

Summary
Digital cytometry aims to identify different cell types in the tumor microenvironment, with the current focus on immune cells. Yet, identifying how changes in tumor cell phenotype, such as the epithelial-mesenchymal transition, influence the immune contexture is emerging as an important question. To extend digital cytometry, we developed an unsupervised feature extraction and selection strategy to capture functional plasticity tailored to breast cancer and melanoma separately. Specifically, principal component analysis coupled with resampling helped develop gene expression-based state metrics that characterize differentiation within an epithelial to mesenchymal-like state space and independently correlate with metastatic potential. First developed using cell lines, the orthogonal state metrics were refined to exclude the contributions of normal fibroblasts and provide tissue-level state estimates using bulk tissue RNA-seq measures. The resulting metrics for differentiation state aim to inform a more holistic view of how the malignant cell phenotype influences the immune contexture within the tumor microenvironment.
Subject Areas: Stem Cells Research, Bioinformatics, Cancer
Graphical Abstract
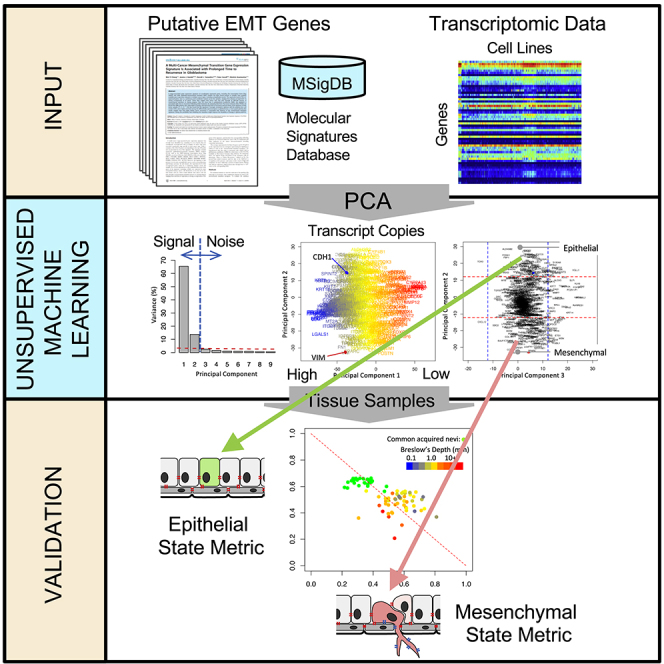
Highlights
-
•
Unsupervised strategy to generate epithelial and mesenchymal state metrics
-
•
Refined metrics for use with bulk RNA-seq data by removing normal fibroblasts genes
-
•
Validated state predictions against independent measures of metastatic potential
-
•
Breast cancer and melanoma share more common genes in de-differentiated metrics
Stem Cells Research; Bioinformatics; Cancer
Introduction
Tissues are composed of a diverse set of different cell types that help maintain homeostasis. Oncogenesis is associated with a shift in the cellular composition of a tissue that can be revealed with increasing confidence through direct measurement, such as single-cell RNA sequencing (scRNA-seq), or using digital methods to deconvolute bulk tissue samples (Newman et al., 2019). Given the correlation with response to immunotherapies, the current focus has been on quantifying immune cell types present within the tumor microenvironment (Thorsson et al., 2018, Tirosh et al., 2016). There is also an increasing appreciation for characterizing the heterogeneity among malignant cells that may arise in the same anatomical location (Shannan et al., 2016, Koren and Bentires-Alj, 2015). Given our interest in understanding functional heterogeneity of malignant cells that originate within a particular anatomical organ rather than uncertainty in etiology, we will focus on breast cancer and melanoma as Li et al. show that melanoma and breast cancer cell lines seem to cluster most uniformly, whereas other cell lines defined by anatomical origin seem to have a more heterogeneous composition (Li et al., 2017).
Although the tumor cells that arise in the skin and breast seem to be most similar, patient treatment strategies and outcomes can be diverse. Initial treatment strategies are guided by specific molecular alterations that can be targeted by drugs: aromatase inhibitors for ER-positive breast cancer, anti-HER2 antibodies for HER2-positive breast cancer, or small molecule inhibitors for BRAF V600E-positive or C-KIT-positive melanoma (Taghian et al., 2019, Sosman, 2019). Luckily, these two cancers are diagnosed in greater than 60% of patients while the malignant cells are confined to the organ of origin (Siegel et al., 2019). However, dissemination of primary tumors to vital organs like liver, brain, and lungs is a key limiter for patient survival in breast cancer and melanoma. Specifically, the 5-year survival rate for patients with localized disease versus distant metastases drops from 98% to 23% and from 99% to 27% for melanoma and breast cancer, respectively (American Cancer Society, 2019). In contrast, patient survival for tumors that originate in vital organs is limited by the degree to which malignant cells locally disrupt organ function. Thus, the importance of distal dissemination in determining patient outcomes can vary based on the tissue of origin.
The distal dissemination and growth of malignant cells—metastasis—is a complex process thought to involve dynamic re-engagement of biological processes used during development that enable migrating cells to form tissues. For carcinomas, initiating metastasis is thought to occur through a process called the epithelial-mesenchymal transition (EMT). EMT is the functional consequence of engaging a genetic regulatory network (GRN) that downregulates the expression of genes associated with an epithelial phenotype and upregulates genes associated with a mesenchymal phenotype. Breast carcinoma primarily originates from either luminal epithelial cells or basal myoepithelial cells within the mammary gland (Zhang et al., 2017). In contrast to breast cancer, melanoma arises from the oncogenic transformation of melanocytes, which follow a different developmental trajectory along the neural crest than epithelial cells and also involves a process similar to EMT (Regad, 2013). Although much of cell specification is imprinted epigenetically via DNA methylation and histone modifications, significant functional changes, such as modifications in cell state due to EMT, may occur within these epigenetic constraints. To characterize cell state based on gene expression, supervised methods have been predominantly used for developing gene signatures that characterize the EMT. Although effective, supervised methods can perform poorly if the strategy is based on misinformation, such as sample misclassification or prior biases as to the number of cell states or defining genes. Although used less frequently, unsupervised methods for feature extraction and selection are advantageous as they can be data driven (Taguchi, 2017). Here, our objective was to use an unsupervised strategy to develop a gene signature capturing this functional plasticity that is tailored to the specific cellular context of breast cancer and melanoma individually, as an illustrative example.
