

Abstract
Alveolar type II (ATII) cells are essential to lung function and a primary site of influenza A virus (IAV) replication. Effects of IAV infection on ATII cell microRNA (miR) expression have not been comprehensively investigated. Infection of C57BL/6 mice with 10,000 or 100 pfu/mouse of IAV A/WSN/33 (H1N1) significantly altered expression of 73 out of 1,908 mature murine miRs in ATII cells at 2 days post-infection (d.p.i.) and 253 miRs at 6 d.p.i. miR-155-5p (miR-155) showed the greatest increase in expression within ATII cells at both timepoints and the magnitude of this increase correlated with inoculum size and pulmonary edema severity. Influenza-induced lung injury was attenuated in C57BL/6-congenic miR-155-knockout mice without affecting viral replication. Attenuation of lung injury was dependent on deletion of miR-155 from stromal cells and was recapitulated in ATII cell-specific miR-155-knockout mice. These data suggest that ATII cell miR-155 is a potential therapeutic target for IAV-induced ARDS.
Keywords: Influenza A virus, microRNA, alveolar type II cell, acute respiratory distress syndrome, mouse
Introduction
Currently, influenza management is based around prophylactic vaccination and use of antiviral drugs early in infection (Uyeki, 2017). However, approximately 20% of patients with severe influenza A virus (IAV) infection develop acute respiratory distress syndrome (ARDS), which is associated with poor prognosis. Once ARDS has developed, the only treatment option is non-specific supportive management in an intensive care unit, which can be of limited efficacy in some cases (Westall and Paraskeva, 2011). In order to develop new drugs that can ameliorate potentially lethal IAV-induced ARDS, we therefore need a better understanding of the pathogenesis of this syndrome.
microRNAs (miRs) are short non-coding RNAs that can regulate the expression of multiple genes simultaneously at the post-transcriptional level by inducing either mRNA degradation or translational repression (Brown et al., 2014). Importantly, a single miR can simultaneously regulate the expression of multiple transcripts. Likewise, multiple different miRs can regulate expression of a single transcript (Martinez-Nunez et al., 2014). miRs therefore play a key role in shaping the transcriptome and consequently the proteome of cells, and provide an additional layer of orchestration to control host immunity and cell survival. Generally speaking, individual miRs are conserved and play comparable roles in mice and humans. Importantly, several miRs have been implicated in the pathogenesis of ARDS and have been shown to either promote or attenuate disease in mouse models (Rajasekaran et al., 2016).
We have shown previously that infection of mice with H1N1 influenza A/WSN/33 (WSN) virus results in development of ARDS, and that is associated with significant impairment of alveolar type II (ATII) cell ion transport, surfactant protein synthesis, and de novo phospholipid synthesis (Hofer et al., 2015; Traylor et al., 2013; Wolk et al., 2008; Woods et al., 2016). We hypothesized that, in order to identify those miRs that are most relevant to the pathogenesis of IAV-induced ARDS (and therefore the most promising therapeutic targets), we should take a reductive approach and focus on changes in the miRnome in a single, functionally important lung cell type following WSN infection. Because they are responsible for both surfactant synthesis and regulation of the depth of the alveolar lining fluid, ATII cells are essential for normal lung function (Mason, 2006). Importantly, ATII cells are also the primary site of IAV infection and replication in the distal lung and participate in the innate immune response to this virus (Ibricevic et al., 2006; Thompson et al., 2006; Wang et al., 2011). We therefore elected to focus on the effects of WSN infection on the miRnome of this important cell type.
Using a targeted array we found that WSN induced progressive changes in the ATII cell miRnome. Interestingly, significantly fewer miRs were induced than repressed, which might reflect inhibition of host miR transcript poly-adenylation by the viral NS-1A protein in infected cells (Noah et al., 2003). Of all identified miRs, miR-155-5p (miR-155) showed the greatest increase in expression within ATII cells at both 2 and 6 d.p.i. Furthermore, the magnitude of the increase in ATII cell miR-155 expression correlated with inoculum size and severity of pulmonary edema. In addition, we found that that, relative to IAV-infected WT controls, influenza-induced ARDS was attenuated in C57BL/6-congenic global miR-155-knockout (155-KO) mice. Moreover, adoptive transfer studies and experiments using ATII cell-specific miR-155-knockout (155-KOATII) mice demonstrated that ARDS attenuation was dependent on deletion of miR-155 from ATII cells but not from myeloid cells.
Overall, our data show for the first time that in vivo IAV infection progressively alters the ATII cell miRnome. Moreover, they indicate that induction of miR-155 in ATII cells at high levels plays a significant causal role in development of severe ARDS in influenza-infected subjects.
Materials and Methods
Mice.
C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). C57BL/6-congenic global 155-KO mice (Stock No. 007745) were purchased from Jackson Laboratory (Bar Harbor, ME) and bred in-house. C57BL/6-congenic sp-c/cre+/+ (B6.Cg-Tg(Gfap-cre)77.6Mvs/J) and miR-155fl/fl (B6.Mir155tm1.1Ggard/J) mice were also obtained from Jackson Laboratory (Stock Nos. 028054 and 026700, respectively). F1 crosses were generated then interbred to generate homozygous sp-c/cre+/+ x miR-155fl/fl (155-KOATII) offspring. Genotyping was performed by PCR on tailsnip DNA in accordance with protocols from the vendor. Mice of both KO strains bred normally, had average-sized litters, and displayed no obvious phenotype.
All mice were maintained in ventilated racks with ad libitum food and water as well as environmental enrichment materials. All animal procedures were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of The Ohio State University. All animal experiments and procedures were carried out so as to ensure minimal suffering and in accordance with ARRIVE guidelines on animal research (Kilkenny et al., 2010).
Virus.
All experiments used a single stock of influenza A/WSN/33 (H1N1) virus, which si a mouse-adapted IAV strain, diluted as necessary in PBS with 0.1% BSA. The virus stock was generated in eggs and was the fourth passage after acquisition from ATCC. The stock was tested for endotoxin and Mycoplasma pulmonis contamination and was found to be free from both.
Mouse infection.
As in our previous studies, 8 to 12 week-old mice of both genders were inoculated intranasally with 100 or 10,000 plaque-forming units (pfu) of influenza A/WSN/33 (H1N1) virus in 50 μl PBS with 0.1% BSA under ketamine/xylazine anesthesia, (Hickman-Davis et al., 2006; Hofer et al., 2015) Mice were individually marked and weighed every 2 days. Data for each experimental group were derived from at least 2 independent infections.
ATII cell isolation.
ATII cells were isolated from uninfected and WSN-infected mice by a standard lung digestion protocol using dispase II (Becton Dickinson, San Jose, CA) and pancreatic DNase (Sigma-Aldrich, St. Louis, MO) (Corti et al., 1996). Leukocytes were removed by panning with rat polyclonal anti-murine CD45 and anti-murine CD16/CD32 antibodies (both Becton Dickinson). Non-adherent cells were collected, pelleted by centrifugation, resuspended in normal saline, and counted using a hemocytometer. Purity of isolated cell ATII cell preparations was determined by visualization of lamellar bodies in modified Papanicolaou-stained cytospins.
Bone marrow transfer (BMT).
BMT was performed as in our previous studies (Aeffner et al., 2014; Woods et al., 2015). 24 hours before transplantation, uninfected bone marrow-recipient mice were irradiated in a 137Cs irradiator. Mice received 1,000 cGy split between 2 doses (8 hours apart). Immediately prior to transfer, low-density bone marrow was isolated from euthanized uninfected donor mice. Long bones were removed from the hind limbs and flushed with conditioned medium to extract bone marrow. 5 × 106 bone marrow cells (in 0.5 ml saline) were transplanted by tail vein injection into recipient mice. Recipients were provided with Baytril (via water bottles) for 2 to 3 weeks peri-irradiation. This protocol resulted in no outward clinical signs. Infection studies were performed a minimum of 6 weeks post-BMT.
Measurement of airway resistance and lung compliance.
As in our previous studies, we measured lung function using the forced oscillation technique(Irvin and Bates, 2003). Mice were anesthetized by intraperitoneal injections of valium (5 mg/kg) then ketamine (200 mg/kg, i.p.) 6 minutes later. Once at a surgical plane of anesthesia mice were tracheotomized, inubated (using a trimmed sterile 18-gauge intravenous catheter), and connected to a flexiVent computer-controlled piston ventilator (SciReq, Montreal, Canada). All mice were ventilated with the following parameters: tidal volume 8-ml/kg, frequency 150 breaths/minute, with 2 to 3 cm H2O positive end-expiratory pressure (PEEP). After two total lung capacity maneuvers to standardize volume history, static lung compliance was measured by performing a volume-stepped discontinuous pressure-volume loop. Pressure and flow data were then collected over a series of 10 consecutive repeats of standardized volume perturbation maneuvers. These data were used to calculate total lung resistance using the single-compartment model.
miR arrays.
Total ATII cell RNA was isolated using a miRNEasy Micro Kit (Qiagen; Germantown, MD). 500ng RNA/mouse was analyzed using a GeneChip® miRNA 4.0 Array (Affymetrix; Santa Clara, CA). This array incorporates 30,424 mature miR probe sets and can characterize expression of 1,908 mature murine miRs. The cutoff value for significance between ATII cells from infected mice and uninfected controls at each timepoint was P≤0.01.
qRT-PCR.
To validate changes in expression of miR-155 and miR-4487, we performed qRT-PCR using mmu-miR-specific looped primers (Thermo Fisher Scientific, Waltham, MA). Total snorna u38b, snorna u43, snorna202, 18s rrna, and 7s rrna were used as internal controls (we showed previously that infection does not alter 18s rrna level (Hofer et al., 2015)). Data were normalized to snorna202. Assays were performed in ATII cells from 3 mice per group.
