

SUMMARY
LMNA encodes nuclear Lamin A/C that tethers lamina-associated domains (LADs) to the nuclear periphery. Mutations in LMNA cause degenerative disorders including the premature aging disorder Hutchinson-Gilford progeria, but the mechanisms are unknown. We report that Ser22-phosphorylated (pS22) Lamin A/C was localized to the nuclear interior in human fibroblasts throughout the cell cycle. pS22-Lamin A/C interacted with a subset of putative active enhancers, not LADs, at locations co-bound by the transcriptional activator c-Jun. In progeria-patient fibroblasts, a subset of pS22-Lamin A/C-binding sites were lost whereas new pS22-Lamin A/C-binding sites emerged in normally quiescent loci. New pS22-Lamin A/C binding was accompanied by increased histone acetylation, increased c-Jun binding, and upregulation of nearby genes implicated in progeria pathophysiology. These results suggest that Lamin A/C regulates gene expression by enhancer binding. Disruption of the gene regulatory rather than LAD tethering function of Lamin A/C may underlie the pathogenesis of disorders caused by LMNA mutations.
Graphical Abstract
Blurb for Table of Contents
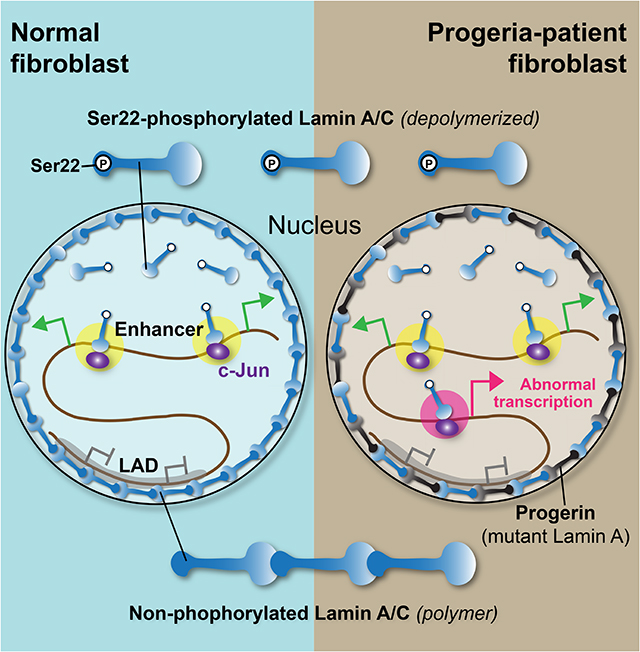
Nuclear lamins bind heterochromatin domains at the nuclear periphery. Ikegami et al. now show that a phosphorylated form of nuclear lamins bind to active enhancers in euchromatin in the nuclear interior. They provide evidence that suggests disruption of phosphorylated lamin function at enhancers contributes to the pathogenesis of progeria.
INTRODUCTION
Nuclear lamins polymerize to form the nuclear lamina, a protein meshwork that underlies the nuclear membrane (Aebi et al., 1986; Goldman et al., 1986). There are two major nuclear lamin types, A-type and B-type (Dittmer and Misteli, 2011). A-type lamins (Lamin A and Lamin C; Lamin A/C) are specific to vertebrates, expressed in differentiated cells, and encoded by LMNA in humans (Dittmer and Misteli, 2011). Lamin A and Lamin C have different C-terminal tails due to alternative splicing but are otherwise identical. Point mutations in the LMNA gene cause a spectrum of human degenerative disorders including cardiomyopathy, muscular dystrophy, and the premature aging disorder Hutchinson-Gilford progeria (Worman et al., 2009), although the underlying molecular mechanisms remain unclear.
Nuclear lamins including Lamin A/C interact with large heterochromatin domains called lamina-associated domains (LADs), which contain mostly transcriptionally inactive genes (Pickersgill et al., 2006; Guelen et al., 2008; Ikegami et al., 2010; Meuleman et al., 2012; Lund et al., 2014). By interacting with LADs, nuclear lamins are implicated in the spatial organization of chromosomal regions at the nuclear envelope (van Steensel and Belmont, 2017). However, whether nuclear lamins play a direct role in transcriptional silencing of genes located at the nuclear periphery remains unclear. Artificially tethering genes to the nuclear periphery or inserting gene promoters into LADs does not always result in transcriptional repression (Finlan et al., 2008; Reddy et al., 2008; Leemans et al., 2019). In addition, many gene promoters in LADs remain inactive when their activities are examined outside of LADs (Leemans et al., 2019). While one study reported that tethering of Lamin A/C to gene promoters resulted in transcriptional downregulation (Lee et al., 2009), depletion of all nuclear lamins did not de-repress many of genes within LADs (Amendola and van Steensel, 2015; Zheng et al., 2018). Thus, whether nuclear lamins including Lamin A/C have direct roles in transcriptional regulation has remained unclear.
Lamin A/C has been observed in the interior of the nucleus, in addition to its localization at the nuclear lamina (Dechat et al., 2010a). Initial descriptions engendered a model in which Lamin A/C in the nuclear interior represented a long-sought “nuclear scaffold” protein (Hozák et al., 1995; Barboro et al., 2002). However, subsequent studies demonstrated that nuclear-interior Lamin A/C was soluble and highly mobile (Broers et al., 1999; Shimi et al., 2008), thus present as a non-polymerized form and not constituting a scaffold structure. The specific function of Lamin A/C in the nuclear interior has been difficult to ascertain, mainly due to a lack of understanding about how Lamin A/C is directed to the nuclear interior and technical challenges isolating nuclear-interior Lamin A/C.
Depolymerization of nuclear lamins, required for nuclear envelope breakdown in mitosis, is regulated by phosphorylation of specific serine residues. Ser22 (S22) and Ser392 (S392) of Lamin A/C are phosphorylated at the late G2 cell-cycle phase by CDK1/Cyclin B, leading to Lamin A/C depolymerization during mitosis (Gerace and Blobel, 1980; Heald and McKeon, 1990; Peter et al., 1990; Ward and Kirschner, 1990; Georgatos et al., 1997). S22/S392 phosphorylation has also been reported in the nuclear interior of interphase cells (Kochin et al., 2014). Separate studies proposed that S22/S392 phosphorylation is increased upon changes in the mechanical environment of the cell and promote Lamin A/C disassembly and degradation (Swift et al., 2013; Buxboim et al., 2014). Therefore, Lamin A/C S22/S392 phosphorylation has been associated with mitotic nuclear lamina disassembly, but also with alternate cellular contexts in which its function remains unclear.
Hutchinson-Gilford progeria is a rare, fatal, childhood syndrome caused by heterozygous LMNA mutations (Eriksson et al., 2003). Progeria patients invariably develop arteriosclerosis that ultimately causes death from myocardial infarction, heart failure, or stroke by the second decade of life (Merideth et al., 2008). Progeria mutations result in a mutant Lamin A protein called “progerin” that lacks an internal part of the C-terminal tail domain and have been postulated to function as a dominant negative allele (Eriksson et al., 2003). The altered C-terminal domain in progerin promotes nuclear peripheral localization of progerin (Goldman et al., 2004) and has reduced DNA and chromatin binding affinity (Bruston et al., 2010). The prevailing models for progerin’s pathological activity include altered function at LADs (Gordon et al., 2014). The effect of progeria mutations on the function of Lamin A/C in the nuclear interior has not been explored.
In this study, we report that nuclear-interior, S22-phosphorylated (pS22) Lamin A/C binds to genomic sites characteristic of active enhancers. New pS22-Lamin A/C binding sites emerged at abnormal locations in progeria-patient fibroblasts, with concomitant upregulation of nearby genes relevant to progeria pathogenesis. We propose that Lamin A/C in the nuclear interior positively modulates enhancer activity, separate from its role at LADs, and that Lamin A/C’s role at enhancers contributes to progeria pathogenesis.
RESULTS
Phospho-S22-Lamin A/C is localized to the interior of the nucleus throughout the cell cycle
We investigated Ser22-phosphorylated (pS22) Lamin A/C as a candidate functional non-polymerized Lamin A/C in the nuclear interior. We identified an anti-pS22-Lamin A/C monoclonal antibody that specifically recognized the S22-phosphorylated synthetic Lamin A/C peptide, but not non-phospho-S22 peptide in ELISA (Fig. S1A, B). We also identified a reference anti-pan-N-terminal-Lamin A/C (aa2–29) monoclonal antibody that recognized both the phospho-S22 and non-phospho-S22 peptides, with approximately four-fold higher reactivity toward the non-phospho peptide (Fig. S1A, B). Immunofluorescence microscopy of TERT-immortalized human BJ-5ta fibroblasts using these antibodies revealed pS22-Lamin A/C signals localized to the nuclear interior but not at the nuclear periphery, whereas pan-N-terminal-Lamin A/C signals localized predominantly to the nuclear periphery with weak signals in the nuclear interior (Fig. 1A). The pS22-Lamin A/C and pan-N-terminal-Lamin A/C immunofluorescence signals were absent in BJ-5ta-derived LMNA−/− cells, confirming the specificity of the Lamin A/C signals (Fig. 1A). During interphase, pS22-Lamin A/C was detectable in the nuclear interior but not at the nuclear periphery while pan-N-terminal-Lamin A/C signals were observed predominantly at the nuclear periphery (Fig. 1B). At late G2, pS22-Lamin A/C signals increased dramatically, consistent with the model that S22 phosphorylation triggers Lamin A/C depolymerization in preparation for mitosis (Georgatos et al., 1997) (Fig. 1B). During mitosis, when the nuclear envelope is absent, strong pS22-Lamin A/C signal was observed throughout the cytoplasm (Fig. 1B). Interestingly, the anti-pan-N-terminal-Lamin A/C antibody did not produce signals during mitosis (Fig. 1B), suggesting that other mitotic modifications, such as phosphorylation at Ser12, Ser18, or Thr19 (Dephoure et al., 2008), may have affected the reactivity of this antibody during mitosis. Flow cytometry of asynchronous BJ-5ta cells using the anti-pS22-Lamin A/C antibody confirmed that pS22-Lamin A/C was present in G0/G1, S, and G2/M phases (Fig. 1C; Fig. S1C). Western blotting of cell-cycle synchronized BJ-5ta cells further confirmed persistent S22-phosphorylated Lamin A/C throughout the cell cycle, with apparently stronger pS22-Lamin C signals than pS22-Lamin A signals (Fig. 1D, E; Fig. S1D, E). Thus, in human fibroblasts, pS22-Lamin A/C exists in the nuclear interior throughout the cell cycle.
Figure 1. pS22-Lamin A/C is localized to the interior of the nucleus throughout interphase.
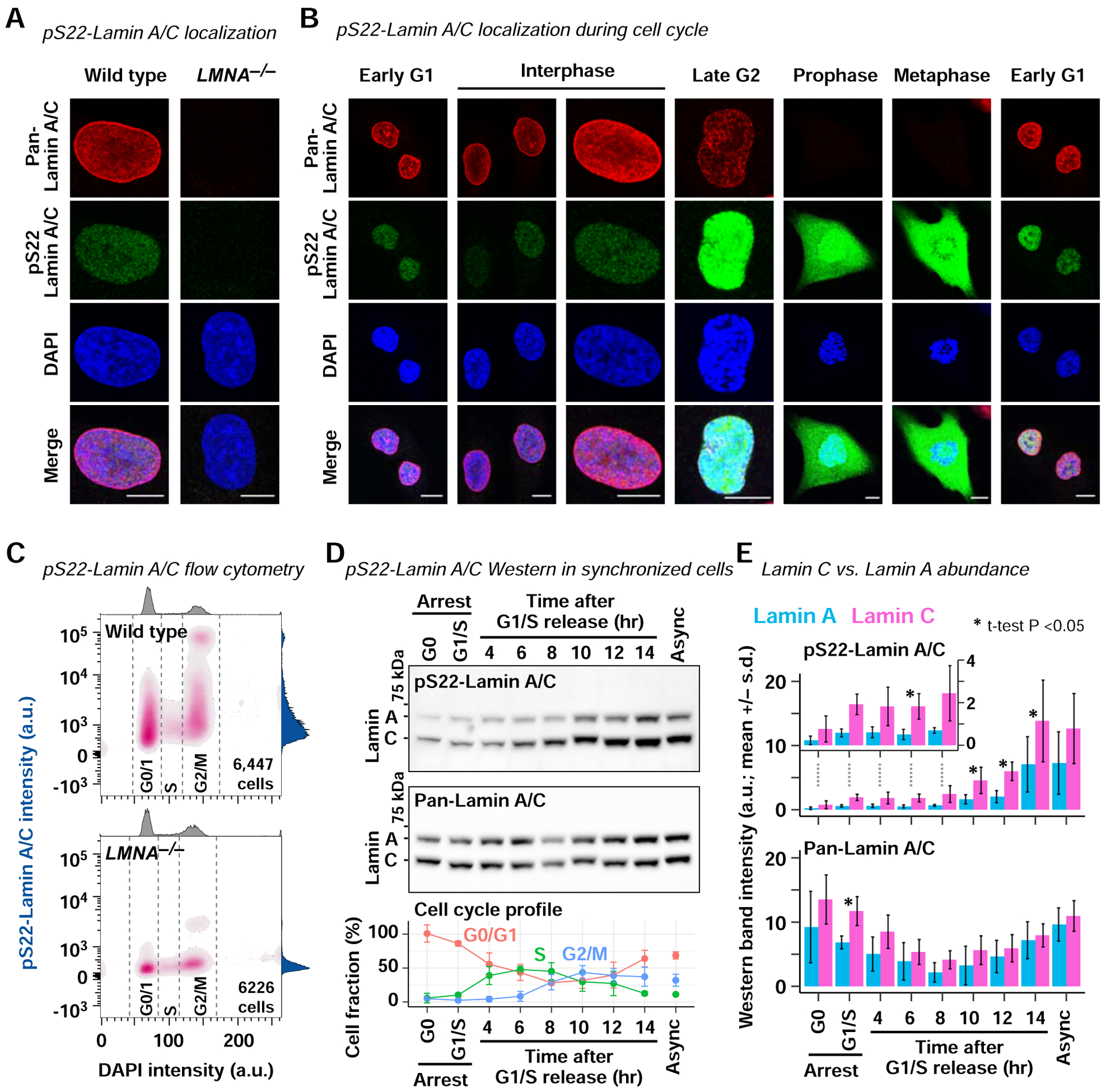
(A) Immunofluorescence using anti-pan-N-terminal-Lamin A/C and anti-pS22-Lamin A/C antibodies. Wild-type, BJ-5ta fibroblast. LMNA−/−, BJ-5ta-derived LMNA−/− fibroblast. Bar, 10 μm. Representative images from 3 biological replicates. See also Fig. S1.
(B) Same as (A), but BJ-5ta cells at specific cell-cycle stages are shown.
(C) Per-cell pS22-Lamin A/C and DNA (DAPI) levels of asynchronous BJ-5ta (top) or LMNA−/− cells (bottom) measured by flow cytometry. Per-cell signal distribution is shown as bivariate density. a.u., arbitrary unit. Representative analysis of 3 biological replicates.
(D) (Top and middle) Western blot of cell-cycle synchronized BJ-5ta. (Bottom) Fraction of cells at indicated cell-cycle phase determined by flow cytometry (mean of 3 biological replicates +/− standard deviation).