Results
RNA Sequencing Provides an Estimate of Protein Abundance
As extracting metrics to quantify cellular state using bioinformatic approaches depends on the quantity and quality of the underlying information, combining repositories of microarray- and RNA-sequencing (RNA-seq)-based transcriptomic profiling of tissue samples could provide a rich trove of data to mine. We first asked whether assaying the same genes using different transcriptomics profiling platforms provides the same information. To do this, we compared gene expression levels assayed by either Agilent microarray or by Illumina RNA-seq for the same samples (Figure 1). Expression values obtained by RNA-seq are in units of transcripts per million (TPM), whereas the Agilent microarray results are in terms of intensity units. Using samples obtained as part of the breast cancer arm of the TCGA, we focused on genes that have been associated with host immunity, as these genes are likely to span a broad dynamic range within these samples. As the TCGA samples are obtained from homogenized bulk samples of tumor and matched normal breast tissue, expression of these genes could be from the malignant cells, like GATA3 expression by breast cancer cells, or from immune cell infiltrates, like the potential expression of IL4 and IL5 by infiltrating T helper type 2 cells.
Figure 1.

Comparison of Gene Expression within the Same Samples Assayed using RNA Sequencing and Oligonucleotide Microarray
Heatmaps for the expression of a subset of genes in the breast cancer arm of the TCGA study assayed using Illumina RNA-seq (A) and using Agilent microarray (B). Color bar shown at the bottom of the heatmaps indicates samples obtained from tumor tissue (black) versus matched normal tissue (yellow). The genes and samples are similarly ordered in both panels. Values were log2 normalized.
Generally, comparing the same row across the two panels illustrates the poor correspondence between transcript abundance assayed using Agilent microarrays and read counts (TPM) obtained by RNA-seq. A subset of genes, like HLA-DRA and HLA-DPA1, exhibit both high microarray intensity units and read counts, whereas other genes, like TBX21 and FASLG, exhibit high microarray intensity units but have low read counts. In addition, the dynamic range observed among these samples is different depending on the platform used, as illustrated in the heatmap by TBX21 and IL17F. Using Illumina RNA-seq, TBX21 is constrained to the low end of the color spectrum (dark to royal blue), whereas the dynamic range spans the middle to upper end of the color spectrum (green to red) when assayed using Agilent microarray. Similarly, IL17F transcripts were not detected by RNA-seq in 87% of the samples but the Agilent microarray shows a rather high average intensity with variation among the samples. The difference in average intensities among genes and in variance among samples assayed by these two platforms suggest that the information provided by these two platforms is not entirely the same. The poor correspondence between Agilent two-channel microarray and RNA-seq data has been attributed to differences in ratio (Agilent two-channel microarray) versus non-ratio (RNA-seq) representations of transcript abundance by the platforms (Guo et al., 2013).
We next asked whether the assayed transcript abundance corresponds to protein abundance. First, we compared RNA-seq counts reported for cell lines associated with the Cancer Cell Line Encyclopedia (CCLE) with protein abundance for the same cell lines measured using Reverse Phase Protein Array (RPPA). We filtered the respective datasets to those cell lines that were reported in both datasets and for genes where there was a positive correlation coefficient greater than 0.36 between read counts in TPM and normalized RPPA values. From the initial datasets, 288 cell lines and 99 genes were retained for analysis after filtering. Next, we determined whether the pairs of mRNA and protein measurements share a common value for steady-state transcript abundance that corresponds to steady-state protein abundance measured above background. To do this, we applied a protein expression model to each gene measured across the cell lines where protein abundance was assumed to be a saturable function of transcript abundance (Figure 2A). Using the fitted curve, the threshold of transcript abundance for detecting a change in protein abundance 2.5% above background was back calculated. Example datasets and the corresponding curve fits for the genes CLDN7, AXL, JAG1, and CDH1 are shown in Figure 2B. Interestingly, the median value in the distribution of calculated threshold values was around 1 TPM (1.47, Figure 2C). We repeated this analysis for transcript abundance assayed by Affymetrix U133+2 microarray, a single-channel approach, using robust multi-array average (RMA)-normalized expression values, where 149 genes were retained for analysis after filtering. Qualitatively, data obtained using the single-channel platform (Affymetrix) exhibited better correspondence with RPPA values than data obtained using Agilent's two-channel platform. However, the distribution in calculated threshold values were more broadly distributed compared with the RNA-seq results (see red line in Figure 2C, F-test p value = 6.92 × 10−7). These results imply that the transcript abundance assayed by RNA-seq provides a higher-quality estimate of protein abundance, that is the signal-to-noise ratio is improved, compared with data obtained using a single-channel microarray platform. Moreover, simple strategies for combining data acquired using different platforms, such as centering across genes, or applying gene signatures cross-platform to interpret new samples warrants caution.
Figure 2.

RPPA Measurements Were Used to Determine a Threshold for Biologically Significant Changes in Gene Expression
(A and B) The model for protein dependence on gene expression (A) where representative data (black circles) and model fits (dotted black line) are shown for CLDN7, AXL, JAG1, and CDH1 (B).
(C) The distribution in threshold values calculated for all genes assayed by RNA-seq (black curve, n = 99) and by Affymetrix microarray (red curve, n = 149) meeting the inclusion criteria. Transcript abundance units for RNA-seq corresponds to TPM and intensity units (I.U.) for Affymetrix microarray. In (B), the vertical red dotted line indicates the threshold value and the melanoma and breast cancer cell lines are highlighted by red and blue circles.