Other methods.
All other methods were performed as previously described (Aeffner et al., 2013; Aeffner et al., 2012).
Statistical Analyses.
Descriptive statistics (mean and standard error) were calculated using Instat software (GraphPad, San Diego, CA). Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. Statistical analyses of datasets were made by unpaired ANOVA, with a post hoc Tukey-Kramer multiple comparison post-test. All data are presented as mean ± S.E.M. P<0.05 was considered statistically significant.
Results
Infection of C57BL/6 mice with IAV induces significant alterations in the ATII cell miRNome.
As in our previous studies, C57BL/6 mice were infected with an acutely lethal dose of WSN IAV. ATII cells were isolated to >97% purity from uninfected mice and at 2 and 6 days post-infection (d.p.i.) by a standard lung digestion protocol. As previously (Hofer et al., 2015), WSN infection resulted in a progressive decline in ATII cell yields, which were significantly reduced at 6 d.p.i. (Fig. 1a). Mock infection for 6 days had no such effect.
Figure 1. Infection of C57BL/6 mice with IAV induces significant alterations in the ATII cell miRNome.
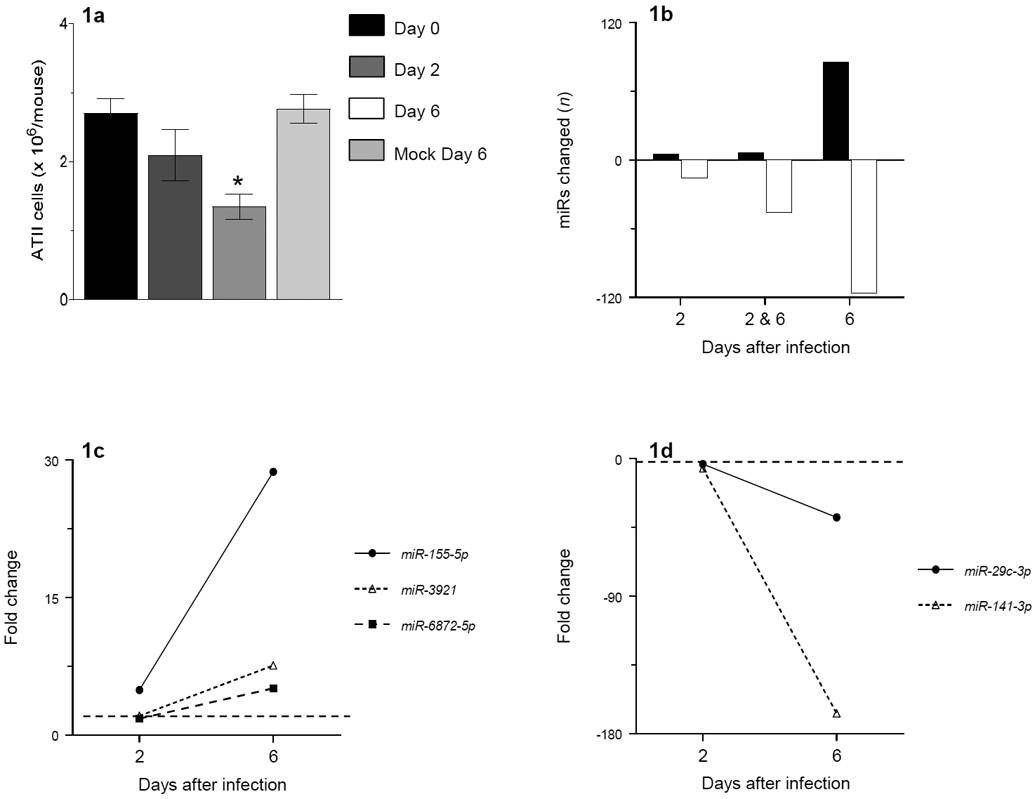
(a) ATII cell yields in uninfected C57BL/6 mice, C57BL/6 mice infected with H1N1 influenza A/WSN/33 (10,000 pfu/mouse) for 2 and 6 days, and C57BL/6 mice mock-infected for 6 days; (b): Number of ATII cell miRs for which level of expression increased (black bars) or decreased (white bars) > 2-fold at 2 and/or 6 days post-infection (d.p.i.), relative to ATII cells from uninfected control mice; (c) Post-infection fold increases in expression of miRs that were most elevated at 6 d.p.i.; and (d) Post-infection fold decreases in expression of miRs that were most reduced at 6 d.p.i.. n=3 per timepoint. Broken horizontal line indicates 2-fold cut-off for significance.
Following ATII cell isolation, total RNA was extracted for analysis of miR expression using GeneChip miRNA 4.0 Arrays. Overall, WSN infection resulted in statistically-significant (more than 2-fold; P<0.01) changes in expression of 73 out of 1,908 mature murine miRs in ATII cells at 2 d.p.i. (approximately 4%), relative to uninfected controls (Table 1). The number of miRs that changed significantly increased to 253 at 6 d.p.i. (approximately 13%; Table 2).
Table 1.
ATII cells miRs that change significantly (>2-fold; P<0.05, relative to ATII cells from uninfected mice) at 2 d.p.i.
| miR I.D. | Fold increase |
|---|---|
| hsa-miR-155-5p | 4.91 |
| hsa-miR-4487 | 3.44 |
| hsa-let-7f-5p | 3.27 |
| hsa-miR-200b-3p | 3.06 |
| hsa-miR-98-5p | 2.99 |
| hsa-miR-4443 | 2.63 |
| hsa-mir-6722 | 2.16 |
| hsa-miR-375 | 2.11 |
| hsa-miR-15b-5p | 2.10 |
| hsa-let-7g-5p | 2.05 |
| hsa-miR-10a-5p | 2.01 |
| miR I.D. | Fold decrease |
| hsa-miR-937-3p | 0.50 |
| hsa-miR-4498 | 0.50 |
| hsa-miR-145-5p | 0.50 |
| hsa-miR-130a-3p | 0.50 |
| hsa-miR-5708 | 0.48 |
| hsa-miR-1538 | 0.48 |
| hsa-mir-4732 | 0.48 |
| hsa-miR-1298-3p | 0.48 |
| hsa-miR-31-5p | 0.48 |
| hsa-let-7i-3p | 0.46 |
| hsa-miR-4657 | 0.45 |
| hsa-miR-16-2-3p | 0.45 |
| hsa-miR-199a-5p | 0.43 |
| hsa-miR-16-5p | 0.43 |
| hsa-miR-200a-3p | 0.43 |
| hsa-miR-3907 | 0.42 |
| hsa-miR-181c-3p | 0.42 |
| hsa-mir-4704 | 0.42 |
| hsa-miR-1298-5p | 0.42 |
| hsa-miR-188-5p | 0.41 |
| hsa-miR-22-5p | 0.40 |
| hsa-miR-138-5p | 0.40 |
| hsa-miR-10a-3p | 0.40 |
| hsa-miR-15a-5p | 0.40 |
| hsa-miR-27a-3p | 0.39 |
| hsa-miR-4306 | 0.39 |
| hsa-miR-451a | 0.39 |
| hsa-miR-34c-5p | 0.39 |
| hsa-mir-4800 | 0.38 |
| hsa-miR-1184 | 0.38 |
| hsa-miR-671-5p | 0.38 |
| hsa-miR-4454 | 0.37 |
| hsa-miR-143-3p | 0.37 |
| hsa-miR-224-3p | 0.37 |
| hsa-miR-30e-5p | 0.36 |
| hsa-miR-19a-3p | 0.36 |
| hsa-miR-6882-3p | 0.35 |
| hsa-miR-497-5p | 0.35 |
| hsa-miR-455-5p | 0.35 |
| hsa-miR-3064-5p | 0.34 |
| hsa-miR-339-3p | 0.34 |
| hsa-mir-6876 | 0.34 |
| hsa-miR-8063 | 0.32 |
| hsa-miR-6753-3p | 0.31 |
| hsa-miR-8084 | 0.30 |
| hsa-miR-93-3p | 0.30 |
| hsa-miR-330-3p | 0.30 |
| hsa-miR-22-3p | 0.30 |
| hsa-miR-503-5p | 0.28 |
| hsa-miR-4485 | 0.28 |
| hsa-miR-29c-3p | 0.27 |
| hsa-miR-297 | 0.27 |
| hsa-miR-6868-3p | 0.27 |
| hsa-miR-140-5p | 4 |
| hsa-miR-181a-3p | 0.24 |
| hsa-miR-221-5p | 0.24 |
| hsa-miR-7975 | 0.23 |
| hsa-miR-29b-3p | 0.21 |
| hsa-miR-652-5p | 0.19 |
| hsa-miR-708-5p | 0.19 |
| hsa-miR-301a-3p | 0.16 |
| hsa-miR-141-3p | 0.16 |
Expression of miRs in bold font is changed at both 2 and 6 d.p.i.
Table 2.
