by increasing epithelial tight junctions via the transcription factor HIF-1 and independent of bacterial burden[77]. Thus, restoration of SCFA pools with FMT therapy may act in concert with anti-C. difficile mechanisms to promote epithelial barrier and cytokine defenses.
Lastly, the SCFAs butyrate and propionate limit inflammatory responses in the intestine by supporting peripheral T-reg development via their histone deacetylase (HDAC) activity[78–80]. The direct addition of SCFAs to ex vivo T-cell cultures inhibits TH-17 differentiation and IL-6 production, while increasing T-reg differentiation [81]. Thus, SCFAs dampen overactive immune responses and suppress colitis through their regulation of the intestinal T-reg:TH-17 ratio. Given recent findings that CD4+ TH-17 cells drive severe C. difficile disease after a previous gut insult, it is possible that SCFAs additionally provide protection during relapsing C. difficile through modulation of the intestinal T-reg:TH-17 balance, and preventing an overactive immune response to host-proximal commensals[36].
FMT-restored secondary bile acids interact with host inflammasome defenses.
Bile acids are produced in the liver and metabolized by the microbiota into secondary forms. Secondary bile acids deoxycholate and lithocholate are inhibitors of C. difficile growth and associated with CDI protection whereas the primary bile acids taurocholate and cholate induce spore germination[25,82–84]. Human patients with recurrent CDI had high levels of primary bile acids yet were completely deficient in secondary bile salts prior to FMT therapy[85]. FMT therapy can restore bile-acid metabolizing microbiota, rescuing protective levels of secondary bile acids[85].
While bile acids have direct implications on C. difficile growth, they also have important implications on host immune defenses mediated by the inflammasome. The pyrin inflammasome sensor is activated by the Rho-Glucosylating activity of C. difficile toxins[86]. The role of the pyrin inflammasome sensor in vivo is less clear as Pyrin-deficient mice have no significant differences in C. difficile disease, whereas mice deficient in apoptosis executioner caspases have delayed recovery during infection[87]. That being said, inflammasome signaling is indeed essential for recovery from acute CDI, as complete abolishment of the inflammasome complex in ASC−/− mice or inhibition of caspase-1 in vivo causes severe mortality during infection, higher bacterial burden and impaired CXCL1-mediated neutrophil chemotaxis[88,89]. Interestingly, when putative secondary bile acids were screened for inflammasome activity, 2 deoxy-cholic acid (DCA) bile acid analogs caused IL-1β and IL-18 release from both epithelial cells and myeloid cells via activation of the pyrin inflammasome[90]. Furthermore, another study demonstrated that multiple secondary and primary bile acids had dose-inhibitory activity on the NLRP3 inflammasome with the secondary bile acid lithocholic acid (LCA) having the greatest anti-IL-1β and caspase-1 inhibition[91]. Thus, the bile acid pool transferred by FMT therapy may have profound effects on inflammasome defenses against C. difficile and should be considering when screening for protective rationally-designed cocktails.
Microbial-associated molecular patterns promote immune health.
The expression of pattern recognition receptors (PRRs) such as TLRs and NOD receptors and their subsequent sensing of microbial-derived patterns is not restricted to pathogen encounter. In contrast, PRRs are expressed in the intestinal epithelium at steady-state and are thus uniquely positioned to sense and interact with the luminal intestinal microbiota[92]. In fact, the interaction between PRRs and commensal-derived signals is essential for maintaining intestinal symbiosis and immune function[93,94]. For example, sensing of commensal-derived signals by intestinal TLR-MyD88 signaling protects mice from chemically induced DSS colitis [93,94]. Microbiota-depleted mice displayed increased mortality, colonic bleeding, and intestinal epithelial injury that was rescued by oral administration of the commensal bacterial product, LPS, in a TLR4 dependent manner. Additionally, TLR5-mediated sensing of microbiota-derived flagellin by dendritic cells increases the production of the anti-microbial C-type lectin, RegIIIy, by epithelial cells. Antibiotic-mediated downregulation of RegIIIγ leads to reduced killing of Vancomycin Resistant Enterococcus (VRE)[95,96]. RegIIIy has antimicrobial activity against gram-positive bacteria and future studies should test requirement of RegIIIy induction by FMT in the setting of CDI.