Collectively, the common threshold value observed using RNA-seq data has two implications. First, there are some genes that have a high sensitivity of detection using microarrays such that the observed changes may not be functionally important. From Figure 1, it seems that IL17F, TBX21, FASLG, KLRD1, IFNG, CCL17, and IL10 are but a few examples (i.e., high Agilent microarray intensity but very low read counts) in that dataset. Without knowing the detection sensitivity by microarray, traditional approaches using a Z score metric may give equal weight to changes in gene expression driven by a biological signal as to changes dominated by random noise. Second, the threshold value provides a rationale for filtering genes that are likely to have a low information content when developing gene signatures for phenotypes that are not well defined.
Gene Expression Patterns in Breast Cancer Cells Are Captured by a Single Component
Given the variety of breast cancer subtypes reported in the literature, we next asked how many different GRNs are at work in breast cancer. GRNs associated with development commonly contain transcription factors that interact via positive feedback such that the target genes are either co-expressed or expressed in a mutually exclusive fashion (Alon, 2007). Given the interest in functional responses, we are focusing on patterns of gene expression in response to signal processing by the GRNs rather than trying to identify their topology. In motivating this study, we made four assumptions. First, we assumed that oncogenic mutations alter the peripheral control of GRN but do not alter the core network topology, where signals processed by a GRN change cell phenotype by engaging a unique gene expression pattern. Second, malignant cells derived from a particular anatomically defined cancer represent the diverse ways that hijacking these GRNs can provide a fitness advantage to malignant cells within the tumor microenvironment. Third, culturable tumor cell lines represent a sampling of these ways in which GRNs are hijacked in a particular anatomical location. Fourth, the process of isolating these malignant cells from tumor tissue to generate culturable cell lines does not bias this view. It follows then that the number of different GRNs can be identified by analyzing the transcriptional patterns of genes likely to participate in GRNs among an ensemble of tumor cells lines that share a common tissue of origin. We focused our attention on 780 genes that have been previously associated with the EMT and related gene sets in MSigDB v4.0. (Sarrio et al., 2008, Carretero et al., 2010, Alonso et al., 2007, Cheng et al., 2012, Tan et al., 2014, Kaiser et al., 2016, Deng et al., 2019, Deng et al., 2020) and analyzed the expression of these genes among 57 breast cancer cell lines included in the CCLE database as assayed by RNA-seq using a feature extraction/feature selection workflow summarized in Figure 3. To identify coordinately expressed genes, we used principal component analysis (PCA), a linear statistical approach for unsupervised feature extraction and selection that enables the unbiased discovery of clusters of genes that exhibit coherent patterns of expression (i.e., features) that are independent of other gene clusters (Jolliffe and Cadima, 2016). The relative magnitude of the resulting gene expression patterns can be inferred from the eigenvalues, which represent the extent of the data's covariance captured by a specific principal component. To facilitate comparisons among datasets, we represent the eigenvalues as the percent of total sum over all of the eigenvalues or, simply, percent variance, which is shown in Figure 4. Specifically, PC1 and PC2 captured 66% and 14% of the variance, respectively. Additional principal components each captured less than 3% of the variance.
Figure 3.

Data Workflow for Identifying Epithelial/Differentiated versus Mesenchymal/De-differentiated State Metrics
Workflow contains three decision points: unsupervised feature extraction (FE)/feature selection (FS) based on PCA, a binary fibroblast filter, and a consistency filter based on Ridge logistic regression of annotated samples.
Figure 4.

Two Opposing Gene Signatures Were Identified among the Cohort of Breast Cancer Cell Lines
(A) Scree plot of the percentage of variance explained by each principal component, where the dotted line corresponds to variance explained by the null principal components.
(B) Projection of the genes along PC1 and PC2 axes, where the font color corresponds to the mean read counts among cell lines (blue-yellow-red corresponds to high-medium-low read counts).
(C) Projection of the genes along PC2 and PC3 axes, where the dotted lines enclose 95% of the null PCA distribution along the corresponding axis.
One of the challenges with PCA is that no clear rules exist to determine how many principal components to consider, such as a gap statistic in clustering (Tibshirani et al., 2001). To select an appropriate number of PCs (i.e., features), we established a threshold for determining significance relative to a null distribution. Specifically, we applied the same PCA to a synthetic noise dataset generated from the original data by randomly resampling with replacement the collection of gene expression values and assigning the values to particular gene-cell line combinations. The resulting set of eigenvalues represent the values that could be obtained by random chance if the underlying dataset has no information, which are shown as the dotted red line in Figure 4A. In comparison with the null distribution, only the first two PCs were above the threshold. The variance captured by the remaining PCs were below the null PCA distribution suggesting that any potential biological interpretations of these additional PCs could also be explained by random chance. Therefore, we focused on the first two PCs.
As variance in read counts is proportional to abundance, gene projections along the PC1 axis were proportional to the average read counts of the corresponding gene among the samples. Measured transcript abundance is proportional to the basal gene expression associated with cell specification and technical artifacts associated with RNA-seq. Genes that were expressed above the 1-TPM threshold in more than 5% of the cell lines were retained for further analysis. For the breast cancer cell lines, this eliminated 26 genes from potential inclusion in the state metrics. Next, we focused on the projection of retained genes along principal components 2 and 3. The projections were annotated with horizontal and vertical dotted lines that enclose 95% of the projections from the null distribution. Although the majority of the genes were distributed around the origin, a subset of genes were projected along the extreme of the PC2 axis (outside of the dotted horizontal lines) and had no significant projection along the PC3 axis (inside of the dotted vertical lines). The list of genes associated with either the high PC2/null PC3 or the low PC2/null PC3 groups are listed in Table S1 and contained 143 and 81 genes, respectively. Of note, all of the genes excluded based on the 1-TPM threshold were projected within the null PC2 distribution. As the projection of Vimentin (VIM, red dot in Figure 4C) and E-cadherin (CDH1, blue dot in Figure 4C) was prototypical for these two groups of genes, the high PC2/null PC3 genes were annotated as a mesenchymal signature (i.e., a de-differentiated state) and the low PC2/null PC3 group were annotated as an epithelial signature (i.e., a terminally differentiated state). In contrast to supervised approaches that use Vimentin and E-cadherin as the basis to identify associated genes (e.g., Tan et al., 2014, Rokavec et al., 2017), the approach used here is unsupervised whereby the association of Vimentin and E-cadherin with these two opposite groups of genes emerges naturally from the data.