ATII cells miRs that change significantly (>2-fold; P<0.05, relative to ATII cells from uninfected mice) at 6 d.p.i.
| miR I.D. | Fold increase |
|---|---|
| hsa-miR-155-5p | 28.70* |
| hsa-miR-3921 | 7.57 |
| hsa-miR-6872-5p | 5.06 |
| hsa-miR-4656 | 4.82 |
| hsa-miR-5094 | 4.64 |
| hsa-miR-7847-3p | 4.56 |
| hsa-miR-4298 | 4.48 |
| hsa-miR-4487 | 4.41 |
| hsa-mir-320e | 4.34 |
| hsa-mir-6722 | 4.34* |
| hsa-miR-6732-5p | 4.09 |
| hsa-miR-8075 | 3.98 |
| hsa-miR-6778-5p | 3.7 |
| hsa-miR-4749-5p | 3.69 |
| hsa-miR-6816-5p | 3.53 |
| hsa-miR-2467-3p | 3.49 |
| hsa-miR-6779-5p | 3.19 |
| hsa-mir-3154 | 3.09 |
| hsa-miR-6085 | 3.08 |
| hsa-miR-6775-5p | 2.99 |
| hsa-miR-337-3p | 2.93 |
| hsa-miR-15b-5p | 2.90 |
| hsa-miR-4697-3p | 2.83 |
| hsa-miR-6132 | 2.74 |
| hsa-miR-6813-5p | 2.74 |
| hsa-miR-149-3p | 2.73 |
| hsa-miR-1587 | 2.72 |
| hsa-miR-6821-5p | 2.70 |
| hsa-miR-3196 | 2.69 |
| hsa-miR-378h | 2.69 |
| hsa-miR-6791-5p | 2.66 |
| hsa-miR-328-5p | 2.64 |
| hsa-miR-5581-3p | 2.58 |
| hsa-miR-1237-5p | 2.55 |
| hsa-miR-6794-5p | 2.53 |
| hsa-miR-6858-5p | 2.49 |
| hsa-miR-762 | 2.47 |
| hsa-miR-4497 | 2.46 |
| hsa-miR-6848-5p | 2.45 |
| hsa-miR-4443 | 2.45 |
| hsa-mir-6776 | 2.45 |
| hsa-miR-4281 | 2.45 |
| hsa-mir-6872 | 2.44 |
| hsa-miR-937-5p | 2.44 |
| hsa-miR-6756-5p | 2.42 |
| hsa-miR-1233-5p | 2.41 |
| hsa-mir-3658 | 2.36 |
| hsa-miR-6722-3p | 2.34 |
| hsa-miR-663a | 2.34 |
| hsa-miR-4507 | 2.34 |
| hsa-miR-6509-3p | 2.33 |
| hsa-miR-6088 | 2.31 |
| hsa-miR-5787 | 2.31 |
| hsa-miR-6886-3p | 2.30 |
| hsa-miR-4530 | 2.29 |
| hsa-miR-6765-5p | 2.28 |
| hsa-miR-1908-5p | 2.27 |
| hsa-miR-4734 | 2.25 |
| hsa-miR-204-5p | 2.25 |
| hsa-miR-4763-3p | 2.24 |
| hsa-miR-1228-5p | 2.24 |
| hsa-miR-455-3p | 2.22 |
| hsa-miR-3620-5p | 2.22 |
| hsa-miR-1207-5p | 2.20 |
| hsa-miR-4516 | 2.20 |
| hsa-miR-6727-5p | 2.19 |
| hsa-let-7f-5p | 2.18 |
| hsa-miR-3960 | 2.17 |
| hsa-miR-3665 | 2.16 |
| hsa-mir-5703 | 2.16 |
| hsa-miR-132-3p | 2.15 |
| hsa-miR-6724-5p | 2.14 |
| hsa-miR-4463 | 2.13 |
| hsa-mir-4281 | 2.12 |
| hsa-miR-3940-5p | 2.11 |
| hsa-miR-638 | 2.11 |
| hsa-miR-4488 | 2.11 |
| hsa-miR-4787-5p | 2.10 |
| hsa-miR-1250-5p | 2.10 |
| hsa-miR-8069 | 2.09 |
| hsa-miR-6803-5p | 2.09 |
| hsa-miR-6786-5p | 2.06 |
| hsa-miR-3663-5p | 2.06 |
| hsa-miR-6090 | 2.06 |
| hsa-miR-4651 | 2.04 |
| hsa-miR-4492 | 2.04 |
| hsa-miR-1915-3p | 2.03 |
| hsa-miR-4505 | 2.02 |
| hsa-miR-6125 | 2.02 |
| hsa-miR-4466 | 2.02 |
| hsa-miR-8072 | 2.01 |
| miR I.D. | Fold decrease |
| hsa-miR-5708 | 0.50 |
| hsa-miR-214-5p | 0.50 |
| hsa-miR-640 | 0.49 |
| hsa-miR-3064-5p | 0.49 |
| hsa-miR-125a-3p | 0.48 |
| hsa-miR-5002-5p | 0.47 |
| hsa-miR-557 | 0.46 |
| hsa-miR-7977 | 0.46 |
| hsa-miR-187-5p | 0.45 |
| hsa-miR-100-5p | 0.45 |
| hsa-miR-134-5p | 0.45 |
| hsa-miR-185-5p | 0.45 |
| hsa-miR-133b | 0.45 |
| hsa-miR-93-5p | 0.45 |
| hsa-miR-30a-3p | 0.44 |
| hsa-miR-4286 | 0.44 |
| hsa-miR-34c-3p | 0.44 |
| hsa-miR-181a-5p | 0.43 |
| hsa-miR-23c | 0.43 |
| hsa-miR-1226-5p | 0.42 |
| hsa-miR-3646 | 0.42 |
| hsa-miR-151a-5p | 0.42 |
| hsa-miR-23b-3p | 0.42 |
| hsa-miR-455-5p | 0.41 |
| hsa-miR-23a-3p | 0.41 |
| hsa-miR-224-5p | 0.41 |
| hsa-miR-195-5p | 0.40 |
| hsa-miR-151b | 0.40 |
| hsa-miR-2392 | 0.40 |
| hsa-miR-23b-5p | 0.39 |
| hsa-miR-26a-5p | 0.39 |
| hsa-miR-183-3p | 0.39 |
| hsa-miR-106b-5p | 0.39 |
| hsa-miR-28-5p | 0.38 |
| hsa-miR-574-5p | 0.38 |
| hsa-miR-99b-5p | 0.38 |
| hsa-miR-1184 | 0.37 |
| hsa-miR-4668-5p | 0.37 |
| hsa-miR-188-5p | 0.37 |
| hsa-miR-181c-3p | 0.37 |
| hsa-miR-96-5p | 0.37 |
| hsa-miR-1298-5p | 0.36 |
| hsa-miR-328-3p | 0.36 |
| hsa-miR-30c-5p | 0.36 |
| hsa-miR-10a-3p | 0.36 |
| hsa-miR-6734-5p | 0.36 |
| hsa-miR-16-2-3p | 0.35 |
| hsa-miR-182-5p | 0.35 |
| hsa-miR-330-5p | 0.35 |
| hsa-miR-1260b | 0.34 |
| hsa-miR-24-3p | 0.34 |
| hsa-miR-411-5p | 0.34 |
| hsa-miR-320e | 0.33 |
| hsa-miR-382-5p | 0.33 |
| hsa-miR-642b-3p | 0.32 |
| hsa-miR-126-3p | 0.32 |
| hsa-miR-1229-5p | 0.32 |
| hsa-miR-99a-5p | 0.32 |
| hsa-miR-6862-3p | 0.32 |
| hsa-miR-138-5p | 0.32 |
| hsa-miR-4657 | 0.32 |
| hsa-miR-3154 | 0.31 |
| hsa-miR-6870-5p | 0.31 |
| hsa-miR-30c-2-3p | 0.31 |
| hsa-miR-542-5p | 0.31 |
| hsa-miR-3613-3p | 0.31 |
| hsa-miR-532-5p | 0.31 |
| hsa-miR-339-3p | 0.31 |
| hsa-miR-30b-5p | 0.30 |
| hsa-miR-18b-5p | 0.29 |
| hsa-miR-652-3p | 0.29 |
| hsa-miR-29c-5p | 0.29 |
| hsa-miR-107 | 0.29 |
| hsa-miR-335-5p | 0.29 |
| hsa-miR-181b-5p | 0.29 |
| hsa-miR-203a | 0.28 |
| hsa-miR-199a-5p | 0.28 |
| hsa-miR-19b-3p | 0.28 |
| hsa-miR-27b-3p | 0.28 |
| hsa-miR-224-3p | 0.27 |
| hsa-miR-103a-3p | 0.27 |
| hsa-mir-34c | 0.27 |
| hsa-miR-16-5p | 0.26 |
| hsa-miR-130a-3p | 0.26 |
| hsa-miR-135a-3p | 0.25 |
| hsa-miR-29a-3p | 0.25 |
| hsa-miR-7-1-3p | 0.24 |
| hsa-miR-193a-5p | 0.24 |
| hsa-miR-6891-5p | 0.24 |
| hsa-miR-200b-5p | 0.23 |
| hsa-miR-143-3p | 0.22 |
| hsa-miR-5100 | 0.22 |
| hsa-miR-4306 | 0.22 |
| hsa-miR-4454 | 0.22 |
| hsa-miR-378e | 0.22 |
| hsa-miR-181a-3p | 0.21 |
| hsa-miR-27b-5p | 0.21 |
| hsa-miR-221-5p | 0.21 |
| hsa-miR-874-3p | 0.21 |
| hsa-miR-151a-3p | 0.21 |
| hsa-miR-27a-5p | 0.20 |
| hsa-miR-378a-3p | 0.20 |
| hsa-miR-4485 | 0.20 |
| hsa-miR-324-5p | 0.20 |
| hsa-miR-22-5p | 0.20 |
| hsa-miR-425-3p | 0.19 |
| hsa-miR-30e-3p | 0.19 |
| hsa-miR-145-5p | 0.19 |
| hsa-miR-30a-5p | 0.19 |
| hsa-miR-338-5p | 0.18 |
| hsa-miR-330-3p | 0.18 |
| hsa-miR-15a-5p | 0.18* |
| hsa-miR-22-3p | 0.18 |
| hsa-miR-500a-3p | 0.178 |
| hsa-miR-27a-3p | 0.178* |
| hsa-miR-6825-5p | 0.17 |
| hsa-miR-19a-3p | 0.16* |
| hsa-miR-7975 | 0.16 |
| hsa-miR-93-3p | 0.16 |
| hsa-miR-127-3p | 0.15 |
| hsa-miR-181d-5p | 0.15 |
| hsa-miR-34a-5p | 0.15 |
| hsa-miR-326 | 0.14 |
| hsa-miR-652-5p | 0.14 |
