

Abstract
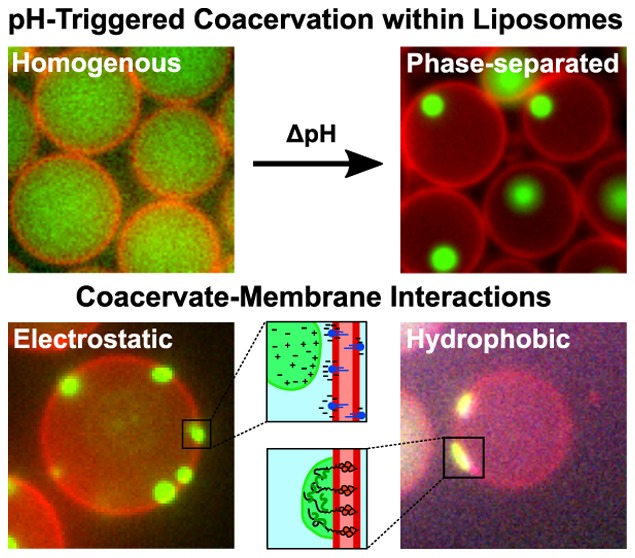
Membraneless organelles formed by liquid–liquid phase separation are dynamic structures that are employed by cells to spatiotemporally regulate their interior. Indeed, complex coacervation-based phase separation is involved in a multitude of biological tasks ranging from photosynthesis to cell division to chromatin organization, and more. Here, we use an on-chip microfluidic method to control and study the formation of membraneless organelles within liposomes, using pH as the main control parameter. We show that a transmembrane proton flux that is created by a stepwise change in the external pH can readily bring about the coacervation of encapsulated components in a controlled manner. We employ this strategy to induce and study electrostatic as well as hydrophobic interactions between the coacervate and the lipid membrane. Electrostatic interactions using charged lipids efficiently recruit coacervates to the membrane and restrict their movement along the inner leaflet. Hydrophobic interactions via cholesterol-tagged RNA molecules provide even stronger interactions, causing coacervates to wet the membrane and affect the local lipid-membrane structure, reminiscent of coacervate–membrane interactions in cells. The presented technique of pH-triggered coacervation within cell-sized liposomes may find applications in synthetic cells and in studying biologically relevant phase separation reactions in a bottom-up manner.
Keywords: liquid−liquid phase separation, coacervates, membranes, liposomes, microfluidics
Compartmentalization, which is evident in the form of cells and many intracellular organelles, is an essential feature that allows organisms to regulate a myriad of biological functions. Many of these organelles, such as the nucleus, mitochondria, or the Golgi body, are separated from the cytoplasm by a lipid membrane and were among the first to be discovered in the early days of light microscopy.1 However, in addition to dozens of such membrane-encompassed organelles,2 a completely new class of subcellular structures has recently gained tremendous interest, viz., membraneless organelles (MOs).3,4 MOs represent a rich and still poorly understood variety of phase-separated subcellular structures such as the nucleolus and germ granules.5−8 These indispensable organelles are formed as a result of liquid–liquid phase separation (LLPS), primarily by the process of complex coacervation, i.e., interactions between charged polyelectrolytes such as proteins and nucleic acids.8 MOs exhibit liquid-like material properties9 and tend to be highly dynamic, as there is a continuous internal diffusive rearrangement of the coacervate material as well as an exchange of components with the surroundings.7,10
LLPS is widely employed by cells to regulate their internal organization,3,4,8,11 as is clear from the variety of MOs such as Cajal bodies,12 pyrenoids,13 and numerous ribonucleic acid (RNA)–protein droplets.14,15 These organelles play versatile roles in regulating the cellular biochemistry, and their malfunctioning is associated with protein-aggregation diseases including Alzheimer’s disease.16−19 With new examples being discovered at a rapid pace, it is increasingly becoming clear that LLPS plays a crucial role in an especially wide variety of cellular processes such as DNA compaction and chromatin organization,20−23 selectively filtering specific biomolecules,24 stress regulation,5,25 transcription regulation,26−29 polarity establishment,7 photosynthesis,13 endocytosis,30 cell signaling,31 and cell adhesion.32 While some functionalities such as sequestering and concentrating specific molecules to assist biochemical reactions are recurring and established themes, many other questions are just starting to get investigated. For example, it is as of yet quite unclear whether, and if so how, MOs physically manipulate their local environment, e.g., mechanically remodel membranes.
The interaction between MOs and membranes is gathering interest but has not yet been widely studied. Recent work has indicated the role of coacervates in endocytosis30 and cell adhesion,32 pointing out the potential of MOs in exerting forces on lipid membranes. In C. elegans, the liquid-like P granules that act as mRNA exporters have been reported to directly wet the nuclear membrane,7,33 possibly enhancing transport. Membrane-bound phase-separated protein clusters have also been shown to be involved in a variety of signaling pathways, modulating signal transduction as well as recruiting cytoskeletal elements.31,34−37 These recent studies indicate previously unknown roles served by MOs, including that of mechanical work.38
Next to in vivo studies of LLPS in cells, powerful in vitro approaches have been developed to study the coacervation process through minimal systems comprising essential biological components or synthetic counterparts.39−41 Various control parameters such as temperature, pH, or enzymatic means have been used to induce and analyze coacervate formation.42−44 While such in vitro experiments are useful tools to pinpoint the interactions responsible for phase separation,45 they are often performed in bulk environments that require large sample volumes and pose limitations to the experimental control that can be exerted. Recent efforts showed that it is possible to reconstitute MOs in cell-sized microcontainers such as liposomes42,46 or water-in-oil droplets.29 With a volume of a few picoliters and a phospholipid bilayer at the outer surface, liposomes serve as ideal reaction vessels that can emulate the cellular environment. Indeed, coacervates-in-liposome structures serve as an excellent model system for multiple reasons: (i) The formed MOs are of similar size as the natural MOs (from a few hundred nm to a few μm). (ii) They provide control of the influx and efflux of solvent and solutes such as biomolecules, salts, etc. (iii) They are spatially restricted and allow for prolonged observation times. Recently, we reported a method that captures the above-mentioned attributes and allows the study of LLPS within liposomes by forming coacervate-in-liposome structures in a controlled manner.46 The method used octanol-assisted liposome assembly (OLA), an on-chip liposome production technique,47 and the liposomes were rendered porous by inserting α-hemolysin protein pores in the membrane. The approach relied on the encapsulation of some of the essential coacervate component(s) inside the liposome and subsequent provision of additional component(s) by diffusion through the membrane-embedded protein pores.
Here, we report another approach, that of controlling the external pH, to induce coacervation inside liposomes. LLPS can thus be tuned by an easily accessible external chemical parameter, which completely eliminates the need for dedicated membrane transporters as well as for any restrictions on the size of the components needed to form MOs. We show that the native proton permeability of liposomes suffices to drive coacervation reactions through the transmembrane proton flux. We monitor pH changes within the liposomal lumen by encapsulating a pH-sensitive fluorophore, sTG, a derivative of Tokyo Green with an increased solubility over a wide pH range.48 Using two model coacervate systems, namely, poly-l-lysine (pLL)/adenosine triphosphate (ATP) and RNA/spermine, we demonstrate pH-induced coacervation reactions within liposomes. While all the coacervate components were already encapsulated inside the liposomes as a homogeneous dispersion at a pH that is unsuitable (either highly acidic or highly basic) for coacervation, a step in the outside pH induced an internal pH change over a course of minutes, resulting in LLPS inside the liposomes.