(E) Quantification of Lamin A/C Western blot signal intensities (mean of 3 biological replicates +/− standard deviation). Inset, narrower y-axis range for the selected samples. P, one sample t-test assessing distribution of log2[Lamin C]/[Lamin A] signal ratios.
pS22-Lamin A/C interacts with genomic sites outside of lamina-associated domains
We hypothesized that pS22-Lamin A/C interacts with the genome. To test this hypothesis, we performed ChIP-seq in asynchronous BJ-5ta cells using the anti-pS22-Lamin A/C antibody and using the anti-pan-N-terminal-Lamin A/C antibody as a comparison. pS22-Lamin A/C ChIP-seq exhibited point-source enrichment at discrete sites located outside of lamina-associated domains (LADs) and not with LADs themselves, in sharp contrast to pan-N-terminal-Lamin A/C, which were strongly enriched at LADs (Guelen et al., 2008) (Fig. 2A–C; Fig. S2A–D; Key Resources Table). We identified 22,966 genomic sites bound by pS22-Lamin A/C genome-wide. The pS22-Lamin A/C ChIP-seq signals were abolished in the BJ-5ta-derived LMNA−/− cell line, confirming the specificity of the pS22-Lamin A/C ChIP-seq signals in wild-type BJ-5ta (Fig. 2B, D). Pan-N-terminal-Lamin A/C ChIP-seq also detected weak signals at pS22-Lamin A/C-binding sites (Fig. 2B, D). To examine the chromatin localization of pS22-Lamin A/C with a different approach, we expressed Lamin A or Lamin C either with phospho-mimetic mutations (S22D/S392D), phospho-deficient mutations (S22A/S392A), or without mutations from a transgene in LMNA−/− cells. We then performed immunofluorescence and ChIP-seq in these cells using the anti-full-length-Lamin A/C antibody. Phospho-mimetic Lamin C (S22D/S392D) was highly abundant in the nuclear interior (Fig. S2E, F) and strongly bound to pS22-Lamin A/C-binding sites (Fig. 2E, F; Fig. S2H). Phospho-mimetic Lamin C was far more enriched at pS22-Lamin A/C-binding sites than phospho-mimetic Lamin A (one-way ANOVA with post-hoc Tukey test P=1×10−9; Fig.2F; Fig. S2H), consistent with the higher abundance of pS22-Lamin C compared with pS22-Lamin A in BJ-5ta (Fig. 1D, E). Phospho-mimetic Lamin C was still weakly localized at the nuclear periphery (Fig. S2E) unlike endogenous pS22-Lamin A/C (Fig. 1A), and correspondingly, was also enriched at LADs (Fig. S2G, I). In contrast, wild-type Lamin A and phospho-deficient Lamin A were strongly localized at the nuclear periphery (Fig. S2E, F) and strongly enriched at LADs (Fig. S2G, I), but showed no enrichment at pS22-Lamin A/C-binding sites (Fig. 2E, F; Fig. S2H). Together, these experiments revealed that S22/S392-phosphorylated Lamin A/C, most prominent in the form of S22/S392-phosphorylated Lamin C, has strong affinity to genomic sites outside of LADs.
Figure 2. pS22-Lamin A/C associates with putative enhancers genome-wide.
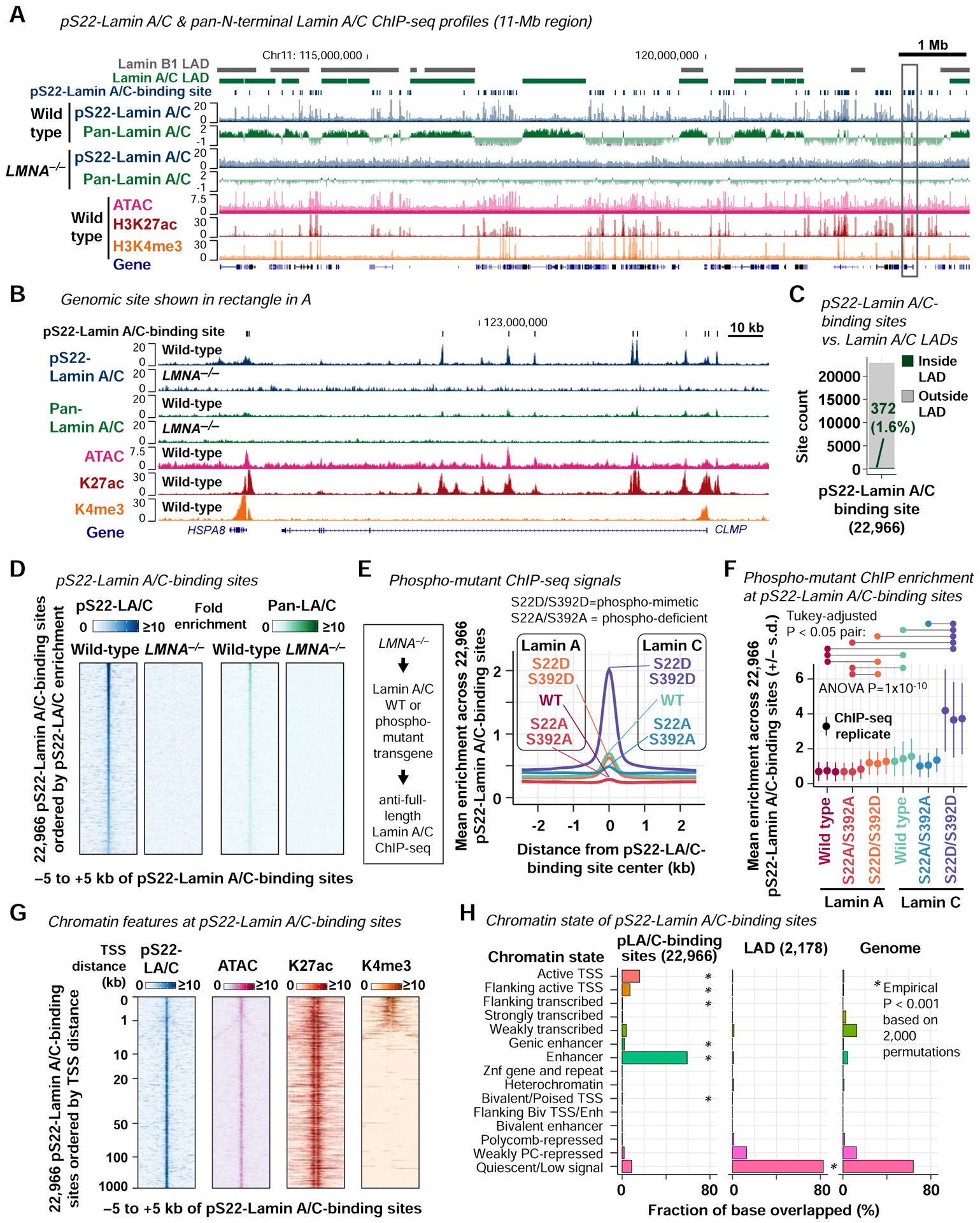
(A) Representative pS22-Lamin A/C ChIP-seq and pan-N-terminal-Lamin A/C ChIP-seq profiles. Histone ChIP-seq and ATAC-seq are shown for comparison. Signals are fold-enrichment (FE) scores (see Methods). Pan-N-terminal-Lamin A/C profiles are in the log2 scale to visualize LAD pattern. Lamin B1 LADs are defined by DamID in lung fibroblast (Guelen et al., 2008). Lamin A/C LADs are defined by pan-N-terminal-Lamin A/C ChIP-seq in BJ-5ta (this study). All performed in ≥2 biological replicates. See also Fig. S2 and S3.
(B) Region shown in rectangle in (A). Pan-N-terminal-Lamin A/C ChIP-seq profiles are in the linear scale.
(C) Positions of pS22-Lamin A/C-binding sites with respect to 2,178 Lamin A/C LADs.
(D) pS22-Lamin A/C and pan-N-terminal-Lamin A/C ChIP-seq FE scores at pS22-Lamin A/C-binding sites.
(E) ChIP-seq FE scores for transgene-driven Lamin A/C with or without S22/S392 mutations, expressed in LMNA−/− cells. ChIP was performed with the anti-full-length-Lamin A/C antibody in 3 biological replicates.
(F) ChIP-seq FE scores at pS22-Lamin A/C-binding sites for each biological replicate of (E). One-way ANOVA compares means of all isoforms, with post-hoc Tukey analysis for pairwise comparison (pairs with P<0.05 are indicated).
(G) ATAC-seq and histone ChIP-seq FE scores at pS22-Lamin A/C-binding sites. pS22-Lamin A/C ChIP-seq signals are shown again for comparison.
(H) Chromatin states of pS22-Lamin A/C-binding sites and Lamin A/C LADs. Chromatin states are defined in normal dermal fibroblasts (Roadmap Epigenomics Consortium et al., 2015).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa-647-conjugated
anti-phospho- Ser22-LMNA antibody D2B2E |
Cell Signaling | Cat # 97262BC, Lot 1 |
| Anti-phospho-Ser22-LMNA antibody D2B2E |
Cell Signaling | Cat # 13448S, Lot 1; RRID:AB_2798221 |
| Anti-pan-N-terminal-LMNA antibody E1 | Santa Cruz Biotechnology |
Cat # sc-376248, Lot H2812; RRID:AB_10991536 |
| Anti-full-length-LMNA antibody 4C4 | Abcam | Cat # ab190380, Lot GR201137–1; RRID:AB_2747781 |
| Anti-c-Jun antibody H-79 | Santa Cruz Biotechnology |
Cat # sc-1694, Lot D1014; RRID:AB_631263 |
| Anti-H3K27ac antibody MABI0309 | Wako | Cat # 306–34849, MABI0309, Lot
14007; RRID:AB_11126964 |
| Anti-H3K4me3 antibody MABI0304 | Wako | Cat # 305–34819, MABI0304, Lot
14004; RRID:AB_11123891 |
| Anti-H3K9me3 antibody ab8898 | Abcam | Cat # ab8898, Lot GR232099–3; RRID:AB_306848 |
| Anti-H3K27me3 antibody MABI0323 | Active Motif | Cat # MABI0323, Lot 17019020; RRID:AB_11123929 |
| Anti-BrdU antibody 3D4 | BD Biosciences | Cat # 555627, Lot 7033666; RRID:AB_395993 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LMNA aa2–30 peptide: ETPSQRRATRSGAQASSTPLSPTRITRL Q |
GenScript | GenScript U2312EI090–1/PE2183 |
| LMNA aa2–30 peptide with
phospho-S22: ETPSQRRATRSGAQASSTPL[phospho- S]PTRITRLQ |
GenScript | GenScript U2312EI090–3/PE2186 |
| Phosphatase inhibitor Nodularin | Enzo | Cat # ALX-350–061 |
| Deposited Data | ||
| Genomic dataset IDs with sample description |
This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md |
| Blacklisted regions used in this study | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/blacklist.bed |
| pS22-Lamin A/C binding sites in BJ-5ta | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/D2B2E_BJ5ta.narrowPeak |
| Lamin A/C LADs in BJ-5ta | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/E1_BJ5ta_LAD.bed |
| ATAC-seq-defined accessible chromatin sites in BJ-5ta |
This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/ATAC_BJ5ta.narrowPeak |
| H3K27ac-enriched sites in BJ-5ta | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/H3K27ac_BJ5ta.narrowPeak |
| H3K4me3-enriched sites in BJ-5ta | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/H3K4me3_BJ5ta.narrowPeak |
| c-Jun-binding sites in BJ-5ta | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/Jun_BJ5ta.narrowPeak |
| Genes linked to pS22-Lamin A/C sites with GRO-seq coverage in BJ-5ta |
This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/gene_pS22-LMNA.bed |
| Differentially-expressed genes in progeria | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/progeria_gene.bed |
| Gained and lost pS22-Lamin A/C-binding sites in progeria |
This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/progeria_gained_lost_pS22-LMNA.bed |
| Gained and lost LADs in progeria | This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/progeria_gained_lost_LAD.bed |
| Progeria-up genes linked to gained
pS22- Lamin A/C-binding sites |
This paper | https://github.com/kohta-ikegami/pS22-LMNA/blob/master/progeria-up_gene.txt |
| Human reference sequence used | UCSC genome browser |
hg19/GRCh37 |
| Gene annotation | Gencode/UCSC genome browser |
Version 19; http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/wgEncodeGencodeBasicV19.txt.gz |
| High-throughput sequencing data | This paper | GEO accession number GSE113354 |
| Hg19 assembly gaps | UCSC genome browser |
http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/gap.txt.gz |
| ENCODE blacklist | UCSC genome browser |
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz |
| Chromatin annotation | NIH Roadmap Epigenomics |
http://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/coreMarks/jointModel/final/E126_15_coreMarks_stateno.bed.gz |
| Lamin B1 LADs | Gluelen et al., 2008 | http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/laminB1Lads.txt.gz |
| Progeria fibroblast RNA-seq datasets | Fleischer et al., 2018 | GEO accession number GSE113957 |
| Experimental Models: Cell Lines | ||
| BJ-5ta fibroblast cell line | ATCC | Cat# CRL-4001; RRID:CVCL_6573 |
| BJ-5ta-derived LMNA–/– cell line | This paper | Ikegami lab ID: cc1170–1AD2 |
| GM07492 normal primary fibroblast | Coriell Cell Repository |
GM07492; RRID:CVCL_7467 |
| GM08398 normal primary fibroblast | Coriell Cell Repository |
GM08398; RRID:CVCL_7481 |
| AG11498 HGPS primary fibroblast | Coriell Cell Repository |
AG11498; RRID:CVCL_H766 |
| HGADFN167 HGPS primary fibroblast | Progeria Research Foundation |
HGADFN167; RRID:CVCL_1Y92 |
| Oligonucleotides | ||
| DNA oligonucleotide KI223 for sgRNA1 targeting exon1 of LMNA: CACCGCATCGACCGTGTGCGCTCGC |
IDT | Ikegami lab ID: KI223 |
| DNA oligonucleotide KI224 for sgRNA1 targeting exon1 of LMNA: AAACGCGAGCGCACACGGTCGATGC |
IDT | Ikegami lab ID: KI224 |
| DNA oligonucleotide KI227 for sgRNA3 targeting exon1 of LMNA: CACCGCGGCATCAAGGCCGCCTACG |
IDT | Ikegami lab ID: KI227 |
| DNA oligonucleotide KI228 for sgRNA3 targeting exon1 of LMNA: AAACCGTAGGCGGCCTTGATGCCGC |
IDT | Ikegami lab ID: KI228 |
| DNA oligonucleotide KI361 for S22A Lamin A/C N-terminus: GGGGACAAGTTTGTACAAAAAAGCAG GCTGCCATGGCTTGTCCTAAAGATCC AGCCAAACCTCCGGCCAAGGCACAAG TTGTGGGATGGCCACCGGTGAGATCA TACCGGAAGAACGTGATGGTTTCCTG CCAAAAATCAAGCGGTGGCCCGGAGG CGGCGGCGTTCGTGAAGGAGACCCC GTCCCAGCGGCGCGCCACCCGCAGC GGGGCGCAGGCCAGCTCCACTCCGC TGGCTCCCACCCGCATCACCCGGCTG CAGGAGAAGGAGGACCTGCAGGAGC TCAATGATCGCTTGGCGGTCTATATTG ATAGGGTCCGGAGTCTCGAAACGGAG AACGCAGGGCTGCGCCTTCGCATCAC CGAGTCTGAAGAGGTGGTCAGCCGCG AGGTGTCTGGAATTAAAGCTGCTTATG AAGCCGAGCTCGGGGATGCCCGCAA GACCCTTGACTCAGTAGCCAAGGAGC GCGCCCGCCTGCAGCTGGAGCTGAG CAAAGTGCGT |
IDT | Ikegami lab ID: KI361 |
| DNA oligonucleotide KI362 for S22D Lamin A/C N-terminus: GGGGACAAGTTTGTACAAAAAAGCAG GCTGCCATGGCTTGTCCTAAAGATCC AGCCAAACCTCCGGCCAAGGCACAAG TTGTGGGATGGCCACCGGTGAGATCA TACCGGAAGAACGTGATGGTTTCCTG CCAAAAATCAAGCGGTGGCCCGGAGG CGGCGGCGTTCGTGAAGGAGACCCC GTCCCAGCGGCGCGCCACCCGCAGC GGGGCGCAGGCCAGCTCCACTCCGC TGGACCCCACCCGCATCACCCGGCTG CAGGAGAAGGAGGACCTGCAGGAGC TCAATGATCGCTTGGCGGTCTATATTG ATAGGGTCCGGAGTCTCGAAACGGAG AACGCAGGGCTGCGCCTTCGCATCAC CGAGTCTGAAGAGGTGGTCAGCCGCG AGGTGTCTGGAATTAAAGCTGCTTATG AAGCCGAGCTCGGGGATGCCCGCAA GACCCTTGACTCAGTAGCCAAGGAGC GCGCCCGCCTGCAGCTGGAGCTGAG CAAAGTGCGT |
IDT | Ikegami lab ID: KI362 |
| DNA oligonucleotide KI393 for S392A Lamin A/C C-terminal cloning: TGGAGCTGAGCAAAGTGCGT |
IDT | Ikegami lab ID: KI393 |
| DNA oligonucleotide KI385 for S392A Lamin A C-terminal cloning: TCGTGGCTCCGGAACCGGTGTTACAT GATGCTGCAGTTCT |
IDT | Ikegami lab ID: KI385 |
| DNA oligonucleotide KI395 for S392A Lamin C C-terminal cloning: TCGTGGCTCCGGAACCGGTGTCAGCG GCGGCTACCACTCACGTGGTGGTGAT GGAGCAGG |
IDT | Ikegami lab ID: KI395 |
| Recombinant DNA | ||
| LentiCRISPRv2 | Sanjana et al., 2014 | Addgene plasmid #52961 |
| psPAX2 | Didier Trono lab | Addgene plasmid #12260 |
| pCMV-VSV-G | Stewart et al., 2003 | Addgene plasmid #8454 |
| pCW57-MCS1-P2A-MCS2-PGK-Blast | Barger et al. 2019 | Addgene plasmid #80921 |
| Wild-type Lamin A cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI218 |
| Lamin A-S22A/S392A cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI226 |
| Lamin A-S22D/S392D cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI241 |
| Wild-type Lamin C cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI247 |
| Lamin C-S22A/S392A cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI242 |
| Lamin C-S22D/S392D cDNA in
pCW57- MCS1-P2A-MCS2-PGK-Blast |
This paper | Ikegami lab ID: bKI245 |
| Software and Algorithms | ||
| R | The R foundation | R 3.3.2 |
| Bowtie aligner | Langmead and Salzberg, 2012 | Version 2.1.0 |
| Bedtools | Quinlan and Hall, 2010 | Version 2.26.0 |
| DESeq2 | Love et al., 2014 | Version 1.14.1 |
| MACS2 | Zhang et al., 2008 | Version 2.1.0 |
| MEME | Machanick and Bailey, 2011 | Version 4.10.0 |
| R package stats | The R foundation | Version 3.3.2 |
| R package preprocessCore | Bolstad et al., 2003 | Version 1.36.0 |
| Metascape | Tripathi et al., 2015 | http://metascape.org/ |
pS22-Lamin A/C physically associates with putative active enhancers
pS22-Lamin A/C-binding sites exhibited genomic features of active enhancers. First, a vast majority of the 22,966 pS22-Lamin A/C-binding sites in BJ-5ta fibroblasts were located distal to the transcription start sites (TSSs) of genes (89% outside of −1000 kb to +500 bp of TSSs) (Fig. 2G). Second, we performed ATAC-seq in BJ-5ta and observed that 88% of pS22-Lamin A/C-binding sites coincided with regions of accessible chromatin, a feature of regulatory regions (Fig. 2B, G; Fig. S3A; Key Resources Table). Third, we performed H3K27ac ChIP-seq in BJ-5ta and observed that 82% of pS22-Lamin A/C-binding sites coincided with regions enriched for H3K27ac, a histone modification associated with active enhancers and promoters (Fig. 2B, G; Fig. S3A; Key Resources Table). Fourth, we performed H3K4me3 ChIP-seq in BJ-5ta and observed that only 13% of pS22-Lamin A/C-binding sites coincided with regions enriched for H3K4me3, a histone modification associated with active promoters (Fig. 2B, G; Fig. S3A; Key Resources Table). Together, the strong association of pS22-Lamin A/C-binding sites with H3K27ac but not H3K4me3 suggested a binding preference of pS22-Lamin A/C for enhancers over promoters. Fifth, comparison of the 22,966 pS22-Lamin A/C-binding sites with the chromatin state annotations in dermal fibroblasts (Roadmap Epigenomics Consortium et al., 2015) found that 59% of pS22-Lamin A/C-binding sites were located in regions annotated as “Enhancers” (empirical P<0.001 based on 2,000 permutations), whereas a smaller fraction of 16% were in “Active TSSs” (P<0.001) (Fig. 2H). Finally, although nearly all pS22-Lamin A/C-binding sites corresponded to accessible chromatin and H3K27ac-marked sites, pS22-Lamin A/C-binding sites corresponded to a minor subset of all accessible chromatin sites (26%) or all H3K27ac-marked sites (14%) in BJ-5ta (Fig. S3A, B). Thus, pS22-Lamin A/C physically associates with a specific subset of promoter-distal, enhancer-like elements.