The Epithelial and Mesenchymal State Measures Stratify Intrinsic Subtypes of Breast Cancer and Metastatic Potential
Using these two sets of genes, we developed a state metric to quantify the extent of a gene expression signature associated with epithelial differentiation and mesenchymal de-differentiation using a normalized sum over all of the genes associated with a signature. Although the PCA results suggest that these two sets of genes are inversely related, the metrics were designed to represent each state independently such that cells that exhibit a pure phenotype would have values of 1 and 0 associated with their respective state metrics and cells with mixed phenotypes could potentially have values of 1 for both state metrics. Next, we calculated the state metric values for all of the breast cancer cell lines, where their projections in state space are shown in Figure 5. Interestingly, the breast cancer cell lines largely followed a linear reciprocal relationship between epithelial (E) and mesenchymal (M) states (dotted line in Figure 5) and were segregated by intrinsic PAM50 subtype (Parker et al., 2009). Although HER2, Luminal A, Luminal B, and Basal subtypes all have a high E signature, they progressively increased in their M signature. The Claudin Low subset spanned the greatest range with some expressing a high E and moderate M signatures (e.g., HCC1569, MDAMB361, HMEL) and others with a low E and high M signatures (e.g., BT549 and HS578T). Of note, a subset of the Claudin Low cell lines (e.g., HS742T, HS343T, HS281T, HS606T, and HS274T) with high M and very low E signatures have been suggested by the CCLE to be fibroblast-like (see Cell_lines_annotations_20181226.txt). Functionally, cells with low E and high M signatures had a high propensity for metastasis, whereas the propensity for metastasis was low in cell lines with high E and low M signatures (Yankaskas et al., 2019). This functional annotation also provided an external validation of the state metrics for breast cancer.
Figure 5.

The Different Subsets of Breast Cancer Were Clustered Along a Reciprocal Epithelial to Mesenchymal State Axes
(A and B) Log2 projections along the epithelial () and mesenchymal () state axes for each breast cancer cell line included in the CCLE (A) and primary breast cancer cells (B and C). Values for and were estimated by bulk RNA-seq data for cell lines associated with the CCLE and by scRNA-seq data for primary tumor cells (Chung et al., 2017).
(C) Log2 state projections are compared for primary breast cancer cells as originally reported and with dropout values imputed using the values averaged over the rest of the sample population, where gray lines connect the original state values to state values determine after imputation. Symbols were colored based on previously annotated breast cancer PAM50 subtypes: basal, red; claudin low, yellow; HER2, pink; luminal (A), blue; luminal (B), black. In (A), the metastatic potential of a subset of cell lines was annotated based on a recent study (Yankaskas et al., 2019): low metastatic potential, gray circle; high metastatic potential, red circle. The dotted line corresponds to a reciprocal relationship between the and state metrics (i.e., = 1 - ).
We next assessed the epithelial and mesenchymal state metrics in breast cancer cells assayed using scRNA-seq (Chung et al., 2017) (see Figure 5B). Similar to the cell lines, the samples were spread across the epithelial to mesenchymal spectrum roughly ordered by their corresponding intrinsic subtype, where HER2 subtype had a high E/low M signature and the basal subtype had the highest M signature without much of a reduction in their E signature. Overall, the state values were farther below the reciprocal trendline than any of the cell lines sampled. As gene-level reads by scRNA-seq are frequently missing (i.e., a dropout read) (Andrews and Hemberg, 2019), we imputed missing values to assess whether the distribution in the E/M state values were a result of read dropouts (see Figure 5C). Although read imputation shifted the cell state metrics toward the reciprocal trendline, the heterogeneity among the cell measurements was lost. Overall, it is unclear whether scRNA-seq measurements can be used to identify biological heterogeneity separately from heterogeneity introduced by technical limits of the assay.
Although single-cell methods are rapidly emerging as tool to assay human tissue samples, bulk transcriptomic assays of tumor tissue samples, like those acquired as part of the Cancer Genome Atlas (TCGA), are more abundant. More samples increase the statistical power for identifying clinical, cellular, and genetic correlates of the EMT. However, applying the epithelial/mesenchymal state metrics to interpret RNA-seq assays of bulk tumor tissue samples requires some additional filtering steps as bulk RNA-seq measurements averages over the heterogeneous normal and malignant cell types present within the tissue. In terms of a gene signature for the EMT, many of the genes commonly associated with acquiring mesenchymal function are also associated with fibroblasts, a relatively common cell type in epithelial tissues. Thus, an enrichment of genes associated with the EMT may be explained solely by a shift in the prevalence of fibroblasts within the tissue sample. Although the functional plasticity of fibroblasts within the tumor microenvironment is of increasing interest (e.g., Vickman et al., 2020), our goal here was to remove the contribution of normal fibroblasts from the state metrics. To deconvolute fibroblast genes from the state metrics, we obtained a list of 2,500 genes that were uniquely associated (Area under receiver operating characteristic curve >0.5) with a cluster annotated as fibroblasts using scRNA-seq data obtained from a digested normal skin sample obtained from a human female. This cluster contained about one-third of the cell samples measured within the CD45-negative population of the digested skin sample (see Figure S1). Although the majority of cells associated with cluster 1 were annotated as fibroblasts, a minor fraction of samples were annotated as “other.” The similarity scores and associated hierarchical clustering dendrogram suggest that the gene expression among cells in cluster 1 are very similar and that the cells included in cluster 1 can be considered as a uniform population. The interpretation of this is that, given the variability in scRNA-seq data, it is likely that some true fibroblasts assayed had no reads of COL1A1 or COL1A2. So, some of the cells annotated as “other” in cluster 1 are likely fibroblasts. Using this fibroblast gene list, overlapping genes were removed from the state metrics and highlighted in yellow in Table S1. All but one of the genes removed were contained within the mesenchymal gene list. In developing the overall approach, removing the contribution of normal fibroblasts was critical as projections of tissue samples in the EMT space without removing the contribution of fibroblasts were clustered around values of 1 for both state metrics.