Commensal sensing by PRRs is also critical for maintaining neutrophil defenses. Microbiota-derived peptidoglycan enhances neutrophil killing of the pathogens Streptococcus pneumoniae and Staphylococcus aureus[97]. Recognition of peptidoglycan by the pattern recognition receptor, NOD1, was sufficient to restore neutrophil function[97]. Thus, PRRs may be important downstream targets of FMT-mediated efforts to restore gut homeostasis during CDI. Interestingly, mice lacking the pattern-recognition receptors TLR4, MYD88, and NOD-1 are more susceptible to CDI and have deficiencies in their granulocyte responses. However whether a lack of host/microbiota immune development contributes to disease severity has not been addressed[31,97,98]. Further in vivo studies of FMT therapy to treat C. difficile in the context of genetic ablation of TLRs or NLRs will be critical to understand whether these sensors play an important role in FMT-mediated recovery.
Concluding Remarks
Many recent studies have refined our approach and application of FMT therapy in the clinic. Freezing of FMT material enables many advances such as pre-screening, banking, and re-testing of donor stool in addition to the possibility of delivery to other hospitals. An additional advance in making FMT therapy more accessible is the demonstration that oral FMT capsules are an effective way to administer donor material relative to other procedures such as enemas[99]. The ideal gold-standard FMT treatment would be the identification of a defined, purified, microbial cocktail that is sufficient to transfer FMT protection.
Likely, there is not just one commensal or one community architecture that contributes to colonization resistance, but multiple community structures exist that can transfer protection from C. difficile colitis. Given the vast complexity of the microbiota, its expansive influence on the metabolic state of the gut, and its numerous interactions with the mucosal interface and the host immune response, it is likely that not one magic bullet exists to combat CDI. This idea is supported by a recent study demonstrating that multiple CDI-resistant states of the microbiota exist with differing community structures however their functional outputs are similar[73]. Further studies defining mechanistic and functional outputs of commensals, such as their metabolomes and their interactions with the host, are key to the advancement of purified, next-generation bacteriotherapy to treat both C. difficile and other diarrheal diseases (see Outstanding Questions box).
Outstanding Questions:
Research has mostly focused on antimicrobial mechanisms by which FMT protects during C. difficile infection. Will targeting of tissue-recovery pathways using defined microbes add additional value when designing rational bacteriotherapies?
Can host cytokine biomarkers improve the clinical decision-making of treatment with antibiotics vs. FMT therapy?
In the context of FMT to treat C. difficile, do microbe-derived metabolites such as SCFAs and secondary bile acids improve disease severity through additional modulation of the host immune defenses?
How can microbes be screened for immune-stimulating or immunosuppressive activity robustly and quickly, to aid in rational design of next-generation bacteriotherapy?
Towards the goal of refining FMT therapy, and defining a consortium of commensals that could be produced under good manufacturing practices, multiple factors should be taken into consideration: limited antibiotic-resistance; durability in passing through the gastrointestinal tract; engraftment stability; and anti-C. difficile competition (e.g. secondary bile acid production). While reducing C. difficile growth via competition is of key importance, the work outlined in this review indicates that the additional ability of microbes to modulate the immune system should be considered as well. Thus, work to identify microbes that increase protective immune signaling within the intestine will add an important layer of host-protection into next-generation bacteriotherapy in the face of C. difficile colitis.
Highlights:
Fecal Microbiota Transplantation (FMT) is highly effective at the treatment of recurrent and severe C.difficile infection (CDI), with 90% relapse-free cure rates reported.
The immune response is a key driver of recovery and pathogenesis during CDI. Antimicrobial defenses including the inflammasome, MYD88-activated neutrophils, and IFNy+ ILC1s are essential for killing invading C.difficile and other pathobionts during infection.
Overly robust inflammation via IL-23, TLR2 signaling, and TH-17 cells increase tissue damage and can lead to more severe tissue damage during CDI.
Type-2 barrier cytokines IL-33 and IL-25 aid in repair of toxin-mediated epithelial damage via intestinal eosinophil and ILC2 activation.
The microbiota impacts various immune pathways that aid in recovery from C.difficile colitis, thus underscoring the importance of monitoring the immune responses before and after FMT therapy.
Glossary:
- NAP1/B1/027
Epidemic, hypervirulent C. difficile strain associated with increased severity and rates of C. difficile infections and linked with global outbreaks over the past 15 years.
- Commensal
bacteria co-existing in a non-harmful and mutualistic manner with host.
- Next-generation sequencing
modernized high-throughput sequencing technologies allowing for quicker and less expensive sequencing of DNA and RNA.
- Dysbiosis
an imbalance of the microbiome that disrupts the symbiotic balance between host and host-resident commensal organisms.
- Fecal Microbiota Transplant (FMT)
procedure whereby healthy donor stool material is delivered into the gastrointestinal tract of a recipient with the aim to restore health.
- Homeostasis
Healthy and balanced equilibrium reached between the microbiota and host.
- Recurrence in C. difficile infection
Failure of initial antibiotic treatment to cure C. difficile resulting in relapse or reinfection of patient.
- Innate Lymphoid Cells (ILCs)
Lymphoid cells lacking antigen-specific receptors that have important effector and regulatory functions during the innate immune response. ILCs are categorized into three groups based on their function, cytokine profile, and transcription factor expression.
- Toll-like receptor (TLR)
a class of proteins that sense conserved structural motifs expressed by micro-organisms and subsequently stimulate the host-immune system.
- Germ-free mice
mice raised in isolators so that they are devoid of any microorganisms.
- Microbe-associated molecule patterns (MAMPs)
Microbe-derived molecules that share patterns or structural motifs which can be recognized by the immune system.
- Short-Chain Fatty Acids (SCFA)
Metabolites derived from bacterial fermentation of dietary fiber that have essential roles in signaling to the immune system.
- 16S rRNA sequencing
Method used for bacterial identification and taxonomy through detection of sequence differences within the hypervariable regions of the 16S rRNA gene.
- Microbiota-accessible carbohydrate (MAC)
non-digestible carbohydrates that are metabolized by the microbiota.
- Bile Acid
End products of cholesterol metabolism in the liver.
- Inflammasome
A multiprotein intracellular complex that orchestrates pro-inflammatory IL-1β and IL-18 activation and release. Activation of the inflammasome complex requires assembly of three essential proteins: 1) An inflammasome sensor containing a pyrin domain 2) the adaptor protein ASC (apoptosis-associated speck-like protein) and 3) the protease caspase-1.
- Pyrin
an intracellular innate immune sensor that triggers inflammasome activation after sensing C. difficile toxin-mediated inactivation of RhoA GTPases.
- NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3)
an intracellular innate immune sensor that triggers inflammasome activation after sensing of cellular stress.
- NOD receptor (Nucleotide-binding oligomerization domain-containing protein)
Cytosolic pattern-recognition receptors that recognize peptidoglycan constituents of bacteria leading to immune activation.
- RegIIIy (Regenerating islet-derived protein 3 gamma)
An antimicrobial peptide produced by intestinal Paneth cells that provides defense against Gram-positive bacteria.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: Disclaimer Statement:
WP is a consultant for TechLab, Inc. which manufactures diagnostic tests for CDI. The authors declare no other competing interests.
Resources:
References
- 1.Bartlett JG (2006) Annals of Internal Medicine Review Narrative Review : The New Epidemic of Clostridium difficile –. An. Intern. Med [DOI] [PubMed] [Google Scholar]
- 2.Rupnik M et al. (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol 7, 526–36 [DOI] [PubMed] [Google Scholar]
- 3.Kelly CP and LaMont JT (2008) Clostridium difficile--more difficult than ever. N. Engl. J. Med 359, 1932–1940 [DOI] [PubMed] [Google Scholar]
- 4.Lessa FC et al. (2015) Burden of Clostridium difficile Infection in the United States. N. Engl. J. Med DOI: 10.1056/NEJMoa1408913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magill SS et al. (2018) Changes in Prevalence of Health Care–Associated Infections in U.S. Hospitals. N. Engl. J. Med DOI: 10.1056/NEJMoa1801550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Institute of Allergy and Infectious Diseases (2014) NIAID ‘ s Antibacterial Resistance Program : Current Status and Future Directions. at <https://www.niaid.nih.gov/sites/default/files/arstrategicplan2014.pdf>
- 7.Deshpande A et al. (2013) Community-associated clostridium difficile infection antibiotics: A meta-analysis. J. Antimicrob. Chemother DOI: 10.1093/jac/dkt129 [DOI] [PubMed] [Google Scholar]
- 8.Slimings C and Riley TV Antibiotics and hospital-acquired Clostridium difficile infection: Update of systematic review and meta-analysis., Journal of Antimicrobial Chemotherapy. (2014) [DOI] [PubMed] [Google Scholar]
- 9.Jernberg C et al. (2010) Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology DOI: 10.1099/mic.0.040618-0 [DOI] [PubMed] [Google Scholar]
- 10.Buffie CG et al. (2012) Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun DOI: 10.1128/IAI.05496-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu S et al. (2016) Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. DOI: 10.1016/j.micinf.2015.09.008 [DOI] [PubMed] [Google Scholar]
- 12.Antharam VC et al. (2013) Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol DOI: 10.1128/JCM.00845-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang JY et al. (2008) Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis DOI: 10.1086/525047 [DOI] [PubMed] [Google Scholar]
- 14.Rupnik M et al. (2009) Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Microbiol DOI: 10.1038/nrmicro2164 [DOI] [PubMed] [Google Scholar]
- 15.Shields K et al. (2015) Recurrent clostridium difficile infection: From colonization to cure. Anaerobe DOI: 10.1016/j.anaerobe.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buffie CG and Pamer EG (2013) Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol DOI: 10.1038/nri3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenman B, Silen W, Bascom GS, K. A (1958) Fecal enema as an adjunct in the treatment of pseudomembranous. Surgery DOI: 10.1067/S0039-6060(03)00474-4 [DOI] [PubMed] [Google Scholar]
- 18.van Nood E et al. (2013) Duodenal Infusion of Donor Feces for Recurrent Clostridium difficile. N. Engl. J. Med DOI: 10.1056/NEJMoa1205037 [DOI] [PubMed] [Google Scholar]
- 19.Tvede M and Rask-Madsen J (1989) Bacteritherapy for chronic relapsing Clostridium difficile diarrhea in six patients. Lancet DOI: 10.1016/S0140-6736(89)92749-9 [DOI] [PubMed] [Google Scholar]
- 20.Surawicz CM et al. (2000) The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin. Infect. Dis DOI: 10.1086/318130 [DOI] [PubMed] [Google Scholar]
- 21.McFarland LV (2009) Evidence-based review of probiotics for antibiotic-associated diarrhea and Clostridium difficile infections. Anaerobe DOI: 10.1016/j.anaerobe.2009.09.002 [DOI] [PubMed] [Google Scholar]
- 22.Lawley TD et al. (2012) Targeted Restoration of the Intestinal Microbiota with a Simple, Defined Bacteriotherapy Resolves Relapsing Clostridium difficile Disease in Mice. PLoS Pathog. DOI: 10.1371/journal.ppat.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeves AE et al. (2011) The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes DOI: 10.4161/gmic.2.3.16333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeves AE et al. (2012) Suppression of Clostridium difficile in the Gastrointestinal Tracts of Germfree Mice Inoculated with a Murine Isolate from the Family Lachnospiraceae. Infect. Immun DOI: 10.1128/iai.00647-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buffie CG et al. (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature DOI: 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steiner TS et al. (1997) Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin. Diagn. Lab. Immunol 4, 719–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang ZD et al. (2006) A common polymorphism in the interleukin 8 gene promoter is associated with Clostridium difficile diarrhea. Am. J. Gastroenterol 101, 1112–1116 [DOI] [PubMed] [Google Scholar]
- 28.El Feghaly RE et al. (2013) Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in clostridium difficile infection. Clin. Infect. Dis DOI: 10.1093/cid/cit147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu H et al. (2017) Cytokines are markers of the Clostridium difficile-induced inflammatory response and predict disease severity. Clin. Vaccine Immunol DOI: 10.1128/CVI.00037-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulaylat AS et al. (2018) Development and Validation of a Prediction Model for Mortality and Adverse Outcomes among Patients with Peripheral Eosinopenia on Admission for Clostridium difficile Infection. JAMA Surg. DOI: 10.1001/jamasurg.2018.3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jarchum I et al. (2012) Critical role for myd88-Mediated Neutrophil recruitment during Clostridium difficile colitis. Infect. Immun 80, 2989–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monticelli LA et al. (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol DOI: 10.1038/ni.2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buonomo EL et al. (2016) Microbiota-Regulated IL-25 Increases Eosinophil Number to Provide Protection during Clostridium difficile Infection. Cell Rep. DOI: 10.1016/j.celrep.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frisbee AL et al. (2019) IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun DOI: 10.1038/s41467-019-10733-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowardin CA et al. (2016) The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat. Microbiol DOI: 10.1038/nmicrobiol.2016.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saleh MM et al. (2019) Colitis-Induced Th17 Cells Increase the Risk for Severe Subsequent Clostridium difficile Infection. Cell Host Microbe DOI: 10.1016/j.chom.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buonomo EL et al. (2013) Role of IL-23 signaling in Clostridium difficile Colitis. J. Infect. Dis DOI: 10.1093/infdis/jit277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDermott AJ et al. (2016) Interleukin-23 (IL-23), independent of IL-17 and IL-22, drives neutrophil recruitment and innate inflammation during Clostridium difficile colitis in mice. Immunology 147, 114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanna S et al. (2016) A Novel Microbiome Therapeutic Increases Gut Microbial Diversity and Prevents Recurrent Clostridium difficile Infection. J. Infect. Dis DOI: 10.1093/infdis/jiv766 [DOI] [PubMed] [Google Scholar]
- 40.Jafari NV et al. (2013) Clostridium difficile Modulates Host Innate Immunity via Toxin-Independent and Dependent Mechanism(s). PLoS One DOI: 10.1371/journal.pone.0069846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abt MC et al. (2015) Innate immune defenses mediated by two ilc subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe 18, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishida Y et al. (2004) Essential Involvement of IFN-in Clostridium difficile Toxin A- Induced Enteritis. J. Immunol DOI: 10.4049/jimmunol.172.5.3018 [DOI] [PubMed] [Google Scholar]
- 43.Xu W et al. (2015) NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep. 10, 2043–2054 [DOI] [PubMed] [Google Scholar]
- 44.Mazmanian SK et al. (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell DOI: 10.1016/j.cell.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 45.Lucas M et al. (2007) Dendritic Cells Prime Natural Killer Cells by trans-Presenting Interleukin 15. Immunity DOI: 10.1016/j.immuni.2007.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganal SC et al. (2012) Priming of Natural Killer Cells by Nonmucosal Mononuclear Phagocytes Requires Instructive Signals from Commensal Microbiota. Immunity DOI: 10.1016/j.immuni.2012.05.020 [DOI] [PubMed] [Google Scholar]
- 47.Nussbaum JC et al. (2013) Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider C et al. (2018), A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling., Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maggi L et al. (2017) Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J. Allergy Clin. Immunol DOI: 10.1016/j.jaci.2016.06.032 [DOI] [PubMed] [Google Scholar]
- 50.De Salvo C et al. (2016) IL-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am. J. Pathol DOI: 10.1016/j.ajpath.2015.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasegawa M et al. (2014) Interleukin-22 Regulates the Complement System to Promote Resistance against Pathobionts after Pathogen-Induced Intestinal Damage. Immunity DOI: 10.1016/j.immuni.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jose S and Madan R (2016) Neutrophil-mediated inflammation in the pathogenesis of Clostridium difficile infections. Anaerobe DOI: 10.1016/j.anaerobe.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castagliuolo I et al. (1998) Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J. Immunol DOI: 10.4049/jimmunol.172.5.3018 [DOI] [PubMed] [Google Scholar]
- 54.Kelly CP et al. (1994) Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J. Clin. Invest DOI: 10.1172/JCI117080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoon YK et al. (2014) Predictors of mortality attributable to Clostridium difficile infection in patients with underlying malignancy. Support. Care Cancer DOI: 10.1007/s00520-014-2174-7 [DOI] [PubMed] [Google Scholar]
- 56.Solomon K et al. (2013) Mortality in patients with Clostridium difficile infection correlates with host pro-inflammatory and humoral immune responses. J. Med. Microbiol DOI: 10.1099/jmm.0.058479-0 [DOI] [PubMed] [Google Scholar]
- 57.Ivanov II et al. (2008) Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe DOI: 10.1016/j.chom.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ivanov II et al. (2009) Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell DOI: 10.1016/j.cell.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaboriau-Routhiau V et al. (2009) The Key Role of Segmented Filamentous Bacteria in the Coordinated Maturation of Gut Helper T Cell Responses. Immunity DOI: 10.1016/j.immuni.2009.08.020 [DOI] [PubMed] [Google Scholar]
- 60.Lee YK et al. (2010) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci DOI: 10.1073/pnas.1000082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Atarashi K et al. (2015) Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell DOI: 10.1016/j.cell.2015.08.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sano T et al. (2015) An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell DOI: 10.1016/j.cell.2015.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castellanos JG et al. (2018) Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity DOI: 10.1016/j.immuni.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mazmanian SK et al. (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature DOI: 10.1038/nature07008 [DOI] [PubMed] [Google Scholar]
- 65.Atarashi K et al. (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science (80-.) DOI: 10.1126/science.1198469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Atarashi K et al. (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature DOI: 10.1038/nature12331 [DOI] [PubMed] [Google Scholar]
- 67.Burrello C et al. (2018) Therapeutic faecal microbiota transplantation controls intestinal inflammation through IL10 secretion by immune cells. Nat. Commun DOI: 10.1038/s41467-018-07359-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei YL et al. (2018) Fecal microbiota transplantation ameliorates experimentally induced colitis in mice by upregulating AhR. Front. Microbiol DOI: 10.3389/fmicb.2018.01921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yacyshyn MB et al. (2014) Clostridium difficile recurrence is characterized by pro-inflammatory peripheral blood mononuclear cell (PBMC) phenotype. J. Med. Microbiol DOI: 10.1099/jmm.0.075382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Britton RA and Young VB (2014) Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology DOI: 10.1053/j.gastro.2014.01.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hove H et al. (1996) Antibiotic-associated diarrhoea, Clostridium difficile, and short-chain fatty acids. Scand. J. Gastroenterol DOI: 10.3109/00365529609009151 [DOI] [PubMed] [Google Scholar]
- 72.Høverstad T et al. (1986) Influence of ampicillin, clindamycin, and metronidazole on faecal excretion of short-chain fatty acids in healthy subjects. Scand. J. Gastroenterol DOI: 10.3109/00365528609003109 [DOI] [PubMed] [Google Scholar]
- 73.Theriot CM et al. (2014) Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun DOI: 10.1038/ncomms4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hryckowian AJ et al. (2018) Microbiota-Accessible carbohydrates suppress Clostridium difficile infection in a murine model. Nat. Microbiol DOI: 10.1038/s41564-018-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karlsson S et al. (2000) Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect. Immun DOI: 10.1128/IAI.68.10.5881-5888.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim MH et al. (2013) Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology DOI: 10.1053/j.gastro.2013.04.056 [DOI] [PubMed] [Google Scholar]
- 77.Fachi JL et al. (2019) Butyrate Protects Mice from Clostridium difficile-Induced Colitis through an HIF-1-Dependent Mechanism. Cell Rep. DOI: 10.1016/j.celrep.2019.03.054 [DOI] [PubMed] [Google Scholar]
- 78.Arpaia N et al. (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature DOI: 10.1038/nature12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Furusawa Y et al. (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature DOI: 10.1038/nature12721 [DOI] [PubMed] [Google Scholar]
- 80.Smith PM et al. (2013) The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (80-.) DOI: 10.1126/science.1241165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asarat M et al. (2016) Short-chain fatty acids regulate cytokines and Th17/treg cells in human peripheral blood mononuclear cells in vitro. Immunol. Invest DOI: 10.3109/08820139.2015.1122613 [DOI] [PubMed] [Google Scholar]
- 82.Sorg JA and Sonenshein AL (2009) Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J. Bacteriol DOI: 10.1128/JB.01260-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sorg JA and Sonenshein AL (2008) Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol DOI: 10.1128/JB.01765-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giel JL et al. (2010) Metabolism of bile salts in mice influences spore germination in clostridium difficile. PLoS One DOI: 10.1371/journal.pone.0008740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weingarden AR et al. (2014) Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Liver Physiol DOI: 10.1152/ajpgi.00282.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu H et al. (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–41 [DOI] [PubMed] [Google Scholar]
- 87.Saavedra PHV et al. (2018) Apoptosis of intestinal epithelial cells restricts Clostridium difficile infection in a model of pseudomembranous colitis. Nat. Commun DOI: 10.1038/s41467-018-07386-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miller LS et al. (2007) Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol 179, 6933–6942 [DOI] [PubMed] [Google Scholar]
- 89.Liu YH et al. (2018) The ATP-P2X 7 signaling axis is an essential sentinel for intracellular Clostridium difficile pathogen-induced inflammasome activation. Front. Cell. Infect. Microbiol DOI: 10.3389/fcimb.2018.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alimov I et al. (2019) Bile acid analogues are activators of pyrin inflammasome. J. Biol. Chem DOI: 10.1074/jbc.RA118.005103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo C et al. (2016) Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity DOI: 10.1016/j.immuni.2016.09.008 [DOI] [PubMed] [Google Scholar]
- 92.Peterson LW and Artis D (2014) Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol DOI: 10.1038/nri3608 [DOI] [PubMed] [Google Scholar]
- 93.Abreu MT (2010) Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol DOI: 10.1038/nri2707 [DOI] [PubMed] [Google Scholar]
- 94.Rakoff-Nahoum S et al. (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell DOI: 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 95.Brandl K et al. (2008) Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature DOI: 10.1038/nature07250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brandl K et al. (2007) MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. J. Exp. Med DOI: 10.1084/jem.20070563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clarke TB et al. (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med DOI: 10.1038/nm.2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ryan A et al. (2011) A role for TLR4 in clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Youngster I et al. (2016) Oral, frozen fecal microbiota transplant (FMT) capsules for recurrent Clostridium difficile infection. BMC Med. DOI: 10.1186/s12916-016-0680-9 [DOI] [PMC free article] [PubMed] [Google Scholar]