pS22-Lamin A/C-binding sites are strongly bound by the c-Jun transcription factor
We investigated whether pS22-Lamin A/C-binding sites are co-occupied by specific transcription factors (TFs). We performed de novo motif analysis and found that TF binding motifs for AP-1, FOX, and RUNX were overrepresented within pS22-Lamin A/C-binding sites, relative to the sequence composition of accessible chromatin sites in BJ-5ta (Fig. 3A). Among those motifs, the AP-1 motif was present at the highest frequency at pS22-Lamin A/C-binding sites (9,250 sites, 41%), with a peak frequency located at the center of pS22-Lamin A/C-binding sites (Fig. 3A). To test whether AP-1 transcription factors bind pS22-Lamin A/C-binding sites, we performed ChIP-seq in BJ-5ta for c-Jun, a core protein of the AP-1 dimeric transcription factors (Key Resources Table). c-Jun was strongly enriched at almost all pS22-Lamin A/C-binding sites (92%; Fisher’s exact test P<2×10−16; Fig. 3B–D). Furthermore, c-Jun binding at pS22-Lamin A/C-binding sites was much stronger than that outside of pS22-Lamin A/C-binding sites (Mann-Whitney U test, P<2×10−16; Fig. 3E). We assessed the possibility that the strong co-association between pS22-Lamin A/C binding and c-Jun binding might be due to the high local chromatin accessibility of these co-associated sites. We stratified all 73,933 accessible sites defined by ATAC-seq in BJ-5ta into deciles by accessibility (i.e. ATAC-seq enrichment), and from each decile, randomly selected 100 pS22-Lamin A/C-bound accessible sites and 100 pS22-Lamin A/C-unbound accessible sites. In every accessibility decile, we observed more frequent c-Jun binding by ChIP-seq (Fisher’s exact test, P < 7×10−4; Fig. S3C) and stronger c-Jun ChIP-seq enrichment levels (Mann-Whitney U test P=4×10−13 to 2×10−23; Fig. 3F) at pS22-Lamin A/C-bound accessible sites, compared with unbound sites. An analogous analysis with all 87,988 c-Jun-bound sites stratified into deciles by accessibility showed that, in every decile, c-Jun enrichment levels at pS22-Lamin A/C-bound sites was stronger than at unbound sites (Mann-Whitney U test P=1×10−9 to 5×10−20) (Fig. S3D). H3K27ac levels were also higher at accessible sites or c-Jun-binding sites with pS22-Lamin A/C binding versus without pS22-Lamin A/C binding, within the same accessibility decile (Mann-Whitney U test P < 6×10−5 among ATAC sites; P < 2×10−14 among c-Jun-binding sites; Fig. 3F; Fig. S3D). Corroborating these observations, pS22-Lamin A/C ChIP signals were strongly positively correlated with c-Jun ChIP signals (Pearson correlation coefficient r=0.63) at the 22,966 pS22-Lamin A/C-binding sites, which, for comparison, were stronger than the correlation with H3K27ac signals (r=0.31) or ATAC-seq signals (r=0.28) (Fig. 3G). Thus, strong pS22-Lamin A/C binding, rather than high chromatin accessibility, predicted strong c-Jun-binding, and vice versa. The strong association between pS22-Lamin A/C binding and c-Jun binding suggests that c-Jun and pS22-Lamin A/C may function together at putative enhancers.
Figure 3. pS22-Lamin A/C association and c-Jun association are strongly correlated at pS22-Lamin A/C-binding sites.
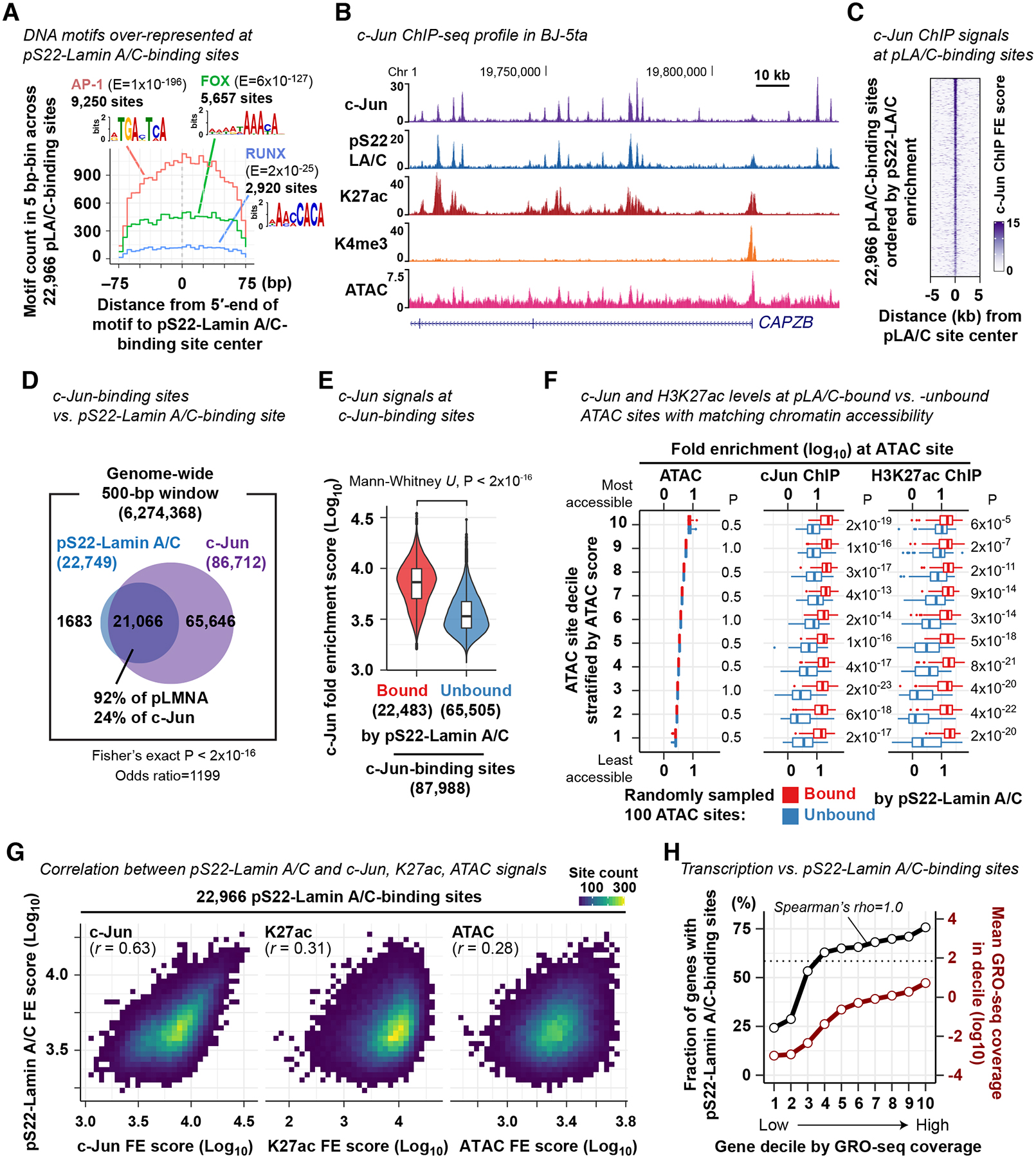
(A) DNA motif frequency at pS22-Lamin A/C-binding sites. E, motif occurrence probability score in de novo DNA motif search.
(B) Representative c-Jun ChIP-seq FE profile in BJ-5ta cells (derived from 3 biological replicates). Other profiles are shown for comparison.
(C) c-Jun ChIP-seq FE scores at pS22-Lamin A/C-binding sites.
(D) Overlap between pS22-Lamin A/C-binding sites and c-Jun-binding sites. Numbers indicate the number of 500-bp windows that overlap pS22-Lamin A/C-binding sites and/or c-Jun-binding sites.
(E) c-Jun ChIP FE scores at c-Jun-binding sites bound or unbound by pS22-Lamin A/C. Box, interquartile range. Violin, kernel density (see Methods).
(F) c-Jun ChIP and H3K27ac ChIP-seq FE scores at 100 pS22-Lamin A/C-bound and 100 unbound ATAC sites. The analyzed 100 sites were randomly selected from each decile of all ATAC sites stratified by accessibility. Mann-Whitney U-test compares FE scores between pS22-Lamin A/C-bound and -unbound sites, and P-values are adjusted for multiple comparisons by the Benjamini-Hochberg method. See also Fig. S3.
(G) Two-dimensional histogram of pS22-Lamin A/C-binding sites by ChIP-seq and ATAC-seq FE scores. One square, one bin, with color grade representing the number of sites. r, Pearson correlation coefficient.
(H) Black line, fraction of genes harboring pS22-Lamin A/C-binding sites within gene body or 100 kb upstream. Genes are stratified by the transcription levels defined by GRO-seq read coverage. Red line, mean GRO-seq coverage in gene decile. Horizontal dotted line, fraction of all genes harboring pS22-Lamin A/C-binding sites for reference. GRO-seq was performed in BJ-5ta in 2 biological replicates.
pS22-Lamin A/C-binding sites are located near highly transcribed genes
We hypothesized that pS22-Lamin A/C-binding sites are located near genes undergoing active transcription. We therefore performed GRO-seq (Core et al., 2008) to quantify transcriptional activity of genes in BJ-5ta cells (Key Resources Table) and linked pS22-Lamin A/C-binding sites to gene transcription levels. We associated genes with pS22-Lamin A/C-binding sites that resided in the gene body or the 100-kb upstream region. The fraction of genes linked to pS22-Lamin A/C-binding sites was highest among the top 10% of highly transcribed genes (76%) and lowest among the bottom 10% of transcribed genes (24%), with strong positive monotonic relationship between the transcription levels and the fraction of genes with pS22-Lamin A/C-binding sites (Spearman’s rank correlation coefficient rho=1.0; Fig. 3H). The strong correlation remained in analysis of genes specifically located outside of LADs (rho=0.98; Fig. S3E), excluding the possibility that the observed positive correlation was driven by the localization of pS22-Lamin A/C binding outside of transcriptionally-inactive LADs. We further excluded the potential confounding effects of gene lengths and gene density (Fig. S3F). Together, these analyses revealed that pS22-Lamin A/C binding is associated with high transcriptional activity of local genes.
Genes abnormally up-regulated in progeria-patient fibroblasts are relevant to progeria phenotypes
The heterozygous LMNA mutations that cause Hutchinson-Gilford progeria encode an altered Lamin A protein, entitled progerin, which interacts with wild-type Lamin A/C and is thought to alter normal Lamin A/C functions (Lee et al., 2016). We hypothesized that progerin may alter pS22-Lamin A/C function at enhancers. We therefore investigated the possibility that altered pS22-Lamin A/C binding to enhancers contributed to gene expression changes in progeria. We performed transcriptome analysis to identify genes dysregulated in fibroblast cell lines from progeria patients. We performed RNA-seq on primary dermal fibroblasts from two progeria patients (AG11498 and HGADFN167) and two normal individuals with similar ages (GM07492 and GM08398) (Fig. S4A–C). In addition, we obtained public RNA-seq data sets for primary dermal fibroblasts from ten progeria patients and ten normal individuals with similar ages (Fleischer et al., 2018) (Fig. S4A). By principal component analysis, RNA-seq datasets of the progeria patients were separated from those of the normal individuals, indicating a common gene expression signature among progeria-patient fibroblasts distinct from that of normal individuals (Fig. 4A). Comparison between the 14 progeria RNA-seq data sets (for 11 distinct cell lines) and the 12 normal RNA-seq data sets (for 10 distinct cell lines after removing 2 outlier datasets; Fig. S4D) identified 1,117 dysregulated genes, 615 up-regulated in the progeria cell lines (“progeria-up” genes) and 502 down-regulated in the progeria cell lines (“progeria-down” genes) (Fig. 4B, C; Key Resources Table). The progeria-up genes were strongly over-represented for specific DisGeNET-curated disease ontology terms (Piñero et al., 2017) (17 terms with P<0.001) that were well-documented progeria phenotypes, such as “infraction, middle cerebral artery” (P=7×10−6) (Silvera et al., 2013), “coronary artery disease” (P=2×10−5) (Olive et al., 2010), “cardiomegaly” (P=4×10−5) (Prakash et al., 2018), and “hypertensive disease” (P=1×10−4) (Merideth et al., 2008), and that were closely related to progeria phenotypes, such as “juvenile arthritis” (P=1×10−5) (Gordon et al., 2007) and “Congenital hypoplasia of femur” (P=1×10−4) (Gordon et al., 2007) (Fig. 4D). The progeria-down genes were associated with only three DisGeNet disease terms (P<0.001), including two terms that might be related to progeria phenotypes including “hip joint varus deformity” (Gordon et al., 2007) (P=6×10−4) and “short stature, mild” (P=9×10−4) (Gordon et al., 2011) (Fig. 4D). Thus, genes dysregulated in progeria-patient fibroblast lines were associated with the clinical components of progeria, with stronger association observed for genes upregulated in progeria fibroblasts.
Figure 4. Genes abnormally upregulated in progeria fibroblasts are relevant to progeria phenotypes.
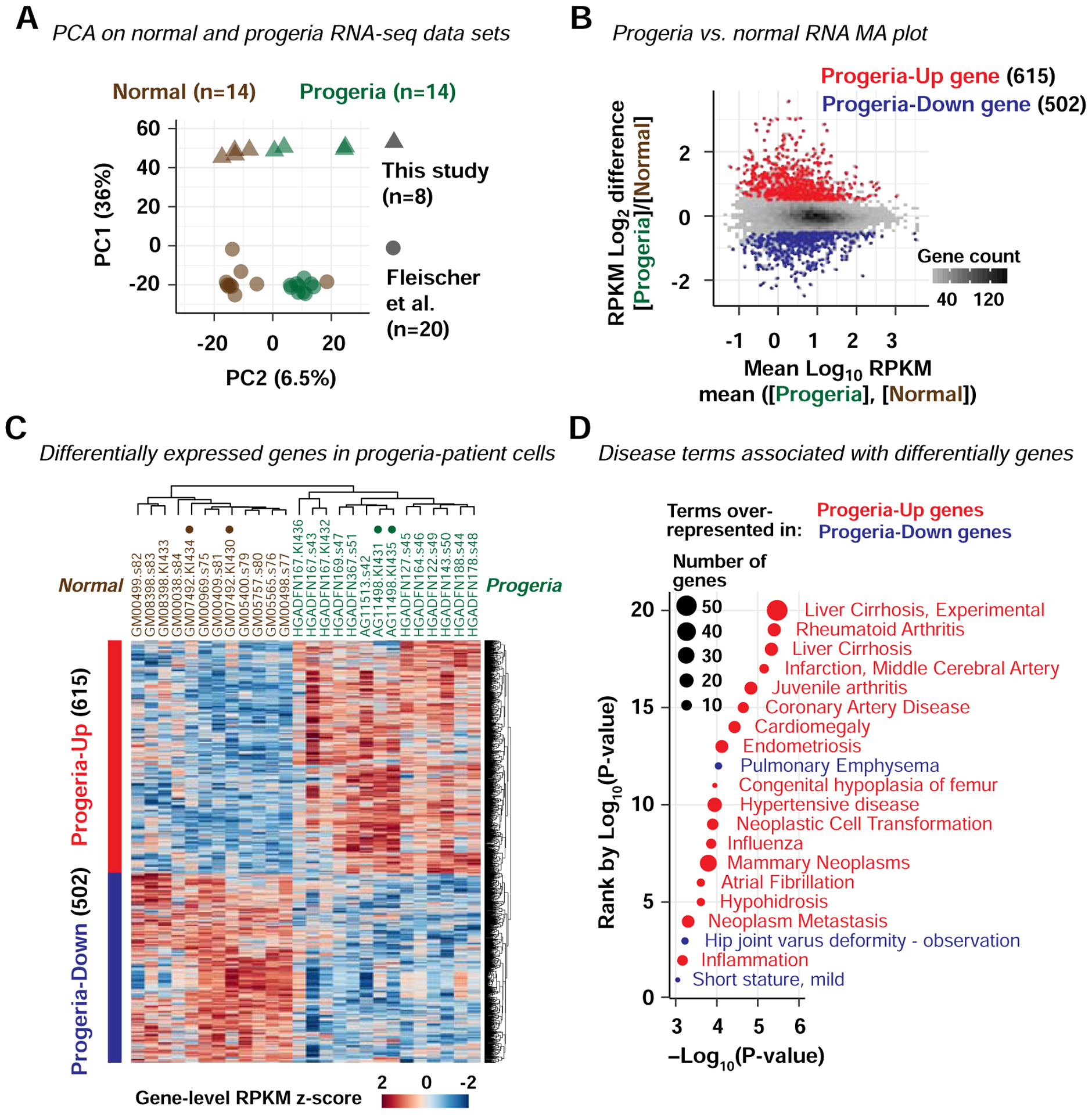
(A) Principal component analysis (PCA) on RNA-seq data sets of primary fibroblasts derived from normal individuals and progeria patients. Percentage, proportion of variance explained. See also Fig. S4.
(B) MA plot comparing the per-gene mean of RNA-seq RPKM scores across progeria-patient fibroblast cell lines and that across normal-individual fibroblast cell lines.
(C) Normalized RPKM scores for genes upregulated in progeria (“progeria-up”) and genes downregulated in progeria (“progeria-down”). Dendrograms represent hierarchical clustering. Solid circles, fibroblast cell lines used in LAD and pS22-Lamin A/C analyses.
(D) DisGeNet-curated disease terms overrepresented among progeria-up (red) and progeria-down genes (blue). Shown are terms with P-value < 0.001.
LAD alterations do not explain most gene expression alterations in progeria
We next investigated the cause of the gene expression alterations in the progeria-patient fibroblasts. A prevailing model for progeria pathogenesis is that disruption of LADs causes loss of heterochromatin-associated histone modifications, which in turn alters gene expression (Shumaker et al., 2006; McCord et al., 2012; Gordon et al., 2014). To determine the extent to which LAD alterations in progeria fibroblasts could explain the observed gene expression changes, we performed ChIP-seq using anti-pan-N-terminal-Lamin A/C antibody in the progeria-patient fibroblast cell line AG11498 and the normal-individual fibroblast cell line GM07492 (Key Resources Table); these cell lines demonstrated transcriptomes representative of progeria versus normal fibroblasts, respectively (Fig. 4C). We identified 2,735 total or ‘union’ LADs, i.e., present in either the progeria or the normal fibroblast or both. Of those, 635 LADs (23%) were unique to either the normal or progeria fibroblasts: 353 LADs (13%) were present in normal fibroblasts but absent in progeria fibroblasts (called “lost LADs”), whereas 282 LADs (10%) were absent in normal fibroblasts but present in progeria fibroblasts (called “gained LADs”) (Fig. 5A, B; Fig. S5A, B). To determine if gained or lost LADs were associated with changes of the heterochromatin-associated histone modifications H3K9me3 and H3K27me3, we performed ChIP-seq for H3K9me3 and H3K27me3 in the progeria cell line AG11498 and the normal cell line GM07492. H3K9me3 levels were reduced in the progeria cell line at lost LADs (Mann-Whitney U test P=3×10−12) but increased at gained LADs (P=3×10−12) (Fig. 5C), showing a positive correlation with the direction of LAD changes. H3K27me3 levels were reduced in progeria at both lost and gained LADs compared with the union LADs (P=1×10−5 for lost LADs and P=5×10−7 for gained LADs) (Fig. 5D), consistent with the previous report that H3K27me3 levels are globally reduced at gene-poor regions in progeria-patient fibroblasts (McCord et al., 2012). These observations confirm the previous findings that the LAD profile was altered in progeria and that LAD alterations in progeria are associated with changes of heterochromatin-associated histone modifications (Shumaker et al., 2006; McCord et al., 2012).
Figure 5. LAD alterations do not explain the majority of gene expression changes in progeria.
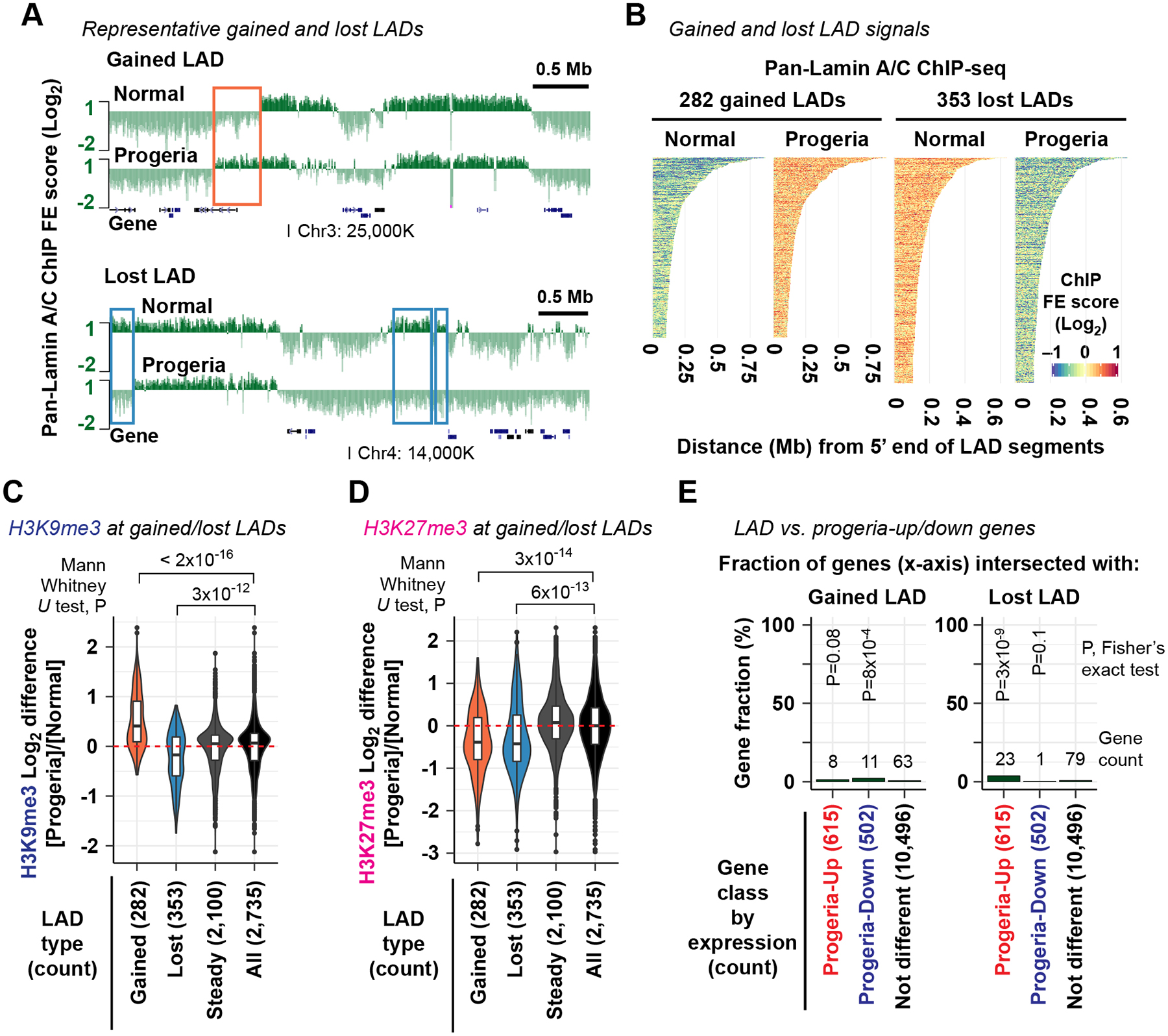
(A) Representative pan-N-terminal-Lamin A/C ChIP-seq profiles in normal (GM07492) and progeria-patient (AG11498) fibroblasts (derived from 2 biological replicates). Log2 scale on the y axis. Rectangle, representative gained LADs (top) and lost LADs (bottom). See also Fig. S5.
(B) Pan-N-terminal-Lamin A/C ChIP-seq FE scores in the normal GM07492 and progeria AG11498 fibroblasts at gained and lost LADs in progeria.
(C) Log2 difference of H3K9me3 ChIP-seq FE scores at LADs between normal (GM07492) and progeria (AG11498) fibroblasts (derived from 2 biological replicates). Red dashed line, Log2 difference equals 0 for reference.
(D) Same as (C), but H3K27me3 ChIP-seq scores are analyzed.
(E) Fraction of progeria-up and progeria-down genes whose gene body or upstream 100-kb region intersected with gained LADs (left) or lost LADs (right). P, Fisher’s exact test P value for association between being differentially expressed and being affiliated with gained/lost LADs.
We next asked if genes upregulated in progeria were affiliated with lost LADs or genes downregulated in progeria were affiliated with gained LADs by examining genes whose gene body or upstream 100-kb region intersected with lost or gained LADs (Key Resources Table). Only 23 out of 615 genes up-regulated in progeria (3.7%) intersected with lost LADs (Fisher’s exact test P=3×10−9; Fig. 5E). Similarly, only 11 out of 502 genes downregulated in progeria (2.2%) intersected with gained LADs (P=8×10−4; Fig. 5E). Furthermore, these progeria-up genes in lost LADs (23 genes) or progeria-down genes in gained LADs (11 genes) were not over-represented for disease ontology terms linked to progeria phenotypes (Fig. S5C, D). These data revealed that local LAD alterations could not explain the vast majority of gene expression changes in progeria, either upregulation of gene expression by LAD losses or downregulation of gene expression by LAD gains.
New pS22-Lamin A/C-binding sites emerge in normally quiescent loci in progeria-patient fibroblasts
We hypothesized that the gene expression changes observed in the progeria-patient fibroblasts were associated with alterations of pS22-Lamin A/C-binding sites. We performed immunofluorescence for pS22-Lamin A/C in progeria (AG11498) and normal (GM07492) fibroblasts and observed that pS22-Lamin A/C was present in the interior of interphase nuclei but not at the nuclear periphery of the progeria fibroblasts, similar to normal fibroblasts (Fig. S6A). We found that the major isoform phosphorylated at S22 in the progeria-patient and normal fibroblast cells was Lamin C, while S22-phosphorylation of progerin was not detectable in Western blotting in the progeria-patient fibroblasts (Fig. S6B). We performed pS22-Lamin A/C ChIP-seq in the progeria-patient fibroblast AG11498 and the normal-individual fibroblast GM07492 (the same cell lines used in pan-N-terminal-Lamin A/C ChIP-seq). We identified a union set of 15,323 pS22-Lamin A/C-binding sites found in either the normal-individual fibroblast GM07492 or the progeria-patient fibroblast AG11498 or both (Key Resources Table). We observed significant alteration of the pS22-Lamin A/C-binding site profile in the progeria fibroblasts: of the 15,323 union pS22-Lamin A/C-binding sites, 2,796 pS22-Lamin A/C-binding sites (18%) were specific to the progeria fibroblast line (termed “gained pS22-Lamin A/C-binding sites”) whereas 2,425 pS22-Lamin A/C-binding sites (16%) were specific to the normal fibroblast line (termed “lost” pS22-Lamin A/C-binding sites) (Fig. 6A, B; Fig. S6C, D). Gained pS22-Lamin A/C-binding sites were highly over-represented within the “quiescent” chromatin-state annotation derived from normal dermal fibroblasts (Roadmap Epigenomics Consortium et al., 2015) (Fisher’s exact test P=2×10−144), whereas lost pS22-Lamin A/C-binding sites were over-represented in “enhancer” regions (P=8×10−30) (Fig. 6C). Thus, new pS22-Lamin A/C-binding sites emerged in locations in progeria, a subset of which possessed chromatin features of quiescence in normal fibroblasts, while a subset of the wild-type enhancer pS22-Lamin A/C-binding sites were lost in progeria. The absence of detectable pS22-progerin (Fig. S6B) suggests that progerin disrupted the binding specificity of wild-type pS22-Lamin A/C in progeria fibroblasts.
Figure 6. New pS22-Lamin A/C-binding sites emerged in progeria are associated with up-regulation of genes relevant to progeria phenotypes.
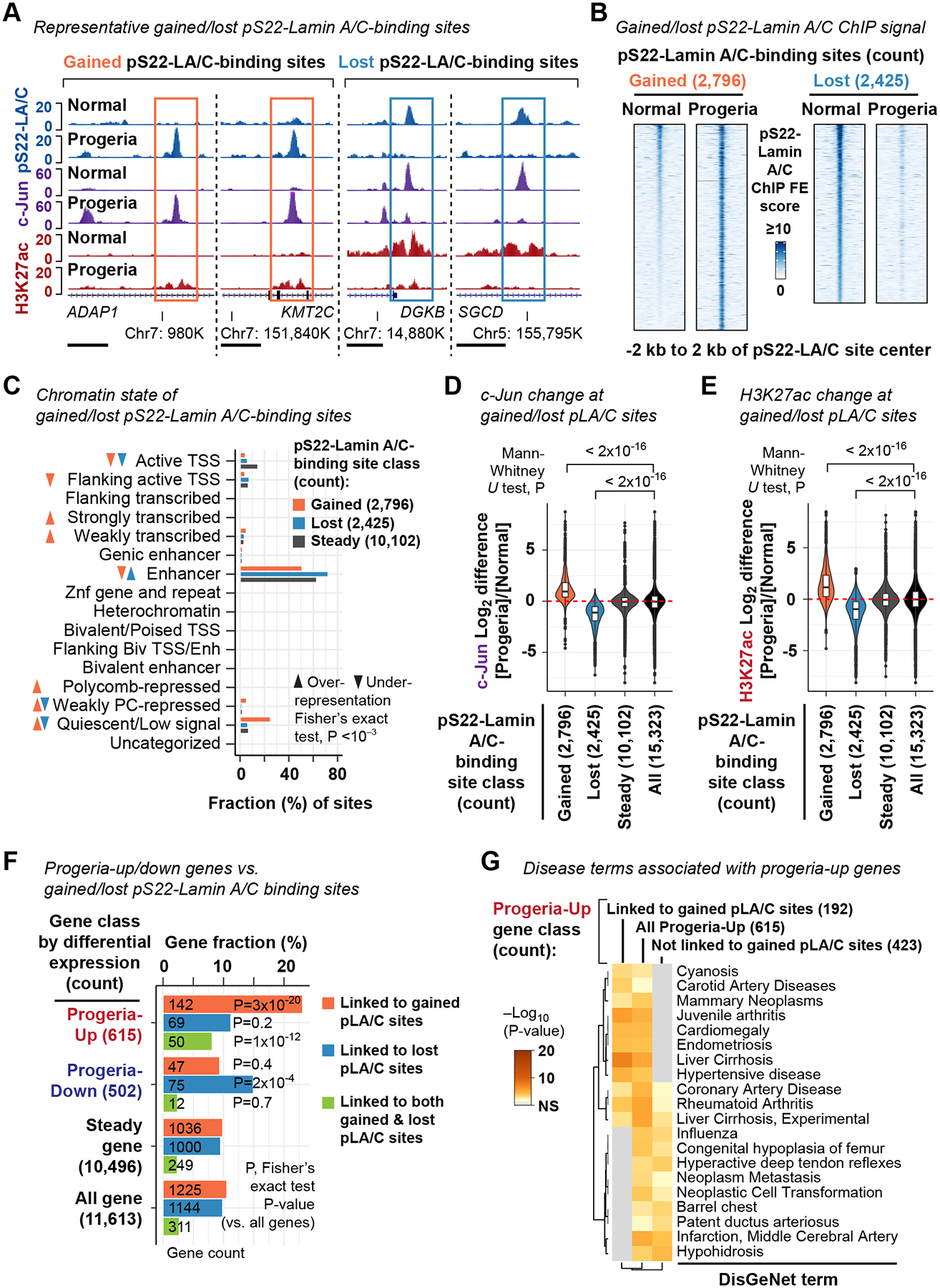
(A) pS22-Lamin A/C ChIP-seq profiles (derived from 2 biological replicates) at representative gained and lost pS22-Lamin A/C-binding sites. Normal, GM07492 fibroblast. Progeria, AG11498 fibroblast. Horizontal bar, 2 kb. See also Fig. S6.
(B) pS22-Lamin A/C ChIP-seq FE scores at gained and lost pS22-Lamin A/C-binding sites in the normal GM07492 and progeria AG11498 fibroblasts.
(C) Chromatin states of gained and lost pS22-Lamin A/C-binding sites. Fisher’s exact test assesses association between being called as gained or lost and being affiliated with each state.
(D) Log2 difference of c-Jun ChIP-seq FE scores between normal (GM07492) and progeria (AG11498) fibroblasts (derived from 2 biological replicates). Red dashed line, log2 difference equals 0 for reference.
(E) Same as (D), but H3K27ac ChIP-seq scores are analyzed.
(F) Fraction of progeria-up and progeria-down genes with gained and/or lost pS22-Lamin A/C-binding sites (within gene body or 100 kb upstream). P, Fisher’s exact test P value for association between being differentially expressed and being affiliated with gained/lost pS22-Lamin A/C-binding sites.
(G) DisGeNet-curated disease terms over-represented among progeria-up genes linked to gained pS22-Lamin A/C-binding sites, or those not linked to gained pS22-Lamin A/C-binding sites, or all progeria-up genes.
We hypothesized that changes in pS22-Lamin A/C-binding sites between progeria and normal fibroblasts reflected changes in the activity level of enhancers. To test this hypothesis, we performed c-Jun and H3K27ac ChIP-seq in the progeria fibroblast AG11498 and normal fibroblast GM07492. At the 2,796 gained pS22-Lamin A/C-binding sites, c-Jun and H3K27ac levels were strongly elevated in the progeria cell line relative to the normal cell line, as compared with all 15,323 union pS22-Lamin A/C-binding sites (Fig. 6D, E; Fig. S6E, F; Mann-Whitney U test P<2×10−16 for both c-Jun and H3K27ac). In contrast, at the 2,425 lost pS22-Lamin A/C-binding sites, c-Jun and H3K27ac levels were strongly diminished in the progeria cell line relative to the normal cell line (Fig. 6D, E; Fig. S6E, F; P<2×10−16 for both c-Jun and H3K27ac). Thus, the alteration of the pS22-Lamin A/C binding profile in progeria fibroblasts was accompanied by local alterations of c-Jun binding and H3K27ac levels at the pS22-Lamin A/C-binding sites.
Gains of pS22-Lamin A/C binding in progeria accompany abnormal transcriptional activation of genes clinically important to progeria pathophysiology
We hypothesized that the gains and losses of pS22-Lamin A/C-binding sites in progeria, and the associated changes in c-Jun and H3K27ac levels at those sites, affected transcription in progeria fibroblasts. To test this, we linked pS22-Lamin A/C-binding sites to genes, associating pS22-Lamin A/C-binding sites that reside in the gene body or the 100-kb upstream region. By this metric, 1,225 genes were linked only to gained pS22-Lamin A/C-binding sites (11% of all genes), and 1,144 genes were linked only to lost pS22-Lamin A/C-binding sites (9.9% of all genes) (Fig. S6G; Key Resources Table). Genes linked only to gained pS22-Lamin A/C-binding sites were highly over-represented among genes up-regulated in progeria (23% of progeria-up genes, P=3×10−20), but not among genes down-regulated in progeria (9.3% of progeria-down genes, P=0.4) (Fig. 6F). Genes linked only to lost pS22-Lamin A/C-binding sites were over-represented among genes down-regulated in progeria (15% of progeria-down genes, P=2×10−4), but not among genes upregulated in progeria (11% of progeria-up genes, P=0.2) (Fig. 6F). Thus, in progeria-patient fibroblasts, gains of pS22-Lamin A/C binding were associated with up-regulation of genes, and losses of pS22-Lamin A/C binding were associated with down-regulation of genes. Interestingly, 311 genes were linked to both gained and lost pS22-Lamin A/C-binding sites (2.7% of all genes), but were only over-represented among genes upregulated in progeria (8.1% of progeria-up genes, P=1×10−12), but not among genes downregulated in progeria (2.3% of progeria-down genes, P=0.7) (Fig. 6F), suggesting a dominant association between gains of pS22-Lamin A/C-binding and up-regulation of genes in progeria.
We hypothesized that gained or lost pS22-Lamin A/C-binding sites linked to genes dysregulated in progeria may be relevant to progeria phenotypes. The progeria-up genes linked to gained pS22-Lamin A/C-binding sites (192 genes) were highly over-represented for DisGeNet disease ontology terms relevant to progeria phenotypes such as carotid artery disease (P=0.007) (Gerhard-Herman et al., 2012), juvenile arthritis (P=1×10−5) (Gordon et al., 2007), hypertensive disease (P=0.0001) (Merideth et al., 2008), and cardiomegaly (P=4×10−5) (Prakash et al., 2018) (Fig. 6G). None of these terms were overrepresented among the progeria-up genes not linked to a pS22-Lamin A/C-binding site (423 genes) (Fig. 6G). Consistent with the ontology analysis, progeria-up genes with gained pS22-Lamin A/C binding included important cardiovascular genes (Key Resources Table). Examples include FHL1, overexpression of which causes myopathies (Schessl et al., 2008) and mutations of which causes Emery-Dreifuss muscular dystrophy also caused by LMNA mutations (Windpassinger et al., 2008; Gueneau et al., 2009) (Fig. S6D). The progeria-down genes linked to lost pS22-Lamin A/C-binding sites were not associated with disease ontology terms relevant to progeria phenotypes (Fig. S6H). Thus, gained pS22-Lamin A/C-binding sites mark a subset of abnormally activated genes in progeria that are highly relevant to progeria phenotypes.
DISCUSSION
Nuclear lamins have been extensively studied in the context of the nuclear lamina (Dechat et al., 2010b; van Steensel and Belmont, 2017). In this paper, we investigated the genomic localization and function of Ser22-phosphorylated (pS22) nuclear lamin A/C localized to the interior of the nucleus. pS22-Lamin A/C associated with kilobase-wide sites characteristic of active enhancers, in stark contrast to nuclear-peripheral Lamin A/C, which associates with megabase-wide heterochromatin domains (Meuleman et al., 2012; Lund et al., 2014). The existence of Lamin A/C in the nuclear interior has been documented for decades (Goldman et al., 1992; Bridger et al., 1993; Moir et al., 1994; Hozák et al., 1995; Barboro et al., 2002; Naetar et al., 2008; Shimi et al., 2008; Swift et al., 2013; Buxboim et al., 2014; Kochin et al., 2014; Gesson et al., 2016), yet the specific function of nuclear-interior Lamin A/C had been elusive. Our results provide evidence that nuclear-interior pS22-Lamin A/C functions as a positive modulator of enhancer activity by direct enhancer binding and that the enhancer function of pS22-Lamin A/C may be altered in Hutchinson-Gilford progeria and contribute to progeria pathogenesis (Fig. 7).
Figure 7. Model.
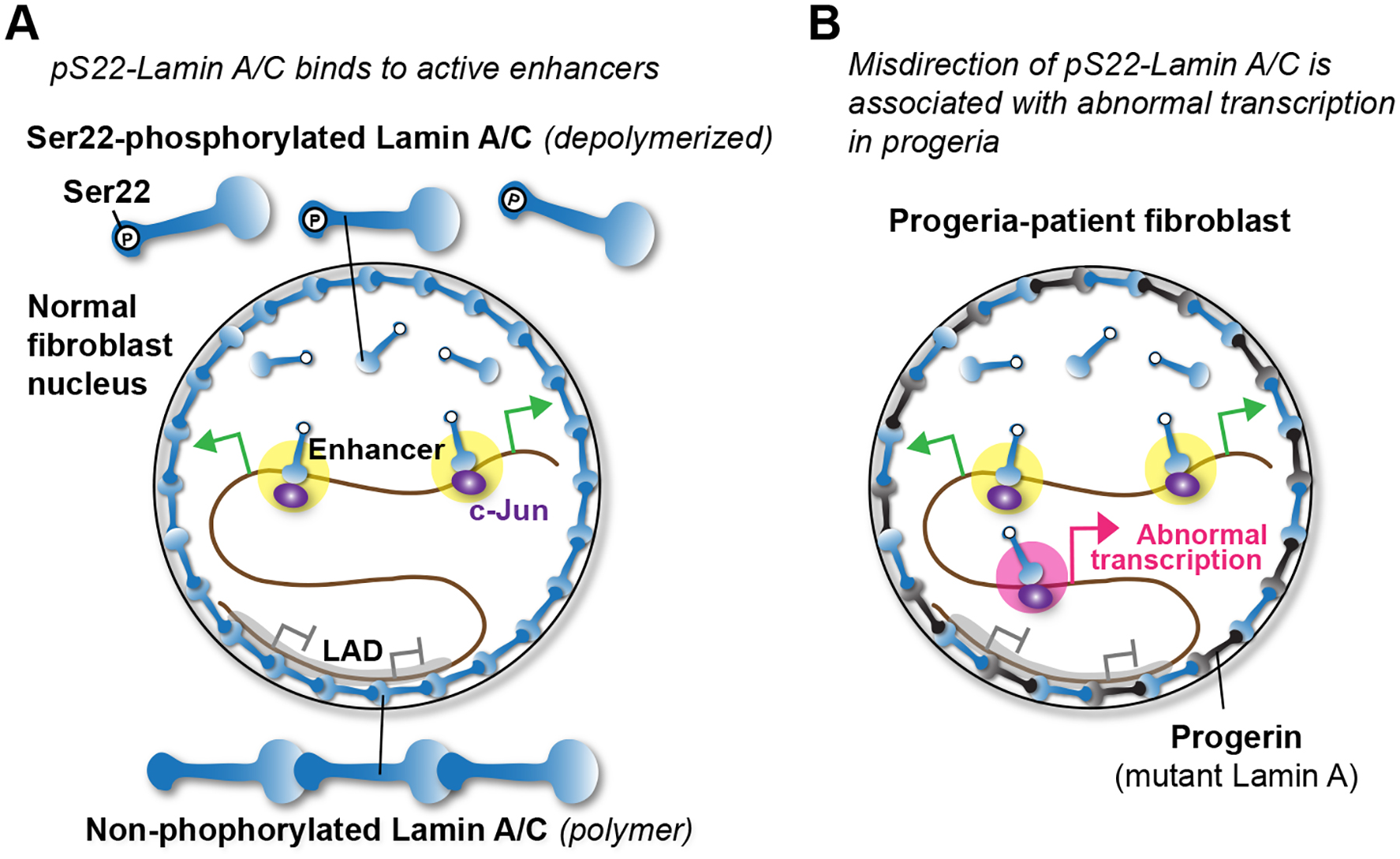
(A) pS22-Lamin A/C binds to putative enhancers near transcriptionally active genes.
(B) In progeria fibroblasts, gains of pS22-Lamin A/C-binding sites in abnormal locations accompany transcription of genes relevant to progeria phenotypes.
Decades of work have established that nuclear lamins at the nuclear lamina are associated with transcriptionally-inactive regions (Pickersgill et al., 2006; Guelen et al., 2008; Ikegami et al., 2010; Meuleman et al., 2012; van Steensel and Belmont, 2017). In this respect, the data showing that pS22-Lamin A/C-binding sites exhibit the characteristics of active enhancers and are associated with active transcription is striking. Our observations that Lamin C was the major S22-phosphorylated isoform in interphase and that phospho-mimetic Lamin C, but not phospho-mimetic Lamin A, strongly bound putative enhancers suggest that enhancer binding may be a distinctive feature of Lamin C as compared with Lamin A. Unlike Lamin A, Lamin C does not undergo farnesylation, a modification thought to promote nuclear lamina incorporation of the Lamin A precursor (Dechat et al., 2008). Thus, the lack of farnesylation in combination with S22/S392 phosphorylation may promote nucleoplasmic Lamin C that binds to putative enhancers. Our finding that chromatin accessibility is not a strong predictor of the location of pS22-Lamin A/C-binding sites suggests that distinct targeting mechanisms exist to direct pS22-Lamin A/C to putative enhancers. Recently, nuclear-interior Lamin A/C has been studied in the context of its interaction with LAP2ɑ (Naetar et al., 2008; Dechat et al., 2010a). LAP2ɑ-interacting Lamin A/C binds megabase-wide genomic regions that lie at euchromatin regions without specific localization at promoters or enhancers (Gesson et al., 2016). Thus, pS22-Lamin A/C bound to putative enhancers is likely distinct from LAP2ɑ-interacting Lamin A/C. Instead, we observed that pS22-Lamin A/C enrichment is strongly correlated with the enrichment of the AP-1 transcription factor c-Jun. Lamin A/C is known to interact with c-Fos, the binding partner of c-Jun in the AP-1 complex (Ivorra et al., 2006; González et al., 2008). The AP-1 transcription factor complex is therefore a candidate for functioning to target pS22-Lamin A/C to specific putative enhancers.
A prevailing hypothesis for the pathogenesis of progeria suggested that progerin accumulates at the nuclear lamina due to its permanent farnesylation and disrupts normal interactions between Lamin A/C and LADs, causing heterochromatin disorganization, and, in turn, altered expression of genes located in LADs (Shumaker et al., 2006; McCord et al., 2012). One limitation of this model is that it does not explain the specific and abundant gene expression changes that occur outside of LADs. We propose an alternative hypothesis, in which misdirection of pS22-Lamin A/C to otherwise unbound enhancer regions or quiescent regions results in abnormal direct transcriptional activation of genes relevant to progeria pathogenesis (Fig. 7B). Because progerin itself did not appear to be phosphorylated at S22, the direct interaction between progerin and Lamin A/C (Lee et al., 2016) may contribute to mis-direction of pS22-Lamin A/C in progeria.
pS22-Lamin A/C has been regarded as a byproduct of mitotic nuclear envelope breakdown (Gerace and Blobel, 1980) or as a pool of disassembled lamins to be degraded when the nuclear lamina is compromised by mechanical stress (Swift et al., 2013; Buxboim et al., 2014). Our data that pS22-Lamin A/C is present throughout the cell cycle during unperturbed cellular conditions, localized at a specific subset of putative active enhancers, and associated with transcriptional alterations in progeria, suggested that pS22-Lamin A/C is a previously-unrecognized functional species of Lamin A/C in the interior of the nucleus. Much like the function of nuclear lamins in transcriptional repression at the nuclear lamina remains under active investigation (Leemans et al., 2019), the causal relationship between pS22-Lamin A/C binding at putative enhancers and transcriptional regulation remains to be determined. Regardless, the characteristics of pS22-Lamin A/C unveiled in this study build a foundation for investigating the functions of Lamin A/C, its role in transcriptional regulation, and the mechanisms underlying human degenerative disorders caused by LMNA mutations.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kohta Ikegami (ikgmk@uchicago.edu). All unique and stable reagents generated in this study will be made available on request but we may require a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
BJ-5ta (ATCC catalog # CRL-4001) is a TERT-immortalized BJ skin fibroblast cell line (source: foreskin of male neonate) that retains normal fibroblast cell growth phenotypes and does not exhibit transformed phenotypes (Jiang et al., 1999). Generation of the BJ-5ta-derived LMNA−/− cell line and LMNA−/− cells expressing wild-type and mutant LMNA transgenes is described in the following sections. Primary dermal fibroblasts used are GM07492 (source: thigh of 17-year-old male normal individual, Coriell Cell Repository), GM08398 (source: inguinal area of 8-year-old male normal individual, Coriell Cell Repository), AG11498 (source: thigh of 14-year-old male Hutchinson-Gilford progeria patient, Coriell Cell Repository), and HGADFN167 (source: posterior lower trunk of 8-year-old male Hutchinson-Gilford progeria patient, Progeria Research Foundation). We verified that, at the time of cell harvest, the progeria and control cells were not undergoing senescence (beta-galactosidase positive cells < 5%), a feature that late-passage progeria cells could manifest (Sieprath et al., 2015) (Fig. S4B, C). All cells were cultured in standard cell-culture-treated plastic dishes unless otherwise noted. BJ-5ta and its derivatives were cultured in high-glucose DMEM (Gibco, 11965–092) containing 9% fetal bovine serum (FBS), 90 U/mL penicillin, 90 μg/mL streptomycin streptomycin at 37°C under 5% CO2. Primary skin fibroblasts were cultured in MEM Alpha (Gibco, 12561–056) containing 9% fetal bovine serum (FBS), 90 U/mL penicillin, 90 μg/mL streptomycin streptomycin at 37°C under 5% CO2.
METHOD DETAILS
Cell synchronization by thymidine block
For synchronization, BJ-5ta cells in the DMEM growth medium were maintained at confluency for 2 days (G0 arrest by contact inhibition) and then passaged to a culture plate at a low density in the DMEM growth medium supplemented with 2 mM thymidine (Sigma-Aldrich T9250) for 17 hours. This allowed cells to re-enter into the G1 phase of the cell cycle and become arrested at the G1/S boundary. Cells were then washed and cultured in the growth medium containing 2.5 μM deoxycytidine (without thymidine) and harvested at 0 (i.e. G1/S-arrested cells), 4, 6, 8, 10, 12, and 14 hours later. As a reference, G0-arrested cells were released in the growth medium without thymidine and harvested 14 hours later (“asynchronous” cells).
Generation of LMNA−/− cell line
We cloned DNA sequences for sgRNA1 (annealing product of oligonucleotides KI223 and KI224, targeting the PAM site on the forward strand at chr1:156,084,863–156,084,866 in hg19; Key Resources Table) or sgRNA3 (annealing product of oligonucleotides KI227 and KI228, targeting the PAM site on the forward strand at chr1:156,084,953–156,084,956 in hg19; Key Resources Table), located in the exon 1 of LMNA, into the all-in-one lentivirus vector LentiCRISPRv2 (a gift from Feng Zhang; Addgene plasmid # 52961) (Sanjana et al., 2014). The cloned lentiCRISPR vectors were individually transfected to HEK293FT cells with the lentivirus packaging vectors psPAX2 (gift from Didier Trono; Addgene plasmid #12260) and pCMV-VSV-G (a gift from Bob Weinberg; Addgene plasmid #8454) (Stewart et al., 2003) to produce lentivirus. A mixture of the lentiviral tissue-culture supernatant for sgRNA1 and sgRNA3 (each at 0.25 dilution) was applied to BJ-5ta cells in the presence of 7.5 μg/mL polybrene for transduction. Successfully transduced cells were selected by 3 μg/mL puromycin and seeded to 10-cm dishes with a density of 100 cells per dish. Clonal populations were expanded and analyzed by western blotting for Lamin A and Lamin C protein expression. The clone cc1170–1AD2 lacks Lamin A and Lamin C protein expression, has nullizygous frameshift mutations, and is used in this study.
Wild-type and phospho-mutant Lamin A/C expression in LMNA−/− cells
We cloned Lamin A or Lamin C cDNAs with S22 and S392 mutations or without mutations into the allin-one doxycycline inducible lentivirus vector pCW57-MCS1-P2A-MCS2-PGK-Blast (gift from Adam Karpf; Addgene plasmid #80921) (Barger et al., 2019) using HiFi assembly (New England Biolabs E2621). DNA fragments containing S22D (TCG to GAC) and S22A (TCG to GCT) mutations were chemically synthesized (Integrated DNA Technologies, Inc; Oligonucleotides KI361 and KI362; Key Resources Table). DNA fragments containing S392D (AGC to GAC) and S392A (AGC to GCC) mutations were amplified by PCR using published Lamin A/C cDNA plasmids containing these mutations as a template (Kochin et al., 2014) (gifts from Drs. Robert Goldman and John E Eriksson) and primers KI393 and KI385 for Lamin A and KI393 and KI395 for Lamin C (Key Resources Table) . In addition, silent mutations at the sgRNA1 and sgRNA3 CRISPR targeting sites (see Generation of LMNA−/− cell line) were introduced to prevent the transgenes from being targeted by Cas9 in LMNA−/− cells. Lentivirus was produced as described above and transduced to BJ-5ta-derived LMNA−/− cells. The transduced cells were selected under 10 μg/mL blasticidin. The cell population IDs are cc1499–1 (wild-type Lamin A); cc1499–2 (Lamin A with S22A/S392A); cc1499–3 (Lamin A with S22D/S392D); cc1499–4 (wild-type Lamin C); cc1499–5 (Lamin C with S22A/S392A); and cc1499–6 (Lamin C with S22D/S392D). The transgene expression was induced by 20 μg/mL doxycycline for 72 hours. This doxycycline concentration did not affect overall growth of the generated cell lines.
ELISA
Lamin A/C N-terminal aa2–30 peptides (ETPSQRRATRSGAQASSTPLSPTRITRLQ) with phosphorylated Ser22 (Lot U2312EI090–3/PE2186; purity 90.0%; MW 3234.45) or non-phosphorylated Ser22 (Lot U2312EI090–1/PE2183; purity 96.1%; MW 3154.47) were synthesized by Genscript (New Jersey, USA), and the quality was verified by the manufacturer. Peptides were immobilized to maleic anhydride-activated plastic wells (Pierce catalog number 15100). Coated wells were blocked with 5% non-fat milk and 0.1% Tween 20 and then incubated with anti-phospho-Ser22-Lamin A/C antibody (Cell Signaling 13448S, Lot 1, 1:1000 dilution) or the anti-pan-N-terminal-Lamin A/C antibody (Santa Cruz Biotechnology sc-376248, Lot H2812, 1:5000) for 1 hour at 37°C. After washing, wells were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (GE Healthcare NA934V, Lot 9636020) or HRP-conjugated anti-mouse IgG (GE Healthcare NA931V, Lot 9648752). HRP activity was detected by 3,3′,5,5′-tetramethylbenzidine (TMB)-based colorimetric reaction (Pierce catalog #34022). The reaction was treated with 3N HCl, and the absorbance at 450 nm (reaction) and 550 nm (reference) was measured by a microplate reader. See Quantification and Statistical Analysis for downstream analyses.
Western blot
Protein extract was separated by 4–12% Bis-Tris SDS-PAGE with MOPS buffer and transferred to a PVDF membrane. After blocking, the membranes were incubated for 12 hours or longer at 4°C with rabbit monoclonal anti-phospho-Ser22-Lamin A/C antibody D2B2E (Cell Signaling 13448S, Lot 1; 1:1000 dilution) or mouse monoclonal anti-pan-N-terminal-Lamin A/C antibody E1 (Santa Cruz Biotechnology sc-376248, Lot H2812; 1:1000 dilution). Primary antibodies were detected by HRP-conjugated anti-rabbit IgG (GE Healthcare NA934V, Lot 9636020; 1:10000 dilution) or HRP-conjugated anti-mouse IgG (GE Healthcare NA931V, Lot 9648752; 1:10000 dilution). Signals were produced by enhanced chemiluminescence (ECL) and detected digitally in a Bio-Rad ChemiDoc imager. The gel after protein transfer was counter-stained by coomassie to evaluate the loaded protein amount. See Quantification and Statistical Analysis for downstream analyses.
Flow cytometry
For cell-cycle analysis by DAPI staining, cells in suspension were incubated with 70% cold ethanol for 12 hours or longer at −20°C for fixation. The fixed cells were incubated with FACS buffer (2% FBS, 1 mM EDTA, and 0.1% Tween 20 in PBS) for 10 min at room temperature. Cells were then incubated with PBS supplemented with 0.1% Triton and 1 μg/mL DAPI for 10 min at room temperature. The DAPI-stained cells were resuspended in FACS buffer and then analyzed by Fortessa 4–15 HTS or Fortessa X20 5–18 flow cytometry analyzers (BD Biosciences).
To stain cells for pS22-Lamin A/C, cells were fixed in PHEM buffer (60 mM PIPES-KOH pH7.5, 25 mM HEPES-KOH pH7.5, 10 mM EGTA, 4 mM MgSO4) containing 4% formaldehyde, 0.5% Triton, and 100 nM phosphatase inhibitor Nodularin (Enzo ALX-350–061) for 15 min at 37°C. Cells were blocked with Blocking buffer (FACS buffer supplemented with 5% normal goat serum) and incubated with Alexa-647-conjugated rabbit monoclonal anti-phospho-Ser22-Lamin A/C antibody D2B2E (labeled at Cell Signaling, product ID 97262BC, Lot 1, 1:30 dilution) in Blocking buffer for 1 hour at 37°C. Cells were counter-stained with 1 μg/mL DAPI in FACS buffer. The stained cells were analyzed by Fortessa 4–15 HTS or Fortessa X20 5–18 flow cytometry analyzers. See Quantification and Statistical Analysis for downstream analyses.
Immunofluorescence
Cells were grown on uncoated glass coverslips under the standard culture condition (see Cell Culture). Cells were fixed in PHEM buffer (60 mM PIPES-KOH pH7.5, 25 mM HEPES-KOH pH7.5, 10 mM EGTA, 4 mM MgSO4) containing 4% formaldehyde, 0.5% Triton, and 100 nM phosphatase inhibitor Nodularin (Enzo ALX-350–061) for 10 min at 37°C. Cells on coverslips were blocked with Blocking buffer (1% skim milk and 5% goat serum in PBS), and then incubated with primary antibodies in Blocking buffer. Antibodies used in immunofluorescence are: Alexa 647-conjugated rabbit monoclonal anti-phospho-Ser22-Lamin A/C antibody D2B2E (labeled at Cell Signaling, product ID 97262BC, Lot 1, 1:100 dilution), mouse monoclonal anti-pan-N-terminal-Lamin A/C antibody E1 (Santa Cruz Biotechnology sc-376248, Lot # H2812, 1:5000), or mouse monoclonal anti-full-length-Lamin A/C antibody 4C4 (Abcam ab190380, Lot GR201137–1; 1:1000). Cells were incubated with secondary antibodies, counterstained by DAPI, and cured in ProLong Gold mounting medium (Molecular Probes, P36930). Cells were imaged using a Leica SP8 confocal microscope with a 63x or 100x objective. See Quantification and Statistical Analysis for downstream analyses.
Senescence-associated beta-galactosidase assay
Cells were grown on coverslips and fixed for 5 min in 2% formaldehyde and 0.2% glutaraldehyde in PBS. For positive controls of the assay, cells were incubated for 2 hours with media containing 300 μM (WI38) or 400 μM (GM07492) H2O2 before replacement with the normal growth media. Cells were washed with PBS and incubated in X-gal staining solution (40 mM citric acid/sodium phosphate buffer, 5 mM K4[Fe(CN)6] 3H2O, 5 mM K3[Fe(CN)6], 150 mM NaCl, 2 mM MgCl2, and 1 mg/mL X-gal) for 16 h at 37°C. The coverslips were washed with PBS and mounted for microscopy. To determine the staining percentages, cells from 10 randomly selected areas at 20x magnification were counted.
ChIP-seq
Cells in a culture dish were crosslinked in 1% formaldehyde for 15 min at room temperature, and the reaction was quenched with 125 mM glycine. Cross-linked cells were washed with LB1 (50 mM HEPES-KOH pH7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 0.25% Triton X-100) and LB2 (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and 10 mM Tris-HCl pH 8.0). Cells were resuspended in LB3-Triton (1 mM EDTA, 0.5 mM EGTA, 10 mM Tris-HCl pH 8, 100 mM NaCl, 0.1% Na-Deoxycholate, 0.5% N-lauroyl sarcosine, 1% Triton) containing 1x protease inhibitor cocktail (Calbiochem 539131) and 100 nM phosphatase inhibitor Nodularin (Enzo ALX-350–061), and chromatin was extracted by sonication. The cell extract was cleared by 14,000 g centrifugation for 10 min. An aliquot of cell extract was saved for input DNA sequencing. Cell extract from one million cells was incubated with antibodies in a 200-μL reaction for 12 hours or longer at 4°C. Antibodies used in ChIP are: rabbit monoclonal anti-phospho-Ser22-Lamin A/C antibody D2B2E (Cell Signaling 13448S, Lot 1; 5 μL per IP); mouse monoclonal anti-pan-N-terminal-Lamin A/C antibody E1 (Santa Cruz Biotechnology sc-376248, Lot H2812; 10 μL per IP); mouse monoclonal anti-full-length-Lamin A/C antibody 4C4 (Abcam ab190380, Lot GR201137–1; 4 μL per IP); rabbit polyclonal anti-c-Jun antibody (Santa Cruz Biotechnology sc-1694, Lot D1014; 20 μL per IP); mouse monoclonal anti-H3K27ac antibody (Wako MABI0309, Lot 14007; 2 μL per IP); mouse monoclonal anti-H3K4me3 antibody (Wako MABI0304, Lot 14004; 2 μL per IP); rabbit polyclonal anti-H3K9me3 antibody (Abcam ab8898, Lot GR232099–3; 2 μL per IP); and mouse monoclonal anti-H3K27me3 antibody (Active Motif MABI0323, Lot 17019020; 2 μL per IP). Immunocomplex was captured by Protein A-conjugated sepharose beads (for rabbit antibodies) or Protein G-conjugated magnetic beads (for mouse antibodies) and washed. Immunoprecipitated DNA was reverse-crosslinked and used to construct high-throughput sequencing libraries using NEBNext Ultra DNA Library Prep Kit (New England Biolabs, E7370). DNA libraries were processed on a Illumina HiSeq machine for single-end sequencing. See Quantification and Statistical Analysis for downstream analyses. ChIP-seq experiments are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md.
ATAC-seq
One hundred thousand trypsinized cells were incubated with ATAC hypotonic buffer (10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2) for 10 min at 4°C during 500 g centrifugation. Cells were incubated in Tagmentation mix (Tagmentation DNA buffer Illumina 15027866; Tagmentation DNA enzyme Illumina 15027865) for 30 min at 37°C. Purified DNA was used to construct high-throughput sequencing libraries using NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs M0541). DNA libraries were processed on a Illumina NextSeq machine for paired-end 41-nt sequencing. See Quantification and Statistical Analysis for downstream analyses. ATAC-seq experiments are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md.
RNA-seq
Total RNAs were purified by Trizol LS (Invitrogen 10296028) and treated with DNase I (Invitrogen Turbo DNase AM2238). mRNAs were isolated using NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs E7490) and fragmented using Fragmentation Buffer (Ambion AM8740). cDNAs were synthesized using SuperScript II (Invitrogen 18064014), and non-directional high-throughput sequencing libraries were prepared using NEBNext Ultra DNA Library Prep Kit (New England Biolabs, E7370). Libraries were processed on the Illumina HiSeq platform for single-end 50-nt sequencing. See Quantification and Statistical Analysis for downstream analyses. RNA-seq experiments are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md.
GRO-seq
Nuclei were isolated by incubating cells in hypotonic NP40 lysis buffer (10 mM NaCl, 3 mM MgCl2, 0.5% MP-40, 10 mM Tris, pH 7.5) supplemented with RNase Inhibitor on ice and resuspended in Nuclear Storage buffer (50 mM Tris pH 8.0, 0.1 mM EDTA, 5 mM MgCl2, 40% glycerol, RNase inhibitor). The nuclear suspension was mixed with an equal volume of 2x NRO buffer (10 mM Tris pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 0.5 mM ATP, 0.5 mM GTP, 0.5 mM BrUTP, 2 μM CTP). The sample was incubated without sarkosyl for 4 min at 30°C and then with 0.5% sarkosyl for 4 min at 30°C (total 8 min). RNAs were purified from the reaction by Trizol LS (Invitrogen 10296028) followed by isopropanol precipitation. RNAs were treated with TurboDNase (Ambion AM18907) and fragmented by Fragmentation Buffer (Ambion AM8740). BrU-incorporated RNA fragments were immunoprecipitated with mouse monoclonal anti-BrdU antibody 3D4 (BD Biosciences 555627 Lot 7033666) and used to construct DNA sequencing libraries using NEBNext Ultra II Directional RNA Library Prep kit (New England Biolabs E7760). DNA libraries were processed on a Illumina NextSeq machine for paired-end 42-nt sequencing. See Quantification and Statistical Analysis for downstream analyses. GRO-seq experiments are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md.
QUANTIFICATION AND STATISTICAL ANALYSIS
Number of data points, number of replicates, and the type of statistical tests employed are indicated in figures and/or figure legends.
ELISA quantification
The reaction absorbance (450 nm) minus the reference absorbance (550 nm) was plotted using software R (v3.3.2). Loess fit was computed using geom_smooth function in ggplot2 package (version 2.2.1).
Western blot quantification
Intensities of Western blot bands and coomassie staining bands were obtained using the Analyze Gels function in Fiji (v1.0). Western blot Intensities were normalized to coomassie staining intensities. The mean and standard deviation of three biological replicates were computed using software R (v3.3.2).
Flow cytometry quantification
Flow cytometry data were processed using FlowJo (v10.5.3). Forward and side scatter gatings were used to identify single cells. To obtain the fraction of cells at G1/G0, S, and G2/M, DAPI signals were processed with the Watson Pragmatic algorithm (Watson et al., 1987) in FlowJo with manually constrained G1/G0 and G2/M signal ranges with the coefficient of variation set to 10%.
Immunofluorescence quantification
Quantification of immunofluorescence signals for LMNA transgene products was performed on the image at a z-axis position with the highest nuclear periphery-to-interior Lamin A/C signal ratio in a given cell. In this image, a 5-μm line segment that crossed the nuclear periphery at position 0 μm (−2.5 to 2.5 μm with negative coordinates indicating positions outside the nucleus) was drawn. An array of pixel intensities along the segment (averaged across the 10-pixel width) was obtained using Fiji (v1.0). From this array, an average pixel intensity in every 0.1 μm bin along the line was computed (50 bins). To visualize signals along the segment, the binned intensities from the same cell type were quantile normalized using normalize.quantiles function in the preprocessCore package (v1.36.0) in software R (Bolstad et al., 2003) to account for overall intensity differences between cell types. To compute nuclear interior-to-periphery ratios, the mean unnormalized signal intensities between +4 to +5 μm of the segment was assigned as the nuclear-interior signal, and the maximum unnormalized signal intensities between −0.5 to +0.5 μm was assigned as the nuclear-peripheral signal.
Reference genome
The February 2009 human reference sequence hg19/GRCh37 was used throughout in this paper.
Blacklisted regions
Before performing genomic data analyses, we excluded all genes and genomic features located in blacklisted regions which are genomic regions that may cause misinterpretation due to high sequence redundancy, uncertain chromosomal locations, high signal background, haplotypes, or potential copy number variations (CNVs) induced in the process of CRISPR-mediated generation of LMNA−/− cells (Aguirre et al., 2016). The collection of such genomic regions were constructed from the following datasets (see Data and Code Availability): assembly gaps in the hg19 reference genome, ENCODE-defined hg19 blacklisted regions, mitochondria sequence (chrM), haplotype chromosomes (chr*_*_hap*), unplaced contigs (chrUn_*), unlocalized contigs (chr*_*_random), and potential CNVs described below. To identify potential large CNVs between wild-type BJ-5ta and BJ-5ta-derived LMNA−/− cells, input sequencing data for wild-type BJ-5ta (ID KI481) and LMNA−/− cells (ID KI489) were processed with CNV-seq (Xie and Tammi, 2009) with the following parameters: [–genome human – global-normalization −log2-threshold 0.5 –minimum-windows-required 3]. After removing windows with low sequence coverage, candidate CNV windows that were overlapping or spaced within 500 kb were merged, and isolated windows smaller than 500 kb in size were removed. This yielded 5 candidate large CNVs (24 Mb in chr1; 15 Mb in chr 4; 2.7 Mb in chr19; 683 kb in chr2; and 528 kb in chrX). The blacklisted regions are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/blacklist.bed.
Gene annotation
The Gencode V19 “Basic” gene annotation was downloaded from the UCSC genome browser. Of the total 99,901 transcripts in the list, we retained transcripts that met all of the following requirements: 1) “gene type” equals “transcript type”; 2) “transcript type” is either protein_coding or antisense or lincRNA; and 3) “transcript ID” appears only once in the list. This processing yielded 75,968 transcripts. To select one transcribed unit per gene locus, the 75,968 transcripts were grouped by “gene symbol,” and within the group, transcripts were sorted by the “exon count” (largest first), then by “length” of transcribed region (largest first), then by the alphanumeric order of the “transcript ID” (smallest first), and the transcript that appeared first in the group was chosen to represent the transcribed unit of that gene. In this processing, in general, a transcript with a largest number of annotated exons among other associated transcripts represented the gene. After removing genes located within the blacklisted regions (see Blacklisted regions), we obtained 31,561 genes, which included 19,469 “protein_coding” genes.
ChIP-seq data processing
ChIP-seq experiments and sequencing depth are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md. ChIP-seq reads were mapped to the hg19 human reference genome using Bowtie2 (Langmead and Salzberg, 2012) with the default “--sensitive” parameter. Reads with MAPQ score greater than 20 were used in downstream analyses. Reads from biological replicates of ChIP and the corresponding input were processed by MACS2 (v2.1.0) (Zhang et al., 2008). MACS2 removed duplicated reads and generated then “fold enrichment score”, which was input-normalized per-base coverage of 200 nt-extended reads. We used the fold-enrichment scores throughout the paper for quantitative analysis of ChIP-seq enrichment. In addition, for ChIP-seq with point-source enrichment profiles, MACS2 was used to identify statistically overrepresented peak regions and peak summits using the following parameter set: [call peak -g hg --nomodel --extsize 200 --call-summits]. Peaks overlapping blacklisted regions (see Blacklisted regions section) were removed. We defined a summit as a unit of a protein binding site. MACS2 identified 22,966 pS22-Lamin A/C-binding sites in BJ-5ta; 79,799 H3K27ac-enriched sites in BJ-5ta; 18,100 H3K4me3-enriched sites in BJ-5ta; 87,988 c-Jun-enriched sites in BJ-5ta (Key Resources Table). The codes used are publicly available (see Data and Code Availability).
ATAC-seq data processing
ATAC-seq experiments and sequencing depth are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md. For alignment, the first 38 nt of the 41-nt reads in the 5′ to 3′ direction were used. The rationale of this trimming is that the minimum size of DNA fragments that can be flanked by Tn5 transposition events has been estimated to be 38 bp (Reznikoff, 2008; Picelli et al., 2014), and therefore, a 41-nt read could contain a part of read-through adaptors. We aligned 38-nt reads to the hg19 reference genome using bowtie2 with following parameters: [-X 2000 --no-mixed --no-discordant --trim3 3]. The center of the active Tn5 dimer is estimated to be located +4–5 bases offset from the 5′-end of the transposition sites (Reznikoff, 2008; Picelli et al., 2014). To place the Tn5 loading center at the center of aligned reads, the 5′-end of the plus-strand read was shifted 4 bp in the 5′-to-3′ direction and that of the minus-strand reads was shifted 5 bp in the 5′-to-3′ direction, and the shifted end (1 nt) was extended +/−100 bp. To generate background datasets that capture local bias of read coverage, the shifted read ends were extended +/−5,000 bp and used to construct local lambda background file. We processed the Tn5 density file and the local lambda file (background) with MACS2 function bdgcomp to generate the fold-enrichment scores for Tn5 density from as a background. We also used MACS2 function bdgpeakcall to identify regions with statistically significant Tn5 enrichment (ATAC peaks). At P-value cutoff of 1×10−10, we obtained 73,933 ATAC peaks (Key Resources Table). The codes used are publicly available (see Data and Code Availability).
RNA-seq data processing
RNA-seq experiments and sequencing depth are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md. Public RNA-seq raw data files (fastq) for normal and progeria-patient fibroblasts (Fleischer et al., 2018) (see Data and Code Availability) were retrieved using fastq-dump (version 2.9.3). All RNA-seq reads were aligned to the hg19 human reference genome using Tophat2 with the default parameter set (Kim et al., 2013). Reads with MAPQ score greater than 50 were used in downstream analyses. For each of the total 31,561 genes (see Gene annotation) for each replicate, we computed (1) unnormalized RNA-seq read coverage, which was the sum of per-base read coverage in exons; and (2) RPKM (Reads Per Kilobase of transcript per Million mapped reads), which was the unnormalized RNA-seq read coverage divided by the read length (50 nt) and then by the sum of the exon size in kilobase and then by the total number of reads in million. The RPKM scores were Log2-transformed (Log2(RPKM+0.001)), z-normalized (for each sample), and then used in Principal Component Analysis (PCA) using prcomp function (stats package version 3.3.2) in R. The PCA found that two data sets, s78 (normal fibroblast GM05381 by Fleischer et al.) and KI429 (normal fibroblast GM08398 by this study), did not cluster with other normal-fibroblast datasets (Fig. S4D). A retrospective assessment of the RNA quality of KI429 found a sign of RNA degradation. The datasets s78 and KI429 were excluded from the subsequent analyses. The codes used are publicly available (see Data and Code Availability).
GRO-seq data processing
GRO-seq experiments and sequencing depth are listed in https://github.com/kohta-ikegami/pS22-LMNA/blob/master/datasets.md. GRO-seq read pairs with maximum fragment length of 2000 bp were aligned to the hg19 human reference genome using Bowtie2 with the parameter set of -X 2000 --no-mixed --no-discordant. Reads with MAPQ score greater than 20 were used in downstream analyses. For each of the total 31,561 genes (see Gene annotation), we computed “normalized GRO-seq base coverage,” which was the sum of GRO-seq fragment per-base coverage normalized to gene lengths and sequencing depths. The codes used are publicly available (see Data and Code Availability).
LADs
LADs in BJ-5ta were defined using pan-N-terminal-Lamin A/C ChIP-seq data in BJ-5ta. The hg19 genome was segmented into 5-kb non-overlapping windows, and for each window, the sum of perbase read coverage (from replicate-combined reads) was computed for pan-N-terminal-Lamin A/C ChIP-seq and the corresponding input. The coverage was normalized by sequencing depth. The depth-normalized coverage was used to compute per-window log2 ratios of ChIP over input. We then created 100 kb windows with a 5-kb step genome-wide. For each 100-kb window, if every one of the 20 constituting 5-kb windows had a positive log2 ratio and the mean log2 ratios of the constituting 5-kb windows was greater than 0.5, this 100-kb window was further processed. The qualified 100-kb windows were merged if overlapping or touching. After filtering regions overlapping blacklisted regions (see Blacklisted regions), we obtained 2,178 regions which we defined as Lamin A/C LADs in BJ-5ta (Key Resources Table). LADs defined by Lamin B1 Dam ID in lung fibroblasts are reported previously (Guelen et al., 2008) (see Data and Code Availability).
Analysis on deciles
Genes, ATAC-seq sites, or c-Jun-binding sites were stratified into deciles by scores specified in figure legends using ntile function in dplyr (version 0.7.4) in R.
DNA motif analysis
To find DNA motifs de novo, 150-bp sequences centered around the summit of the top 500 high-confident (defined by p-values) pS22-Lamin A/C-binding sites were analyzed by MEME (v4.10.0) (Machanick and Bailey, 2011) with the following parameters: minimum motif size, 6 bp; maximum motif size, 12 bp; and the expected motif occurrence of zero or one per sequence (-mod zoops) and with the 1st-order Markov model (i.e. the dinucleotide frequency) derived from the 73,933 ATAC-seq sites as the background. The top five overrepresented motifs were then processed by TOMTOM (v4.11.3) (Gupta et al., 2007; Tanaka et al., 2011) to identify known motifs that corresponded to the de novo identified motifs from human HOCOMOCOv10 motif database (Kulakovskiy et al., 2018). At the q-value cutoff of less than 0.001, the motifs #1 (AP1), #2 (FOX) and #4 (RUNX) found the corresponding known motifs in the database. The location and frequency of these motifs within the all pS22-Lamin A/C-binding sites (total 22,966) were determined by FIMO (v4.10.0) with p-value threshold less than 0.0001.
Upregulated and downregulated genes in progeria
To identify differentially expressed genes between progeria-patient fibroblasts and normal-individual fibroblasts, we applied DESeq2 (version 1.14.1) (Love et al., 2014) on the unnormalized RNA-seq read coverage for the total 31,561 genes (see RNA-seq data processing). To account for the variables due to the study origins, we included the study origin annotation as an additive term in the DESeq2 model. We applied the cutoff of DESeq2-computed adjusted p-value smaller than 0.05 and absolute DESeq2-adjusted log2-fold change greater than 0.5. Among 11,613 protein coding genes that had the minimum RPKM score greater than 0.01 across all analyzed samples (“expressed genes”), 615 genes were defined as upregulated and 502 genes were defined as downregulated in progeria-patient fibroblasts compared with normal-individual fibroblasts (Key Resources Table). The codes used are publicly available (see Data and Code Availability).
Gained and lost pS22-Lamin A/C-binding sites in progeria
We first generated a union set of pS22-Lamin A/C-binding sites in normal-individual fibroblast GM07492 and progeria-patient fibroblast AG11498 by collecting pS22-Lamin A/C-enriched sites from the two cell lines and then merging neighboring summits if the distance between the summits was equal to or smaller than 200 bp. When summits were merged, the center of the region generated by the merged summits was assigned as the new summit. This resulted in 15323 union pS22-Lamin A/C-binding site summits (Key Resources Table). The summits were then extended +/−500 bp, and the sum of per-base read coverage of pS22-Lamin A/C ChIP-seq (reads extended to 200 bp) within the 1000-bp regions was computed for each replicate of the normal and progeria fibroblasts (two replicates each). This coverage matrix was processed using DESeq2 (version 1.14.1) (Love et al., 2014) to identify pS22-Lamin A/C-binding sites with statistically-significant difference between the normal and progeria-patient fibroblast cell lines. At DESeq2-computed adjusted p-value < 0.05 and absolute log2-fold change > 0.5, we identified 2,796 sites whose pS22-Lamin A/C signals were higher in progeria fibroblasts than in normal fibroblasts (gained pS22-Lamin A/C-binding sites) and 2,425 sites whose pS22-Lamin A/C signals were higher in normal fibroblasts than in progeria fibroblasts (lost pS22-Lamin A/C-binding sites) (Key Resources Table).
Box plot and Violin plot
Throughout the paper, box plots with or without overlaid “violins” were used to visualize distribution of numeric data. The box indicates interquartile (IQR) range with the bar inside the box indicating the median. The upper and lower whiskers indicate maximum and minimum data points within 1.5x IQR from the box, respectively. The circles outside the whisker range indicate data points outside of this range. The overlaid violin indicates the kernel density of the data. Box and violin plots were generated using ggplot2 package (version 2.2.1) in R (version 3.2.2).
Gained and lost LADs in progeria
We first generated 5-kb windows genome-wide, and for each window and for each cell type (normal-individual GM07492 and progeria-fibroblast AG11498), we computed the mean of the replicate-combined pan-N-terminal-Lamin A/C ChIP-seq log2-fold-enrichment scores within the window. The data was then quantile-normalized using normalize.quantiles function in the preprocessCore package (v1.36.0) in software R (Bolstad et al., 2003). From the normalized data, we identified: (1) “seeds” of lost LADs, which were 5-kb windows whose normal-fibroblast score was a positive number and whose progeria-fibroblast score was a negative number; (2) “seeds” for gained LADs, which were 5-kb windows whose normal-fibroblast score was a negative number and whose progeria-fibroblast score was a positive number; and (3) “seeds” for steady LADs, which were 5-kb windows whose normal-fibroblast and progeria-fibroblast scores were both positive. For gained and lost LADs, the neighboring aforementioned seeds were merged (individually for gained and lost LAD seeds) if they were located within 10 kb. For steady LADs, seeds were merged if the neighboring seeds were located within 5 kb. Finally, the merged windows greater than 100 kb in size were isolated, and then those intersecting blacklisted regions (see Blacklisted regions) were removed. These processing resulted in 282 gained LADs, 353 lost LADs, and 2,100 steady LADs (Key Resources Table).
ChIP-seq and ATAC-seq signal analysis
A ChIP-seq or ATAC-seq fold-enrichment score for pS22-LMNA-binding sites, or cJun-binding sites, or ATAC-defined accessible sites was the sum of the perbase fold-enrichment scores (i.e. “area”) within +/−250 bp of the site center (total 500 bp).
To generate aggregate plots and heatmaps for features, two data files were first generated: (a) a window file, which consists of, for each genomic feature, a set of fixed-size genomic windows that cover genomic intervals around the feature; and (b) a genome-wide signal file (in bedgraph format). For each genomic window in the window file, all signals within that window were obtained from the signal file, and either mean or max of the signals or sum of the perbase signal (“area”) were computed.
For the heatmaps and aggregate plots around pS22-Lamin A/C-binding sites or ATAC sites, a set of 250-bp windows with a 50-bp offset that covered a 10-kb region centered around the summit of these sites was generated for each site. For each window, the mean of fold-enrichment score was computed from replicate-combined input-normalized fold enrichment bedgraph files.
For the heatmaps of LADs, a set of 5-kb windows (without an offset) that covered the LAD body or the LAD body plus 250 kb downstream region was generated for each LAD. For each window, the mean of fold-enrichment score was computed from replicate-combined input-normalized fold enrichment bedgraph files.
The computed scores were plotted using ggplot2 package (version 2.2.1) in R (version 3.2.2). The codes used are publicly available (see Data and Code Availability).
RNA-seq signal analysis
For the heatmap of the differentially expressed genes, RPKM scores for the 11,613 expressed genes for the 26 RNA-seq data sets (12 normal fibroblasts and 14 progeria-patient fibroblasts) were log10-transformed and quantile-normalized using normalize.quantiles function in the preprocessCore package (v1.36.0) in R (Bolstad et al., 2003). These normalized scores were then processed using ComBat function in the sva package (v3.22.0) in R with the sample origin annotation as the batch to remove (Johnson et al., 2007). The batch-normalized RPKM scores for the total 1,117 dysregulated genes in progeria fibroblasts were clustered and visualized using pheatmap function in the pheatmap package (v1.0.12) in R with the “correlation” clustering distance measurement method between sample clusters.
The computed scores were plotted using ggplot2 package (version 2.2.1) in R (version 3.2.2). The codes used are publicly available (see Data and Code Availability).
Gene ontology analysis
Gene ontology (GO) analyses were performed using Metascape (Tripathi et al., 2015). The input data type was Gencode 19 Gene Symbol (see Gene annotation). For background, the 11,613 protein-coding genes with reliable sequencing coverage were used (see Differentially expressed genes). Enrichment for “DisGeNet” terms (Piñero et al., 2017) was analyzed under the default parameter settings (minimum gene count 3, P <0.01, enrichment over background >1.5). P-values were derived from cumulative hypergeometric statistical tests and computed in Metascape (Zhou et al., 2019).
One-way ANOVA
One-way ANOVA was used to assess statistical difference of numeric scores among cells expressing wild-type or phospho-mutant Lamin A/C (6 cell types). The ANOVA tests were performed using aov function with default parameters in R (stats package v3.3.2) under the null hypothesis that the population mean of the numeric scores was the same across all cell types, with the alternative hypothesis that at least one cell type had a different population mean. We then applied post-hoc Tukey’s test to perform pairwise comparison. The Tukey’s test was performed using TukeyHSD function on the ANOVA result above with default parameters in R (stats package v3.3.2) under the null hypothesis that the population mean of the numeric scores between the two cell types being compared was the same, with the alternative hypothesis that they were different.
Mann-Whitney U test
Throughout the paper, Mann-Whitney U test was used to assess statistical difference of numeric scores between two groups. The tests were performed using wilcox.test function with default parameters in R (stats package v3.3.2) under the null hypothesis that numeric scores of the two groups were selected from one population, with the alternative hypothesis that they came from different populations.
Fisher’s exact test
Throughout the paper, Fisher’s exact tests were used to assess the association between two features that were unambiguously and independently assigned to each data point in one data set. The tests were performed on a 2-by-2 contingency table using fisher.test function with default parameters in R (stats package v3.3.2) under the null hypothesis that the odds ratio is equal to 1, with alternative hypothesis that the odds ratio is not equal to 1.
One-sample Student’s t test
One-sample t-tests were performed using t.test function with default parameters in R (stats package v3.3.2) under the null hypothesis that the mean of the numeric vector equal to zero, with the alternative hypothesis that the mean is not equal to zero.
Permutation test for chromatin state analysis
For the 22,966 pS22-Lamin A/C-binding site summits (1 bp), a random set of 22,966 genomic locations was selected from the blacklisted-region-filtered genome using shuffle function in Bedtools (v2.25.0) (Quinlan and Hall, 2010) such that the chromosome distribution of the original 22,966 pS22-Lamin A/C-binding sites was maintained. This process was iterated 2,000 times. For each iteration, the total number of bases overlapped with a given chromatin state was computed. For each chromatin state, the number of iterations in which the base coverage of permutated pS22-Lamin A/C-binding sites exceeded the actual base coverage of the 22,966 pS22-Lamin A/C-binding sites was counted. If this number is 0 (one-sided test for over-representation), we assigned empirical P-value of < 0.001. Essentially, the same computation was performed for 2,178 LADs, except that the random sets of LADs maintained the distribution of the original feature sizes.
Correlation analysis
Pearson correlation coefficient was used to assess correlation between two fold-enrichment scores at 22,966 pS22-Lamin A/C-binding sites (sum of fold-enrichment scores within +/−500 bp of the site center). The computation of Pearson’s r was performed using cor function in R with method=”pearson” (stats package v3.3.2).
Spearman’s rank correlation coefficient was used to assess the monotonic relationship between gene deciles by GRO-seq coverage and the fraction of genes linked to pS22-Lamin A/C-binding sites within each of the deciles. The computation of Spearman’s rho was performed using cor function in R with method=”spearman” (stats package v3.3.2).
DATA AND CODE AVAILABILITY
Data and Code Availability Statement
The genomic datasets generated during this study are available at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE113354. Custom computational scripts used in this study are available at: https://github.com/kohta-ikegami/pS22-LMNA/
Public datasets
Assembly gaps
http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/gap.txt.gz
Blacklisted Regions
Chromatin annotation
Genecode Release 19 Gene Model
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/wgEncodeGencodeBasicV19.txt.gz
Lamin B1 LADs
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/laminB1Lads.txt.gz
Progeria fibroblast RNA-seq datasets from Fleischer et al.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113957
Supplementary Material
HIGHLIGHTS.
Phospho-Ser22-Lamin A/C (pLamin) is localized to the nuclear interior
pLamin binds to enhancers near active genes, not lamina-associated domains
pLamin-bound enhancers are co-bound by transcriptional activator c-Jun
New pLamin binding in progeria accompanies upregulation of clinically relevant genes
ACKNOWLEDGEMENTS
We thank technical assistance from the Functional Genomics, Cytometry and Antibody Technology, and Light Microscopy core facilities at the University of Chicago, and the Genomics core facility at Princeton University. We thank Sebastian Pott for discussion. We acknowledge Life Science Editors for editorial assistance. This work was funded by NIH R21/R33 AG054770 (K.I. and I.P.M.); NIH R01 HL147571, R01 HL148719, and R01 HL126509 (I.P.M.); and the Progeria Research Foundation grant #2009-0028 (K.I. and J.D.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aebi U, Cohn J, Buhle L, and Gerace L (1986). The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323, 560–564. [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola M, and van Steensel B (2015). Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep. 16, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboro P, D’Arrigo C, Diaspro A, Mormino M, Alberti I, Parodi S, Patrone E, and Balbi C (2002). Unraveling the organization of the internal nuclear matrix: RNA-dependent anchoring of NuMA to a lamin scaffold. Exp. Cell Res 279, 202–218. [DOI] [PubMed] [Google Scholar]
- Barger CJ, Branick C, Chee L, and Karpf AR (2019). Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, and Speed TP (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. [DOI] [PubMed] [Google Scholar]
- Bridger JM, Kill IR, O’Farrell M, and Hutchison CJ (1993). Internal lamin structures within G1 nuclei of human dermal fibroblasts. J. Cell Sci 104 (Pt 2), 297–306. [DOI] [PubMed] [Google Scholar]
- Broers JL, Machiels BM, van Eys GJ, Kuijpers HJ, Manders EM, van Driel R, and Ramaekers FC (1999). Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins. J. Cell Sci 112 (Pt 20), 3463–3475. [DOI] [PubMed] [Google Scholar]
- Bruston F, Delbarre E, Ostlund C, Worman HJ, Buendia B, and Duband-Goulet I (2010). Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 584, 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PCDP, Athirasala A, Kao Y-RC, Cho S, Harada T, Shin J-W, et al. (2014). Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol 24, 1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, and Goldman RD (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T, Gesson K, and Foisner R (2010a). Lamina-independent lamins in the nuclear interior serve important functions. Cold Spring Harb. Symp. Quant. Biol 75, 533–543. [DOI] [PubMed] [Google Scholar]
- Dechat T, Adam SA, Taimen P, Shimi T, and Goldman RD (2010b). Nuclear lamins. Cold Spring Harb. Perspect. Biol 2, a000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, and Gygi SP (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer TA, and Misteli T (2011). The lamin protein family. Genome Biol. 12, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, and Bickmore WA (2008). Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4, e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer JG, Schulte R, Tsai HH, Tyagi S, Ibarra A, Shokhirev MN, Huang L, Hetzer MW, and Navlakha S (2018). Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 19, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgatos SD, Pyrpasopoulou A, and Theodoropoulos PA (1997). Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane. J. Cell Sci 110 (Pt 17), 2129–2140. [DOI] [PubMed] [Google Scholar]
- Gerace L, and Blobel G (1980). The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19, 277–287. [DOI] [PubMed] [Google Scholar]
- Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, Schwartzman A, Giobbie-Hurder A, Neuberg D, and Gordon LB (2012). Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 59, 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesson K, Rescheneder P, Skoruppa MP, von Haeseler A, Dechat T, and Foisner R (2016). A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 26, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AE, Maul G, Steinert PM, Yang HY, and Goldman RD (1986). Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl. Acad. Sci. U. S. A 83, 3839–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AE, Moir RD, Montag-Lowy M, Stewart M, and Goldman RD (1992). Pathway of incorporation of microinjected lamin A into the nuclear envelope. J. Cell Biol 119, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A 101, 8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JM, Navarro-Puche A, Casar B, Crespo P, and Andrés V (2008). Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J. Cell Biol 183, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, et al. (2011). Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Miner. Res 26, 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, and Kieran MW (2007). Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics 120, 824–833. [DOI] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, López-Otín C, and Misteli T (2014). Progeria: a paradigm for translational medicine. Cell 156, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. [DOI] [PubMed] [Google Scholar]
- Gueneau L, Bertrand AT, Jais J-P, Salih MA, Stojkovic T, Wehnert M, Hoeltzenbein M, Spuler S, Saitoh S, Verschueren A, et al. (2009). Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet 85, 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol. 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R, and McKeon F (1990). Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589. [DOI] [PubMed] [Google Scholar]
- Hozák P, Sasseville AM, Raymond Y, and Cook PR (1995). Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J. Cell Sci 108 (Pt 2), 635–644. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Egelhofer TA, Strome S, and Lieb JD (2010). Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol. 11, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivorra C, Kubicek M, González JM, Sanz-González SM, Alvarez-Barrientos A, O’Connor J-E, Burke B, and Andrés V (2006). A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 20, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, et al. (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet 21, 111–114. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack C-G, Melo-Cardenas J, Imanishi SY, Goldman RD, and Eriksson JE (2014). Interphase phosphorylation of lamin A. J. Cell Sci 127, 2683–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA, et al. (2018). HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Welton KL, Smith ED, and Kennedy BK (2009). A-type nuclear lamins act as transcriptional repressors when targeted to promoters. Exp. Cell Res 315, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-J, Jung Y-S, Yoon M-H, Kang S-M, Oh A-Y, Lee J-H, Jun S-Y, Woo T-G, Chun H-Y, Kim SK, et al. (2016). Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Invest 126, 3879–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans C, van der Zwalm MCH, Brueckner L, Comoglio F, van Schaik T, Pagie L, van Arensbergen J, and van Steensel B (2019). Promoter-Intrinsic and Local Chromatin Features Determine Gene Repression in LADs. Cell 177, 852–864.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, and Collas P (2014). Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 42, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, and Cao K (2012). Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 23, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith ACM, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, et al. (2008). Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med 358, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman W, Peric-Hupkes D, Kind J, Beaudry J-B, Pagie L, Kellis M, Reinders M, Wessels L, and van Steensel B (2012). Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 23, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Montag-Lowy M, and Goldman RD (1994). Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J. Cell Biol 125, 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naetar N, Korbei B, Kozlov S, Kerenyi MA, Dorner D, Kral R, Gotic I, Fuchs P, Cohen TV, Bittner R, et al. (2008). Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat. Cell Biol 10, 1341–1348. [DOI] [PubMed] [Google Scholar]
- Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, et al. (2010). Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol 30, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Nakagawa J, Dorée M, Labbé JC, and Nigg EA (1990). In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591–602. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, and van Steensel B (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet 38, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, and Furlong LI (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A, Gordon LB, Kleinman ME, Gurary EB, Massaro J, D’Agostino R, Sr, Kieran MW, Gerhard-Herman M, and Smoot L (2018). Cardiac Abnormalities in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA Cardiol 3, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, and Singh H (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247. [DOI] [PubMed] [Google Scholar]
- Reznikoff WS (2008). Transposon Tn5. Annu. Rev. Genet 42, 269–286. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, Sewry C, Battini R, Hu Y, Cottle DL, et al. (2008). Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J. Clin. Invest 118, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T, Pfleghaar K, Kojima S-I, Pack C-G, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, et al. (2008). The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 22, 3409–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U. S. A 103, 8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprath T, Corne TDJ, Nooteboom M, Grootaert C, Rajkovic A, Buysschaert B, Robijns J, Broers JLV, Ramaekers FCS, Koopman WJH, et al. (2015). Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus 6, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvera VM, Gordon LB, Orbach DB, Campbell SE, Machan JT, and Ullrich NJ (2013). Imaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am. J. Neuroradiol 34, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin J-W, Tewari M, et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104–1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka E, Bailey T, Grant CE, Noble WS, and Keich U (2011). Improved similarity scores for comparing motifs. Bioinformatics 27, 1603–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LCF, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward GE, and Kirschner MW (1990). Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 61, 561–577. [DOI] [PubMed] [Google Scholar]
- Watson JV, Chambers SH, and Smith PJ (1987). A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 8, 1–8. [DOI] [PubMed] [Google Scholar]
- Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, Lohberger B, Farra N, Petek E, Schwarzbraun T, Ofner L, et al. (2008). An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am. J. Hum. Genet 82, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Fong LG, Muchir A, and Young SG (2009). Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Invest 119, 1825–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, and Tammi MT (2009). CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hu J, Yue S, Kristiani L, Kim M, Sauria M, Taylor J, Kim Y, and Zheng Y (2018). Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell 71, 802–815.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and Code Availability Statement
The genomic datasets generated during this study are available at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE113354. Custom computational scripts used in this study are available at: https://github.com/kohta-ikegami/pS22-LMNA/
Public datasets
Assembly gaps
http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/gap.txt.gz
Blacklisted Regions
Chromatin annotation
Genecode Release 19 Gene Model
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/wgEncodeGencodeBasicV19.txt.gz
Lamin B1 LADs
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/laminB1Lads.txt.gz
Progeria fibroblast RNA-seq datasets from Fleischer et al.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113957
The genomic datasets generated during this study are available at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE113354. Custom computational scripts used in this study are available at: https://github.com/kohta-ikegami/pS22-LMNA/