Gene expression assayed from a bulk tissue sample reflects the combined contributions of non-malignant cells plus the changes induced by oncogenic transformation and reciprocal changes due to de-differentiation among malignant cells. Observable changes depend on the relative contributions of each cell source. As the unsupervised PCA analysis of the cell line data suggested that genes associated with EMT can be revealed by identifying a reciprocal pattern of gene expression, we performed Ridge logistic regression using the sample annotation to obtain regression coefficients for the list of EMT genes that passed the fibroblast filter (n = 151). The regression coefficients were used to filter the list of EMT genes for consistency with the reciprocal gene signature identified in the CCLE analysis. Genes that passed the consistency filter were used to define the epithelial and mesenchymal state metrics for bulk tissue samples. Of note, E-cadherin (CDH1) was still associated with the epithelial state metric, whereas N-cadherin (CDH2), Wnt-inducible signaling pathway protein 1 (WISP1/CCN4), and matrix metallopeptidase 3 (MMP3) were also retained in the mesenchymal state metric. The list of genes associated with the corresponding state metrics is given in Table S2.
Next, we projected the tissue samples obtained as part of the breast cancer arm of the TCGA in EMT space using the two tissue-based state metrics. Similar to the CCLE analysis, all samples clustered along the reciprocal versus line but exhibited greater dispersion. Samples obtained from normal breast tissue clustered separately from breast cancer samples (Figure 6), with normal breast tissue samples having the highest values for the epithelial state and lower values, on average, for the mesenchymal state. Among the different clinical breast cancer subtypes, the median value for progressively decreased from HER2+, triple negative (TN: ER-/PR-/HER2-), and ER/PR+ (luminal) subtypes. The mesenchymal projections were about equal for both TN and ER/PR+ subsets and higher than the HER2+ samples. The higher values for the HER2+ samples along the axis align with clinical observations. For instance, patients with HER2+ subtype of breast cancer are at increased risk for developing metastatic lesions compared with TN and luminal subtypes (Kennecke et al., 2010). The two different state metrics seem to capture gene signatures that help anchor a cell to its designated location within the tissue and that promote active migration, respectively. In other words, reducing corresponds to raising the anchor and increasing corresponds to hoisting the sail. In summary, both cell-level and tissue-level EMT state metrics provide an estimate of metastatic potential and a digital measure of malignant cell differentiation state in the context of breast cancer.
Figure 6.

The Samples from Normal Breast Tissue and Breast Cancer Were Clustered Separately Along a Reciprocal Epithelial to Mesenchymal State Axes
Using EMT genes that passed the gene filter workflow, each sample contained within the breast cancer (BrCa) arm of the TCGA was projected along the epithelial () versus mesenchymal () state axes using the corresponding bulk RNA-seq data. Symbols were colored based on normal breast tissue (green) or clinical breast cancer subtype: ER/PR +, blue; HER2, pink; triple negative (TN), red. The dotted line corresponds to a reciprocal relationship between the and state metrics (i.e., = 1 - ).
Gene Expression Patterns in Melanoma Cells Are Also Captured by a Single Component
Using the same feature extraction/feature selection workflow as the breast cancer analysis (Figure 3), we applied PCA to the expression of EMT-related genes assayed in an ensemble of 56 melanoma cell lines associated with the CCLE (Figure 7). We focused on the first two principal components, PC1 and PC2, that captured 80% and 6% of the variance, respectively. Additional principal components each captured less than 4% of the variance. PC1 captured the variance associated with read abundance, as gene projections along the PC1 axis were proportional to the average read counts among the samples. Vimentin (VIM) and fibronectin (FN1) were two of the most highly expressed genes, whereas members of the Wnt family were some of the genes with low expression (e.g., WNT1, WNT6, WNT8B, WNT3A, WNT8A, WNT9B). Genes retained for further analysis were expressed above the 1-TPM threshold in more than 5% of the cell lines, which eliminated 78 genes from potential inclusion in the state metrics. These excluded genes were also projected within the null PC2 space.
Figure 7.

Two Opposing Gene Signatures Were Identified among the Cohort of Melanoma Cell Lines
(A) Scree plot of the percentage of variance explained by each principal component, where the dotted line corresponds to variance explained by the null principal components.
(B) Projection of the genes along PC1 and PC2 axes, where the font color corresponds to the mean read counts among cell lines (blue-yellow-red corresponds to high-medium-low read counts).
(C) Projection of the genes along PC2 and PC3 axes, where the dotted lines enclose 95% of the null PCA distribution along the corresponding axis.
Similar to the analysis of the breast cancer data, we focused on the projection of retained genes along PC2 and PC3 axes. Specifically, we developed state metrics around a subset of genes that were projected along the extreme of the PC2 axis and had no significant projection along the PC3 axis. The genes associated with either the high PC2/null PC3 or the low PC2/null PC3 groups are listed in Table S1 and contained 26 and 90 genes, respectively. In contrast to the breast cancer results, the projection of Vimentin (VIM, red dot in Figure 7C) and E-cadherin (CDH1, blue dot in Figure 7C) was not associated with either of these two groups of genes. As the high PC2/null PC3 group included MITF, a master regulator of melanocyte differentiation, and the low PC2/null PC3 group included a number of EMT-related genes (e.g., FN1, TCF4, ZEB1, TWIST2, and WISP1), these two gene sets were annotated as a terminally differentiated signature (i.e., an epithelial-like state) and a de-differentiated signature (i.e., a mesenchymal-like state), respectively. We noted that MITF was projected in the null PC2/null PC3 space in the breast cancer analysis.
Projections of the melanoma cell lines in differentiation state space were calculated using the two state metrics (Figure 8). Similar to the breast cancer cell lines, the melanoma cell lines largely followed a linear reciprocal relationship between terminally differentiated () and de-differentiated () states (dotted line in Figure 8). The majority of cell lines exhibited primarily a terminally differentiated signature with some expression of de-differentiated genes, whereas only a small subset of the cell lines exhibited primarily a de-differentiated signature. The gene signatures for single melanoma cells were also highly heterogeneous owing to dropout of gene reads.