| hsa-miR-331-3p | 0.14 |
| hsa-miR-122-5p | 0.14 |
| hsa-miR-378a-5p | 0.14 |
| hsa-miR-152-3p | 0.13 |
| hsa-miR-6780b-5p | 0.13 |
| hsa-miR-24-2-5p | 0.13 |
| hsa-miR-671-5p | 0.13* |
| hsa-miR-148b-3p | 0.12 |
| hsa-miR-29b-2-5p | 0.12 |
| hsa-miR-497-5p | 0.12* |
| hsa-miR-181c-5p | 0.12 |
| hsa-miR-378c | 0.11 |
| hsa-miR-26b-5p | 0.11 |
| hsa-miR-193a-3p | 0.11 |
| hsa-miR-339-5p | 0.11 |
| hsa-miR-378g | 0.11 |
| hsa-miR-503-5p | 0.11* |
| hsa-miR-379-5p | 0.10 |
| hsa-miR-99b-3p | 0.10 |
| hsa-miR-31-5p | 0.10 |
| hsa-miR-324-3p | 0.10 |
| hsa-miR-378d | 0.09 |
| hsa-miR-194-5p | 0.09 |
| hsa-miR-708-5p | 0.09* |
| hsa-miR-200a-5p | 0.09 |
| hsa-miR-378f | 0.08 |
| hsa-miR-422a | 0.08 |
| hsa-miR-148a-3p | 0.08 |
| hsa-miR-378i | 0.07 |
| hsa-miR-34c-5p | 0.07* |
| hsa-miR-200a-3p | 0.06 |
| hsa-miR-301a-3p | 0.06* |
| hsa-miR-362-5p | 0.05 |
| hsa-miR-451a | 0.05* |
| hsa-miR-31-3p | 0.05 |
| hsa-miR-29b-3p | 0.04* |
| hsa-miR-210-3p | 0.04 |
| hsa-miR-30e-5p | 0.03* |
| hsa-miR-140-5p | 0.03* |
| hsa-miR-192-5p | 0.03 |
| hsa-miR-187-3p | 0.03 |
| hsa-miR-486-5p | 0.03 |
| hsa-miR-29c-3p | 0.03* |
| hsa-miR-141-3p | 0.01* |
Expression of miRs in bold font is changed at both 2 and 6 d.p.i.
: Greater than 2-fold change in expression from 2 d.p.i. to 6 d.p.i.
Expression of 11 miRs significantly increased at 2 d.p.i., while 91 increased at 6 d.p.i. (Fig. 1b). For 6 of these 91 miRs, expression increased at both 2 and 6 d.p.i. (miRs-155, -4487, -let-7f-5p, -4443, -6722, and -15b-5p). Moreover, expression of all 6 was higher at 6 d.p.i. than at 2 d.p.i. Out of all 91 miRs that increased after infection, miR-155 underwent the greatest fold-change in expression at both timepoints: expression increased 5-fold at 2 d.p.i. (relative to uninfected mice; Fig. 1c). miR-155 expression increased a further 5-fold from 2 to 6 d.p.i. Only 2 other miRs (miR-3921 and miR-6872-5p) showed greater than a 5-fold increase in expression by ATII cells at 6 d.p.i. Interestingly, WSN infection did not significantly alter expression of these 2 miRs at 2 d.p.i.
WSN infection resulted in significant decreases in expression of 62 miRs at 2 d.p.i. and 162 miRs at 6 d.p.i. (Fig. 1b). The expression of 46 of these 162 miRs was reduced at both 2 and 6 d.p.i. The extent of reduction in expression of 14 of these miRs was significantly greater at 6 d.p.i. than 2 d.p.i. Moreover, 7 of the 10 most decreased miRs at 6 d.p.i. were also significantly decreased at 2 d.p.i. Overall, miR-29c-3p and miR-141-3p showed the greatest decreases in expression in ATII cells at both 2 d.p.i. (4- and 6-fold, respectively) and 6 d.p.i. (39- and 175-fold, respectively; Fig. 1d).
In order to validate results of the array studies, we performed qRT-PCR for miR-155 and miR-4487, which were among the most significantly increased miRs at 6 d.p.i. We found that, relative to ATII cells from uninfected control mice, miR-155 and miR-4487 were induced by >21-fold and >4-fold, respectively, which confirms the validity of our Affymetrix data. Moreover, mock infection for 6 days did not increase ATII cell miR-155 expression relative to uninfected mice (not shown).
miR-155 expression in ATII cells is dependent on inoculum size and correlates with severity of hypoxemia and pulmonary edema.
Because miRnome analysis indicated that miR-155 underwent the greatest increase in expression at both timepoints following WSN infection, we chose to focus subsequent functional studies on this miR. In order to determine the impact of viral inoculum size on miR-155 expression, we established a non-lethal infection model by challenging C57BL/6 mice with 100 pfu/mouse WSN. Compared to animals infected with the acutely lethal dose of WSN (10,000 pfu/mouse), infection with 100 pfu/mouse WSN caused relatively mild weight loss (approximately 10%; Fig. 2a), minimal hypoxemia (Fig. 2b), and only mild pulmonary edema (lung wet:dry weight ratio; Fig. 2c) at 6 d.p.i. However, infection with 100 pfu/mouse WSN for 10 days resulted in comparable weight loss, hypoxemia, and pulmonary edema to that found in mice infected with 10,000 pfu/mouse WSN for 6 days (Figs. 2a, 2b, and 2c, respectively). Mock infection did not cause weight loss or hypoxemia and mock-infected mice continued to grow normally.
Figure 2. miR-155 expression in ATII cells is dependent on inoculum size and correlates with severity of hypoxemia and pulmonary edema.
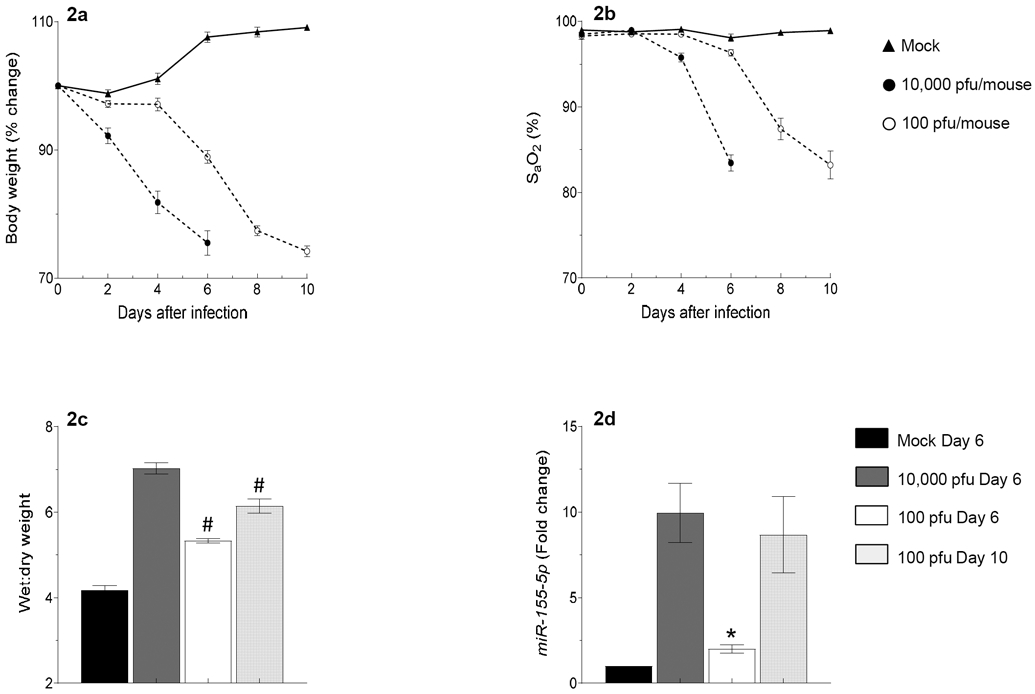
Effects of intranasal infection of C57BL/6 mice with 10,000 or 100 pfu/mouse of influenza A/WSN/33 (H1N1) virus on: (a) Body weight (BWT; % change from day 0; n>9 per group); (b) Carotid arterial oxygen saturation (% SaO2; n>9 group); (c) Lung water content (wet:dry weight ratio; n=5-6 per group); and (d) miR-155-5p expression (fold change from day 0; n=3-4 per group). All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM. *P<0.05, #P<0.001, vs. WT mice infected with 10,000 pfu/mouse WSN virus at 6 d.p.i.
Relative to mice inoculated with the acutely lethal dose of WSN, ATII cell miR-155 expression was significantly reduced in mice inoculated with the non-lethal dose of virus at 6 d.p.i. (Fig. 2d). However, ATII cell miR-155 expression in mice challenged with the non-lethal inoculum was significantly higher at 10 d.p.i. than at 6 d.p.i. Moreover, ATII cell miR-155 expression in animals infected with 100 pfu/mouse WSN did not significantly differ at 10 d.p.i. from miR-155 expression in ATII cells from mice infected with 10,000 pfu/mouse WSN at 6 d.p.i. This indicates that there is a correlation between disease severity at any given timepoint (which is dependent on the size of the inoculum) and ATII cell miR-155 expression levels.
Influenza severity is most attenuated in influenza-infected mice lacking miR-155 in stromal cells.
To further investigate the role of ATII cell miR-155 in IAV pathogenesis we compared responses to high-dose WSN infection (10,000 pfu/mouse) between wild-type (WT) C57BL/6 mice and C57BL/6-congenic, constitutive, global miR-155-knockout (155-KO) mice. Because IAV has previously been shown to induce miR-155 in macrophages (O'Connell et al., 2007; Tili et al., 2007), NK cells (Burocchi et al., 2015), and B cells (Vigorito et al., 2007) we also employed a reciprocal WT/155-KO bone marrow transfer (BMT) stratagem to differentiate the role(s) of stromal (epithelial) and myeloid (immune cell) miR-155-5p in IAV-induced ARDS. In this model, any difference in outcomes between the two bone marrow recipient strains is a consequence of which cell type(s) lack miR-155. KO recipients of WT bone marrow will only lack miR-155 in stromal cells (e.g., ATII cells, other respiratory epithelial cells, fibroblasts, and endothelial cells). In contrast, WT recipients of KO bone marrow retain miR-155 in stromal cells but do not express this miR in their immune cells.
Uninfected 155-KO mice appeared phenotypically normal. miR-155 expression was undetectable by qRT-PCR in ATII cells isolated from WSN-infected 155-KO mice at 6 d.p.i. (not shown). Post-infection weight loss was moderately attenuated in 155-KO mice at 4 and 6 d.p.i. (Figure 3a). Carotid arterial oxygen saturation (SaO2) and heart rate did not differ between WT controls and 155-KO animals prior to infection. However, WSN-induced hypoxemia and bradycardia were both attenuated in 155-KO mice at 6 d.p.i., as compared to WSN-infected WT animals (Figs. 3b and 3c, respectively). As in prior studies, influenza infection of WT mice for 6 days resulted in a large increase in lung water content, indicating development of severe pulmonary edema (Figure 3d). Pulmonary edema was significantly reduced in influenza-infected 155-KO mice at 6 d.p.i. Importantly, however, whole lung homogenate WSN titers did not differ between strains at either 2 d.p.i. (5.1 ± 0.2 log pfu/g in WT controls vs. 5.0 ± 0.2 log pfu/g in 155-KO mice) or 6 d.p.i. (5.4 ± 0.3 log pfu/g in WT controls vs. 5.7 ± 0.1 log pfu/g in 155-KO mice). Together, these data indicate that induction of miR-155 plays an important role in the pathogenesis of IAV-induced ARDS.
Figure 3. Influenza severity at 6 d.p.i. is most attenuated in influenza-infected mice lacking miR-155 in stromal cells.

Effects of intranasal mock and IAV infection (with 10,000 pfu/mouse WSN virus) of wild-type (WT) C57BL/6 mice, C57BL/6-congenic miR-155-knockout (155-KO) mice, or mice that have undergone reciprocal bone marrow transfer on: (a) Body weight (BWT; % change from day 0; n=10-20 per group); (b) Carotid arterial oxygen saturation (SaO2; n=10-20 per group); (c) Heart rate (beats per minute; n=10-20 per group); and (d) Lung water content (wet:dry weight ratio; n=4-6 per group). All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM. *P<0.05, **P<0.005, #P<0.001, vs. WSN-infected WT mice.
We found that transfer of WT bone marrow to irradiated 155-KO mice (6 weeks after irradiation) generally recapitulated the phenotype of WSN-infected 155-KO mice. Relative to WSN-infected WT controls, WT to 155-KO BMT decreased WSN-induced weight loss at 4 but not 6 d.p.i. (Fig. 3a), and also resulted in attenuation of WSN-induced hypoxemia (Fig. 3b), bradycardia (Fig. 3c), and pulmonary edema at 6 d.p.i., (Fig. 3d). In contrast, 155-KO to WT BMT recapitulated the phenotype of WT mice after WSN infection. Again, BMT did not alter lung homogenate viral titers (not shown). These findings indicate that increased miR-155 expression in stromal cells plays a greater role in the pathogenesis of IAV-induced ARDS than does increased expression in infiltrating bone marrow-derived myeloid cells.
Severity of IAV-induced lung dysfunction and cellular inflammation is dependent on expression of miR-155 in stromal cells.
Static lung compliance, which is an index of lung stiffness, did not differ between uninfected WT and 155-KO mice (not shown) and was reduced to a comparable degree in both strains at 6 d.p.i. (Fig. 4a). In contrast, while total lung resistance to airflow at baseline also did not differ between uninfected WT and 155-KO mice, the WSN infection-induced increase in total lung resistance at 6 d.p.i. was significantly attenuated in 155-KO animals (Figure 4b).
Figure 4. Severity of IAV-induced lung dysfunction and cellular inflammation at 6 d.p.i. is dependent on expression of miR-155 in stromal cells.
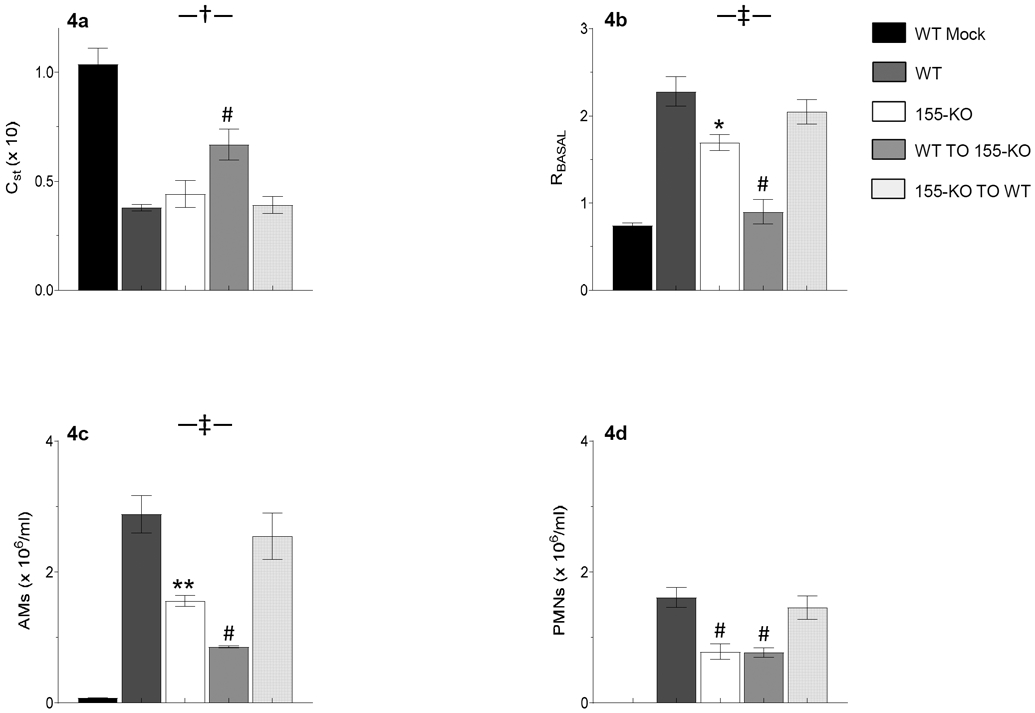
Effects of intranasal mock and IAV infection (with 10,000 pfu/mouse WSN virus) of wild-type (WT) C57BL/6 mice, C57BL/6-congenic global 155-KO mice, or mice that have undergone reciprocal bone marrow transfer on: (a) Static lung compliance (CST; ml/cmH2O, x 10); (b) Baseline total lung resistance (RBASAL; cmH2O.s/ml); (c) Bronchoalveolar lavage fluid (BALF) alveolar macrophage (AM) counts (x 106/ml); and (d) BALF neutrophil (PMN) counts (x 106/ml). n>5 per group. No neutrophils were found in BALF from mock-infected WT mice. All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM. *P<0.05, **P<0.005, #P<0.001, vs. WSN-infected WT mice. †P<0.05, ‡P<0.005, vs. WSN-infected 155-KO mice.
Interestingly, WT to 155-KO BMT resulted in a much greater decrease in infection-induced lung dysfunction than that observed in 155-KO mice. At 6 d.p.i., these animals had significantly higher static lung compliance than both WSN-infected WT controls and WSN-infected 155-KO mice (Fig. 4a) and baseline airway resistance was decreased to normal (Fig. 4b). In contrast, there were no differences in either lung compliance or airway resistance between WSN-infected WT and WSN-infected 155-KO to WT BMT mice at 6 d.p.i.
BALF from mock-infected WT and 155-KO mice contained comparable and low numbers of alveolar macrophages (6 x 104/ml and 4 x 104/ml, respectively) and no neutrophils. BALF alveolar macrophage counts were significantly lower in WSN-infected 155-KO mice than WSN-infected WT controls at 6 d.p.i. (Fig. 4c). Likewise, neutrophil infiltration was reduced in WSN-infected 155-KO mice (Fig. 4d). As in our other experiments, WSN effects were also less severe in WT to 155-KO BMT mice and in fact alveolar macrophages were present at significantly lower levels in WT to 155-KO BMT BALF than in 155-KO BALF at 6 d.p.i. (Fig. 4c). However, transfer of 155-KO bone marrow to WT mice had no impact on BALF cellularity relative to WT animals after WSN infection. Again, this emphasizes the importance of stromal, rather than myeloid (immune cell), miR-155 to the pathogenesis of IAV-induced ARDS.
BALF inflammatory mediator levels are altered in influenza-infected 155-KO mice.
Minimal levels of inflammatory cytokines and chemokines were detectable in BALF from mock-infected WT or 155-KO mice (not shown). However, at 6 d.p.i. 155-KO mouse BALF contained 4-fold less IFN-γ than WT (Fig. 5a). BALF from WSN-infected 155-KO mice also contained significantly lower amounts of IL-5, IL-10, MIP-1α, and MIP-1β, than WT BALF at 6 d.p.i., although overall the magnitude of these responses was relatively low (not shown). In contrast, BALF IL-6 and IL-12 [p40] levels were significantly higher in 155-KO mice at 6 d.p.i. (Figs. 5b and 5c, respectively). Interestingly, and despite differences in BALF neutrophil counts, levels of the neutrophil chemoattractant chemokine KC/CXCL-1 did not differ between the two strains at this timepoint (Fig. 5d). We also found no difference in BALF levels of IL-1β, MCP-1, and RANTES/CCL-5 between the two mouse strains at 6 d.p.i. (not shown). Finally, we did not detect significant amounts of other measured cytokines (IL-2, IL-3, IL-4, IL-9, IL-12[p70], IL-13, IL-17, and TNF-α) and chemokines in BALF from either WT or 155-KO mice at 6 d.p.i. (not shown).
Figure 5. BALF inflammatory mediator levels are altered in influenza-infected 155-KO mice at 6 d.p.i.

Effects of intranasal IAV infection of wild-type (WT) C57BL/6 mice, C57BL/6-congenic global 155-KO mice, or mice that have undergone reciprocal bone marrow transfer on bronchoalveolar lavage fluid levels of: (a) Interferon-γ (IFN-γ; ng/ml); (b) Interleukin-6 (IL-6; ng/ml); (c) p40 subunit of interleukin-12 (IL-12 [p40]; ng/ml); and (d) Keratinocyte cytokine/CXCL-1 (KC; ng/ml). n>5 per group. All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM. *P<0.05, **P<0.005, #P<0.001, vs. WSN-infected WT mice. †P<0.05, ¶P<0.001, vs. WSN-infected 155-KO mice.
Interestingly, the altered cytokine and chemokine response to WSN infection observed in 155-KO mice was not fully recapitulated in WT to 155-KO BMT animals: relative to WSN-infected WT controls, IFN-γ was only modestly decreased in the latter group, and remained significantly higher than in WSN-infected 155-KO mice (Fig. 5a). Likewise, IL-6 and KC/CXCL-1 levels were significantly higher in these mice than in both WT and 155-KO groups after WSN infection (Figs. 5b and 5d, respectively). However, the IL-12 response did not differ between 155-KO and WT to 155-KO BMT mice at 6 d.p.i. (Fig. 5c). In contrast, the cytokine responses of 155-KO to WT mice did not follow an obvious pattern. BALF IFN-γ levels did not differ between WT and 155-KO to WT groups at 6 d.p.i. However, BALF IL-6, IL-12, and KC/CXCL-1 content were all significantly higher than in WSN-infected WT mice. Moreover, BALF IL-6 and KC/CXCL-1 were also significantly higher in 155-KO to WT mice than 155-KO mice at 6 d.p.i.
Influenza severity is reduced in ATII cell-specific 155-KO mice.
To confirm our findings, we generated constitutive, ATII cell-specific, 155-KO (155-KOATII) mice. To do so, we crossed C57BL/6-congenic sp-c/cre+/+ mice, which express Cre recombinase under control of an SP-C promoter, with C57BL/6-congenic miR-155fl/fl mice. To confirm that miR-155 was not expressed in ATII cells from 155-KOATII mice but was present in other cells from the same lung, we performed qRT-PCR on the initial lung digest (which contains all lung cell types) and purified ATII cells from that same digest. Assays were performed at 6 d.p.i. in order to ensure maximal miR-155 induction. We found that miR-155 was detectable at high levels in whole lung digests from IAV-infected mice at 6.d.p.i. (approximately 13-fold increased relative to mock-infected controls), but was undetectable in purified ATII cells from the same lungs.
Carotid SaO2 values (Fig. 6a) and heart rates (not shown) were significantly higher in WSN-infected 155-KOATII mice than WSN-infected WT controls at 6 d.p.i., and lung water content was significantly decreased (Fig. 6b). ATII cell-specific deletion of miR-155 also resulted in improved static lung compliance (Fig. 6c) and reduced basal airway resistance at 6 d.p.i. (Fig. 6d). Importantly, there were no significant differences in SaO2, heart rates, and wet:dry weights between WSN-infected 155-KOATII and WSN-infected WT to 155-KO BMT mice (not shown). Hence, 155-KOATII mice reproduced the functional phenotype of WT to 155-KO BMT mice, confirming that induction of miR-155 at high levels in ATII cells plays an important role in the pathogenesis of IAV-induced lung dysfunction and injury.
Figure 6. Influenza severity is reduced in ATII cell-specific 155-KO mice.

Effects of intranasal mock and IAV infection of wild-type (WT) C57BL/6 mice and C57BL/6-congenic, ATII cell-specific 155-KO (155-KOATII) mice for 6 days on: (a) Carotid arterial oxygen saturation (% SaO2); (b) Lung water content (wet:dry weight ratio); (c) Static lung compliance (CST; ml/cmH2O, x 10); and (d) Baseline total lung resistance (RBASAL; cmH2O.s/ml). n>10 per group. Broken horizontal lines indicate values in uninfected WT mice. All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM. #P<0.001, vs. WSN-infected WT mice.
In our adoptive transfer studies, we found that both alveolar macrophages and neutrophil counts were significantly lower in WT to 155-KO BMT BALF than WT BALF at 6 d.p.i. (Fig. 4c). In contrast, there were no differences in BALF alveolar macrophage and neutrophil numbers between WSN-infected WT and 155-KOATII mice at 6 d.p.i. (Fig. 7a). Likewise, ATII cell-specific miR-155 deletion did not alter BALF IFN-γ, IL-6, and KC levels (Fig. 7b). This discrepancy between WT to 155-KO BMT and 155-KOATII mice suggests that miR-155 expression in other stromal cells (e.g., endothelium) contributes to the inflammatory response to IAV infection.
Figure 7. ATII cell-specific deletion of mir-155 does not attenuate influenza-induced pulmonary inflammation at 6 d.p.i.
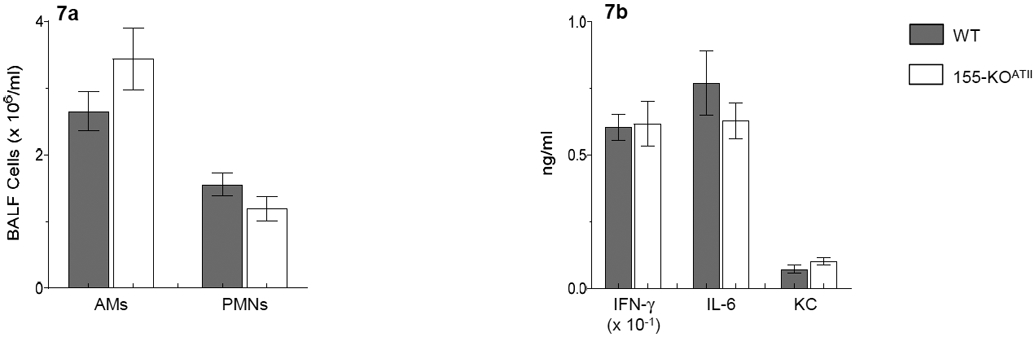
Effects of intranasal infection of wild-type (WT) C57BL/6 mice and 155-KOATII mice with 10,000 pfu/mouse of influenza A/WSN/33 (H1N1) virus on bronchoalveolar lavage fluid: (a) Alveolar macrophage (AM) and neutrophil (PMNs) counts; and (b) Interferon-γ (IFN-γ; ng/ml), interleukin-6 (IL-6; ng/ml), and keratinocyte cytokine (KC; ng/ml). n>6 per group. All data were analyzed by ANOVA with a post hoc Tukey-Kramer multiple comparison post-test and are presented as mean ± SEM.
Discussion
Given the significance of influenza to public health and the potential for pandemic spread of novel IAVs, we urgently need to develop new host-directed therapies that can ameliorate potentially lethal IAV-induced ARDS. Because individual miRs can control multiple regulatory pathways simultaneously, modulators of miR expression constitute a potent, new class of “one for all” host-directed ARDS therapeutics (Nana-Sinkam et al., 2009). Clearly, however, miR-directed therapies cannot be developed for influenza until suitable target miRs have been identified.
The impact of infection with both seasonal and highly-pathogenic IAV strains on the miRnome has also been characterized in vivo in whole lung homogenates from experimentally-infected mice (Choi et al., 2014; Li et al., 2010; Peng et al., 2011; Tan et al., 2014; Vela et al., 2014; Wu et al., 2013), pigs (Skovgaard et al., 2013), and nonhuman primates (Li et al., 2011). However, the normal lung contains more than 40 cell types (Franks et al., 2008), making it impossible to ascribe changes in any one miR to a particular cell type and thereby determine its function. Infiltration of leukocytes as a result of infection further aggravates this problem. To our knowledge, there have been no studies to date that have investigated the impact of IAV infection in vivo on cell-specific miR expression in a specific lung cell type.
ATII cells are both essential to normal lung function and the primary site of IAV infection in the distal lung. At the single cell level, several studies have been performed using human adenocarcinoma epithelial cell lines (primarily A549 cells) to model effects of in vitro IAV infection on the ATII cell miRnome (Bakre et al., 2013; Buggele et al., 2012; Buggele et al., 2013; Guan et al., 2012; Lam et al., 2013; Loveday et al., 2012; Meliopoulos et al., 2012; Terrier et al., 2013). However, the relevance of data derived from continuously growing cell lines to the normal lung is dubious. Unfortunately, both human and murine primary ATII cells rapidly differentiate into alveolar type I cells during short-term culture by most methods (Mao et al., 2015), which is likely to significantly alter their baseline miRnome relative to in vivo even before they are challenged with virus. Moreover, all in vitro respiratory epithelial cell culture systems suffer from the fact that they cannot model the complex milieu of the infected lung. Hence, they provide little relevant information regarding the role of other resident and infiltrating lung cell types in determining pre- and post-infection ATII cell miR expression profiles.
Because of the limitations of in vitro and whole lung studies, we chose to focus on ex vivo miR profiling in ATII cells isolated from IAV-infected mice. We found that infection with an acutely lethal dose of IAV that causes ARDS resulted in statistically-significant changes in expression of 73 miRs in ATII cells isolated from infected mice at 2 d.p.i. and 253 miRs at 6 d.p.i. Although only a minority of miRs were altered (less than 5% and 15% of all miRs analyzed, respectively), such broad changes in the miRnome are likely to have a significant impact on the ATII cell response to influenza, particularly later in infection. Interestingly, we found that at both 2 and 6 d.p.i., infection resulted in decreased expression of far more miRs than were increased. For example, at 2 d.p.i. 4% of significantly altered miRs were increased and 24% decreased. By 6 d.p.i. 35% had increased and 63% decreased. Increased expression of miRs reduces expression of target genes containing seed sequences for those miRs, the end result of which is decreased protein levels for those gene products. Hence, these data showing decreased miR expression are consistent with the overall increase in transcriptional activity and protein synthesis that would be expected to result from viral infection. Furthermore, for several miRs that were differentially expressed relative to uninfected controls at both 2 and 6 d.p.i, fold-changes in expression were significantly greater at 6 d.p.i. Together with the greater number of miRs that were altered at 6 d.p.i., this increase in expression of individual miRs indicates a progressively greater impact of IAV on the ATII cell miRnome as infection proceeded.
We found that, out of all 259 miRs that were significantly altered by infection, only 3 (miR-6872-5p, miR-3921, and miR-155) were more than 5-fold induced at 6 d.p.i. miR-155 underwent the greatest increase in expression in ATII cells at both 2 and 6 d.p.i. By 6 d.pi., miR-155 expression had increased 25-fold, relative to uninfected mice. These data suggested that miR-155 may be an important epigenetic regulator of the transcriptional and translational responses of ATII cells to IAV infection and therefore a potential therapeutic target.
Higher miR-155 expression in whole lung tissue was correlated with greater disease severity in mice infected with influenza A/PR/8/34 virus vs. those infected with a 2009 pandemic H1N1 strain (Wu et al., 2013). Increased whole lung miR-155 was also correlated with higher mortality in mice infected with a highly virulent avian H5N2 virus (Choi et al., 2014). Likewise, Pociask et al. showed recently that infection with A/PR/8/34 induced miR-155 in whole lung tissue from mice at 21 d.p.i., which is far later than in our study (Pociask et al., 2017). They also reported that, despite comparable viral replication, global C57BL/6-congenic 155-KO mice (similar to those used in our study) recovered from IAV infection more rapidly than C57BL/6 controls and exhibited reduced parenchymal injury and ER stress at later post-infection timepoints. Their data suggested an important role for miR-155 in influenza pathogenesis, which is confirmed in this report. However, viruses and TLR ligands (including polyI:C) can induce miR-155 in macrophages (O'Connell et al., 2007; Tili et al., 2007), NK cells (Burocchi et al., 2015), and B cells (Vigorito et al., 2007), so it was not possible to tell from the work of Pociask et al. either which cell types were expressing miR-155 at increased levels. Nor was it possible to tell from this study whether attenuation of lung damage in 155-KO mice resulted from absence of miR-155 in epithelial cells, infiltrating leukocytes, or both.
To define the specific role of miR-155 in ATII cells in influenza pathogenesis, we used mice with global and ATII cell-specific miR-155 gene deletions. Both 155-KO and 155-KOATII mice appeared phenotypically normal prior to infection and did not differ in any respect from Wt controls after mock infection. This suggests that expression of miR-155 is not essential for normal lung or ATII cell function. Nevertheless, our studies using global 155-KO mice confirmed that miR-155 plays an important role in the pathogenesis of IAV-induced ARDS, although deletion of this miR did not fully block ARDS development. To demonstrate that specific induction of miR-155 in ATII cells is important to influenza pathogenesis, we first performed reciprocal BMT experiments, in which irradiated WT and 155-KO mice received bone marrow from the opposite strain. We found that deletion of miR-155 from stromal cells (primarily respiratory epithelial cells, endothelial cells, and fibroblasts) had a far greater impact on influenza pathogenesis than did its deletion from myeloid cells. Indeed, the phenotype of WSN-infected 155-KO mice that received WT bone marrow was very similar to that of WSN-infected 155-KO mice, whereas WSN-infected 155-KO to WT BMT mice, which lack myeloid miR-155, behaved very much like WSN-infected WT controls. Follow-up studies in 155-KOATII mice confirmed these findings: in terms of impact on lung function, there were no significant differences in between WSN-infected 155-KOATII and WT to 155-KO BMT mice.
Overall, therefore, these experiments indicate that induction of miR-155 in ATII cells may play a more important role in pathogenesis of IAV-induced lung dysfunction and injury than its induction in the inflammatory leukocytes that infiltrate the lung after infection. Interestingly, however, in certain cases beneficial effects of miR-155 deletion were more profound in WT to 155-KO mice than in 155-KO animals. For example, detrimental effects of WSN on lung compliance and airway resistance were attenuated to a far greater degree, and alveolar macrophage infiltration was less pronounced. These findings suggest that miR-155 induction in myeloid cells could paradoxically be protective in influenza, at least in the absence of stromal miR-155. Hence, targeting of miR-155 in an ATII cell-specific fashion may well be more effective for treating IAV-induced ARDS than a non-specific whole lung approach. We have shown that RNA-containing nanoparticles can be specifically targeted to ATII cells and these could be used to deliver miR-155-specific antagomiRs (Wu et al., 2015).
Deletion of miR-155 resulted in a large decrease in BALF leukocytes, which suggests that this miR plays a pro-inflammatory role in influenza. Again, this beneficial effect tracked with loss of miR-155 from stromal cells rather than myeloid cells in BMT experiments. Interestingly, however, this effect was not recapitulated in 155-KOATII mice: ATII cell-specific miR-155 deletion did not attenuate cellular infiltration into the lungs, suggesting that recruitment of myeloid cells in response to IAV infection is mediated by miR-155 expressed by one or more other cell types. The latter finding also suggests that development of pulmonary edema after IAV infection is not a consequence of immune cell-mediated damage, since WSN-infected 155-KOATII mice have less edema but comparable immune infiltrates and cytokine responses in their lungs to WSN-infected WT controls.
Effects of altered miR-155 expression on BALF cytokines and chemokines were less clear-cut. For example, attenuation of the IFN-γ response to WSN infection was only seen in 155-KO mice. In contrast, BALF IL-6 was significantly increased by infection in all 155-KO groups, was higher in 155-KO to WT than 155-KO animals, but not altered in 155-KOATII mice: these findings emphasize the lack of strong correlation between severity of IAV-induced ARDS and IL-6 levels which we have reported previously. IL-12 levels were comparably increased in 155-KO, WT to 155-KO, and 155-KO to WT mice by infection. Finally, levels of the neutrophil chemoattractant KC (the mouse homolog of CXCR1) were only elevated in BALF from WSN-infected BMT mice (relative to WT controls). Moreover, KC levels were higher in BALF from 155-KO to WT mice than WT, despite comparable neutrophil infiltration. However, we have shown previously that adenosine can act as a powerful neutrophil chemoattractant in IAV-infected mice, and it is possible that adenosine responses are more comparable between these two groups (Aeffner et al., 2014).
Conclusions
We found that IAV induced significant changes in expression of a relatively large number of miRs in ATII cells, particularly later in infection. Interestingly, significantly fewer miRs were induced than repressed. Furthermore, we showed that ATII cell miR-155 plays an important role in the pathogenesis of IAV-induced ARDS. Taken together, our findings confirm those of Pociask et al. indicating that miR-155 may be an important target for new therapeutics for IAV-induced ARDS. However, they also suggest that ATII cell-specific drug targeting may be more beneficial than non-specific inhibition of miR-155 expression in the lung.
Acknowledgments.
The authors would like to acknowledge Lisa Joseph for her excellent technical assistance.
Funding. Studies were supported by: The C. Glenn Barber Fund (to P.S.W. and L.M.D.), The American Cancer Society (IRG-67-003-50; to E.T.), The National Institute of Neurological Disorders and Stroke at The National Institutes of Health (R03 NS102861; to ET), and The American Thoracic Society (GRT00044898; to I.C.D.).
Footnotes
Declarations of interest. The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aeffner F, Abdulrahman B, Hickman-Davis JM, Janssen PM, Amer A, Bedwell DM, Sorscher EJ, Davis IC, 2013. Heterozygosity for the F508del mutation in the cystic fibrosis transmembrane conductance regulator anion channel attenuates influenza severity. J Infect Dis 208, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeffner F, Bratasz A, Flaño E, Powell KA, Davis IC, 2012. Post-infection A77-1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol 47, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeffner F, Woods PS, Davis IC, 2014. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol 88, 10214–10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakre A, Andersen LE, Meliopoulos V, Coleman K, Yan X, Brooks P, Crabtree J, Tompkins SM, Tripp RA, 2013. Identification of host kinase genes required for influenza virus replication and the regulatory role of microRNAs. PLoS ONE 8, e66796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Rahman M, Nana-Sinkam SP, 2014. MicroRNAs in respiratory disease. A clinician's overview. Annals Am Thorac Soc 11, 1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggele WA, Johnson KE, Horvath CM, 2012. Influenza A virus infection of human respiratory cells induces primary microRNA expression. J Biol Chem 287, 31027–31040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggele WA, Krause KE, Horvath CM, 2013. Small RNA profiling of influenza A virus-infected cells identifies miR-449b as a regulator of histone deacetylase 1 and interferon beta. PLoS ONE 8, e76560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burocchi A, Pittoni P, Tili E, Rigoni A, Costinean S, Croce CM, Colombo M, 2015. Regulated expression of miR-155 is required for iNKT cell development. Front Immunol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EJ, Kim H, Baek Y, Kim EH, Pascua P, Park SJ, Kwon HI, Lim GJ, Kim S, Kim YI, Choi YK, 2014. Differential microRNA expression following infection with a mouse-adapted, highly virulent avian H5N2 virus. BMC Microbiol 14, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti M, Brody AR, Harrison JH, 1996. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14, 309–315. [DOI] [PubMed] [Google Scholar]
- Franks TJ, Colby TV, Travis WD, Tuder RM, Reynolds HY, Brody AR, Cardoso WV, Crystal RG, Drake CJ, Engelhardt J, Frid M, Herzog E, Mason R, Phan SH, Randell SH, Rose MC, Stevens T, Serge J, Sunday ME, Voynow JA, Weinstein BM, Whitsett J, Williams MC, 2008. Resident cellular components of the human lung. Proc Am Thorac Soc 5, 763–766. [DOI] [PubMed] [Google Scholar]
- Guan Z, Shi N, Song Y, Zhang X, Zhang M, Duan M, 2012. Induction of the cellular microRNA-29c by influenza virus contributes to virus-mediated apoptosis through repression of antiapoptotic factors BCL2L2. Biochem Biophys Res Commun 425, 662–667. [DOI] [PubMed] [Google Scholar]
- Hickman-Davis JM, Nicholas-Bevensee C, Davis IC, Ma HP, Davis GC, Bosworth CA, Matalon S, 2006. Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am J Respir Crit Care Med 173, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer CC, Woods PS, Davis IC, 2015. Infection of mice with influenza A/WSN/33 (H1N1) virus alters alveolar type II cell phenotype. Am J Physiol Lung Cell Mol Physiol 308, L628–L638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL, 2006. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80, 7469–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin CG, Bates JH, 2003. Measuring the lung function in the mouse: the challenge of size. Respir Res 4, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG, 2010. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WY, Yeung AC-M, Ngai KL-K, Li MS, To KF, Tsui SK-W, Chan PK-S, 2013. Effect of avian influenza A H5N1 infection on the expression of microRNA-141 in human respiratory epithelial cells. BMC Microbiol 13, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chan EY, Li J, Ni C, Peng X, Rosenzweig E, Tumpey TM, Katze MG, 2010. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J Virol 84, 3023–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li J, Belisle S, Baskin CR, Tumpey TM, Katze MG, 2011. Differential microRNA expression and virulence of avian, 1918 reassortant, and reconstructed 1918 influenza A viruses. Virology 421, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveday EK, Svinti V, Diederich S, Pasick J, Jean F, 2012. Temporal- and strain-specific host microRNA molecular signatures associated with swine-origin H1N1 and avian-origin H7N7 influenza A virus infection. J Virol 86, 6109–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Wu S, Li J, Fu W, He W, Liu X, Slutsky AS, Zhang H, Li Y, 2015. Human alveolar epithelial type II cells in primary culture. Physiol Rep 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Nunez RT, Bondanese VP, Louafi F, Francisco-Garcia AS, Rupani H, Bedke N, Holgate S, Howarth PH, Davies DE, Sanchez-Elsner T, 2014. A microRNA metwork dysregulated in asthma controls IL-6 production in bronchial epithelial cells. PLoS ONE 9, e111659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RJ, 2006. Biology of alveolar type II cells. Respirology 11 Suppl, S12–S15. [DOI] [PubMed] [Google Scholar]
- Meliopoulos VA, Andersen LE, Brooks P, Yan X, Bakre A, Coleman JK, Tompkins SM, Tripp RA, 2012. microRNA regulation of human protease genes essential for influenza virus replication. PLoS ONE 7, e37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana-Sinkam SP, Karsies T, Riscili B, Ezzie M, Piper M, 2009. Lung microRNA: from development to disease. Expert Rev Respir Med 3, 373–385. [DOI] [PubMed] [Google Scholar]
- Noah DL, Twu KY, Krug RM, 2003. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3' end processing of cellular pre-mRNAS. Virology 307, 386–395. [DOI] [PubMed] [Google Scholar]
- O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D, 2007. microRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA 104, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Gralinski L, Ferris MT, Frieman MB, Thomas MJ, Proll S, Korth MJ, Tisoncik JR, Heise M, Luo S, Schroth GP, Tumpey TM, Li C, Kawaoka Y, Baric RS, Katze MG, 2011. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. mBio 2, e00198–00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pociask DA, Robinson KM, Chen K, McHugh KJ, Clay ME, Huang GT, Benos PV, Janssen-Heininger YMW, Kolls JK, Anathy V, Alcorn JF, 2017. Epigenetic and transcriptomic regulation of lung repair during recovery from influenza infection. Am J Pathol 187, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran S, Pattarayan D, Rajaguru P, Sudhakar Gandhi PS, Thimmulappa RK, 2016. MicroRNA regulation of acute lung injury and acute respiratory distress syndrome. J Cell Physiol 231, 2097–2106. [DOI] [PubMed] [Google Scholar]
- Skovgaard K, Cirera S, Vasby D, Podolska A, Breum SØ, Dürrwald R, Schlegel M, Heegaard PM, 2013. Expression of innate immune genes, proteins and microRNAs in lung tissue of pigs infected experimentally with influenza virus (H1N2). Innate Immun 19, 531–544. [DOI] [PubMed] [Google Scholar]
- Tan K, Choi H, Jiang X, Yin L, Seet J, Patzel V, Engelward B, Chow V, 2014. micro-RNAs in regenerating lungs: an integrative systems biology analysis of murine influenza pneumonia. BMC Genomics 15, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrier O, Textoris J, Carron C, Marcel V, Bourdon JC, Rosa-Calatrava M, 2013. Host microRNA molecular signatures associated with human H1N1 and H3N2 influenza A viruses reveal an unanticipated antiviral activity for miR-146a. J Gen Virol 94, 985–995. [DOI] [PubMed] [Google Scholar]
- Thompson CI, Barclay WS, Zambon MC, Pickles RJ, 2006. Infection of human airway epithelium by human and avian strains of influenza A virus. J Virol 80, 8060–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM, 2007. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-a stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 179, 5082–5089. [DOI] [PubMed] [Google Scholar]
- Traylor ZP, Aeffner F, Davis IC, 2013. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses 7, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeki TM, 2017. Influenza. Ann Intern Med 167, ITC33–ITC48. [DOI] [PubMed] [Google Scholar]
- Vela EM, Kasoji MD, Wendling MQ, Price JA, Knostman KAB, Bresler HS, Long JP, 2014. microRNA expression in mice infected with seasonal H1N1, swine H1N1 or highly pathogenic H5N1. J Med Microbiol 63, 1131–1142. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, Smith KGC, Rada C, Enright AJ, Toellner KM, MacLennan ICM, Turner M, 2007. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 27, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Nikrad MP, Phang T, Gao B, Alford T, Ito Y, Edeen K, Travanty EA, Kosmider B, Hartshorn K, Mason RJ, 2011. Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am J Respir Cell Mol Biol 45, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westall G, Paraskeva M, 2011. H1N1 influenza: Critical care aspects. Semin Respir Crit Care Med 32, 400–408. [DOI] [PubMed] [Google Scholar]
- Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, Durbin JE, Davis IC, 2008. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med 178, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods PS, Doolittle LM, Rosas LE, Joseph LM, Calomeni EP, Davis IC, 2016. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am J Physiol Lung Cell Mol Physiol 311, L1160–L1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods PS, Tazi MF, Chesarino NM, Amer AO, Davis IC, 2015. TGF-b-induced IL-6 prevents development of acute lung injury in influenza A virus-infected F508del CFTR-heterozygous mice. Am J Physiol Lung Cell Mol Physiol 308, L1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Ma J, Woods PS, Chesarino NM, Liu C, Lee LJ, Nana-Sinkam SP, Davis IC, 2015. Selective targeting of alveolar type II respiratory epithelial cells by anti-surfactant protein-C antibody-conjugated lipoplexes. J Control Release 203, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Hao R, Li P, Zhang X, Liu N, Qiu S, Wang L, Wang Y, Xue W, Liu K, Yang G, Cui J, Zhang C, Song H, 2013. microRNA expression profile of mouse lung infected with 2009 pandemic H1N1 influenza virus. PLoS ONE 8, e74190. [DOI] [PMC free article] [PubMed] [Google Scholar]