We employ this technique to induce and study interactions between coacervates and the lipid membrane. We explore two different types of interactions, electrostatic and hydrophobic. Electrostatic interactions are induced by doping the lipid membrane with charged lipids, causing the charge-dense coacervates to bind and diffuse along the inner surface of the lipid bilayer. Hydrophobic interactions can be realized by encapsulating cholesterol-tagged RNA molecules, causing coacervates to nucleate at the membrane. These coacervates wet the membrane at low contact angles and, interestingly, affect the local lipid membrane structure. We thus present a useful on-chip methodology to study spatiotemporally controlled LLPS triggered via pH change within cell-sized confinements and show the usefulness of our technique by investigating coacervate–membrane interactions. The approach can be expected to facilitate controlled studies of biologically relevant LLPS phenomena in a bottom-up manner as well as aid the development of synthetic cells.
Results
External Control of the Internal pH of Liposomes
In order to eliminate the need of any dedicated membrane protein pores to induce coacervation inside liposomes,46 we decided to use pH as the controlling parameter. Since coacervation is driven by complexation, our idea was to initially inhibit the process by rendering one of the involved polyelectrolytes effectively uncharged by setting the pH to a value beyond the isoelectric point (pI) of that molecule. In this manner, a homogeneous solution containing both coacervate components could be initially encapsulated inside the liposome without inducing phase separation. The internal conditions, we hypothesized, could later be adjusted to favor complexation and thus induce coacervation, by means of an externally applied pH change.
To test this hypothesis, we first investigated the proton permeability of the lipid-bilayer surface of our liposomes, which is essential to convey an external pH shift to the liposomal lumen. We generated unilamellar liposomes (10–15 μm in diameter, with an initial internal pH of 4.0) using OLA47 and separated them from the waste product (1-octanol droplets) using a previously reported technique.46 For all the experiments, care was taken to maintain the isotonicity between the liposomal lumen and the environment; if needed, osmolarity was balanced by addition of glucose. The lumen of the liposomes carried 25 μM sTG, a pH-sensitive fluorescent dye that is structurally similar to fluorescein.48,49 Initially, the pH of the surrounding environment was set at the same value as that of the liposomal lumen (Figure 1a, first panel). We then increased the external pH in a stepwise fashion by adding a concentrated solution buffered at pH 9.0 to the solution in which the liposomes were suspended. The concentration of this feed solution was always chosen such that osmotic conditions were not significantly changed upon application of the pH jump. The moment of local mixing was monitored by an observable increase in the fluorescence of the outside solution (due to the presence of some residual sTG in the external solution as a result of liposome production;50 see also Supplementary Figure 1). This was taken to be the starting point (t0) where the pH gradient over the membrane was applied (Figure 1a, second panel). We analyzed the fluorescence intensity of multiple liposomes of approximately the same radius (5.6 ± 0.4 μm, mean ± standard deviation, = 20) over time. Representative time-lapse fluorescence images are shown in Figure 1b. By seperately measuring the fluorescence of solutions containing sTG at known pH values in a bulk assay, we obtained a calibration curve (Figure 1c, see Methods for further details) that showed a strong transition from a low to a high fluorescence occurring at a pH of around 6.
Figure 1.

External control over the pH level inside the liposomes. (a) Schematic representation of the pH-trajectory during the experiment. As the outer pH is increased to 9, the inside pH starts to increase due to the leakage of protons and hydroxyl ions across the lipid bilayer. Over time, the inner and the outer pH levels equilibrate. (b) Time-lapse fluorescence images showing how liposomes containing sTG fluorescent dye respond to an increased outer pH. Top row shows the lipid bilayer fluorescence; bottom row shows the sTG fluorescence, which clearly increases upon the pH change. The images are presented at the same contrast and imaging settings and show the equatorial cross sections of the liposomes. (c) Bulk calibration curve of the sTG fluorescence intensity over a wide range of pH. The intensity plateaus for pH levels above 7.5, indicating the maximum pH that we can monitor in the liposomes from the fluorescence time traces. (d) Change in the mean liposome sTG signal (blue line, n = 20) over time. The blue shaded area indicates one standard deviation. Top axis shows approximate pH values determined from the sTG calibration.
Upon the pH change in the external environment, protons and hydroxyl ions started to leak across the liposome, thus increasing the internal pH in the liposome. The mean internal fluorescence intensity over time (Figure 1d) followed a sigmoidal path, similar to the change in the fluorescence intensity of sTG as a function of pH (Figure 1c). A plot of the fluorescence trajectory over the entire experiment is provided in Supplementary Figure 2. Two plateaus can be distinguished in the graph of Figure 1d: one where the pH is far below the transition value and one where the fluorescent intensity has reached a maximum. The time required for the liposomal pH to change from 4.0 to >7.5 was on the order of a few minutes. This time span required for equilibrating the liposomal lumen with the external pH is very practical for the purpose of inducing and monitoring coacervation within the liposomes.
pH-Induced Coacervation in Liposomes
Since complex coacervation is driven by charge-matching of polyelectrolytes, a change in the degree of ionization of one or both components can have a strong effect on the components’ mutual affinity and thus induce or dissolve phase separation51 (Figure 2a). In order to make the components charge-neutral, we found it more practical to set the pH to a value beyond the pI for the smaller component, i.e., ATP or spermine, rather than to try to neutralize the larger polymer (Supplementary Figure 3). This possibly relates to the fact that large polyelectrolytes have many ionizable groups, thus requiring more extreme conditions before reducing the charge to a low enough value52 (and further complicated for polynucleotides, which have an extremely low pI).53
Figure 2.
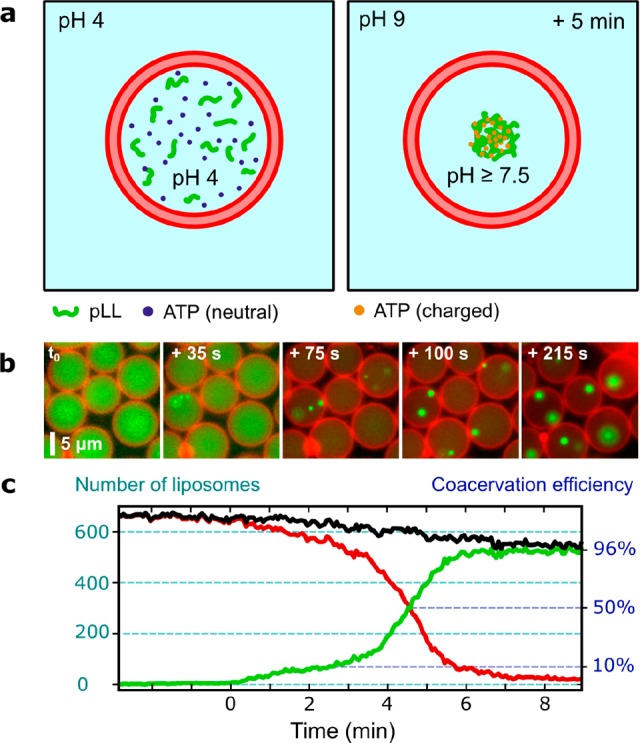
pH-controlled coacervation of pLL/ATP within liposomes. (a) Schematic representation of the initial and final conditions in the experiment. Before adjusting the pH, the acidic environment renders the molecular charge of ATP to be neutral, and as a result, coacervation is inhibited. When the pH inside the liposomes rises in response to an externally applied pH jump, ATP is deprotonated and gains negative charge, upon which coacervation occurs. (b) Fluorescence time-lapse images of the liposomes (equatorial cross sections). After the external pH is raised, the pH level inside the liposomes equilibrates to it over the course of minutes and coacervation begins to take place. t0 was chosen as the time just before the first coacervation event occurred. (c) The number of liposomes that exhibit coacervation versus time, for a large population of liposomes (n = 660 initially). Black line indicates the total number of liposomes; red line denotes the number of liposomes without a coacervate; green line those with a coacervate. In the end, over 96% of liposomes contained coacervates, indicating a very high efficiency of the process. The slight decrease in the total liposome count over time was due to some liposomes drifting out of the field-of-view. About 5 min passed between the first and last coacervation events.
We tested our method of pH-controlled coacervation by inducing coacervation between ATP and pLL (MW 15–30 kDa) within liposomes. The membrane was made up of DOPC (1,2-dioleyl-sn-glycero-3-phosphocholine) and a small fraction of fluorescent lipids (Rh-PE:1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl); 1:1000 molar ratio) for visualization. Importantly, it did not bear any protein pores; that is, the liposomes were sealed off from the environment except for water and proton transport through the bilayer membrane. The initial population of liposomes contained 15 mM ATP, 5 mg/mL pLL, 0.5 mg/mL FITC-pLL, and 15 mM citrate-HCl and showed a completely homogeneous fluorescence signal of the liposome volumes (first panel in Figure 2b). A large population of liposomes (n > 600) was imaged after increasing the pH from an initial value of 4.0 to a final value of 9.0. Once the pH value inside the liposomes had risen sufficiently, the acidic ATP molecules lost protons and became negatively charged. This rendered conditions favorable for coacervation with the positively charged pLL. Indeed, within a few minutes after stepping up the pH, the initial homogeneously distributed pLL was seen to condense into small nucleates that grew into multiple distinct droplets that further merged over time, eventually forming a single coacervate (latter panels in Figure 2b; see also Supplementary Video 2, while Supplementary Figure 4 provides an entire field-of-view of the initial and final populations). The initiation of phase separation was observed to be highly efficient. Figure 2c shows the number of liposomes that underwent coacervation over time. Normalizing against the total liposome count, it can be seen a coacervate is formed in over 95% of liposomes. There was a moderate spread in the time of onset of coacervation, as about 5 min passed between the first and the last liposome exhibiting LLPS within its lumen. This can be attributed to the pH increasing diffusively along the sample, by small differences in the surface-area-to-volume ratio between liposomes, and possibly by transient membrane defects that may affect the permeability.54,55
Inducing Coacervate–Membrane Interactions
Having established a method of inducing and monitoring coacervation in liposomes without the need of transmembrane transport of coacervate components, we decided to probe the interaction between coacervates and lipid membranes. It is known that the interfacial tension between dense coacervates and the surrounding liquid is generally very low (<mN/m).56,57 Therefore, we hypothesized that if the interaction between the coacervate and lipid bilayer is strong enough, the coacervate might be recruited to the membrane and either adhere to it while still maintaining its spherical shape or wet the membrane partially or fully, depending on the strength of the interaction. To explore such scenarios, we used coacervates with two different types of interactions, electrostatic and hydrophobic.
Inspired by complexation as the driving force for phase separation, we first experimented with electrostatic interactions and anticipated that an interaction might occur between charge-dense coacervates and multivalent lipid molecules (Figure 3a). We used phosphatidylinositol (3,4,5)-triphosphate (PIP3), an anionic lipid with a charge density as high as −7 per molecule, as a charged lipid that would recruit coacervate droplets to the membrane. Liposomes with a surface charge density similar to the liposomes under consideration were reported to have a zeta potential on the order of −20 mV.58 Since we measured the zeta potential of pLL/ATP coacervate droplets to be positive with a value of 14.1 ± 1.6 mV (see Methods for details), the resulting potential difference can be expected to be >30 mV, suggesting that the coacervates and membranes will adhere to each other.
Figure 3.

Coacervate–membrane interactions in charged (using PIP3) liposomes. (a) Conceptual sketch showing coacervate formation for the case of charge-based interactions between the coacervate and lipid membrane, due to charge matching of the polyanionic PIP3 and polycationic pLL. (b) Fluorescent time-lapse images showing the initial state, nucleation, and end result of coacervation in both pure DOPC and PIP3-doped liposomes. Note that the images were taken by focusing at the equatorial plane of the liposomes. Membrane-bound coacervates are observed to locate and diffuse mainly along the 2D surface of the vesicle (bottom), whereas coacervates without lipid interactions show 3D Brownian motion (top). (c) Heat maps overlaying all the observed positions of coacervates in a single liposome during the time of observation. The top one is for pure DOPC, while the bottom one is for PIP3-doped liposome. The circular arc in the latter is indicative of the membrane-bound diffusion of the coacervate(s). (d) Coacervate position as a function of liposome radius. Heat maps were generated for multiple liposomes (DOPC: n = 203, PIP3-doped: n = 43) and transformed into a radial distribution, where the radius was normalized from 0 to 1. These radial plots were summed to obtain a distribution for the average coacervate location within the liposomes. For the PIP3-doped liposomes, a significant fraction of coacervates resides at the membrane. Shaded areas indicate the standard deviation. (e) Coacervation dynamics in a PIP3-doped liposome. Once bound, the coacervates were observed to reside at the membrane, diffusing along the surface and merging into larger coacervate clusters (white arrows).
We compared the behavior of coacervates in pure DOPC liposomes and in liposomes doped with a small fraction of PIP3 (0.4% w/w or a molar ratio of DOPC:PIP3 = 370:1). The liposomes produced in both experiments contained 15 mm citrate-HCl buffer, 15 mM ATP, 5 mg/mL pLL, and 0.5 mg/mL FITC-pLL and had an initial internal pH value of 4.0. Phase separation was induced as before by adding a solution containing Tris-HCl at pH 9.0 to the otherwise acidic well solution. The dynamics of coacervation were imaged over the course of the entire phase separation process, where again a transition was observed from a homogeneous solution to phase-separated clusters of ATP/pLL (Figure 3b).
The effect of doping the membrane with the polyanionic lipid was distinct: coacervates in the charged liposomes were recruited to the membrane and stayed bound, diffusing around the surface of the membrane but not back into the bulk solution (see Supplementary Video 3). By contrast, coacervates in the pure DOPC vesicles always diffused randomly within the vesicle. The difference is particularly apparent in the heat maps showing the coverage of the coacervate fluorescence signal within inividual vesicles (Figure 3c) or by plotting the coacervate fluorescence signal as a function of radius, averaged over multiple liposomes (Figure 3d). Time-lapse images of the coacervation process in charged liposomes clearly show that upon touching the membrane, coacervates remained adhered to it (Figure 3e, white arrows). This observation may indicate a nonuniform distribution of PIP3 molecules, where they are concentrated at the coacervate–membrane interface, with transient interactions between the PIP3 molecules and the coacervate. While membrane doping with charged lipids clearly resulted in coacervate–membrane interaction, the lack of any morphological changes in the liposomes or coacervates suggests that the interaction was of relatively low strength.
To induce a stronger interaction, we decided to physically anchor the coacervate into the membrane using a cholesterol-tagged coacervate component, i.e., using hydrophobic interactions. For this purpose, we chose to use the lipophilic molecule cholesterol, as it is known to spontaneously insert into lipid bilayers. We used spermine and cholesterol-tagged RNA as the coacervate components. The RNA of choice, 5′-cholesterol-polyU (chol-polyU), was generated enzymatically using the enzyme polynucleotide phosphorylase (PNPase)46,59 (see Methods for details). We expected the cholesterol moiety to recruit the RNA molecules to the inner leaflet of the membrane, allowing coacervation to occur only at the membrane (Figure 4a). We generated liposomes containing spermine and chol-polyU, along with fluorescently tagged auxiliary polymers cy5-U20 (shown in the SI) and FITC-pLL for visualization of the coacervate. Since chol-polyU and spermine were observed to phase-separate even at exceedingly low values of the pH (<1), we switched to a high rather than a low pH as the initial condition: with the initial pH set to approximately 13 (and thus well above the highest pKa of spermine),60 the solution remained homogeneous.
Figure 4.

Membrane wetting by cholesterol-RNA/spermine coacervates. (a) Conceptual sketch showing the initial, intermediate, and final states of the liposome in the experiment. At highly basic pH, the contents of the liposome are homogeneous, with chol-polyU molecules covering the inner surface of the membrane. Upon lowering the pH, coacervation is seen to occur only at the membrane, owing to the anchored chol-polyU molecules. Due to the strong coacervate–membrane interaction, wetting of the membrane is seen as well as what appears as remodeling of the lipid membrane (rightmost panel shows a zoom-in). (b) Liposomes (images show equatorial cross sections) are imaged as time progresses. Five time points are presented, showing the development of membrane-bound coacervation. The resulting condensate is rich in cholesterol and has a strong affinity for the lipid membrane, causing the coacervate to wet the membrane and even disrupt the membrane structure to some extent. Note the increased lipid fluorescence intensity at the coacervate positions, indicating that lipids are concentrated at those spots. (c) Kymographs of the angular (top) and radial (bottom) fluorescence distribution in the liposome shown in (c). Each vertical line represents one frame in the time lapse. The time points corresponding to the above fluorescence microscopy images are indicated. (d) Mean pLL fluorescence versus normalized radius across multiple liposomes (n = 15). Shaded area indicates one standard deviation. (e) Liposome radius versus time. Upon coacervation, the radius of some liposomes is observed to suddenly decrease in a single step, with a concomitant increase in the lipid fluorescence at the coacervate patch. This is possibly due to local disruption of the membrane because of the membrane-bound nucleation events and subsequent absorption of lipids in the coacervate patch. The red bars in the radius versus time plot indicate the moment when the start of the phase separation was observed. The fluorescence images show a liposome at a time point shortly before and after coacervation occurred, indicating the radius drop that occurred in between.
Lowering the pH by the addition of Tris-HCl buffer induced coacervation. The effect of the addition of cholesterol was immediately clear: nucleation of coacervates occurred predominantly at the membrane, and the formed coacervates remained membrane-bound and wetted the lipid membrane (Figure 4b; Supplementary Video 4; Supplementary Figure 5). The coacervates were detected via the fluorescent signal from FITC-pLL as well as cy5-U20, both of which partitioned into the coacervate. Unexpectedly, the signal from fluorescent Rh-PE lipids also increased at the site where the coacervate wetted the membrane. Moreover, as the coacervates diffused around the surface of the membrane, this bright membrane patch moved along. This colocalization suggests that the coacervate wetting the liposome locally affected the membrane structure. Furthermore, in cases where neighboring liposomes were physically touching each other, the coacervates were generally present at the contact points, indicating membrane modulations induced transmembrane interactions in the form of bridging sites (Supplementary Figure 6). These observations can be explained in multiple ways. For instance, the coacervate material wetting the membrane might lead to membrane reconfigurations at the interaction site (Figure 4a, panel 4), resulting in an increased lipid fluorescence. Alternatively, it is possible that the cholesterol-rich coacervate enables absorption of lipid-conjugated fluorophores.
We further plotted kymographs to depict the kinetics of coacervates within the liposomes. The upper plot in Figure 4c shows the coacervate fluorescent signal plotted as the angular position versus time, while the lower plot shows the same signal plotted as the normalized radius versus time. The kymographs illustrate the transition from a homogeneous (left) to inhomogeneous (right) fluorescence, indicating the onset of phase separation (for more examples of kymographs, see Supplementary Figure 6). The angular plot illustrates how coacervates diffuse along the membrane and fuse over time. After nucleation, multiple fluorescence tracks can be seen in the kymograph, which correspond to coacervates residing at different sites on the membrane. Over time, as coacervates touch and merge, a single track is obtained. For the radial plot, a bright fluorescent signal at a value of R just under 1, i.e., near the membrane, clearly shows the propensity of the coacervates to reside at the membrane. This is also shown in the mean FITC-pLL fluorescent intensity plotted against normalized liposome radius for multiple liposomes (Figure 4d; n = 15).
Another interesting consequence of coacervation seemed to affect the liposome itself. In many cases, liposomes appeared to suddenly decrease in size, shortly before the onset of coacervation became apparent. Figure 4e shows a few examples, where clearly a discrete drop can be seen in a plot of liposome radius versus time. The decrease in the radius was small (∼5%) but permanent and was concomitant with the formation of a bright patch of lipid fluorescence at the site of the membrane-bound coacervate. The shrinkage occurred concomitantly with a marked decrease in the fluorescence intensity of the lumen, indicating the onset of phase separation. While coacervates appeared shortly after the liposome shrinkage (within ∼30 s), this corroborates well with our recent observation that nucleation precedes the formation of coacervates (by ∼30 s) as observable via fluorescence microscopy.46 Such a drop in the liposome radius was not observed in any of the previous experiments (with comparable osmotic conditions), thus discarding artifacts, nor upon addition of feed solution in a control experiment in which membrane-interacting components were present but coacervation did not occur (see Supplementary Figure 7). The observed decrease in the radius is likely explained by a local disruption of the membrane during coacervate formation due to the nucleation events happening at the membrane via locally residing chol-polyU molecules. These nucleation events may be caused by local membrane remodeling, destabilization of the lipid bilayer, or incorporation of lipids into the coacervate.
Conclusions
In this paper, we have presented two main findings regarding LLPS within membrane-bound microcompartments. First, we observed that the internal pH of a liposome can be tuned by changing the external pH, without the need of any membrane pores, and accordingly that pH regulation can be employed to induce LLPS within liposomes. This method facilitates the study of coacervate systems, especially those that are otherwise hard to control, for example, due to the large size of the components or limited diffusion rates through protein pores. Second, using pH-induced coacervation, we could successfully induce interactions between lipid membrane and liquid condensates and study their effects. Via electrostatic interactions (by doping the lipid membrane with a charged lipid) or hydrophobic interactions (by using a cholesterol-tagged coacervate component), we were able to induce interactions that resulted in varying levels of affinity between the coacervate and the membrane. Due to such interactions, coacervates preferentially resided at the surface of the liposomes, lose their regular spherical shape by wetting the membrane, and even locally affect the structure of the lipid bilayer.
pH control is advantageous to previous work,46 where coacervation was induced by diffusive addition of small components, for three important reasons: (i) It discards the need for membrane pores: Protein pores such as α-hemolysin can be used to diffusively transport components across the membrane, but such transport lacks selectivity for small molecules and also puts a limit on the maximum size of the components that can be transported. (ii) Practicality: pH is a parameter that can be easily changed during an experiment. Multiple options exist to refine the bulk administering of solution for a pH change, e.g., the use of chemical compounds that reversibly emit hydroxyl groups upon illumination and thereby induce pH jumps of over 4 units,61 or proteins such as bacteriorhodopsin that allow for reversible pH changes in liposomes.62 Moreover, since pH changes are reversible and can cause redissolution of coacervates,44 the system described here can potentially be extended to allow for repeated cycles of coacervation within the same experiment. This could be done by facilitating efficient external buffer exchange without physically disturbing the liposomes by, for example, using a combination of a dial-a-wave design63 along with microfluidic traps64 to confine the liposomes. (iii) Versatility: pH is an important parameter not just in dictating the energetics of phase separation but also in many enzymatic reactions. Regulation of protein activity by pH may allow the study of diverse biochemical reactions in the presented coacervate-in-liposome system. The controlled formation and redissolution of coacervates in a cyclic fashion are also critical steps toward adding functionality to synthetic cells, and pH control may be a useful tool to achieve it.65
Reported values for the proton permeability (P) of phosphatidylcholine membranes vary over a broad range (from 10–4 to 10–7 cm/s),66 yielding a characteristic permeation time (R/3P, where R is the liposome radius) for a 10 μm diameter vesicle ranging from seconds to tens of minutes. Since the pH equilibrates over a few minutes in our case, the liposomes under consideration show a permeability within this range. While a possible trace amount of 1-octanol left in the bilayer can in principle affect the membrane permeability,67 the recently measured permeability of OLA-based liposomes to antibiotic molecules is in very good agreement with the established literature.68 On the other hand, particular experimental conditions can affect the proton permeability, especially if the ionic components of the solutions are not in balance, which is the case in our experiments with various polyelectrolytes inside the liposomes.69 While it is hard to quantify the innate proton permeability of liposomes under consideration, we here effectively used the proton leakage as a convenient tool to induce coacervation.
We employed pH control of the coacervation to study membrane interactions in two different coacervate–liposome systems: pLL/ATP in liposomes containing a small fraction of negatively charged PIP3 lipids, and chol-polyU/spermine in pure DOPC vesicles. While the former system is often used as a model system to study liquid–liquid phase separation, the latter is biologically more relevant due to the many ways in which RNA can be enzymatically manipulated. We showed different degrees of coacervate–membrane interactions: from simple bound states to significant deformation of the coacervates to even local restructuring of the lipid bilayer. Indeed, coacervates appeared to be able to affect the membrane of the containers: liposomes were observed to lose surface area, with a concomitant increase in the lipid fluorescence at the site where the coacervate interacted with the membrane, suggesting sequestering of lipids into the coacervate.
Our work thus provides a versatile method to study the dynamics of phase separation within picoliter confinements and enables a more sophisticated control over the formation of functional organelles in cell-sized containers. Importantly, it allows investigating the interactions between coacervates and the confining membranes. While LLPS is ubiquitously used by cells for a variety of purposes, recent studies have particularly hinted at the vital role of coacervate–membrane interactions in endocytosis,30 cell signaling,31 cell adhesion,32etc. While different in their compositions, the interplay between coacervate and membrane in our work shows clear similarities to these biological examples, e.g. the wetting of membranes by coacervates seen in cell signaling complexes or adjacent cells adhering due to a bridging layer of coacervate material. The presented method is therefore highly suitable for reconstituting biologically relevant MOs and studying their interactions with membranes. Furthermore, the driving force behind various coacervate–membrane interactions observed in nature is also partly electrostatic and/or hydrophobic. Simplified systems, such as those shown here, can help provide a mechanistic understanding of the formation of membrane-bound coacervates observed in cells.
These effects also provide possibilities for the employment of coacervates in the bottom-up construction of synthetic cells. For example, the “polarization” of a liposome with a coacervate perched at one site on the membrane could allow for setting up gradients of reactants for the internal spatial organization. Membrane-bound coacervates could serve as localized sites for the production of lipids or membrane proteins and could, due to their strong interaction with the membrane, perhaps even be engineered for transmembrane transport that would otherwise require complicated machinery. Future research on coacervate–membrane interactions could produce more refined manifestations of the interplay between cell-sized compartments and condensates. By varying experimental parameters in, for example, the chol-polyU/spermine system, such as cholesterol-to-UDP ratio or cholesterol concentration, the strength of the interaction could be tuned to a desired level. A clearer picture of the molecular structure at the coacervate–membrane site and a broader knowledge of the parameters that are important in dictating their mutual behavior would allow for the use of coacervate–membrane interactions as a general tool in the construction of artificial cells.
Methods
Materials
Poloxamer 188 (P188), 1-octanol, glycerol, poly(vinyl alcohol) (PVA, MW 30–70 kDa, 87–90% hydrolyzed), KCl, NaCl, MgCl2, NaOH, HCl, Tris-HCl, trisodium citrate, EDTA, glucose, dextran (MW 6 kDa), (FITC)-pLL hydrobromide (MW 15–30 kDa), ATP disodium salt, UDP disodium salt, PNPase (polynucleotide phosphorylase from Synechocystis Sp.), and spermine tetrahydrochloride were purchased from Sigma-Aldrich. sTG and 5′ cy5-U20 were gifted by Rikiya Watanabe (Molecular Physiology Laboratory, RIKEN, Saitama, Japan) and Marileen Dogterom (Kavli Institute of Nanoscience Delft), respectively. DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), PIP3 (1,2-dioleoyl-sn-glycero-3-phospho-(1′-myoinositol-3′,4′,5′-trisphosphate) (ammonium salt)), and Rh-PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt)) were purchased from Avanti Polar Lipids. Microfluidic devices were prepared from the materials provided in the SYLGARD 184 silicone elastomer kit purchased from Dow Corning. 5′-Cholesterol-U20 was purchased from biomers.net GmbH. pH paper used was bought from Carl Roth, Art. H913.2.
Production of Liposomes by OLA
Liposomes in all experiments discussed in this work were produced by an altered version of OLA.46,47 Briefly, masks needed to cast the microfluidic devices were made out of silicon wafers by e-beam lithography, etching, and surface silanization. PDMS and curing agent were mixed in a 10:1 ratio and poured on the wafers to form a roughly 3–4 mm thick layer and cured in the oven at 80 °C for at least 4 h. Inlet holes (0.75 mm) and an exit hole (3–4 mm) were pierced in the devices using a biopsy punch (World Precision Instruments) before bonding the devices to glass slides covered with a thin layer of PDMS. The outer aqueous channels as well as the post-junction part of the device were treated with a 5% w/v poly(vinyl alcohol) (PVA) solution for 5 min, and the solution was subsequently removed by vacuum suction. After drying in the oven at 120 °C for 15–30 min, the devices were ready for experimentation. During experimentation, depending on solution components, the inner aqueous solution was allowed to flow freely for up to 15 min to ensure that any possible initial absorption of IA components to the PDMS walls of the microfluidic channels would not affect the concentration of the solution to be encapsulated. In case this was done, the exit well was thoroughly washed by repeated addition and removal of exit solution before collecting a fresh batch of liposomes.
Solution Compositions
OLA, as employed here, requires five different solutions to carry out an experiment: inner aqueous (IA), outer aqueous (OA), lipids in 1-octanol (LO), exit well solution (EX), and a feed solution (FE). IA, OA, and EX always contained 15% v/v glycerol, 150 mM KCl, 5 mM MgCl2, and a pH-regulating buffer or a base, unless otherwise indicated. Additionally, 5 mM dextran and 5% w/v P188 surfactant were always present in IA and OA, respectively. A detailed list of solution compositions for various experiments can be found in Supplementary Table 1. For each experiment, the osmolarity of the aqueous solutions was calculated, and, if needed, the solutions were balanced by addition of glucose. LO was always prepared by mixing lipids (10% w/v in ethanol, 99.9% DOPC + 0.1% Rh-PE, molar ratio) with 1-octanol to a final concentration of 0.2% w/v. Exceptions were experiments that included charged lipids: for PIP3, lipid composition was 99.5% DOPC, 0.4% PIP3, 0.1% Rh-PE (molar ratio). To induce as little movement in the exit well as possible when adding the feed solution, we used a Hamilton syringe (7105, 5 μL volume capacity) for the stepwise addition of submicroliter volumes of feed by fusing a small droplet at the end of the needle with the surface of the exit well solution containing the liposomes (∼3 mm distance between surface and liposomes at the bottom of the well).
Enzymatic Production of chol-polyU
5′-Cholesterol-U20 seeds were elongated by PNPase as follows: 80 mM Tris-HCl pH 9.0, 1 mM EDTA, 5 mM MgCl2, 5 μM PNPase, 60 mM UDP, and 75 μM 5′ cholesterol-U20 were mixed and incubated at 37 °C for 2 h before heating of the solution at 60 °C for 5 min to denature the protein. This solution was considered as a “10× concentrated stock” and was stored at −20 °C.
sTG Fluorescence-versus-pH Calibration
Buffers were prepared at pH values between 4.0 and 9.0, with a step size of 0.5. Citrate-HCl (pH 4.0–6.0), MES-NaOH (pH 6.5), and Tris-HCl (pH 7.0–9.0) were the buffer types of choice. Actual pH values were measured using a benchtop pH probe (resolution 0.01 units). Multiple solutions were then prepared containing 50 mM buffer, 150 mM NaCl, 5 mM MgCl2, 5 mM dextran (MW 6000), 15% v/v glycerol, and 1 mM sTG. The fluorescence (λex = 488 nm, λem = 525 nm) of each of the solutions (in triplicates) and blanks (containing no sTG) were then measured using an Infinite 200 PRO plate reader (Tecan Group Ltd., Switzerland).
Measuring the Net Charge of the Coacervates
Zeta potential measurements were conducted on a Zetasizer Nano-ZS (Malvern Instruments). pLL/ATP coacervates were formed in similar buffer conditions to those for the microfluidic experiments (150 mM KCl, 5 mM MgCl2, 15% v/v glycerol, 50 mM Tris-HCl at pH 7.4, 5.5 mg/mL pLL, 15 mM ATP). The solution was intensely vortexed and then immediately diluted 100-fold, maintaining the buffer conditions (150 mM KCl, 5 mM MgCl2, 15% v/v glycerol, 50 mM Tris-HCl at pH 7.4). The diluted solution was vortexed again, and the zeta potential was measured at 20 °C. Three measurements were taken, each consisting of 100 runs. The average values of the three individual measurements were 15.7, 12.6, and 13.9 mV.
Image Acquisition
A Nikon Ti2 inverted wide-field epifluorescence microscope equipped with Spectra X light engine (Lumencor), filter set LED-DA/FI/TR/CY5-4X-B (Semrock), and Nikon objectives (CFI Plan Apo Lambda 60× with NA 1.4, CFI Plan Fluor 20× (oil) with NA 0.75, and CFI Plan Achro 10× with NA 0.25) was used. Images were acquired using NIS-Elements software (Nikon) in combination with sCMOS camera Prime BSI (Photometrics). Exposure times and frame rates varied between experiments; some typical frame rates were 0.33 Hz (Figure 1), 1.42 Hz (Figure 3-PIP3), and 0.2 Hz (Figures 2, 3-DOPC, 4).
Image Analysis
Raw microscopy data were prepared for analysis using FIJI (ImageJ). Liposomes were detected, tracked, and analyzed using FIJI (Figure 1) and MatLab (Mathworks, all other figures) using self-written scripts based around MatLab’s imfindcircles function. The scripts are available upon request. For the experiment in Figure 1, liposomes were detected in the lipid fluorescence channel and the inside sTG fluorescence was integrated to obtain a total value per frame. Regions of interest (ROIs) around individual liposomes were found by watershed partitioning of liposome images. For each ROI, the standard deviation of sTG fluorescence was measured; if this deviation was higher than 5 units (8-bit), the ROI contained a coacervate. Validity of this method was determined by visual inspection. Heat maps as well as angular and radial plots (Figures 3, 4) were generated as follows: liposomes were detected by lipid fluorescence in the first frame of a time-lapse video and tracked for the length of the time lapse. For the majority of these liposomes (tracks with large gaps or large linking distances were discarded), a coacervate-signal heat map was then generated by cropping a video containing the liposome in the center, thresholding this video at an intensity value in between that of the dilute and coacervate phase, and summing the binary image of coacervate fluorescence over the full length of the video. To generate a plot of mean coacervate position versus the liposome radius, these heat maps were turned into a polar map by sampling along circles of increasing radius centered on the liposome center (Δθ = 1°, ΔR = 0.02, radius normalized from 0 to 1) and subsequently integrated over the angular dimension. The final radial profile was then calculated by normalizing this single-liposome profile and finally taking the average of this profile between all detected liposomes (n = 15 to 203). Polar maps generated for single frames of the cropped time-lapse videos were also used to generate the kymographs in Figure 4c. Summing the polar map over the angular dimension produced a kymograph of fluorescence intensity along the radius of the liposome (Figure 4c, bottom), and summing over the radial dimension resulted in an angular kymograph (Figure 4c, top).
Acknowledgments
We would like to thank M. Dogterom for providing cy5-U20 and R. Watanabe for kindly gifting us sTG and for fruitful discussions. We also acknowledge experimental help from X. Li and K. Ganar. This work was supported by the NWO TOP-PUNT grant (no. 718014001), The Netherlands Organisation for Scientific Research (NWO/OCW) as part of the NanoFront, BaSyC and FOM (no. 110) programs, and European Research Council Advanced Grant SynDiv (no. 669598).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b10167.
Supplementary Table 1 details the solution compositions; Supplementary Figures 1–7 show additional fluorescence microscopy images and/or image analyses (PDF)
Supplementary Video 1 shows pH increase within a liposome, corresponding to Figure 1 (AVI)
Supplementary Video 2 shows pH-induced coacervation within a liposome, corresponding to Figure 2 (AVI)
Supplementary Video 3 shows electrostatic coacervate-membrane interactions within a liposome, corresponding to Figure 3 (AVI)
Supplementary Video 4 shows hydrophobic coacervate-membrane interactions within a liposome, corresponding to Figure 4 (AVI)
Author Contributions
M.G.F.L. and S.D. performed the experiments and analyzed the data. M.G.F.L., S.D., and C.D. conceived the experiments and wrote the paper.
Author Contributions
§ M. G. F. Last and S. Deshpande contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Diekmann Y.; Pereira-Leal J. B. Evolution of Intracellular Compartmentalization. Biochem. J. 2013, 449, 319–331. 10.1042/BJ20120957. [DOI] [PubMed] [Google Scholar]
- Satori C. P.; Henderson M. M.; Krautkramer E. A.; Kostal V.; Distefano M. M.; Arriaga E. A. Bioanalysis of Eukaryotic Organelles. Chem. Rev. 2013, 113, 2733–2811. 10.1021/cr300354g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea D. M.; Kriwacki R. W. Phase Separation in Biology; Functional Organization of a Higher Order. Cell Commun. Signaling 2016, 14, 1–20. 10.1186/s12964-015-0125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S.; Alberti S.; Fawzi N. L.; Mittag T.; Polymenidou M.; Rousseau F.; Schymkowitz J.; Shorter J.; Wolozin B.; Van Den Bosch L.; Tompa P.; Fuxreiter M. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott T. J.; Petsalaki E.; Farber P.; Jervis D.; Fussner E.; Plochowietz A.; Craggs T. D.; Bazett-Jones D. P.; Pawson T.; Forman-Kay J. D.; Baldwin A. J. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Weber C. A.; Nousch M.; Adame-Arana O.; Hoege C.; Hein M. Y.; Osborne-Nishimura E.; Mahamid J.; Jahnel M.; Jawerth L.; Pozniakovski A.; Eckmann C. R.; Jülicher F.; Hyman A. A. Polar Positioning of Phase-Separated Liquid Compartments in Cells Regulated by an MRNA Competition Mechanism. Cell 2016, 166, 1572–1584. 10.1016/j.cell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne C. P.; Eckmann C. R.; Courson D. S.; Rybarska A.; Hoege C.; Gharakhani J.; Jülicher F.; Hyman A. A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Banani S. F.; Lee H. O.; Hyman A. A.; Rosen M. K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A. A.; Weber C. A.; Jülicher F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Li P.; Banjade S.; Cheng H. C.; Kim S.; Chen B.; Guo L.; Llaguno M.; Hollingsworth J. V.; King D. S.; Banani S. F.; Russo P. S.; Jiang Q. X.; Nixon B. T.; Rosen M. K. Phase Transitions in the Assembly of Multivalent Signalling Proteins. Nature 2012, 483, 336–340. 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Intrinsically Disordered Proteins in Overcrowded Milieu: Membrane-Less Organelles, Phase Separation, and Intrinsic Disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. 10.1016/j.sbi.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Hyman A. A.; Brangwynne C. P. Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev. Cell 2011, 21, 14–16. 10.1016/j.devcel.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Freeman Rosenzweig E. S.; Xu B.; Kuhn Cuellar L.; Martinez-Sanchez A.; Schaffer M.; Strauss M.; Cartwright H. N.; Ronceray P.; Plitzko J. M.; Förster F.; Wingreen N. S.; Engel B. D.; Mackinder L. C. M.; Jonikas M. C. The Eukaryotic CO2-Concentrating Organelle Is Liquid-Like and Exhibits Dynamic Reorganization. Cell 2017, 171, 148–162. 10.1016/j.cell.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler M. A.; Bassell G. J. Neuronal RNA Granules: Movers and Makers. Neuron 2006, 51, 685–690. 10.1016/j.neuron.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Weber S. C.; Brangwynne C. P. Getting RNA and Protein in Phase. Cell 2012, 149, 1188–1191. 10.1016/j.cell.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Shin Y.; Brangwynne C. P.. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- Holehouse A. S.; Pappu R. V. Functional Implications of Intracellular Phase Transitions. Biochemistry 2018, 57, 2415–2423. 10.1021/acs.biochem.7b01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. R.; King O. D.; Shorter J.; Gitler A. D. Stress Granules as Crucibles of ALS Pathogenesis. J. Cell Biol. 2013, 201, 361–372. 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A.; Altmeyer M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Biol. 2016, 26, 547–558. 10.1016/j.tcb.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Strom A. R.; Emelyanov A. V.; Mir M.; Fyodorov D. V.; Darzacq X.; Karpen G. H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245. 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbondanzieri E. A.; Meyer A. S. More than Just a Phase: The Search for Membraneless Organelles in the Bacterial Cytoplasm. Curr. Genet. 2019, 65, 691–694. 10.1007/s00294-018-00927-x. [DOI] [PubMed] [Google Scholar]
- Patel A.; Lee H. O.; Jawerth L.; Maharana S.; Jahnel M.; Hein M. Y.; Stoynov S.; Mahamid J.; Saha S.; Franzmann T. M.; Pozniakovski A.; Poser I.; Maghelli N.; Royer L. A.; Weigert M.; Myers E. W.; Grill S.; Drechsel D.; Hyman A. A.; Alberti S. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Altmeyer M.; Neelsen K. J.; Teloni F.; Pozdnyakova I.; Pellegrino S.; Grøfte M.; Rask M. B. D.; Streicher W.; Jungmichel S.; Nielsen M. L.; Lukas J. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6, 8088. 10.1038/ncomms9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott T. J.; Craggs T. D.; Baldwin A. J. Membraneless Organelles Can Melt Nucleic Acid Duplexes and Act as Biomolecular Filters. Nat. Chem. 2016, 8, 569–575. 10.1038/nchem.2519. [DOI] [PubMed] [Google Scholar]
- Molliex A.; Temirov J.; Lee J.; Coughlin M.; Kanagaraj A. P.; Kim H. J.; Mittag T.; Taylor J. P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. K.; Spille J. H.; Hecht M.; Lee C.; Li C.; Grube V.; Cisse I. I. Mediator and RNA Polymerase II Clusters Associate in Transcription-Dependent Condensates. Science 2018, 361, 412–415. 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S.; Dugast-Darzacq C.; Liu Z.; Dong P.; Dailey G. M.; Cattoglio C.; Heckert A.; Banala S.; Lavis L.; Darzacq X.; Tjian R.. Imaging Dynamic and Selective Low-Complexity Domain Interactions That Control Gene Transcription. Science 2018, 361, eaar2555. 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari B. R.; Dall A.; Boija A.; Klein I. A.; Coffey E. L.; Shrinivas K.; Abraham B. J.; Hannett N. M.; Zamudio, A. V; Manteiga J. C.; Li C. H.; Guo Y. E.; Day D. S.; Schuijers J.; Vasile E.; Malik S.; Hnisz D.; Ihn Lee T.; Cisse I. I.; Roeder R. G.. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E.; Spruijt E.; Hansen M. M. K.; Dubuc E.; Groen J.; Chokkalingam V.; Piruska A.; Heus H. A.; Huck W. T. S. Enhanced Transcription Rates in Membrane-Free Protocells Formed by Coacervation of Cell Lysate. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11692–11697. 10.1073/pnas.1222321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron-Sandoval L.-P.; Heris H. K.; Hendricks A. G.; Ehrlicher A. J.; Francois P.; Pappu R. V; Michnick S. W.. Endocytosis Caused by Liquid-Liquid Phase Separation of Proteins. 2018BioRxiv doi.org/10.1101/145664 (accessed: March 29, 2020).
- Chong P. A.; Forman-Kay J. D. Liquid–Liquid Phase Separation in Cellular Signaling Systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. 10.1016/j.sbi.2016.08.001. [DOI] [PubMed] [Google Scholar]
- Beutel O.; Maraspini R.; Pombo-García K.; Martin-Lemaitre C.; Honigmann A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936. 10.1016/j.cell.2019.10.011. [DOI] [PubMed] [Google Scholar]
- Sheth U.; Pitt J.; Dennis S.; Priess J. R. Perinuclear P Granules Are the Principal Sites of MRNA Export in Adult C. Elegans Germ Cells. Development 2010, 137, 1305–1314. 10.1242/dev.044255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. Y. C.; Alvarez S.; Kondo Y.; Kwang Lee Y.; Chung J. K.; Monatrice Lam H. Y.; Biswas K. H.; Kuriyan J.; Groves J. T. A Molecular Assembly Phase Transition and Kinetic Proofreading Modulate Ras Activation by SOS. Science 2019, 363, 1098–1103. 10.1126/science.aau5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L. B.; Zhang X.; Ditlev J. A.; Rosen M. K. Stoichiometry Controls Activity of Phase-Separated Clusters of Actin Signaling Proteins. Science 2019, 363, 1093–1097. 10.1126/science.aau6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X.; Ditlev J. A.; Hui E.; Xing W.; Banjade S.; Okrut J.; King D. S.; Taunton J.; Rosen M. K.; Vale R. D. Phase Separation of Signaling Molecules Promotes T Cell Receptor Signal Transduction. Science 2016, 352, 595–599. 10.1126/science.aad9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditlev J. A.; Vega A. R.; Köster D. V.; Su X.; Tani T.; Lakoduk A. M.; Vale R. D.; Mayor S.; Jaqaman K.; Rosen M. K. A Composition-Dependent Molecular Clutch between T Cell Signaling Condensates and Actin. eLife 2019, 8, 1–44. 10.7554/eLife.42695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron-Sandoval L. P.; Michnick S. W. Mechanics, Structure and Function of Biopolymer Condensates. J. Mol. Biol. 2018, 430, 4754–4761. 10.1016/j.jmb.2018.06.023. [DOI] [PubMed] [Google Scholar]
- Wunder T.; Cheng S. L. H.; Lai S. K.; Li H. Y.; Mueller-Cajar O. The Phase Separation Underlying the Pyrenoid-Based Microalgal Rubisco Supercharger. Nat. Commun. 2018, 9, 1–10. 10.1038/s41467-018-07624-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S.; Kim Y.; Szczepaniak K.; Chen C. C. H.; Eckmann C. R.; Myong S.; Brangwynne C. P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7189–7194. 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm S. A.; Kamenz J.; Nöldeke E. R.; Neu A.; Zocher G.; Sprangers R. In Vitro Reconstitution of a Cellular Phase-Transition Process That Involves the MRNA Decapping Machinery. Angew. Chem., Int. Ed. 2014, 53, 7354–7359. 10.1002/anie.201402885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng N.-N.; Huck W. T. S. Microfluidic Formation of Monodisperse Coacervate Organelles in Liposomes. Angew. Chem., Int. Ed. 2017, 56, 1–6. 10.1002/anie.201703145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumiller W. M.; Keating C. D. Phosphorylation-Mediated RNA/Peptide Complex Coacervation as a Model for Intracellular Liquid Organelles. Nat. Chem. 2016, 8, 129–137. 10.1038/nchem.2414. [DOI] [PubMed] [Google Scholar]
- Koga S.; Williams D. S.; Perriman A. W.; Mann S. Peptide-Nucleotide Microdroplets as a Step towards a Membrane-Free Protocell Model. Nat. Chem. 2011, 3, 720–724. 10.1038/nchem.1110. [DOI] [PubMed] [Google Scholar]
- Alberti S.; Gladfelter A.; Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande S.; Brandenburg F.; Lau A.; Last M. G. F.; Spoelstra W. K.; Reese L.; Wunnava S.; Dogterom M.; Dekker C. Spatiotemporal Control of Coacervate Formation within Liposomes. Nat. Commun. 2019, 10, 1–11. 10.1038/s41467-019-09855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande S.; Caspi Y.; Meijering A. E. C.; Dekker C. Octanol-Assisted Liposome Assembly on Chip. Nat. Commun. 2016, 7, 10447. 10.1038/ncomms10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano Y.; Kamiya M.; Kanda K.; Ueno T.; Hirose K.; Nagano T. Evolution of Fluorescein as a Platform for Finely Tunable Fluorescence Probes. J. Am. Chem. Soc. 2005, 127, 4888–4894. 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- Watanabe R.; Komatsu T.; Sakamoto S.; Urano Y.; Noji H. High-Throughput Single-Molecule Bioassay Using Micro-Reactor Arrays with a Concentration Gradient of Target Molecules. Lab Chip 2018, 18, 2849–2853. 10.1039/C8LC00535D. [DOI] [PubMed] [Google Scholar]
- Deshpande S.; Dekker C. On-Chip Microfluidic Production of Cell-Sized Liposomes. Nat. Protoc. 2018, 13, 856–874. 10.1038/nprot.2017.160. [DOI] [PubMed] [Google Scholar]
- Priftis D.; Laugel N.; Tirrell M. Thermodynamic Characterization of Polypeptide Complex Coacervation. Langmuir 2012, 28, 15947–15957. 10.1021/la302729r. [DOI] [PubMed] [Google Scholar]
- Priftis D.; Tirrell M. Phase Behaviour and Complex Coacervation of Aqueous Polypeptide Solutions. Soft Matter 2012, 8, 9396–9405. 10.1039/C2SM25604E. [DOI] [Google Scholar]
- Hunter T. Why Nature Chose Phosphate to Modify Proteins. Philos. Trans. R. Soc., B 2012, 367, 2513–2516. 10.1098/rstb.2012.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D. W.Proton Flux across Model and Biological Membranes. In Membrane Processes; Springer: New York, 1984; pp 111–120. [Google Scholar]
- Oglęcka K.; Rangamani P.; Liedberg B.; Kraut R. S.; Parikh A. N. Oscillatory Phase Separation in Giant Lipid Vesicles Induced by Transmembrane Osmotic Differentials. eLife 2014, 3, 1–18. 10.7554/eLife.03695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priftis D.; Farina R.; Tirrell M. Interfacial Energy of Polypeptide Complex Coacervates Measured via Capillary Adhesion. Langmuir 2012, 28, 8721–8729. 10.1021/la300769d. [DOI] [PubMed] [Google Scholar]
- Spruijt E.; Sprakel J.; Cohen Stuart M. A.; Van Der Gucht J. Interfacial Tension between a Complex Coacervate Phase and Its Coexisting Aqueous Phase. Soft Matter 2010, 6, 172–178. 10.1039/B911541B. [DOI] [Google Scholar]
- Smith M. C.; Crist R. M.; Clogston J. D.; McNeil S. E. Zeta Potential: A Case Study of Cationic, Anionic, and Neutral Liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. 10.1007/s00216-017-0527-z. [DOI] [PubMed] [Google Scholar]
- Yehudai-resheff S.; Hirsh M.; Schuster G. Polynucleotide Phosphorylase Functions as Both an Exonuclease and a Poly (A) Polymerase in Spinach Chloroplasts. Mol. Cell. Biol. 2001, 21, 5408–5416. 10.1128/MCB.21.16.5408-5416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.; Wang Y.; Yang A.; Yang G. The Effect of PH on Charge Inversion and Condensation of DNA. Soft Matter 2016, 12, 6669–6674. 10.1039/C6SM01344A. [DOI] [PubMed] [Google Scholar]
- Irie M. Light-Induced Reversible PH Change. J. Am. Chem. Soc. 1983, 105, 2078–2079. 10.1021/ja00345a075. [DOI] [Google Scholar]
- Kouyama T.; Kouyama A. N.; Ikegami A. Bacteriorhodopsin Is a Powerful Light-Driven Proton Pump. Biophys. J. 1987, 51, 839–841. 10.1016/S0006-3495(87)83411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M.; Jug F.; Julou T.; Deshpande S.; Pfohl T.; Silander O. K.; Myers G.; Van Nimwegen E. Monitoring Single-Cell Gene Regulation under Dynamically Controllable Conditions with Integrated Microfluidics and Software. Nat. Commun. 2018, 9, 212. 10.1038/s41467-017-02505-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahas K. Microfluidic Platform for the Characterisation of Membrane Active Antimicrobials. Lab Chip 2019, 19, 837–844. 10.1039/C8LC00932E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love C.; Steinkühler J.; Gonzales D. T.; Yandrapalli N.; Robinson T.; Dimova R.; Tang T. -Y. D.. Reversible PH Responsive Coacervate Formation in Lipid Vesicles Activates Dormant Enzymatic Reactions. Angew. Chem. 2020, 10.1002/ange.202081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D. W. Proton Permeation of Lipid Bilayers. J. Bioenerg. Biomembr. 1987, 19, 457–479. [DOI] [PubMed] [Google Scholar]
- Wodzinska K.; Winterhalter M.; Heimburg T.; Blicher A.; Fidorra M. The Temperature Dependence of Lipid Membrane Permeability, Its Quantized Nature, and the Influence of Anesthetics. Biophys. J. 2009, 96, 4581–4591. 10.1016/j.bpj.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaich M.; Cama J.; Al Nahas K.; Sobota D.; Sleath H.; Jahnke K.; Deshpande S.; Dekker C.; Keyser U. F. An Integrated Microfluidic Platform for Quantifying Drug Permeation across Biomimetic Vesicle Membranes. Mol. Pharmaceutics 2019, 16, 2494–2501. 10.1021/acs.molpharmaceut.9b00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D. W.; Nichols J. W. Proton-Hydroxide Permeability of Liposomes. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 165–168. 10.1073/pnas.80.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.