Figure 8.

Melanoma Cell Lines and Primary Single Melanoma Cells Are Distributed Along Path between Extremes in Differentiation States
Projections along the terminally differentiated () versus de-differentiated () state axes for each melanoma cell line included in the CCLE (A) and primary melanoma cells (B). Values for the terminally differentiated and de-differentiated state metrics were estimated by RNA-seq data for cell lines associated with the CCLE and by scRNA-seq data for primary melanoma cells. Symbols for primary melanoma cells were colored differently for each patient sample. The dotted line corresponds to a reciprocal relationship between the and state metrics (i.e., = 1 - ).
Using state metrics refined for use with tissue samples (see Table S2), samples acquired from benign melanocytic nevi and untreated primary melanoma tissue were projected onto the state space. Of note, CEACAM1 and MITF were associated with the differentiated state and no genes were shared with the breast cancer epithelial state metric. The de-differentiated state metric had five genes, WISP1/CCN4, FOXC2, ITGA5, SERPINE1, and SPOCK1, that were shared with the breast cancer mesenchymal state metric. Although samples were more narrowly distributed in state space compared with the cell lines (Figure 9), all of the benign nevi exhibited higher terminally differentiated () and tended to have lower de-differentiated values (). The samples from primary melanoma were color-coded based on the annotated Breslow's depth, where higher values were associated with lower terminal differentiation scores. Using Breslow's depth as a surrogate measure of metastatic potential (Balch et al., 2009), tissue-level EMT state metrics provide an estimate of metastatic potential and a digital measure of malignant cell differentiation state in the context of melanoma. This functional annotation also provided an external validation of the state metrics for melanoma.
Figure 9.

Gene Expression Patterns Associated with Benign Melanocytic Nevi and Primary Melanoma Tissue Samples Are Distributed Along Path between Extremes in Differentiation States
Projections along the terminally differentiated () versus de-differentiated () state axes for 78 tissue samples obtained from common acquired melanocytic nevi (n = 27, green circles) and primary melanoma (n = 51). The primary melanoma samples are colored based on the Breslow's depth (blue: 0.1 mm to red: 10+ mm). The dotted line corresponds to a reciprocal relationship between the and state metrics (i.e., = 1 - ).
Terminal Differentiation Is Associated with Distinct Gene Signatures, whereas De-differentiation Seems to Engage Common Gene Regulatory Networks
The separate gene signatures generated for breast cancer cells and melanoma cells using an unsupervised approach provide an opportunity to identify unique and shared aspects of the genetic regulatory mechanisms underpinning cell specification, as summarized in Figure 10. In terms of shared aspects, the overlap in the genes between melanoma and breast cancer metrics were not explained by random chance, as assessed by a Fisher exact test (p value <2.2 × 10−16 for and p value <4.3 × 10−4 for ). We also found that the extent of overlap in the mesenchymal state metrics was greater than the overlap in the epithelial state metrics (odds ratio: 3.211 [95% confidence interval, 1.114–9.254], p value <2.2 × 10−16) as assessed by an exact hypergeometric test. Considering just the genes that overlap in the state metrics, the Ki values associated with the metric, although generally lower, trend similarly to the Ki values associated with the (see Figure 10B: slope = 0.927 with = 0.841). The Ki values associated with the , although generally higher, seemed to trend differently than the metrics (slope = 1.44 with = 0.878), although there are only eight genes in common. In addition, we used GOnet (Pomaznoy et al., 2018) to identify genes with transcription factor activity using the molecular function Gene Ontology term: DNA_binding (GO:0003677). In the breast cancer cell lines, nine transcription factors were upregulated in cells with a terminally differentiated phenotype, including GRHL2 and OVOL2, that have been associated with enforcing epithelial differentiation (Cieply et al., 2012). Correspondingly, four transcription factors were upregulated in melanoma cells, including MITF, which is essential for melanocyte differentiation (Goding and Arnheiter, 2019). Interestingly, there was no overlap in the genes with transcription factor activity in the two differentiated cell signatures. In contrast, melanoma and breast cancer cell lines that exhibited a de-differentiated phenotype shared seven transcription factors, including TWIST2 and ZEB1. De-differentiation in breast cancer cell lines was also associated with an additional eight transcription factors, including TWIST1 (Yang et al., 2004). Overall, the analysis of these transcription factors is consistent with specificity in phenotype as a consequence of engaging gene regulatory networks unique to a specialized cell subset, whereas de-differentiation seemed to engage common gene regulatory networks that facilitate the loss of cell specificity.
Figure 10.

A Comparison of the Genes Included in the Different State Metrics across Cancers
(A) Venn diagram illustrating overlap in genes contained in the opposing state metrics for terminally differentiated/epithelial versus de-differentiated/mesenchymal extracted from breast cancer (blue circle) and melanoma (red circle) cell lines. The subset of the genes listed below the Venn diagram were annotated with transcription factor GO terms.
(B) A biplot of the Ki values for the overlapping genes in the terminally differentiated/epithelial state metrics (blue circles and blue linear trendline) and in the de-differentiated/mesenchymal state metrics (orange circles and orange linear trendline). A 1:1 correspondence is represented by the black dotted line.
Discussion
Here we used an unsupervised feature extraction and selection approach based on PCA and resampling to identify state metrics for the EMT in breast cancer and melanoma individually. Given the importance for identifying patients with tumors likely to metastasize, a number of gene signatures have been developed to predict the prevalence of tumor cells with an EMT signature (Tan et al., 2014, George et al., 2017, Rokavec et al., 2017, Koplev et al., 2018, Malta et al., 2018). Supervised approaches are most common (Tan et al., 2014, George et al., 2017, Rokavec et al., 2017, Koplev et al., 2018, Malta et al., 2018), where samples are classified a priori. For instance, Koplev et al. (2018) developed gene signatures that average over all anatomical locations, whereas Levine and coworkers (George et al., 2017, Jia et al., 2019) classify training samples a priori into one of three cell states: epithelial, mesenchymal, or hybrid E/M. Rokavec et al. generate features based on co-expression with E-cadherin and Vimentin (Rokavec et al., 2017). Although effective, supervised methods can perform poorly if the strategy is based on misinformation, such as sample misclassification or prior biases as to the number of cell states or defining genes. Moreover, developing metrics to classify the EMT status of tumors purely based on bulk tumor samples without deconvoluting the contribution of fibroblasts has unclear interpretation (Panchy et al., 2019). We also note that state metrics developed using microarray technology (e.g., Tan et al., 2014, Koplev et al., 2018) are not likely relevant for interpreting data based on RNA-seq, given the unclear relation between transcriptome and protein abundance as assayed using microarray technology. Although these methods rarely used, the data-driven nature of unsupervised methods for feature extraction and selection are attractive (Taguchi, 2017). For instance, Umeyama et al. used an unsupervised approach for feature extraction to identify genes associated with metastasis (Umeyama et al., 2014). To illustrate this data-driven approach, we have focused on breast cancer and melanoma separately, where metastatic dissemination to vital organs is a key limiter of patient survival and the cell-of-origin for these cancers have different developmental trajectories. In summary, we hope that our developed state metrics find use alongside other digital cytometry tools to better understand how oncogenic transformation and associated functional plasticity alters the immune contexture within the tumor microenvironment.
Limitations of the Study
Given the focus on breast cancer and melanoma as illustrative examples, the state metrics developed for these two biological contexts may not apply to cancers that originate in other anatomical locations. In terms of the bioinformatic approach, PCA is a linear approach that is used here for identifying genes that vary in expression together. Given that regulatory networks that underpin gene expression can give rise to non-linear behavior, genes that exhibit non-linear dependence with differentiation state are likely to be excluded from the state metrics. In addition, there may be additional patterns in gene expression that are biologically significant but fall below the null threshold due to a bias sampling of cell lines. In terms of limitations of the underlying data used in the study, next-generation RNA-seq of the cell lines included in the CCLE was performed on RNA isolated from frozen cell pellets using Trizol. We noted that the cells were cultured according to vendors' instructions for preservation, which includes adding the cryoprotectant dimethyl-sulfoxide (DMSO) to the cells prior to freezing. DMSO has also been reported to synchronize cells by inducing reversible G1 arrest (Fiore et al., 2002), which might impact transcriptional profiles. Although some work suggests that DMSO has no impact on the transcriptome (Guillaumet-Adkins et al., 2017), differences in whether samples were fresh or frozen when processed could be convoluted with differences in the state metrics compared between cell line versus tissue samples. Ultimately, the resulting state metrics should be revisited for consistency as additional transcriptomic datasets are reported and additional EMT-related genes are identified.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by National Science Foundation (NSF CBET-1644932 to D.J.K.) and National Cancer Institute (NCI R01CA193473 to D.J.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSF or NCI.
Author Contributions
Conceptualization: D.J.K.; Methodology: D.J.K. and A.T.; Software: D.J.K. and A.T.; Formal Analysis: D.J.K. and A.T.; Visualization: D.J.K.; Supervision: D.J.K.; Funding acquisition: D.J.K.; Project administration: D.J.K.; Writing – original draft: D.J.K.; Writing – review & editing: D.J.K. and A.T.
Declaration of Interests
The authors declare no competing financial interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101080.
Data and Code Availability
The code used in the analysis can be obtained from the following GitHub repository:
Supplemental Information
References
- Alon U. volume 10. Chapman & Hall/CRC; 2007. An introduction to systems biology: design principles of biological circuits; pp. 97–104. (Chapman & Hall/CRC Mathematical and Computational Biology Series). [Google Scholar]
- Alonso S.R., Tracey L., Ortiz P., Perez-Gomez B., Palacios J., Pollan M., Linares J., Serrano S., Saez-Castillo A.I., Sanchez L. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007;67:3450–3460. doi: 10.1158/0008-5472.CAN-06-3481. [DOI] [PubMed] [Google Scholar]
- American Cancer Society . American Cancer Society; 2019. Cancer Facts & Figures 2019. [Google Scholar]
- Andrews T.S., Hemberg M. False signals induced by single-cell imputation [version 2; peer review: 4 approved] F1000Res. 2019;7:1740. doi: 10.12688/f1000research.16613.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch C.M., Gershenwald J.E., Soong S.J., Thompson J.F., Atkins M.B., Byrd D.R., Buzaid A.C., Cochran A.J., Coit D.G., Ding S. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero J., Shimamura T., Rikova K., Jackson A.L., Wilkerson M.D., Borgman C.L., Buttarazzi M.S., Sanofsky B.A., McNamara K.L., Brandstetter K.A. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W.Y., Kandel J.J., Yamashiro D.J., Canoll P., Anastassiou D. A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS One. 2012;7:e34705. doi: 10.1371/journal.pone.0034705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W., Eum H.H., Lee H.O., Lee K.M., Lee H.B., Kim K.T., Ryu H.S., Kim S., Lee J.E., Park Y.H. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017;8:15081. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B., Riley P., Pifer P.M., Widmeyer J., Addison J.B., Ivanov A.V., Denvir J., Frisch S.M. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012;72:2440–2453. doi: 10.1158/0008-5472.CAN-11-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Fernandez A., McLaughlin S.L., Klinke D.J. WNT1-inducible signaling pathway protein 1 (WISP1/CCN4) stimulates melanoma invasion and metastasis by promoting the epithelial-mesenchymal transition. J. Biol. Chem. 2019;294:5261–5280. doi: 10.1074/jbc.RA118.006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Fernandez A., McLaughlin S.L., Klinke D.J. Cell communication network factor 4 (CCN4/WISP1) shifts melanoma cells from a fragile proliferative state to a resilient metastatic state. Cell. Mol Bioeng. 2020;13:45–60. doi: 10.1007/s12195-019-00602-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore M., Zanier R., Degrassi F. Reversible G(1) arrest by dimethyl sulfoxide as a new method to synchronize Chinese hamster cells. Mutagenesis. 2002;17:419–424. doi: 10.1093/mutage/17.5.419. [DOI] [PubMed] [Google Scholar]
- George J.T., Jolly M.K., Xu S., Somarelli J.A., Levine H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017;77:6415–6428. doi: 10.1158/0008-5472.CAN-16-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding C.R., Arnheiter H. MITF-the first 25 years. Genes Dev. 2019;33:983–1007. doi: 10.1101/gad.324657.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumet-Adkins A., Rodríguez-Esteban G., Mereu E., Mendez-Lago M., Jaitin D.A., Villanueva A., Vidal A., Martinez-Marti A., Felip E., Vivancos A. Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol. 2017;18:45. doi: 10.1186/s13059-017-1171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Sheng Q., Li J., Ye F., Samuels D.C., Shyr Y. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS One. 2013;8:e71462. doi: 10.1371/journal.pone.0071462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., George J.T., Tripathi S.C., Kundnani D.L., Lu M., Hanash S.M., Onuchic J.N., Jolly M.K., Levine H. Testing the gene expression classification of the EMT spectrum. Phys. Biol. 2019;16:025002. doi: 10.1088/1478-3975/aaf8d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe I.T., Cadima J. Principal component analysis: a review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016;374:20150202. doi: 10.1098/rsta.2015.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J.L., Bland C.L., Klinke D.J. Identifying causal networks linking cancer processes and anti-tumor immunity using Bayesian network inference and metagene constructs. Biotechnol. Prog. 2016;32:470–479. doi: 10.1002/btpr.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennecke H., Yerushalmi R., Woods R., Cheang M.C., Voduc D., Speers C.H., Nielsen T.O., Gelmon K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010;28:3271–3277. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- Koplev S., Lin K., Dohlman A.B., Ma’ayan A. Integration of pan-cancer transcriptomics with RPPA proteomics reveals mechanisms of epithelial-mesenchymal transition. PLoS Comput. Biol. 2018;14:e1005911. doi: 10.1371/journal.pcbi.1005911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S., Bentires-Alj M. Breast tumor heterogeneity: source of fitness, hurdle for therapy. Mol. Cell. 2015;60:537–546. doi: 10.1016/j.molcel.2015.10.031. [DOI] [PubMed] [Google Scholar]
- Li J., Zhao W., Akbani R., Liu W., Ju Z., Ling S., Vellano C.P., Roebuck P., Yu Q., Eterovic A.K. Characterization of human cancer cell lines by reverse-phase protein arrays. Cancer Cell. 2017;31:225–239. doi: 10.1016/j.ccell.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta T.M., Sokolov A., Gentles A.J., Burzykowski T., Poisson L., Weinstein J.N., Kamińska B., Huelsken J., Omberg L., Gevaert O. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018;173:338–354. doi: 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchy N., Azeredo-Tseng C., Luo M., Randall N., Hong T. Integrative transcriptomic analysis reveals a multiphasic epithelial-mesenchymal spectrum in cancer and non-tumorigenic cells. Front. Oncol. 2019;9:1479. doi: 10.3389/fonc.2019.01479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomaznoy M., Ha B., Peters B. GOnet: a tool for interactive Gene Ontology analysis. BMC Bioinformatics. 2018;19:470. doi: 10.1186/s12859-018-2533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad T. Molecular and cellular pathogenesis of melanoma initiation and progression. Cell. Mol. Life Sci. 2013;70:4055–4065. doi: 10.1007/s00018-013-1324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokavec M., Kaller M., Horst D., Hermeking H. Pan-cancer EMT-signature identifies RBM47 down-regulation during colorectal cancer progression. Sci. Rep. 2017;7:4687. doi: 10.1038/s41598-017-04234-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D., Rodriguez-Pinilla S.M., Hardisson D., Cano A., Moreno-Bueno G., Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Shannan B., Perego M., Somasundaram R., Herlyn M. Heterogeneity in melanoma. Cancer Treat. Res. 2016;167:1–15. doi: 10.1007/978-3-319-22539-5_1. [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- Sosman J. UpToDate; 2019. Overview of the Management of Advanced Cutaneous Melanoma. [Google Scholar]
- Taghian A., El-Ghamry M.D., Merajver S.D. UpToDate; 2019. Overview of the Treatment of Newly Diagnosed, Non-metastatic Breast Cancer. [Google Scholar]
- Taguchi Y.H. Principal components analysis based unsupervised feature extraction applied to gene expression analysis of blood from dengue haemorrhagic fever patients. Sci. Rep. 2017;7:44016. doi: 10.1038/srep44016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.Z., Miow Q.H., Miki Y., Noda T., Mori S., Huang R.Y., Thiery J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014;6:1279–1293. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. The immune landscape of cancer. Immunity. 2018;48:812–830. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R., Walther G., Hastie T. Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. B. 2001;63:411–423. [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeyama H., Iwadate M., Taguchi Y.H. TINAGL1 and B3GALNT1 are potential therapy target genes to suppress metastasis in non-small cell lung cancer. BMC Genomics. 2014;15(Suppl 9):S2. doi: 10.1186/1471-2164-15-S9-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickman R.E., Broman M.M., Lanman N.A., Franco O.E., Sudyanti P.A.G., Ni Y., Ji Y., Helfand B.T., Petkewicz J., Paterakos M.C. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate. 2020;80:173–185. doi: 10.1002/pros.23929. [DOI] [PubMed] [Google Scholar]
- Yang J., Mani S.A., Donaher J.L., Ramaswamy S., Itzykson R.A., Come C., Savagner P., Gitelman I., Richardson A., Weinberg R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yankaskas C.L., Thompson K.N., Paul C.D., Vitolo M.I., Mistriotis P., Mahendra A., Bajpai V.K., Shea D.J., Manto K.M., Chai A.C. A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat. Biomed. Eng. 2019;3:452–465. doi: 10.1038/s41551-019-0400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Lee A.V., Rosen J.M. The cellular origin and evolution of breast cancer. Cold Spring Harb. Perspect. Med. 2017;7:a027128. doi: 10.1101/cshperspect.a027128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used in the analysis can be obtained from the following GitHub repository: