

SUMMARY
Genomic studies in African populations provide unique opportunities to understand disease etiology, human diversity, and population history. In the largest study of its kind, comprising genome-wide data from 6,400 individuals and whole-genome sequences from 1,978 individuals from rural Uganda, we find evidence of geographically correlated fine-scale population substructure. Historically, the ancestry of modern Ugandans was best represented by a mixture of ancient East African pastoralists. We demonstrate the value of the largest sequence panel from Africa to date as an imputation resource. Examining 34 cardiometabolic traits, we show systematic differences in trait heritability between European and African populations, probably reflecting the differential impact of genes and environment. In a multi-trait pan-African GWAS of up to 14,126 individuals, we identify novel loci associated with anthropometric, hematological, lipid, and glycemic traits. We find that several functionally important signals are driven by Africa-specific variants, highlighting the value of studying diverse populations across the region.
Graphical Abstract
In Brief
Genome-wide data from Ugandans reveal insights into their ancestry, trait heritability, and loci associated with metabolic parameters, thereby providing a diverse resource for the study of African population genetics.
INTRODUCTION
Africa is central to our understanding of human origins, genetic diversity, and disease susceptibility (Tishkoff et al., 2009). The marked genomic diversity and allelic differentiation among populations in Africa, in combination with the substantially lower linkage disequilibrium (correlation) among genetic variants, has the potential to provide new opportunities to understand disease etiology relevant to African populations but also globally (Tishkoff et al., 2009; Gurdasani et al., 2015). Consequently, there is a clear scientific and public health need to develop large-scale efforts that examine disease susceptibility across diverse populations within the African continent. Such efforts will need to be fully integrated with research-capacity-building initiatives across the region (Rotimi et al., 2014).
Countries in Africa are undergoing epidemiological transitions—with a high burden of endemic infectious disease and growing prevalence of non-communicable diseases (Maher et al., 2010). Importantly, because of varying environments, population history, and adaptive evolution, the distribution of risk factors for a broad range of cardiometabolic and infectious diseases, and their individual contributions, may differ among populations globally (Campbell and Tishkoff, 2010). Differences in allele frequencies among populations, due to either selection or genetic drift, provide unique opportunities to identify novel disease susceptibility loci highlighting the value of conducting such studies in African populations. However, while there has been a recent increase in genetic studies of cardiometabolic traits including African-Americans (Peprah et al., 2015; Lanktree et al., 2015), there have been relatively few investigations of population diversity or the genetic determinants of cardiometabolic or infectious traits and diseases across the continent.
To conduct genetic studies in diverse populations across Africa, appropriate study designs that account for population structure, admixture, and genetic relatedness (overt and cryptic), as well as the development of genetic tools to capture variation in African genomes, are needed (Gurdasani et al., 2015). To leverage the relative benefits of different strategies, we undertook a combined approach of genotyping and low coverage whole-genome sequencing (WGS) in a population-based study of 6,400 individuals from a geographically defined rural community in southwest Uganda (Figures 1 and S1; Table S1; STAR Methods). We present data from 4,778 individuals with genotypes for ~2.2 million SNPs from the Uganda Genome-wide Association Study (UGWAS) resource (STAR Methods), and sequence data (Table S1.1; STAR Methods) on up to 1,978 individuals including 41.5 M SNPs and 4.5 M indels (Uganda 2000 Genomes [UG2G]) (Figure S1; Table S1.1; STAR Methods). Collectively, these data represent the Uganda Genome Resource (UGR). To enhance trait-associated locus discovery, we also include collective data on up to 14,126 individuals from across the African continent for genome-wide association analysis (STAR Methods).
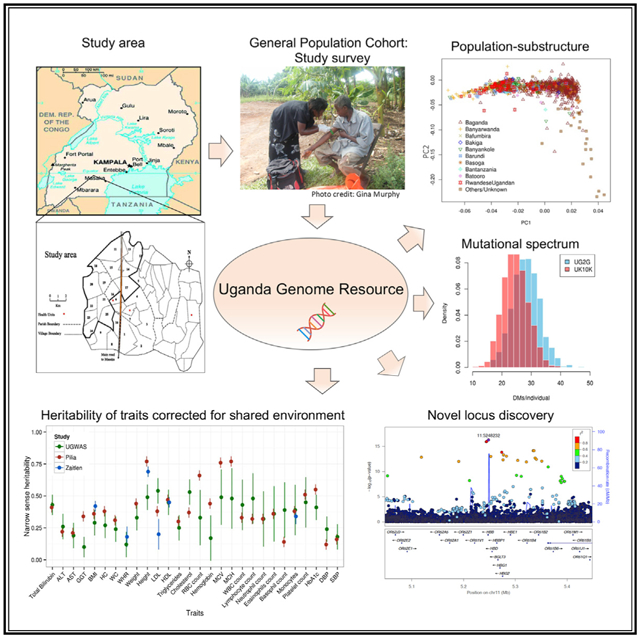
Figure 1. Genetic Substructure and Population Admixture within the General Population Cohort, Uganda.
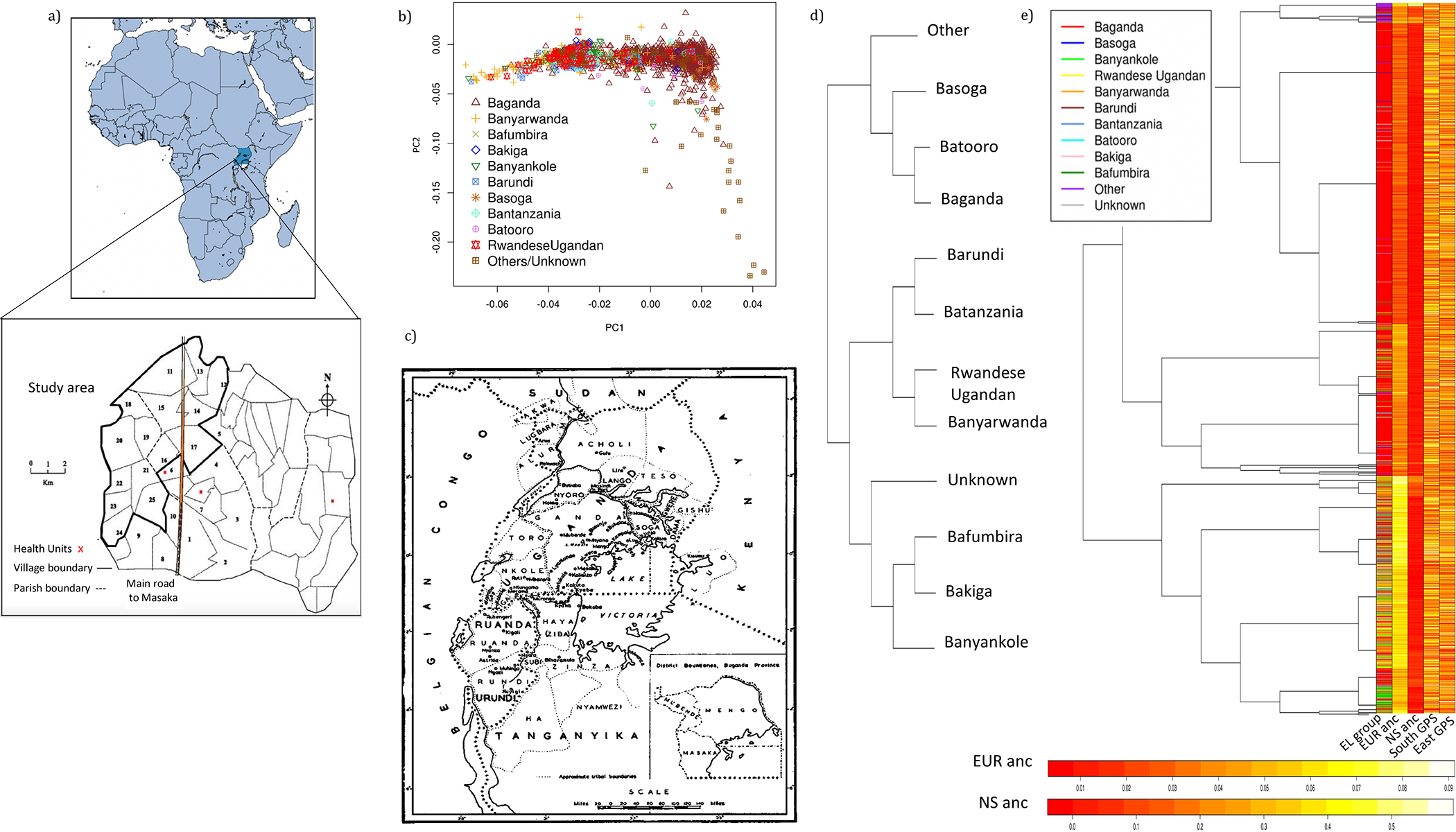
(A) Study area that encompasses 25 villages in the southwestern region of Uganda.
(B) fineSTRUCTURE inferred principal components (PCs) among unrelated individuals with the clines along PC1 and PC2 representative of Eurasian and East African gene flow respectively (n = 1,893). See also Figure S2 for PCA of Ugandans in a regional and global context. Modest structure is observed by ethno-linguistic group.
(C) Map of the district structure of Uganda during the colonial era, representing different districts different ethno-linguistic groups are likely to have migrated from (map reproduced with permission from (Richards, 1954).
(D) Dendrogram tree of population relationships among ethno-linguistic groups inferred by fineSTRUCTURE based on a summary co-ancestry matrix in analysis of unrelated Ugandans. The tree represents the summary of population relationships for ethno-linguistic groups and shows substructure among populations based on their geographical source (see also Tables S2.2–S2.4 for Procrustes analyses). Two major clades are represented, one from central Uganda and the second from populations migrating from western and southwestern Uganda.
(E) Unsupervised tree structuring with fineSTRUCTURE analysis of unrelated Ugandans. The dendrogram shows the inferred tree structure with various panels annotated for additional information below, including ethno-linguistic group (EL group), proportion of Eurasian ancestry as inferred by ADMIXTURE, K = 4 (EUR anc), proportion of Nilo-Saharan ancestry as inferred by ADMIXTURE (NS anc), and transformed latitude (south gps) and longitude (east gps) coordinates for each individual. Prominent clustering of clades is observed by ethno-linguistic group and Eurasian ancestral proportions.
See also Figures S3, S4, and S5.
Using these resources, we conducted a series of analyses to: (1) understand the population structure, admixture, and demographic history in a geographically defined population from Uganda (STAR Methods); (2) describe the spectrum of disease-causing mutations in the UG2G cohort (STAR Methods); (3) highlight the value of the UG2G sequence panel as an imputation resource (STAR Methods); (4) refine estimates of heritability of 34 complex traits, accounting for environmental correlation among individuals (STAR Methods); and (5) assess the spectrum of genetic variants associated with cardiometabolic and other complex traits in populations from sub-Saharan Africa (STAR Methods). Importantly, the UGR was designed to help develop local resources for public health and genomic research, including building research capacity, training, and collaboration across the region. We envisage that data from these studies will provide a global resource for researchers, as well as facilitate genetic studies in African populations.
RESULTS
A History of Ugandan Ethnic Diversity
Uganda has a diverse and complex history of extensive historical migration from surrounding regions over several hundred years. Migration has included economic migration for labor, as well as migration due to conflict in surrounding regions. Uganda is home to several diverse ethno-linguistic groups. The Ganda (“Baganda”) are most common ethno-linguistic group in central Uganda (previously the Kingdom of Buganda). This central region has also seen extensive migration from the surrounding regions of Rwanda, Burundi (formerly Ruanda-Urundi), and Tanzania (formerly the district of Tanganyika) (Figures 1A–1C) identifying as the “Banyarwanda,” “Barundi,” and “Batanzania,” respectively (Richards, 1954). More recent migration has occurred from Rwanda, due to displacement following conflict (identified as “Rwandese Ugandans,” distinct from the “Banyarwanda”). In addition to migration from surrounding regions, there have been large movements of people within Uganda relating to economic incentives during the colonial era. These include the Bakiga from Kigezi (Kiga), the Banyankole (Nkole), and Bafumbira from Kisoro from southwestern Uganda, and the Batooro (Toro), Basoga (Soga) from regions adjacent to central Uganda (Figure 1C; Richards, 1954). There are a number of other ethnic groups that have migrated to Buganda from adjoining areas of South Sudan (the Madi and Acholi), the Democratic Republic of Congo on the northwestern Ugandan border, as well as the from the West Nile region of Uganda (the Lugbara and Alur), and are referred to as “West Nile” migrants (Figure 1C; Richards, 1954). These groups often speak Nilotic languages. In our cohort, these ethno-linguistic groups are collectively classified as “Others,” because fine-scale ethno-linguistic group information was not available for these individuals. In this study, ethnolinguistic groups are based on self-identification and should be considered as representing a broad construct that encompasses shared cultural heritage, ancestry, history, homeland, language, or ideology.
Population Structure in a Rural Ugandan Community
We characterized genetic diversity and fine-scale structure among nine ethno-linguistic population groups from a geographically defined rural community from the Kalungu district in southwest Uganda (Figure 1A; STAR Methods). Principal components (PCs) 1 and 2 explained 0.3% and 0.1% of the genetic variation observed, respectively, with the cline along PC1 (Figure S2A) being strongly correlated with Eurasian admixture (r = −0.98, p < 2 3 10−16) as inferred from ADMIXTURE, K = 4 (Figure 2). This was corroborated in principal component analysis (PCA) of Ugandan ethno-linguistic groups in the context of global populations (Figures S2C and S2E) and our fineSTRUCTURE (Lawson et al., 2012) analysis (Figure 1B). FineSTRUCTURE analysis of the co-ancestry matrix inferred from linked genetic variants showed evidence suggestive of population substructure (Figures 1B; STAR Methods) with PCs 1 and 2 explaining 11.9% and 3.5% of observed variation, respectively. Clines along fineSTRUCTURE PC1 and PC2 were highly correlated with Eurasian (r = −0.90) and East African Nilo-Saharan ancestry (r = −0.98) as delineated by ADMIXTURE, K = 4, respectively (Figure 1B; STAR Methods). Here, Nilo-Saharan ancestry is defined as the ancestral component in ADMIXTURE analysis that was most prominent among the Dinka (Figure 2). The PC2 cline representing Nilo-Saharan ancestry was seen predominantly among the ethnolinguistic group classified as “Others” (Figure 1B), consistent with these representing ethno-linguistic groups that have migrated into Uganda from the northwestern region along the Nile. This suggests that the largest proportion of variation among the cohort was possibly driven by Eurasian and East African Nilo-Saharan gene flow.
Figure 2. Unsupervised ADMIXTURE Analysis of Ugandan Populations in a Global Context (n = 3,904) for clusters K = 2 to K = 18.
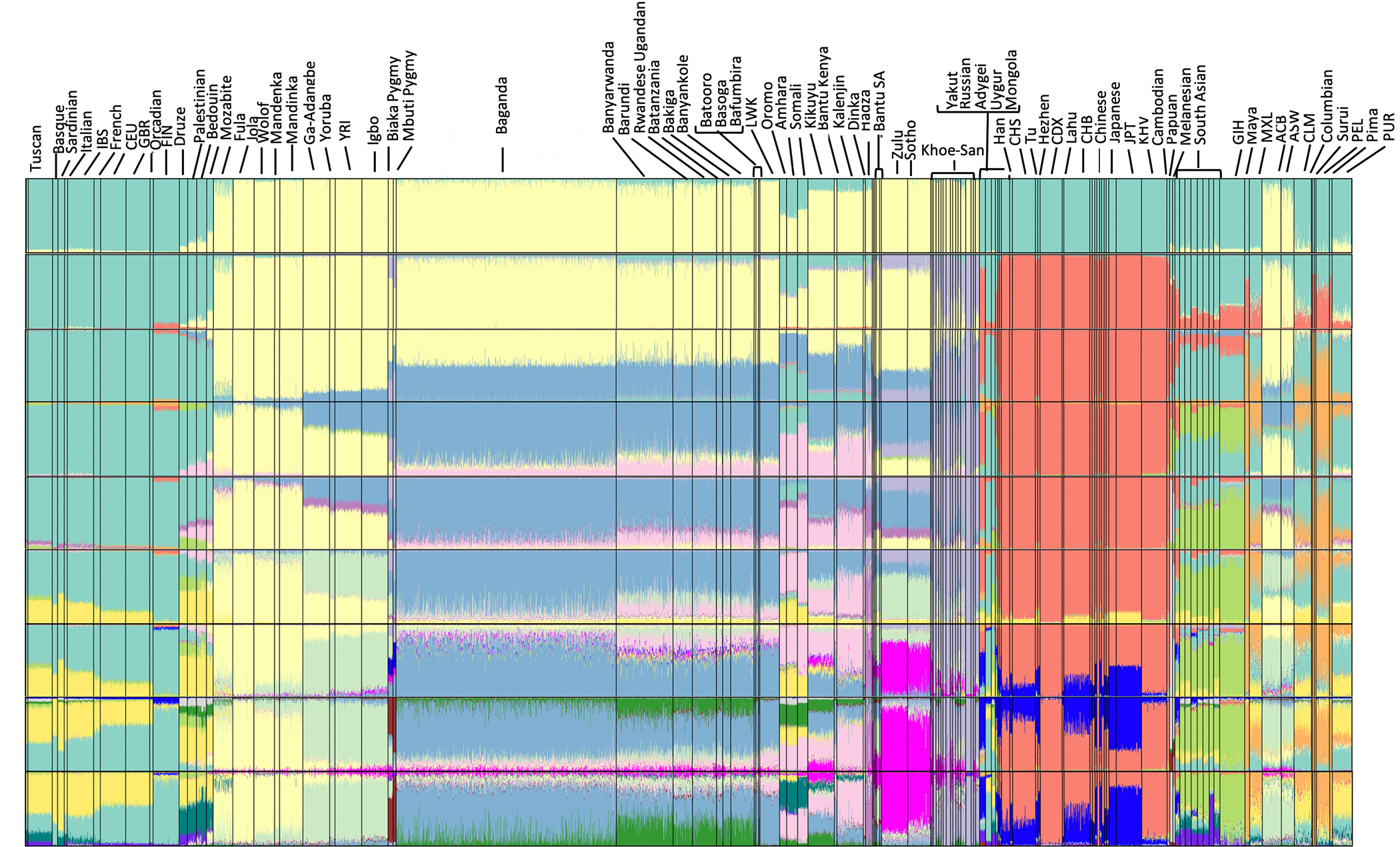
K = 2 represents separation of African, and non-African ancestry. Subsequent clusters show further delineation of Eurasian, East Asian, African hunter-gatherer (light purple ancestry seen in the Khoe-San), and Nilo-Saharan ancestry (light pink component observed predominantly in the Dinka). The Ugandans appear to be represented by multiple ancestral components, including ancestry predominant in East African Bantu populations, Nilo-Saharan populations, as well as different proportions of Eurasian-like components. We confirm these results with formal tests of admixture: QpWave (Tables S3.1, S3.8, and S3.9), f3 tests (Table S3.2), MALDER (Table S3.3), GLOBETROTTER (Figure S3), MT and Y chromosome analysis (Figure S4; Table S3.4), and the double-conditioned site frequency spectrum (Figure S5; Table S3.6).
Using Procrustes analyses, we find that substructure among ethno-linguistic groups in this rural Ugandan community is correlated with the historical geographical origins of these migrant populations (Figure 1; Tables S2.1–S2.3; STAR Methods). This suggests that in spite of extensive migration and mixture, substructure does exist among individuals in regional Uganda, and this substructure shows statistically significant correlation with the historical distribution of population groups across the region. We find no clear association with current geographical coordinates, consistent with extensive movement and mixing following migration within this region (Table S2.4). These findings are corroborated by fineSTRUCTURE tree inference from the co-ancestry matrix that also shows clade structure reflecting historical geographical regions from which these populations have migrated (Figure 1D). Ethno-linguistic groups from the central region of Uganda (the Baganda, Basoga, and Batooro), migrant populations from Rwanda, Burundi, Tanzania (Banyarwanda, Rwandese Ugandans, Barundi, and Batanzania, respectively), and those from southwestern Uganda (Bakiga, Banyankole, and Bafumbira) form separate clades (Figures 1C and 1D; STAR Methods). This clade structure may potentially also reflect the different amounts of Eurasian admixture observed among these populations, as we discuss subsequently.
With unsupervised fineSTRUCTURE analysis, we identify 52 population clusters (Figure 1E; STAR Methods). These clusters appear to represent a combination of factors, including ethno-linguistic group, historical geographical context (Figures 1D and 1E), as well as proportion of Eurasian and Nilo-Saharan ancestry, as estimated by ADMIXTURE, K = 4 (Figure 2). No clear pattern was observed by current GPS coordinate (Figure 1E), consistent with Procrustes analysis (Table S2.4).
Using QpWave, we find evidence for at least three distinct streams of ancestry across the Ugandan populations relative to outgroups (rank 2, p = 0.02) (Table S3.1; STAR Methods). On examining change in rank on removing populations one at a time, we find that the distinct streams of ancestry correspond well with the clade structure inferred in fineSTRUCTURE and historical geographic origins of these groups (Figures 1C and 1D). Specifically, we find that the rank of the matrix drops by one on excluding Rwandese_Ugandan, Banyarwanda, Bakiga, Banyankole, suggesting that these include a distinct source of ancestry potentially not present in other populations (Table S3.1). Another stream of ancestry appears to be contributed by Barundi and Batanzania, consistent with the tree structure inferred by fineSTRUCTURE (Figure 1D). Baganda, Basoga, and Mutooro appear to be relatively homogeneous, with only a single source of ancestry inferred across these populations (Table S3.1).
Inference of Complex Admixture in Uganda
Consistent with the extensive history of migration into this region, unsupervised ADMIXTURE (Alexander et al., 2009) and GLOBETROTTER (Hellenthal et al., 2014) analyses suggest that Ugandans are best represented by a mosaic of East African (Bantu, Nilo-Saharan, Afro-Asiatic, and rf-HG) and Eurasian-like ancestral components among modern global human populations (Figures 1, 2, and S3; STAR Methods). These findings are in keeping with other recent studies among East African populations that have suggested modern East African populations have been subject to complex admixture events over the past 5,000 years (Scheinfeldt et al., 2019; Fan et al., 2019). The proportion of Eurasian admixture appears to be lower in Baganda, Basoga, and Batooro (Figure 1D), suggesting that waves of admixture may have occurred with regional specificity within Uganda.
Delineation of Eurasian-like Ancestry within Uganda
Formal tests for admixture (f3 tests, MALDER, and GLOBETROTTER analyses) (Patterson et al., 2012; Hellenthal et al., 2014) consistently support evidence for Eurasian-like gene flow in Uganda (Figure S3; Tables S3.2 and S3.3; STAR Methods). Eurasian-like gene flow may be inferred by these tests if the source population has allele frequency spectra correlated with modern Eurasians. This does not in itself provide evidence for Eurasian back migration into East Africa. We evaluate the source of this ancestry further. The presence of Eurasian MT (K1a, R0a1a, N1a1a3, HV1b1a, I, J1d1a1, and W8) (Table S3.4) and Y chromosome (R1b and H) haplogroups within Uganda provide support for back-migration, as these haplotypes are thought to have arisen from out-of-Africa (Figure S4; Table S3.4; STAR Methods; Richards et al., 2000; Soares et al., 2010; Mishmar et al., 2003). In order to distinguish Eurasian gene flow from ancient structure within East Africa, we also assessed the double conditioned site-frequency spectrum (dcsfs) among Ugandans, with the sfs being conditioned on alleles being derived in a French sample, and ancestral in Yoruba (YRI) (Figure S5; Table S3.5; STAR Methods). A non-linear L-shaped sfs, enriched for rare derived alleles would be consistent with recent admixture, and not ancient substructure, as discussed previously (Yang et al., 2012). Our results confirm an observed dcsfs enriched for rare derived alleles and consistent with Eurasian gene flow (Figure S5). On assessing the fit of simulated data under different parameters with observed data, we find that gene flow from Eurasian populations into Ugandans is necessary to explain the observed frequency spectra (Figure S5; Table S3.6; STAR Methods). Overall, a dual model of admixture (~7% admixture) and ancient structure outperformed other models, including a model of ancient structure alone (p < 0.005) (Table S3.6). We note, however, that it is possible that fine-scale geographical spatial structure among populations could also explain these findings (Eriksson and Manica, 2014).
Using the conditional random field model (CRF), we assessed the presence of Neanderthal haplotypes among Ugandans (STAR Methods). Because Neanderthal ancestry is restricted to populations outside Africa, any evidence of Neanderthal ancestry among Africans is likely to be due to Eurasian back migration. We show evidence of detectable Neanderthal ancestry in Uganda, providing support for Eurasian admixture resulting from back-to-Africa migration (Table S3.7; STAR Methods). We first validate our approach by confirming enrichment of inferred Neanderthal sites within Eurasian segments, and with known maps of Neanderthal ancestry using simulated data (p < 0.001) (Table S3.7). We find that segments of inferred Neanderthal ancestry among Ugandans show high (95%) overlap with inferred Eurasian haplotype segments in the same individuals (as inferred by ChromoPainter) (Lawson et al., 2012). On assessing the overlap of segments of inferred Neanderthal ancestry among Ugandans with the inferred map of Neanderthal ancestry among Europeans and Asians in the 1000 Genomes project (Sankararaman et al., 2014), we find that 90% of segments identified as Neanderthal in origin (permutation p < 0.001), overlapped with known maps of Neanderthal introgression (STAR Methods; Sankararaman et al., 2014). Furthermore, in line with expectations, we also find evidence of significantly lower background selection in identified regions of Neanderthal ancestry relative to other regions (mean B scores 920 and 799, respectively, permutation p < 0.003) (STAR Methods). Collectively, our analyses support Eurasian back-migration into Uganda, consistent with previous work (Gallego Llorente et al., 2015; Henn et al., 2012; Pickrell et al., 2014; Fan et al., 2019).
Gene Flow between Ugandans and Regions rf-HG Populations
Analysis with MALDER also detects multiple complex admixture events, with the older events inferred as best represented by modern rf-HG-like and Eurasian-like ancestral components having occurred 2,000–4,500 years ago, and more recent Eurasian-like admixture 7–11 generations ago, consistent with previous reports (Figure S3; Gurdasani et al., 2015; Patin et al., 2017). Given the relatively low proportion of rf-HG admixture inferred within Ugandans by ADMIXTURE, GLOBETROTTER, and fineSTRUCTURE analysis, we evaluated this further. ALDER suggests low levels of rf-like admixture in Baganda (lower bound 4.4%), consistent with previous reports (Patin et al., 2017) and our results from ADMIXTURE and GLOBETROTTER analysis (Figures 2 and S3). Inference of rf-HG-like and Eurasian ancestry as primary sources of admixture by MALDER here is likely to reflect the known bias of the algorithm toward identifying source ancestral components that are more drifted, even if they contribute proportionately little to ancestry (Pickrell et al., 2014).
Asymmetrical gene flow has previously been noted between rf-HGs and East Africans, with predominantly Bantu admixture inferred within regional rf-HGs. We recapitulate these findings (Patin et al., 2014, 2017) confirming substantial Bantu admixture in rf-HGs (Mbuti) dating to 760 years ago in ALDER analysis (lower bound admixture 18%). Collectively, our findings suggest that assimilation of eastern rf-HG like ancestry into East African Bantu populations may have occurred during early migrations as part of the Bantu expansion, as these populations expanded into this region (Gurdasani et al., 2015). The route through which this ancestry entered these populations is unclear and may have involved gene flow between Bantu and possibly other regional pastoralist or HG populations. We explore this further by examining ancient East African populations as possible representative sources of ancestry among modern Ugandans.
Ancient Populations Representative of Admixture in Modern Ugandans
QpAdm analysis examining possible sources of admixture in modern Ugandans (STAR Methods) suggests that among global modern and ancient populations, modern Ugandan populations are best represented by ancestral components relating to ancient East African pastoralist populations (Tanzania_Pemba_ 700BP and Tanzania_Luxmanda_3000BP) (Tables S3.8 and S3.9; STAR Methods). These ancient pastoralists have been shown to be represented by multiple ancestral components, including ancient hunter-gatherer (Mota) and Eurasian (Levant-like) ancestry (Skoglund et al., 2017), suggesting that these ancestral components may have entered modern Ugandans proximately through ancient East African pastoralists in the region. Our primary results identify a single source of ancestry represented by Tanzania_Pemba_700BP in Baganda and Basoga, consistent with previous qpWave analyses (Table S3.8). Other populations can be modeled either as a mixture of Tanzania_ Pemba_700BP and Tanzania_Luxmanda_3000BP, or as a mixture of Tanzania_Pemba_700BP and modern or ancient Eurasians. Eurasian admixture in Ugandans varies from 5.8%–10.9% (Table S3.12). Consistent with qpWave results suggesting multiple streams of admixture within Uganda (Table S3.1), we find that Banyarwanda and Rwandese Ugandans cannot be modeled by any combination of two- or three-source populations, reflecting complex ancestry in these ethno-linguistic groups.
We also note that although Tanzania_Pemba_700BP has been shown to be represented well by Mende previously (Skoglund et al., 2017) (a finding we were able to recapitulate in our analyses), replacing Tanzania_Pemba_700BP with Mende as a source population for admixture into Uganda in our models results in a poor model fit (p < 0.01 in all cases). Our findings suggest that West African populations may not reliably represent Bantu ancestry in East African Bantu populations. In order to assess this, we examine the f4 statistic f4(chimp, Ancient South African; YRI/Mende, Uganda) (Table S3.10); we find asymmetry of Ugandan and West African populations relative to ancient South African Khoe-San, inferred from statistically significantly positive f4 statistics. Recent evidence has suggested that West Africans may carry a differential contribution of ancestry from an ancient population basal to ancient South Africans, leading to different West African populations (e.g., YRI and Mende) being asymmetrically related to ancient South Africans (Skoglund et al., 2017). In this context, the asymmetry observed between West and East Africans relative to ancient Khoe-San may be due to lower or absent basal ancestry in East African Bantu populations relative to West Africans (Table S3.10; STAR Methods). Alternatively, this may also be explained by Hadza-like or Khoe-San-related ancestry in modern Ugandans. Further evaluation and interpretation of these findings will require a wider sampling of ancient DNA samples from across Africa.
Demographic History of East Africans
To investigate ancient population size changes and split events, we examined a Ugandan trio sequenced at high depth (303) using MSMC2 (Schiffels and Durbin, 2014; Figure S6; Tables S1.6 and S1.7; STAR Methods). We find that the demographic history of Ugandans is broadly comparable to other Africans such as Yoruba and Luhya (LWK), with an estimated effective population size of ~20,000 over the past 10,000 years (Figures S6A–S6C). However, recent changes in population size of Ugandans seem more similar to LWK, as compared with YRI, and are consistent with patterns described by Schiffels and Durbin (2014) for LWK in the recent past (<10,000 years). Schiffels and Durbin (2014) observed a long “hump” in ancestral population size extending back from 6,000 years ago to beyond 50,000 years ago; we see a similar pattern in Uganda, likely reflecting complex admixture in Uganda, with modern Ugandans being a mosaic of multiple structured populations that were separated for several thousands of years, until recent admixture due to the extensive migration into this region.
On examining cross-coalescence between Uganda, YRI, and LWK, we find that Ugandan populations split from Yoruba, Nigeria (YRI) ~11,500 years ago (ya), with subsequent gene flow between Uganda and LWK in recent times (Figures S6D–S6F; STAR Methods). The Uganda-YRI divergence is older than the Bantu expansion (de Filippo et al., 2012) and may reflect varying patterns of Eurasian, basal, and regional admixture in East and West African populations. It also should be noted that these divergence times are lower bounds and are likely to be affected by gene flow between these populations following divergence, as previously documented (Schiffels and Durbin, 2014). We note that while our cross-coalescence rates (CCR) for Uganda-YRI when using 1000 Genomes Project YRI haplotypes are more in line with trio-based phasing, CCRs from Complete Genomics data are suggestive of more recent split times (Figure S6G). This suggests that statistical phasing of the 1000 Genomes Project high coverage samples may be more reliable than phasing of the same samples sequenced with Complete Genomics when phased using reference-based phasing with our merged reference panel. This is also in line with previous reports that inaccuracies in statistical phasing can impact inferences of split times (Song et al., 2017). Our results support the sequencing of trios in diverse population sets to maximize phasing accuracy, or alternatively using strategies that can greatly improve phasing accuracy, such as linked read sequencing (Zheng et al., 2016), optical nano-technology, or SMRT sequencing, as implemented with the PacBio platform.
Recent Haplotype Sharing between Ugandans and Other Global Populations
We explored more recent population history by examining rare variant sharing between the Baganda and other populations; we examined variants occurring only twice in the entire dataset (designated f2) (Figure S7; STAR Methods). On assessing average f2 sharing on repeatedly subsampled random haplotypes (n = 40) from each population, we see extensive sharing of f2 variants between Ugandan populations and other Niger-Congo language-speaking populations in the 1000 Genomes Project from East and West Africa. We also see extensive sharing with European and Asian populations consistent with Eurasian gene flow into these populations (Figure S7A). Paradoxically, we see little sharing among Ugandan populations; however, it must be noted that this is likely to be a consequence of our ascertainment scheme, with f2 variants being rarer among the Ugandan populations, and therefore, less likely to be sampled in a random set of 40 haplotypes (Figure S7A; STAR Methods).
Dating haplotypes surrounding f2 variants can provide important information about the interrelation among populations, including ancient and recent population divergence (Mathieson and McVean, 2014). Using this approach, we observe a total of 12,477,686 f2 variants in our dataset belonging to 9,875,361 f2 haplotypes. Given our ascertainment of f2 variants in a sample size comprising largely Ugandans, we expect f2 variation within Ugandans to be more recent than within other populations; therefore, we decided only to focus on the relationship of f2 variation between Ugandan and other populations, because this is likely to be relatively unbiased. We find that f2 variants shared between European and Ugandan populations are more recent than those shared between European and West African populations (median f2 dates were ~19,500 ya for Baganda compared with ~51,000 ya for YRI) (Figure S7B). This finding is consistent with back migration (Henn et al., 2012) and Eurasian admixture in the Uganda populations (Gurdasani et al., 2015; Pickrell et al., 2014), however, this may also reflect bias due to ascertainment of f2 variants in a larger population of Ugandans, thereby resulting in f2 variation representing rarer, and therefore more recent variation. Examining Ugandan populations in the context of other African populations, we find that f2 sharing between Ugandan populations and Ethiopians tend to be older (median f2 dating was ~23,000 ya) than Ugandan-West African splits (Figures S7B and S7C), probably reflecting a combination of deeper population splits between Bantu- and Afro-Asiatic-speaking groups, and relatively high Eurasian admixture in the Ethiopian populations. We also find evidence of very ancient divergence (with a median f2 dating of ~29,000 ya) between Baganda and Zulu (Figures S7B and S7C); this could reflect old f2 sharing with highly divergent Khoe-San haplotypes present among Zulu and other Southern African populations (Gurdasani et al., 2015). Our large African sequence resource allows the first such examination of shared rare variation among populations and highlights the complex demographic histories of populations in this region.
A Whole Genome Sequence Resource for Population and Medical Genetics
With the largest whole genome sequence dataset from Africa to date (Figure 3; STAR Methods), we present a unique resource representing the spectrum of human genetic diversity in East Africa, as well as a resource to facilitate medical genetics studies in the region.
Figure 3. Genomic Diversity and Mutational Spectrum within the Uganda Genome Resource.

(A) Discovery of autosomal SNP variation among 1,978 individuals from UGR relative to the 1000 Genomes Project phase 3 project (n = 2,504), the AGVP (n = 320), and UK10K cohorts (n = 3,781).
(B) Number of heterozygous sites per individual for each population in AGVP and the UGR (see Table S1.8 for number of individuals in each population group and Table S4.1 for the mean total number of variants per individual).
(C) Comparative allele frequency spectrum between 379 Europeans from the 1000 Genomes Project phase 1, a random sample of 379 individuals from all Ugandans (Uganda-all-379), and a random sample from only unrelated Ugandans (Uganda-unrel-379).
(D–F) Distribution of different functional classes of HGMD mutations within the UGR and also in comparison with UK10K ALSPAC; disease-causing mutations (DM), mutation reported to be pathogenic but with some degree of uncertainty (DM?), funcional polymorphisms (FP), disease-associated polymorphisms (DP), DPs with supportive functional evidence (DFP), frameshift or truncating variants (FTV). See Table S4.2 for the distribution of Clinvar clinically significant variants across populations. (D) We stratified the variation in four categories depending on allele frequency: common (>5% AF), low frequency (0.5%–5% AF), rare (0.1%–0.5% AF), and very rare (<0.1% AF). We find that while categories (FP, DFP, and DP) are preferentially observed as common variants in the UG2G data, the DM and DM? categories (disease-causing) are mainly observed as low-frequency or rare variants, as expected with deleterious mutations that are prone to purifying selection. In order to better understand the relevance of these mutations, we specifically examine DMs common in Uganda but rare among Europeans (see Figure S9 and Table S4.3). (E) Allele frequency spectrum for different functional classes of HGMD mutations within UGR. Expectedly, DMs are highly enriched for rare variation. (F) Distribution of DM among individuals in UG2G compared to UK10K ALSPAC.
As expected, and consistent with the out-of-Africa model, Africans carry higher levels of variation relative to other continental populations, the overwhelming majority being rare (Figure 3; Table S4.1; STAR Methods). In line with these observations, African populations provide greater opportunities for variant discovery as a function of sample size (Figure S8A; STAR Methods). We find that despite higher sequencing coverage within UK10K, the rate of discovery of genetic variation with increases in sample sizes among the Ugandans is greater than with European individuals from UK10K, at least up to a sample size of 500, after which gains plateau (Figures S8A and S8B). Of 41.5 M SNPs called in UG2G, we identify 9.5 M novel variants that are not present in the 1000 Genomes Project phase 3, African Genome Variation Project (AGVP), and UK10K reference panels (Figure 3A). We find that 28.7% of SNPs discovered in UG2G are not found in the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/), highlighting the importance of assessing diverse populations on a larger scale. Multi-allelic variants represented 0.87% of called SNPs.
The average number of variants/individual in UG2G was greater than variation/individual observed in the UK10K cohorts dataset (4,298,968 and 3,412,214 in UG2G and UK10K cohorts, respectively), consistent with African populations having greater genetic diversity (Table S4.1). Heterozygosity rates among Ugandans were comparable to other African populations, except Ethiopian populations that had lower levels of heterozygosity, consistent with high levels of Eurasian admixture in Ethiopian populations (Figure 3B). We also note a much greater proportion of rare variants among Ugandans, when comparing with an equal number of European individuals from the 1000 Genomes Project phase 3 (Figure 3C), which has comparable depth of coverage. The differences in site frequency spectrum observed are consistent with a historical population bottleneck in Europeans and greater genetic diversity with enrichment of rare variation among African populations.
We also explored the predicted functional consequences of variation in the UG2G population (Figures 3 and S9; Tables S4.2–S4.3; STAR Methods). Consistent with overall diversity, UG2G participants carried more missense variants per individual compared with the UK10K population (12,198 and 10,153 variants/individual, respectively) (STAR Methods). As with previous studies, we find that in spite of the lower absolute number of missense mutations (149,251 in UG2G and 69,761 in UK10K Avon Longitudinal Study of Children and Parent [ALSPAC]) in Europeans, these form a higher relative proportion of total variation (0.4% and 0.5% in UG2G and UK10K, respectively, p < 2×10−16) among Europeans (STAR Methods). For disease-causing mutations (DMs), as annotated by the Human Gene Mutation Database (HGMD) (Figure 3; STAR Methods), we identified a median of 29 DMs/individual in our cohort compared to 25 DMs/individual in UK10K, despite more extensive studies in European populations and potentially biased ascertainment (Figure 3F; Xue et al., 2012). By contrast, in UG2G, we observed a median of 3 homozygous DMs/individual compared to 4 homozygous DMs/individual in UK10K (STAR Methods) (p < 2 × 10−16). In contrast to the Genome of the Netherlands (GoNL) study (Genome of the Netherlands, 2014), where more than half of the DM variants were common (>5% allele frequency [AF]), the Ugandan population shows the opposite pattern, with DM variants predominantly being rare (AF <0.5%) in our cohort (Figures 3D and 3E). A total of 650 out of the 998 DM variants had a frequency lower than 0.5%, whereas only 47 were common (>5% AF) in the UG2G. These findings are consistent with previous reports that suggest a shift toward the higher frequency spectrum for deleterious variants in out-of-Africa populations. However, these differences to some extent may also represent ascertainment of DMs primarily in Europeans.
On examining the number of ClinVar mutations per individual (2015 Clinvar database) in UG2G compared with the UK10K ALSPAC, and 1000 Genomes Project phase 3 African and European populations, we observed greater number of median alleles/individual in the African individuals (UG2G and 1000 Genomes Project phase 3 African populations) compared to Europeans (UK10K ALSPAC and 1000 Genomes Project phase 3) in spite of the higher coverage of the ALSPAC dataset compared to UG2G (Table S4.2). Our results do not support substantial ascertainment bias in either the HGMD or ClinVar database, in contrast with previous reports of ascertainment (Xue et al., 2012; Auton et al., 2015). On comparing results using an older version of the ClinVar database (2014 version), we find clear evidence of ascertainment bias in the older database, with a greater number of clinically significant disease alleles/individual among Europeans compared with Africans, as have been reported before (Table S4.2; Auton et al., 2015). Our findings suggest that generation of larger scale sequence data in more diverse panels have contributed to reduction in ascertainment bias among mutation databases over time.
The distribution of the mutational spectrum in African and European populations is consistent with previous reports (Do et al., 2015; Lohmueller et al., 2008) and the impact of differences in demographic history among these populations. The higher burden of homozygous deleterious variation in Europeans is consistent with previous literature (Lohmueller, 2014; Henn et al., 2016), resulting from a loss of rare alleles following a population bottleneck thereby leading to greater co-occurrence of these mutations in recessive form (Do et al., 2015). The differences observed are unlikely to represent differences in efficiency of selection in European and African populations since the split, but rather non-selective demographic forces of drift and mutation in an expanding population after a bottleneck, as has been suggested previously (Do et al., 2015). The higher frequency of deleterious variation in European populations may also be related to ascertainment bias, with more common recessive variation in European populations more likely to be identified and cataloged (Amorim et al., 2017).
Allele frequency differences between populations along with clinical phenotype data may provide insights into the functional relevance of putative DMs. On assessing 38 DMs that were common in our cohort (AF >5%), but rare or absent in the UK10K data (AF <1%) (Table S4.3) (Walter et al., 2015), we identify established causal loci associated with hematological traits, such as the G6PD and sickle cell (HBB) variants, which are common in UG2G, but absent from the UK10K data, consistent with these loci being under positive or balancing selection and protective against malaria (Table S4.3) (Karlsson et al., 2014). However, we also demonstrate that several putative DMs associated that are common in UG2G, but rare in UK10K, do not show strong evidence for association with relevant cardiometabolic or hematological traits (Figure S10). These variants common in UG2G include rs41264848 in the LPA region (p = 0.40 for association with total cholesterol), rs36220239 in the ADAMTS13 region (p = 0.90 for association with platelet count), and rs115080759 in the HNF1A gene associated with MODY3 showing no association with HbA1C (p = 0.20 in entire cohort and p = 0.29 when only including individuals >40 years age) (Figure S9). Our results for rs115080759 are consistent with reports that suggest this variant is benign (Kleinberger et al., 2018). This emphasizes the need to carefully and comprehensively evaluate the impact of putative functional or disease-causing mutations across global populations, because they may not have any clinical or biological relevance or be readily transferable across populations (Saraf et al., 2014; Xue et al., 2012). The lack of strong associations between these DMs and phenotypes in our cohort indicate that they are unlikely to be causal for the associated traits or may have different or lower penetrance within African populations due to complex factors, including epistasis or gene-environment interplay.
Finally, we assess the impact of the addition of the UG2G panel to existing reference panels on imputation accuracy among populations from sub-Saharan Africa (Figure 4). We show that addition of the UG2G panel to existing sequence panels with African haplotypes, such as the 1000 Genomes Project phase 3 and AGVP (combined n = 3,895), markedly improved imputation accuracy (r2 increase by 0.08 [MAF % ≤ 0.01] and 0.04 [all MAF]) for rare and common variants in Ugandan populations (Figure 4; STAR Methods). Additionally, we observe a substantial increase in imputation accuracy across the allele frequency spectrum generally in East African populations, including Nilo-Saharan linguistic groups such as the Kalenjin (Figure 4), probably reflecting haplotype sharing across the region. The number of variants “successfully” imputed (info ≥ 0.3) substantially increased using the UG2G panel in comparison with the 1000 Genomes Project phase 3 and AGVP panels combined, with an additional 8 M variants being successfully imputed in Baganda and 1.5 M additional variants successfully imputed among other East African populations (Figure 4). These analyses emphasize the importance of building regional sequence-based resources to facilitating genetic studies in Africa, including alongside current initiatives such as the Haplotype Consortium (McCarthy et al., 2016).
Figure 4. Improvement in Imputation Accuracy with Addition of the African Genome Variation Project (AGVP) and Ugandan Sequence (UG2G) Panel to the 1000 Genomes Project Phase 3 (1000Gp3) Imputation Panel (n = 3,895 for the Combined Reference Panel) when Imputation Is Carried Out into the Omni 2.5M Genotype Data for AGVP Population Sets Not Included in the Reference Panel.
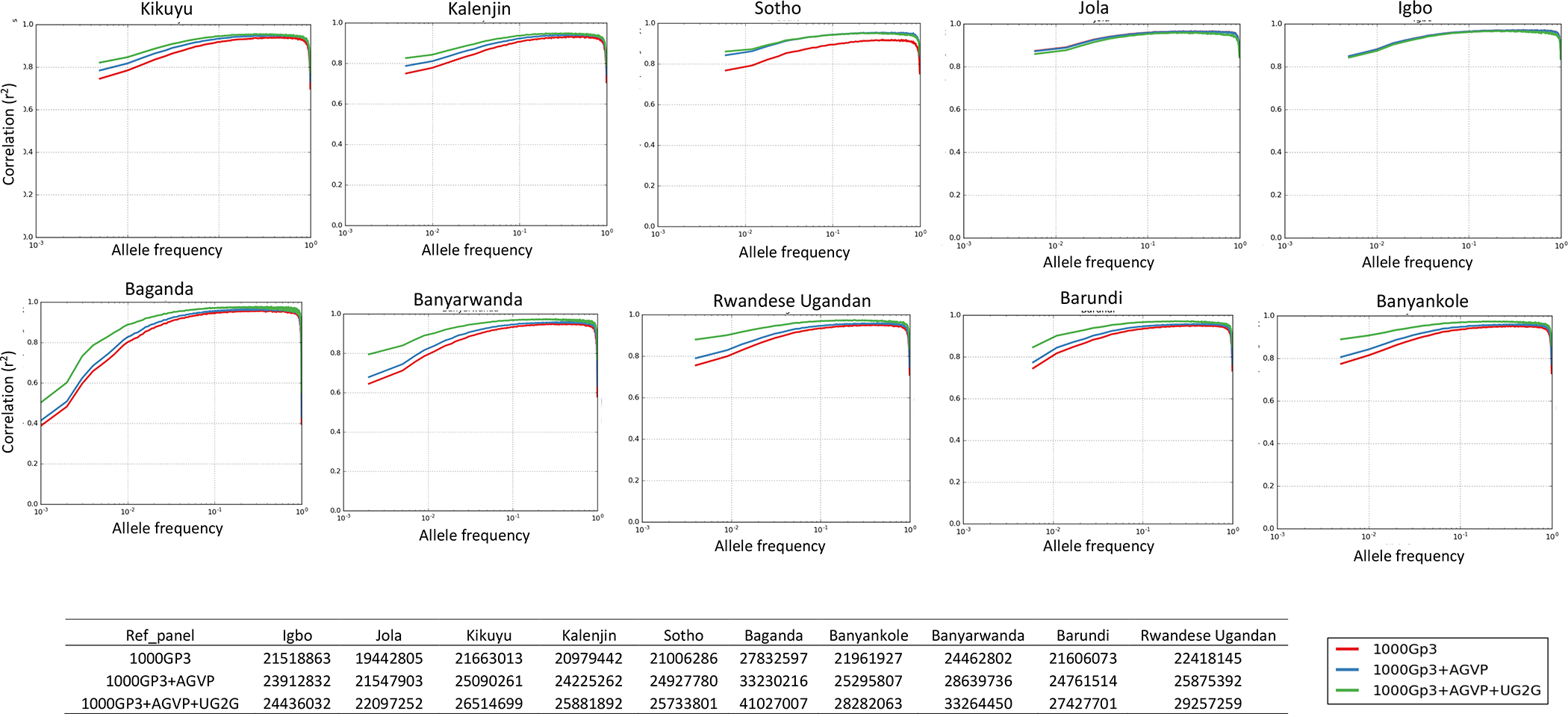
Marked improvements are observed for East African populations such as Kalenjin and Kikuyu across the allele frequency spectrum. We also observe substantial improvements when imputing into the unrelated individuals from different ethno-linguistic groups in UGWAS. The tables below the figure show the number of variants successfully imputed (info score ≥ 0.3) into the Omni 2.5M array data for each population using different reference panels. We see a substantial increase in informatively imputed variants with addition of the UG2G sequence reference panel across all populations.
Heritability of Cardiometabolic Traits in a Rural Ugandan Community
Narrow-sense heritability represents the fraction of phenotypic variation in a population that is due to additive genetic variation. As such, it represents an important metric determining the genetic basis of complex traits and diseases. There have been no comprehensive evaluations of heritability and the interrelation with environment among African populations. We, therefore, assessed heritability for 34 complex cardiometabolic traits using a mixed model approach that also models environmental correlation (Heckerman et al., 2016; Figure 5; STAR Methods).
Figure 5. Heritabilities for 34 Complex Traits within the Ugandan GWAS Cohort (UGWAS, n = 4,778) (Green Markers) Measured Using FAST-LMM (Blue Markers), Compared with Those Estimated in a Sardinian (Red Markers) and Icelandic Population (Blue Markers).
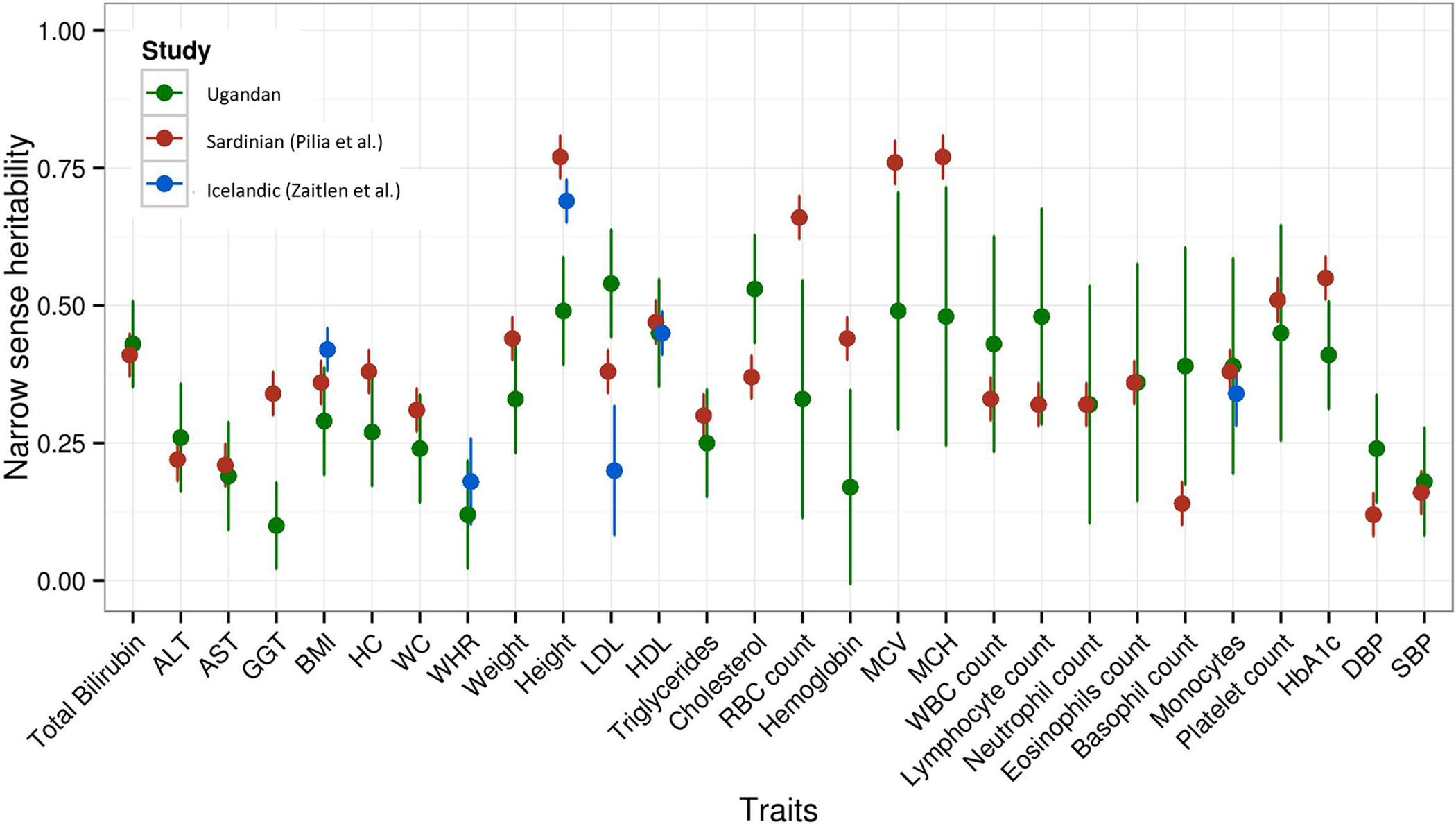
The estimated heritabilities in UGWAS are adjusted for environmental correlation among individuals using GPS coordinates. The heritabilities in Pilia et al. (2006) are also adjusted for shared environment in pedigrees. We observe statistically different heritability for LDL-cholesterol, total cholesterol, height, and serum GGT. See Tables S5.1–S5.4 for raw data.
Estimates of heritability corrected for environmental correlation varied from relatively modest (e.g., 10% for GGT, a liver biomarker) to 55% for traits such as mean platelet volume (MPV) (Figure 5; Table S5.1; STAR Methods) We find clear statistical differences in heritability estimates for several traits, compared to European populations (Figure 5; Tables S5.2–5.4). For example, the narrow-sense heritability for height was 49% in Ugandans, compared with estimates of 70%–80% in European populations (p < 0.0001); by contrast, the heritability estimates for LDL were statistically significantly higher in the Ugandan population (54% versus 20%–43% in European studies, p < 0.002) (Figure 5; Tables S5.2–S5.4; STAR Methods). We speculate that these differences may be due to varying patterns of genetic loci influencing these traits in European and African populations, or perhaps more plausibly due to a larger proportion of environmental variation explaining phenotypic variance. For example, malnutrition or nutritional deficits in rural African populations may attenuate the effects of genetic variance on height, whereas dietary consumption and obesogenic environments in European populations may reduce the impact of genetic factors on the variation in LDL levels (Nalwoga et al., 2010). We note, however, that lower estimates of heritability (e.g., for height) in the Ugandan cohort may also arise from differences in LD (lower LD with causal variants), lack of adjustment for shared environment in previous studies, or gene-environment interactions. While we do not find statistically significant gene-environment interactions for height, we find evidence for statistical gene-environment interaction for waist/hip ratio, red blood cell distribution width (RDW), and hematocrit (permutation p = <0.0001). These statistical interactions may represent interplay between genetic factors and dietary factors, iron stores, and nutritional status (Table S5.1). Reliable assessment of the interrelation between genetic and environmental variation, including specific environmental indices, will require application of these methods in much larger-scale studies with relevant phenotypic information. Examining locus-specific heritability would complement direct assessments of population differences in heritability of population traits.
GWAS of Cardiometabolic Traits in African Populations
To assess the spectrum of genetic variants associated with cardiometabolic traits in African populations, we performed a GWAS of 34 cardiometabolic traits in up to 14,126 individuals from across the African continent, including populations from Ghana, Kenya, Nigeria, South Africa, and Uganda (Tables 1 and S6.1–S6.12; STAR Methods). To maximize opportunities for genomic discovery, we meta-analyzed GWAS data from all study populations imputed with our combined reference panel, using the Han-Eskin random-effect meta-analytic approach implemented in METASOFT (Han and Eskin, 2011) to allow for potential heterogeneity in allelic effects (STAR Methods). We first re-assessed thresholds for genome-wide statistical significance in African populations using several approaches (Gao et al., 2008; Chen and Liu, 2011; Moskvina and Schmidt, 2008; Nyholt, 2004) and found that a statistical threshold of 5.0 × 10−9 is more relevant in populations with high genetic diversity and relatively lower levels of LD (Table S6.1; STAR Methods).
Table 1.
Novel and Distinct Association Signals Discovered in GWAS Meta-Analysis
| Trait | rs ID | chr:pos | A1 | A2 | Number | p_assoc | p het | Gene | MAF_ AFR (%) | MAF_ EUR (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Novel associations with traits | ||||||||||
| Albumin | rs540810730 | 7:19228309 | C | A | 8,995 | 3 × 10~°9 | 0.33 | NA | 2.9 | 0 |
| Bilirubina | rs151330263 | 16:302161 | A | G | 9,326 | 2 × 10~12 | 0.60 | HBA1/HBA2 | 5.6 | 0 |
| BMI | rs7798566 | 7:141549317 | A | G | 13,976 | 3 × 10~15 | 6 × 10~17 | NA | 4.9 | 1.2 |
| HbA1c | rs6724428 | 2:189377509 | A | G | 7,161 | 4 × 10~°9 | 0.12 | GULP1 | 44 | 55 |
| HDL-chol | NA | 19:21749298 | G | A | 6,407 | 4 × 10~°9 | NA | RP11–678G14.3 | 0.7 | 0 |
| RDW | NA | 7:131419316 | CAA | C | 1,119 | 1 × 10~°9 | NA | NA | 38 | NA |
| WBC count | rs4755389 | 11:35115148 | C | T | 2,741 | 4 × 10~°9 | 0.54 | CD44 | 5.9 | 45.2 |
| Novel associations previously associated with similar traits | ||||||||||
| RDW | rs334 | 11:5248232 | T | A | 1,625 | 2 × 10~17 | NA | HBB | 7.7 | 0 |
| BMI | rs12405634 | 1:243102900 | C | T | 13,976 | 3 × 10~10 | 4 × 10~12 | NA | 11.3 | 1.5 |
| neut_count | rs1347767 | 2:136485657 | C | T | 2,671 | 7 × 10~” | 3 × 10~°3 | R3HDM1 | 12.8 | 0 |
| Distinct associations at known loci | ||||||||||
| ALT | NA | 8:145730373 | G | C | 6,407 | 6 × 10~38 | NA | GPT | 0.6 | 0 |
| ALP | 1:21897903 | rs4654971 | T | C | 2,588 | 8 × 10~” | 0.04 | ALPL | 3.3 | 8.3 |
| ALP | 6:24489961 | rs189263035 | G | C | 9,322 | 7 × 10~2° | 6 × 10−4 | GPLD1 | 7.8 | 0 |
| GGT | 22:25084815 | NA | G | A | 8,995 | 1 × 10~6° | 0.12 | NA | 8.3 | 0 |
| HbA1ca | rs148228241 | 16:227187 | G | T | 7,161 | 3 × 10~12 | 0.02 | HBA1/HBA2 | 10.1 | 0 |
| Cholesterol | 5:156378584 | NA | CGGAA | C | 6,407 | NA | NA | TIMD4 | 0.9 | 0 |
| LDL-chol | 5:156378584 | NA | CGGAA | C | 6,407 | NA | NA | TIMD4 | 0.9 | 0 |
| Triglycerides | 19:45422587 | rs12721054 | A | G | 13,115 | 7 × 10~25 | 0.03 | APOC1 | 13 | 0 |
| Triglycerides | 1:63171024 | rs569795903 | C | T | 6,407 | NA | NA | RP11–230B22.1 | 1.4 | 0 |
| MCHC | 16:302161 | rs151330263 | A | G | 2,744 | 8 × 10~13 | 0.17 | ITGF3 | 4.8 | 0 |
A1, effect allele; A2, non-effect allele; neut_count, neutrophil count; NA, not applicable; p_assoc, p value from RE2 (Han-Eskin) METASOFT meta-analysis across cohorts (where relevant); p het, p value for Cochran’s Q heterogeneity statistic. See Tables S6.2–S6.8 for all results, and Figures 6 and S10 for locusview plots.
These associations were found to be driven by the α−3.7 thalassemia deletion on further sensitivity analyses.
In our meta-analysis, we identified 43 distinct signals statistically significantly associated with at least one trait (Table S6.2). Following visual inspection of locusview plots, two association signals were excluded (Figure S10) as likely to be artifactual. More than half of all remaining signals (23/41) were attributable to genetic variants specific to African populations or extremely rare in other populations (Table S6.2; STAR Methods). Among these, we identified ten distinct or secondary signals at previously identified loci (Table 1), of which nine were driven by genetic variants that were specific to Africa or extremely rare in other populations (Tables 1 and S6.2). We also identified ten association signals within novel loci (Table 1). These novel signals included associations with anthropometric indices, lipid, hematological, and blood cell traits (Figures 6 and S10I; Tables 1 and S6.2). Among these novel signals, three were noted to have been previously identified as associated with biologically related traits (Table 1).
Figure 6. Locusview Plots for Selected Novel Association Signals Associated with Specific Traits in a GWAS of up to 14,126 Individuals.
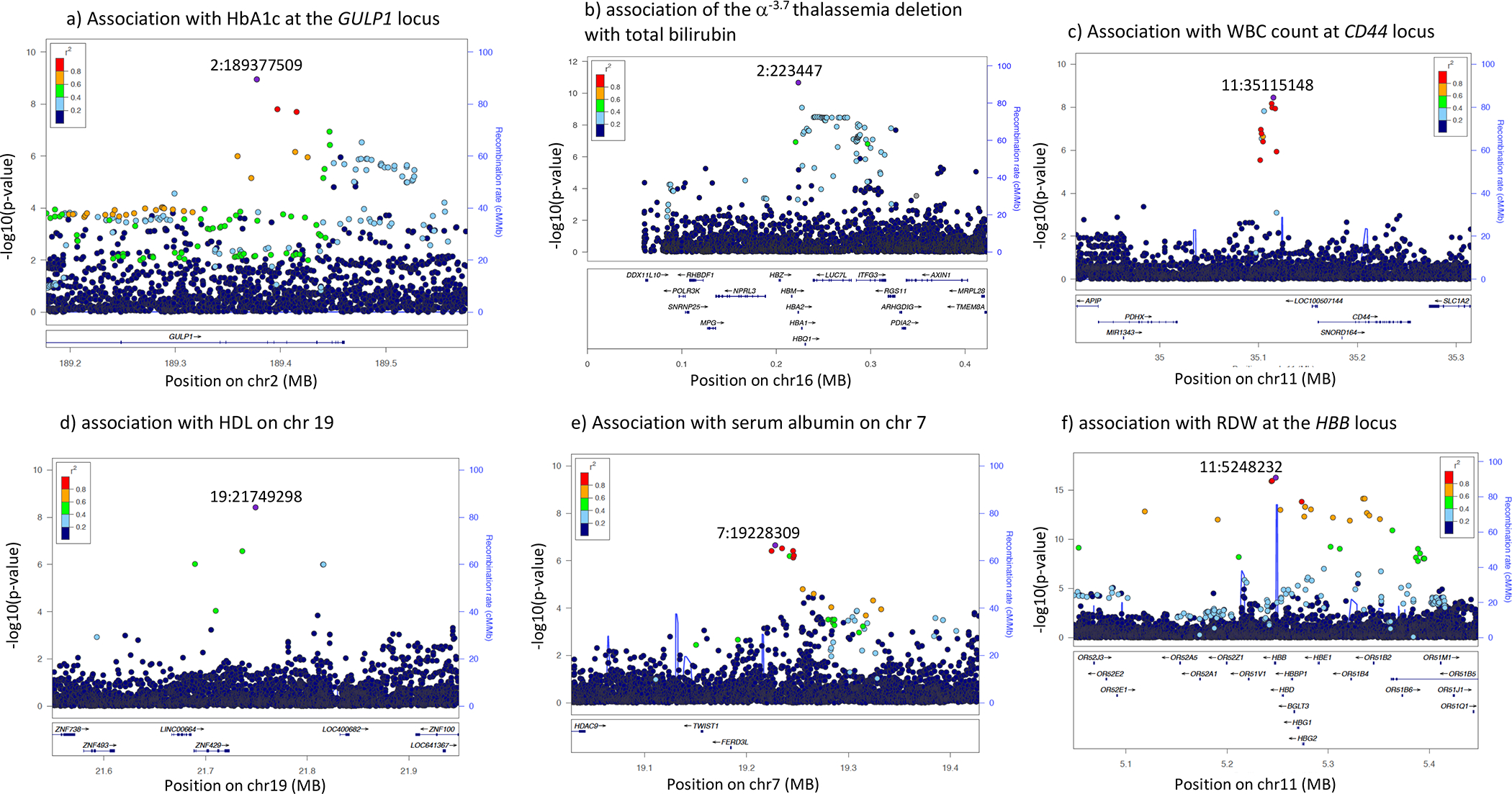
(A) Novel association of the GULP1 locus with HbA1c.
(B) We highlight functionally important and novel associations of the α−3.7 thalassemia deletion with total bilirubin.
(C) We identified a novel association with WBC count at the CD44 locus; CD44 encodes a cell-surface protein that regulates neutrophil adhesion, migration, and apoptosis, among other functions
(D and E) Associations of Africa-specific variants with HDL levels (D) and total albumin (E).
(F) Association of the sickle cell variant with RDW, recapitulating the known pathophysiology of sickle cell disease.
Our novel association signals included a functionally relevant association between a 3.8 Kb deletion (−α3.7), known to cause alpha thalassemia, and total bilirubin levels (p = 2 × 10−12) (Figure 6; Table 1; STAR Methods). The α3.7 variant is thought to have risen to high frequencies in African populations in regions endemic for malaria by virtue of providing resistance to severe malaria (Mockenhaupt et al., 2004).
We also identified a novel association with BMI on chromosome 1 (p = 2.8 × 10−10) in the intergenic region between PLD5 and SDCCAG8 (Tables 1 and S6.2). The SDCCAG8 locus has been previously associated with extreme childhood obesity in Europeans (Scherag et al., 2010). Recent unpublished summary data from Genetic Investigation of Anthropometric Traits (GIANT) and UK Biobank suggests that this locus may be associated with BMI (peak SNP rs11807000, p = 5.7 × 10−11). Our peak SNP is not present in these data or in the GIANT summary data. However, the presence of a comparably statistically significant association at this locus in a relatively small study (with respect to the UK Biobank and GIANT meta-analysis that examined ~700 K individuals) is interesting and needs further exploration. We also identified a novel association signal for the SNP rs7798566 (RE2 p = 3 × 10−15) with BMI on chromosome 7 in the intergenic region within the TAS2R gene family (Tables 1 and S6.2). The TAS2R family of genes expressed within the gastro-intestinal tract are involved in taste sensitivity to bitter-tasting compounds (Bachmanov and Beauchamp, 2007) and regulation of thyroid activity. Both these loci showed significant statistical heterogeneity of effect among African cohorts (Tables 1 and 6.2), with the association being seen only within the AADM cohort. The heterogeneity of effect for the SDCCAG8 locus among African cohorts (Tables 1 and 6.2), and European cohorts may point to differential effects in different environments or genetic backgrounds (epistasis), or differences in demographic makeup of these studies. The significance of these novel discoveries will require further evaluation across diverse population groups.
Among hematological traits, we identified a novel association on chr11 between the PDHX and CD44 region with white blood cell (WBC) count (Figure 6; Table 1). CD44 encodes a cell-surface protein that regulates neutrophil adhesion, migration, and apoptosis (Wang et al., 2002; Khan et al., 2004) among other functions (Figure 6; Table S6.2). We also identified a novel association between rs1347767, an Africa-specific (MAF = 10%) variant, downstream to R3HDM1, associated with neutrophil count (Table S6.2). While this locus has not been previously associated with neutrophil count, this region lies near the LCT locus, known to be associated with WBC count in an exome association study of African-Americans (Auer et al., 2012). The association at this locus was noted to be dependent on ancestry at the LCT locus in this study, suggesting the association may be population-specific (Auer et al., 2012). We also observed an association of the SNP causing sickle cell anemia (rs334) with RDW within our analysis (Figure 6). Notably, this SNP has not been identified as associated with RDW in the UK Biobank analysis of ~171 K individuals (p = 0.006) highlighting the utility of examining diverse cohorts in identifying functionally important associations with disease.
Fine mapping with MANTRA resulted in narrow credible intervals for most traits with 16 of 41 distinct loci being mapped to a single SNP in the credible interval (Table S6.3; Musunuru et al., 2010). We also resolved the previously identified association with HbA1c at the ITFG3 locus to the α−3.7 thalassemia deletion, which explained 3% of variation in HbA1c levels (Figure S10). We note that associations of the α−3.7 thalassemia with both HbA1c and total bilirubin were driven primarily by the Ugandan cohort, and not observed within other cohorts, consistent with the higher allele frequency of the deletion observed in Ugandans and the endemicity of malaria within this region. Our findings recapitulate the need to more fully understand functional variation, including for hemoglobinopathies, that may explain a substantial proportion of variation in HbA1c in African populations. These factors may have a direct impact on the utility of using HbA1c as a clinical tool for detection and diagnosis of diabetes in Africa (Herman and Cohen, 2012).
Given the complex and regionally specific genetic diversity within Africa, we assessed patterns of heterogeneity and transferability of association signals across the four cohorts to inform the design of medical genetics studies as well as understand the utility of European-centric polygenic scores for risk prediction in African populations. While most known associations with data available in >1 cohort were transferable (had nominally statistically significant p values in two or more cohorts) (Table S6.4), we identified several known and functionally important loci—the LIPC locus associated with HDL, the DARC locus encoding the Duffy antigen associated with monocyte count, and the α−3.7 thalassemia variant at the HBA1/A2 locus associated with RBC count and HbA1c that only had statistical support from a single cohort. Limited transferability at some of these loci appears to reflect allele frequency differences among cohorts potentially related to positive selection relating to the endemicity of malaria in some geographical regions and not others (e.g., the DARC and HBA1/A2 loci) (Liu et al., 2013; Hedrick, 2012; Hamblin et al., 2002). However, lack of transferability for other loci (e.g., LIPC) where the candidate SNP is common across all cohorts, may reflect several factors, including allelic heterogeneity (multiple distinct variants at loci) or gene-environment interactions, and will need further investigation in large-scale studies of diverse African populations. Additionally, there were four associations at known loci where the association signal was driven by a single cohort due to population-specificity of the variant examined or rarity of the variant in other cohorts (MAF <0.5%) (Table S6.4). These included the GPT locus associated with ALT, with variants driving the association specific to Uganda (no association was observed at this locus in other cohorts), and TIMD4 locus associated with LDL and total cholesterol levels (Table S6.4).
Expectedly, transferability was observed to be lower among novel association signals. Among nine novel associations with data in >1 cohort identified, 5 were noted to have support only from a single cohort (Table S6.4); among these was the functionally relevant the sickle cell locus associated with RDW and the SDCCAG8 previously associated with childhood obesity (Scherag et al., 2010), associated with BMI in our data. While the reasons for specificity of some of the novel loci to a single cohort relate to allele frequency differences of variants among cohorts (e.g., for the sickle cell locus), reasons for specificity at other loci are less clear and require further exploration.
To systematically examine differences in effect sizes across cohorts, we examined statistical heterogeneity of effect at associated loci among studies (STAR Methods). While most peak-associated SNPs did not show evidence of statistically significant heterogeneity, we found strong evidence of statistical heterogeneity (Cochran Q p < 5 × 10−9) in regions around several peak SNPs within known and biologically important regions associated with total cholesterol, LDL (e.g., the PCSK9 and the APOE regions), bilirubin (UGT1A3–9 genes), GGT (GGT1 locus), MCHC (HBA1/A2 locus), ALT levels (GPT), and neutrophil count (DARC locus). This heterogeneity was partly attributable to differences in LD structure around causal or peak variants across populations or the presence of multiple distinct variants at loci (allelic heterogeneity) (Figure S10; STAR Methods). For example, joint and conditional analysis at the UG1TA3-9 locus associated with bilirubin in UGR showed evidence for three distinct SNPs associated with total bilirubin in joint and conditional analysis in the UGR (Figure S10; Table S6.5), suggesting that statistical heterogeneity at a locus can provide important information about the genetic architecture of traits. Using the same approach, we also identified three distinct association signals at the GGT1 locus in UGR, (Figure S10; Table S6.6), with differences in LD around these distinct signals potentially explaining the statistical heterogeneity observed within this locus between cohorts.
In addition to allelic heterogeneity representing multiple distinct associations at a given locus, we also identified loci where distinct associations were identified as driving the association signal with a given trait among different populations. One example of this is the GPT locus associated with ALT levels (Table 1), where distinct population-specific variants drive the association in Africans and Europeans (Abul-Husn et al., 2018). We also identified a distinct association with ALP levels at the known ALPL locus. Peak-associated SNPs at this locus have been previously noted to be different across large studies of European (Chambers et al., 2011), Chinese (Yuan et al., 2008), and Japanese (Kamatani et al., 2010) cohorts (Table S6.7); these peak SNPs were not in LD with the peak SNP in Uganda, suggesting that multiple signals may be driving these associations at the locus in different populations (Table S6.8). An alternate explanation is that all these SNPs may be differentially tagging an as yet unidentified causal variant.
Collectively, our findings highlight the utility of genetic resources from diverse populations in novel discovery, especially for population-specific and low-frequency association signals. In this context, differences in frequencies of functional alleles, allelic heterogeneity, and differences in LD structure provide unique opportunities for discovery and resolution of causal loci and a better understanding of the genetic architecture of disease.
DISCUSSION
Here, we present the largest whole-genome sequence dataset from an East African population to date, as well as a large genome-wide genotyped and phenotyped dataset from the same population. We provide rich genomic resources for studies of human population history and GWAS and a mechanism to evaluate the clinical relevance of genetic diversity both in African populations and globally.
We present evidence for fine-scale structure and admixture in this Ugandan population, reflecting complex ancient and recent population migrations and expansions in East Africa. Our findings highlight the need for larger-scale deep sequencing, including a systematic assessment of hunter-gatherer populations across Africa, to more fully understand the genetic history and diversity of Africa. Sequencing of DNA from ancient skeletal material across Africa will greatly facilitate such efforts (Pickrell and Reich, 2014)—allowing stronger inferences into the source of genetic diversity and population history in Africa and globally.
Accounting for environmental correlation, we describe statistical differences in heritability for traits between African and European populations; these differences may be suggestive of the interplay between genetic and environmental effects on heritable traits, as well as the impact of differences in genetic architecture as a result of selection, drift, and historical demographic events. Our findings reiterate the dynamic and context-specific nature of heritability, potentially varying among populations, demographic factors, and environmental exposures (Haworth and Davis, 2014).
In combined meta-analyses of pan-African cohorts from five different countries across Africa totaling 14,126 individuals, we present results from trait-association discovery efforts. Our identification of several novel susceptibility loci across a range of complex traits argues for scaling efforts in the region. The continental and population-specificity of a large proportion of these association signals suggests that inclusion of diverse populations across Africa in GWAS may have the greatest potential for discovery and refinement of novel loci. Collectively, these findings provide the first empirical evidence to support theoretical models that suggest that power for discovery increases in meta-analyses of ethnically diverse populations, specifically driven by increased detection of low-frequency and population-specific novel associations (Pulit et al., 2010).
Given high genetic diversity, and regionally specific patterns of admixture, we highlight the need to design GWAS studies to leverage these differences in allele frequency spectrum and LD patterns across the African cohorts, including the creation of more diverse African whole genomic resources. The differences in LD structure observed around peak association signals across African populations will facilitate the refinement of association signals and help identify causal variants. With caveats for rare variant discovery in some scenarios, our analyses emphasize the value of utilizing diverse populations across the region—to maximize opportunities for genomic discovery (Cook and Morris, 2016) and replication, particularly in the context of rare and population-specific associations. Furthermore, understanding differences in heritability, and identifying the full spectrum of genetic variation associated with complex traits and diseases across Africa, will require much larger-scale prospective studies that should include rich genomic and phenotypic data for complex traits and diseases, as well as information on environmental factors. In these contexts, our results provide a framework for undertaking more extensive GWAS in populations from Africa. Our findings also emphasize the need to develop methods to understand and compare heritability acrosspopulations. Recently, methods have been developed to assess heritabilities from summary statistics from GWAS, accounting for LD structure (Finucane et al., 2015); however, these methods will need to be extended to studies of diverse admixed populations with significant tracts of admixture LD and within populations with high levels of relatedness.
Because genetic diversity is greatest in African populations, including a substantial proportion of genetic variation that is continentally and regionally distinct, it will be critical to understand the functional and biological relevance of this diversity. Understanding the biological basis for population-specific association signals, as well as the impact and transferability of putatively functional and disease-causing mutations at the individual and population level, will require representative genomic resources. We emphasize the need for the parallel development of transcriptomic and cellular biological resources at the population level to better reflect global human diversity (Chang et al., 2015).
STAR☆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and information should be directed to and will be fulfilled by the Lead Contact, Dr. Manjinder Sandhu (mss31@cam.ac.uk).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The Uganda Genome Resource (UGR)
We genotyped 5,000 and sequenced 2,000 samples from 9 ethno-linguistic groups from the General Population Cohort (GPC), Uganda (Table S1.1) (Asiki et al., 2013); these constitute the Uganda Genome Resource (UGR). The GPC is a population-based open cohort study established in 1989 by the Medical Research Council (MRC), UK in collaboration with the Uganda Virus Research Institute (UVRI) to examine trends in prevalence and incidence of HIV infection and their determinants. Samples were collected from individuals during a survey from the study area located in south-western Uganda in the Kyamulibwa sub-county of the Kalungu district, approximately 120 km from Entebbe town. The study area is divided into villages defined by administrative boundaries varying in size from 300 to 1,500 residents, and includes several families living within households. Data on health and lifestyle are collected using a standard individual questionnaire, blood samples obtained and biophysical measurements taken, when necessary, as described previously (Asiki et al., 2013).
We chose exactly 5,000 individuals with relatively complete phenotypic data (described in Method Details) for genotyping (UGWAS) and 2,000 individuals who underwent low coverage whole-genome sequencing (UG2G). These included several pedigrees, and individuals with cryptic relatedness, as well as individuals clustered by household and village. Due to extensive migration into and around the region, several ethno-linguistic groups were sampled (Table S1.1). The final quality controlled Uganda Genome Resource included genotype data on 4,778 and sequence data on 1,978 individuals (Table S1.1). We note that there are 343 individuals who have been genotyped and sequenced; for these individuals, we only included the sequence data, and not the genotype data. We also excluded 6 genotyped samples that were found to be potentially contaminated on fineSTRUCTURE analysis. The final dataset, therefore, included 6,407 individuals (4,429 with genotype, and 1,978 with sequence data).
For genome-wide association analyses, we meta-analyzed association statistics from the Uganda Genome Resource, with three additional cohorts: the Durban Diabetes Study (DDS) (n = 1,165), the Diabetes Case control study (n = 1,542), and the AADM study (n = 5,231). Details regarding studies are below.
The Durban Diabetes Study (DDS)
The Durban Diabetes Study (DDS) is a population-based cross-sectional study of individuals aged > 18 years, who were not pregnant, and residing in urban black African communities in Durban (eThekwini municipality) in KwaZulu-Natal (South Africa), conducted between November 2013 and December 2014 (n = 1,204) (Hird et al., 2016). The survey (n = 1,165) combines health, lifestyle and socioeconomic questionnaire data with standardized biophysical measurements, biomarkers for non-communicable and infectious diseases, and genetic data. A detailed description of the survey design and procedures has been previously published (Hird et al., 2016). The DDS was approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference: BF030/12) and the UK National Research Ethics Service (reference: 14/WM/1061).
The Durban Case Control Study (DCC)
The Diabetes Case Control study is a study of individuals with diabetes recruited from a tertiary hospital in Durban (n = 1,542). The Diabetes Case Control (DCC) study was planned as a case control study of type 2 diabetes to examine the epidemiology and genomics of type 2 diabetes and related cardiometabolic traits in a South African population. Collection started in 2009 and finished in 2013, however at the end of the study only cases (n = 1,600) had been recruited. The study includes participants of Zulu descent, resident in KwaZulu-Natal, aged > 40 years and with a diagnosis of T2D (WHO criteria). The DCC was approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference: BF078/08) and the UK National Research Ethics Service (reference: 11/H0305/6).
The Africa America Diabetes Mellitus Study (AADM)
AADM is an ongoing genetic epidemiology study of type 2 diabetes and related traits in Africans which has been described in detail elsewhere (Rotimi et al., 2001, 2004; Adeyemo et al., 2015) (3–5)(100–102)(99–101)(91–93)(95–97)(94–96)(79–81)(80–82). A total number of 5,231 individuals from the Africa America Diabetes Mellitus (AADM) study were included. In brief, ethical approval was obtained from the Institutional Review Boards (IRB) of all participating institutions. Written informed consent was obtained from all participants. Demographic information was collected using standardized questionnaires across the AADM study centers in Nigeria (Ibadan, Lagos, and Enugu), Ghana (Accra and Kumasi), and Kenya (Eldoret). Anthropometric, medical history, and clinical examination parameters were obtained by trained study staff during a clinic visit.
METHOD DETAILS
Laboratory measurements and Phenotype Data
A summary of phenotypic trait information available for the Ugandan resource can be found in Table S1.2, and trait information across all studies can be found in Table S1.3.
Uganda Genome Resource
Detailed information on demographic characteristics, village, household clustering, GPS coordinates, anthropometry was collected. The study comprised three stages: collection of questionnaire data, biophysical measurements, and collection and analysis of venous blood samples.
Prior to data collection, staff were trained using standard operating procedure documents to standardize data collection. The survey questionnaire retained aspects of the previous GPC questionnaire on sexual behavior, marital status, pregnancy, childbirth, education, and occupation. In addition, a non-communicable disease component, based on the WHO STEPs questionnaire, was included (World Health Organization, 2010). The non-communicable disease component of the questionnaire included sections on tobacco use, alcohol consumption, diet, physical activity, and history of non-communicable disease.
The questionnaires were available to interviewers in English and the local language (Luganda). The Luganda versions of the questionnaires were back-translated by a team of bilingual staff and piloted to ensure that original meanings of questions and answers were maintained. The e-questionnaires were validated against paper versions of the questionnaires for 300 participants.
Biophysical measurements
Once the questionnaire was completed, height, weight, hip and waist circumferences, and blood pressure were measured. Pregnant women in their second or third trimester were excluded from anthropometric measurements.
Height
Height was measured, with the head placed in the Frankfort plane, to the nearest 0.1 cm using the Leicester stadiometer. Head pieces and shoes were removed for height measurements. Calibration of the stadiometer was checked weekly.
Weight
Weight was measured to the nearest 1 kg using the Seca 761 class III mechanical flat scales. Shoes and excess clothing were removed before weight measurements. Calibration of the scales was checked weekly.
Hip and waist circumferences
Waist and hip circumferences were measured to the nearest 0.1 cm over one layer of loose clothing using the non-stretch Seca 201 Ergonomic Circumference Measuring Tape. Waist circumference was measured at the mid-point between the lower costal margin and the level of the anterior superior iliac crests. Hip circumference was measured at the greater trochanter of the femur. Waist and hip circumferences were measured twice. In the case where the first and second measurements disagreed by 3 cm or more, a third measurement was taken. A participant’s hip and waist circumference values were calculated as the mean values of measurements taken.
Blood pressure
Blood pressure was measured using the fully automated Omron M6-I. The Omron M6-I has been validated for medical use, including for those who are obese, children, or elderly (Topouchian et al., 2006; Altunkan et al., 2007, 2008). Participants had been resting for at least 15 minutes prior to the measurement and were asked to refrain from eating and drinking for 30 minutes prior to the measurement. Prior to the blood pressure measurements, the arm circumference was determined and the appropriate Omron cut-size used. Blood pressure was measured in the sitting position three times with resting intervals of 3–5 minutes. Blood pressure for a participant was calculated as the mean of the second and third reading.
Blood samples
Once biophysical measurements had been performed, venous blood samples were obtained. An 8.5 mL serum sample was collected in a vacutainer serum separation tube for serological and biochemical analysis. A 6 mL whole blood sample was collected in an EDTA tube for blood counts, HbA1c measurement and genetic analysis.
The 8.5 mL serum and 6 mL whole blood samples were kept at 4°C – 8°C, and protected from sunlight to prevent degradation of bilirubin. The 2 mL whole blood samples for full blood count were kept at ambient temperature. Vacutainer serum separation tubes were centrifuged for 10 minutes at 1,000–13,000 RCF (g) in a swing bucket centrifuge in the field station laboratory. Samples were centrifuged no earlier than 45 minutes and no later than 2 hours after blood sample collection.
Haematological analysis of full blood count took place in the Kyamulibwa field station laboratories, and other samples were transported to MRC/UVRI Central Laboratories in Entebbe, Uganda, every day for immediate biochemical analysis.
Biochemistry
Biochemistry data on lipid levels and liver function were captured digitally using the Cobas Integra 400 Plus Chemistry analyzer (Roche Diagnostics), an advanced integrated system for research and diagnostic clinical chemistry testing. The instrument carries out all test orders automatically and employs four different technologies, namely, absorption photometry, fluorescence polarization immunoassay, immune-turbidimetry, and potentiometry.
Lipids were measured non-fasting. The lipids of interest were cholesterol, high-density lipoprotein (HDL)-cholesterol, low-density lipoprotein (LDL)-cholesterol and triglycerides. The liver function tests comprise of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), gamma glutamyltransferase (GGT), total bilirubin, and albumin. HDL-cholesterol and LDL-cholesterol were measured using the homogeneous enzymatic colorimetric assays, as described by Sugiuchi et al. (1995, 1998). ALT and AST were measured by kinetic assay with photometric detection method according to the International Federation of Clinical Chemistry (IFCC), but without pyridoxal-5′-phosphate (Bergmeyer et al., 1986a, 1986b; ECCLS, 1989a, 1989b). Assays for bilirubin, albumin, and ALP were colorimetric assays with photometric detection. Assays for GGT, cholesterol, and triglycerides were enzymatic colorimetric assays with photometric detection.
The precision of assays was also tested by the manufacturer using both study samples and controls.
HbA1c
HbA1c data were captured digitally using the Roche Cobas Integra 400 Plus Chemistry analyzer (Roche Diagnostics). Total haemoglobin and HbA1c concentrations were determined after haemolysis of the anticoagulated whole blood specimen. Total haemoglobin was measured colorimetrically. HbA1c was determined by turbidimetric inhibition immunoassay-quant Haemoglobin Alc Gen2 for haemolysed whole blood. The Cobas result output was expressed as IFCC percent. HbA1c and was calculated from the IFCC protocol HbA1c/haemoglobin ratio as HbA1c (%) = (HbA1c/haemoglobin) × 100. These results were converted to the DCCT/NGSP percentage units using the following equation: HbA1c % DCCT/NGSP = 0.915 × (HbA1c % IFCC) + 2.15.
This Roche second generation HbA1c assay has been validated for accuracy in the presence of haemoglobinopathies HbS, HbC, and also HbE or HbD (Abadie and Koelsch, 2008; Fleming, 2007; Little et al., 2008). The Cobas Integra 400 Plus assay has also been validated against the high-performance liquid chromatography method (Barrot et al., 2012).
Full blood count
Full blood count and other hematological traits were measured using the Coulter ACT5 Diff CP hematology analyzer. The following information was output: white cell count, red cell count, haemoglobin (Hb), packed cell volume (PCV), mean corpuscular volume (MCV), mean cell haemoglobin (MCH), mean cell haemoglobin concentration (MCHC), mean platelet volume (MPV) and platelet count.
Durban Diabetes Study and Durban Case Control Study
Automated enzymatic assays were used on fasting serum to determine TC, high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), triglyceride, aspartate, amino transferase, alanine amino transferase, alkaline phosphatase, gamma glutamyl-transferase, bilirubin and albumin levels (ABBOTT ARCHITECT 2: CI 8200, Abbott Laboratories, Chicago, IL, USA.).
HbA1c was measured using ion-exchange high-performance liquid chromatography (HPLC) (BIORAD VARIANT II TURBO 2.0, Bio-Rad Laboratories, Inc., Hercules, CA, USA), using an instrument certified by the National Glycohaemoglobin Standardization Program (NGSP) and International Federation of Clinical Chemistry and Laboratory Medicine (IFCC). The BIORAD VARIANT II TURBO 2.0 method is not significantly affected by HbS, HbC, HbE and HbD-trait haemoglobin variants.
For DDS only, a full blood count was completed for all participants using a SYSMEX XT-2000i, including determination of haemoglobin level, mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH).
Africa America Diabetes Mellitus Study
Weight was measured in light clothes on an electronic scale to the nearest 0.1 kg and height was measured with a stadiometer to the nearest 0.1 cm. Body mass index (BMI) was computed as weight (kg) divided by the square of height in meters (m2). Blood pressure was measured while seated using an oscillometric device (Omron Healthcare, Inc., Bannockburn, Illinois). Three readings were taken at 10-minute intervals. Reported readings were the averages of the second and third readings. Blood samples were drawn after an overnight fast of at least 8 hours.
DNA Extraction, Genotyping and Sequencing
Whole blood for DNA extraction was collected in EDTA vacutainer tubes, transferred to cryogenic tubes and stored at 80°C for up to one year. To minimize human error, tubes were barcoded and most of the processing was done using automation. DNA was extracted from 5 to 6 mL of whole blood using NUCLEON® chemistry (Gen-Probe Life Sciences Ltd., now Hologic). A control blood sample was included per operator per day. DNA was resuspended in Standard TE Buffer (10mM EDTA, 1mM EDTA) in a volume of 200–1,000mL depending on the size of the DNA pellet. DNA samples underwent quality control checks including PicoGreen® quantitation (Life Technologies, Thermo Fisher Scientific Inc.), agarose gel electrophoresis and iPLEX genotyping (Sequenom Inc.) of a panel of 30 SNPs including 4 gender markers.
We genotyped 5,000 samples from the Ugandan Survey on the Illumina HumanOmni 2.5M BeadChip array at the Wellcome Trust Sanger Institute (WTSI). These were chosen as a subset of the survey population with the most complete phenotype data on the traits measured. Sequenom QC and gender checks were carried out prior to genotyping. A further 2000 samples were sequenced on the Illumina HiSeq 2000 with 75bp paired end reads, at low coverage, with an average coverage of 4x for each sample.
The DDS (n = 1,165) and DCC (n = 1,542) DNA samples were genotyped on the consortium-driven Illumina HumanOmni Multi-Ethnic GWAS/Exome Array (MEGA pre-commercial v1) using the Infinium Assay. The MEGA array (1.7M SNPs) leverages content discovered in Sequencing Consortia and databases such as the 1000 Genomes Project, the CAAPA Consortium, PAGE, OMIM/Clinvar and NEXTBio. Genotypes were called using the Illumina GenCall algorithm.
Samples from the AADM study were genotyped on high density GWAS arrays: 1,808 samples were genotyped using the Affymetrix® Axiom® Genome-Wide PanAFR Array Set and 3,423 samples were genotyped using the Illumina MEGA array.
Quality Control of Genotype Data
Uganda Genome Resource
A total of 2,314,174 autosomal and 55,208 X chromosome markers were genotyped on the HumanOmni2.5–8 chip. Of these, 39,368 autosomal markers were excluded because they did not pass the quality thresholds for the SNP called proportion (< 97%, 25,037 SNPs) and Hardy Weinberg Equilibrium (HWE) (p < 10−8, 14,331 SNPs). HWE testing was only carried out on the founders for autosomes (defined by an IBD threshold < 0.10 as estimated by PLINK), and female unrelated individuals for the X chromosome (Purcell et al., 2007). Owing to the sampling strategy, there were high levels of cryptic relatedness within the cohort, which have been described previously (Asiki et al., 2013; O’Connell et al., 2014). The average IBD sharing between individuals was 0.0015 with 0.07% of pairs and 5,307 individuals with an IBD > 0.125 with at least one other individual.
A total of 91 samples were dropped during sample QC as they did not pass the quality thresholds for proportion of samples called (> 97%) or heterozygosity (outliers: mean ± 3SD), or the gender inferred from the X chromosome data did not match the supplied gender. Three additional samples were dropped because of high relatedness (i.e., IBD > 0.90). Principal component analysis was carried out on unrelated individuals projecting onto related individuals, for SNPs LD pruned at an r2 threshold of 0.2, with a MAF threshold of > 5%. No samples were identified as population/ancestry outliers based on this. Downstream analyses were carried out on the remaining 2,230,258 autosomal markers and 4,778 samples which passed quality checks. Phasing and imputation, and further filtering of these data for GWAS are described in the section ‘Quantification and Statistical Analysis’.
Durban Diabetes Study (DDS) and Diabetes Case-Control Study (DCC)
Quality control for DDS and DCC genotype data was carried out collectively, with sample QC including filtering for called proportion (< 97%), heterozygosity (> 4SD from mean), sex check fails (F statistic < 0.8 for men, and > 0.2 for women). Sample QC was followed by SNP QC, including filtering for called proportion (< 97%), Hardy Weinberg disequilibrium (p < 1e-06), relatedness (IBD > 0.90). SNPs with stasticially significant difference in missingness (p < 1e06), between the DCC and DDS datasets were also removed from analysis. In total, 1478+1119 samples and 1,395,345 SNPs were retained in the two studies.
The Africa America Diabetes Study (AADM)
For the AADM data, filtering for the Affymetrix and Illumina data were carried out separately. Quality control included appropriate sample- and SNP-level exclusion filtering (individual call rate ≤ 95%, SNP call rates ≤ 95%, Hardy – Weinberg < 10−6, and minor allele frequency (MAF) < 0.01).
Curation of Sequence Data
Following genotyping in 5,000 individuals, we carried out whole-genome sequencing in the General Population Cohort for 2,000 individuals with phenotype data available to provide a resource to better understand genetic diversity in the region, and for better imputation into the remaining individuals, to maximize power for discovery in GWAS. Of these, 343 were overlapping with individuals who had already been genotyped. These samples were sequenced and genotyped for comparison, and assessment of systematic differences between genotype and sequence data.
Read mapping and bam processing
Following generation of raw reads on the sequencing machine, the reads were converted to BAM format using Illumina2BAM. Illumina2BAM was again used to de-multiplex the lanes that had been sequenced so that the tags were isolated from the body of the read, decoded, and could be used to separate out each lane into lanelets containing individual samples from the multiplex library and the PhiX control. Reads corresponding to the PhiX control were mapped and used with Sanger’s spatial filter program to identify reads from other lanelets that contained spatially oriented INDEL artifacts and mark them as QC fail. Mapping of the human samples was carried out using the BWA backtrack algorithm with the GRCh37 1000 Genomes phase II reference (also known as hs37d5). PCR and optically duplicated reads were marked using Picard MarkDuplicates and after manual QC passing data was deposited with the EGA and the Sanger Institute’s internal archive (study/dataset accession numbers EGAS00001001558/EGAD00010000965 and EGAS00001000545/EGAD00001001639, respectively).
One sample from the Genome in a Bottle highly curated set was included for validation of the data processing pipeline (NA12878). PCR-free reads were used for these validation samples, to avoid PCR artifacts. This validation sample was downsampled to 4x coverage, and processed through the same pipeline (see Figure S1), to provide a comparator against high coverage 30x data. This was considered the gold standard for evaluation. The accuracy of called data from a 4x sample would provide a guide to the accuracy of the workflow applied.
Quality control of sequence data
In order to ensure the quality of the large quantity of BAMs produced for the project, an automatic quality control system was employed to reduce the number of data files that required manual intervention. This system was derived from the one originally designed for the UK10K project (Walter et al., 2015) (https://www.uk10k.org) and used a series of empirically derived thresholds to assess summary metrics calculated from the input BAMs. These thresholds included: percentage of reads mapped; percentage of duplicate reads marked; various statistics measuring INDEL distribution against read cycle and an insert size overlap percentage. Any lane that fell below the “fail” threshold for any of the metrics were excluded; any lane that fell below the “warn” threshold on a metric would be manually examined; and any lane that did not fall below either of these thresholds for any of the metrics was given a status of “pass” and allowed to proceed into the later stages of the pipeline. Fourteen samples were excluded at the QC stage.
Re-alignment of reads and base quality score recalibration
Passed lanelets were then merged into BAMs corresponding to sample’s libraries and duplicates were marked again with Picard after which they were then merged into BAMs for each sample. We ran verifyBAMID to identify samples that did not match the frequency distributions of corresponding genotype data, and excluded eight samples as failures. Finally, sample level bam improvement was carried out using GATK and samtools. This consisted of re-alignment of reads around known and discovered INDELs followed by base quality score recalibration (BQSR) both using the GATK. Lastly samtools calmd was applied and indices were created. Known INDELs for realignment were taken from Mills 1000 Genomes indels set and the 1000G phase low coverage set both part of the Broad’s GATK resource bundle version 2.2. Known variants for BQSR were taken from dbSNP 137 also part of the Broad’s resource bundle.
Assessment of calling algorithms
We carried out careful assessment of several calling algorithms before calling our low coverage sequences. In order to explore the sensitivity and specificity of variant callers when applied to low coverage datasets, we carried out an evaluation with 1,986 samples from Uganda sequenced at 4x average coverage with Illumina HiSeq 2000. The downsampled GIAB sample (Zook et al., 2014) (4x) was included in the called set for evaluation of calling accuracy. We calculated the sensitivity and specificity of calls relative to the highly curated variant sites for the NA12878 sample, to identify the caller with greatest area under ROC curve at different filtering thresholds (Figures S1B and S1C). We note that the accuracy of variant calling in this single European sample may not fully reflect the accuracy calls in the African samples; however, it is likely to give an indicator of the relative performance of calling algorithms. We used varied two different filters to generate ROC curves: the SNP quality metric (QUAL), and the VQSLOD score obtained using the VQSR model implemented by GATK for callers. We compared commonly used callers at the time of calling: Unified Genotyper v3.3, Haplotype caller v3.2, samtools v0.2.0-rc12+htslib-0.2.0-rc12 and FreeBayes v0.9.18–3. As the filtering algorithm recommended for data produced from Unified Genotyper and Haplotype caller is VQSR, we presented ROC curves using different VQSLOD thresholds. However, for comparability with other callers, we also present ROC curves using only QC thresholds. We also carried out additional evaluation, generating annotations on samtools based calls using GATK, followed by VQSR to assess using VQSLOD for filtering combined with samtools calling improved calls. With this evaluation, we show that UnifiedGenotyper3.3 produced the best area under ROC curve with the lowest FDR for a given sensitivity for SNPs and indels (Figures S1B and S1C); we therefore used this for variant calling for these data. All callers, however produce very low sensitivity and high FDRs for indel calls (Figure S1C), indicating the need for more stringent filtering downstream. It is likely that the sensitivity and specificity of calls will have improve with genotype refinement. We note that we only assessed these callers using filtering options available at the time, and use of different filtering approaches using these callers could potentially improve the sensitivity for a given FDR.
Data processing workflow
The workflow for data processing is represented in Figure S1A. Variant calling was carried out on the samples that passed QC in the UG2G data along with the sequence data from 320 individuals from the African Genome Variation Project (AGVP) (Gurdasani et al., 2015). Sequence data from the AGVP have been described in detail previously (Gurdasani et al., 2015). These combined data were called together with GATK Unified Genotyper 3.3. During variant calling each sample was by default downsampled to a maximum coverage of 250 (–downsampling_type BY_SAMPLE–downsample_to_coverage 250). Reads with an inferior mapping quality were ignored (–min_mapping_quality_score 20). Duplicate reads were filtered (-rf DuplicateReadFilter). Reads whose mate mapped to a different contig were filtered out (-rf BadMateFilter). Bases with an inferior quality were not considered for calling (–min_base_quality_score 10). No more than 6 alternate alleles were emitted at each site (–max_alternate_alleles 6). In an attempt to provide better input for the subsequent variant filtering step, variants of inferior quality were not called (–stand_call_conf 10 and–stand_emit_conf 10). We carried out variant calling for the autosomes. The X chromosome was called as diploid within PAR1 and PAR2 and also outside the PARs.
Filtering of variants was carried out with GATK VariantRecalibrator 3.2 using variant quality score recalibration (VQSR). To train the Gaussian mixture model and calculate a truth score (log odds ratio) for each variant we used HapMap III and 1000G phase 1 Omni2.5 sites as truth and training sets (prior probabilities of 15 and 12) for SNPs. High confidence 1000G phase 1 SNPs were used as an additional training set (prior 10) for SNPs. For indels we used the Mills 1000 Genomes gold standard as a truth and training set (prior 12). For both SNPs and indels dbSNP138 acted as a set of known sites.
To build our VQSR Gaussian mixture model we used annotations at each site related to coverage (QD = QualByDepth and DP), strand bias (FS = FisherStrand, SOR = StrandOddsRatio) and mapping quality (MQ, MQRankSum, ReadPosRankSum). For indels we use the same annotations, except for MQ being left out, as per GATK Best Practice recommendations at the time. DP is the approximate read depth after filtering reads with poor mapping quality and bad mates. QD is the variant confidence normalized by the unfiltered depth for the variant allele. FS is a Phred-scaled p value using Fisher’s exact test to detect strand bias. SOR is the odds ratio of a 2×2 contingency table (rows and columns are positive/negative strand and reference/alternate allele) to detect strand bias. MQ is the RMS of the mapping qualities, which serves an average across reads and samples. MQRankSum is the Z-score from a Wilcoxon rank sum test of alternate versus reference mapping qualities. ReadPosRankSum is the Z-score from a Wilcoxon rank sum test of alternate versus reference read position biases. We did not use the InbreedingCoeff annotation, which is a likelihood-based Hardy-Weinberg test for the inbreeding among samples, because of the possible deviation from Hardy-Weinberg equilibrium among the cohort due to substructure between ethno-linguistic groups and cryptic relatedness among individuals.
We chose a truth sensitivity threshold based on the ROC curve (Figure S1D). We applied truth sensitivity thresholds of 99.5% and 99.0% to SNPs and indels, respectively. After variant filtering we called 45,309,067 SNPs and 5,483,098 indels, respectively, across the UG2G and AGVP combined datasets. The Ti/Tv ratio was noted to be 2.2 and 1.9 for known and novel SNP variants with respect to dbSNP138, suggesting a high quality of calls.
Following variant filtering, genotype refinement was carried out with Beagle v4.r1274 across all individuals. To evaluate the accuracy of variant calling we calculated the non-reference concordance for chromosome 20. Concordance was calculated by comparison to the GIAB/NIST reference/baseline calls for sample NA12878 (PMID 27578503). Prior to calculating the concordance the indels were left aligned and trimmed with bcftools norm. (http://samtools.github.io/bcftools/). Concordance for SNPs and indels was noted to be 92% and 82%, respectively for SNPs, and indels for the GIAB sample. The script used to calculate the concordance is available from https://github.com/teamSandhu/tc9/blob/master/projects/uganda_gwas/concordance.sh.
We carried out further quality control for analysis, for which additional samples were excluded as heterozygosity outliers (heterozygosity > = 3 SD from mean). Following quality control, 1,978 samples with WGS were included for analysis of sequence data.
Generation of Merged Reference Panel
To generate the reference panel for imputation, we phased combined data from the Uganda Genome Resource and 320 sequences from the African Genome Variation Project (Gurdasani et al., 2015). The merged reference panel was refined with Beagle4 and then phased with SHAPEIT2 release 790 using used an–effective-size of 17,469 as per the recommendations. These haplotypes were then merged with the 1000G phase III database using the -merge_ref_panels_output_ref option with IMPUTE2. The final reference panel included 4,802 individuals and 98,608,172 variants. Following this, we extracted unrelated individuals from the reference panel. For this, we generated an IBD matrix from merged data using an intersection of sites for sequence and genotyped data, and iteratively removed individuals, so that all individuals in the reference panel were related with an IBD < 0.10.
We used a subset of this reference panel for imputation into Ugandan and AGVP genotype sets to compare the accuracy of imputation using different panels. For this, we further extracted 3,895 individuals unrelated to individuals in UGWAS and to other individuals in UG2G, to avoid bias in imputation accuracy due to inclusion of related individuals, and used this panel for imputation.
Phasing, Imputation and filtering
Phasing and Imputation
Phasing and imputation into each study was carried out separately, except for DDS and DCC, which were combined for phasing and imputation, given the homogeneity of ancestry among these two studies. Following imputation, these studies were separated out for meta-analysis, as one group consisted primarily of a diabetic population, whereas DDS was a general population study.
Imputation into the genotype data in UGR, DDS, DCC and AADM was carried out using the merged reference generated by merging whole genome sequence data from the African Genome Variation Project (n = 320), the UG2G sequence resource (n = 2,000), and the 1000 Genomes phase 3 project (n = 2504), as outlined previously.
Imputation for AADM was performed using the African Genome Resources Haplotype Reference Panel (Loh et al., 2016) available from the Sanger Imputation Service (https://imputation.sanger.ac.uk/); this is a more recent version of the panel described above, with the panel being curated by recalling genotype likelihoods across all samples, including from the 1000 Genomes Project Phase 3. The imputation reference panel comprised 4,956 individuals, including all 2,504 from the 1000 Genomes Project Phase 3, ~2,000 individuals from Uganda (Baganda, Banyarwanda, Barundi, and others), and 100 individuals from each of a set of populations from Ethiopia (Gumuz, Wolayta, Amhara, Oromo, and Somali), Egypt, Namibia (Nama/Khoe-San) and South Africa (Zulu), yielding 9,912 haplotypes for 93,421,145 SNPs.
For DDS, DCC and Uganda, phasing was carried out with SHAPEIT2 (O’Connell et al., 2014) using default parameters, followed by imputation with IMPUTE2 (Howie et al., 2012). Pre-phasing for AADM was performed with EAGLE version 2.0.5 (Loh et al., 2016) and imputation was performed using PBWT (Durbin, 2014).
Filtering of Imputed data
Uganda Genome Resource.
We used the info threshold output by IMPUTE2 to identify high quality variants. The info metric produced by IMPUTE2 is a measure of certainty of imputation. This typically takes values between 0 and 1. A value of 1 indicates that there is no uncertainty in the imputed genotypes whereas a value of 0 means that there is complete uncertainty about the genotypes. All of these measures can be interpreted in the context of effective sample size: an information measure of a on a sample of N individuals indicates that the amount of data at the imputed SNP is approximately equivalent to a set of perfectly observed genotype data in a sample size of axN. For SNPs overlapping between imputed and directly genotyped data (type 2 SNPs), we also used an r2 threshold for quality control. Here, r2 represents the squared correlation between the input genotypes and the best-guess imputed genotypes calculated, where the input genotypes at that SNP have been masked internally and then imputed as if the SNP were present in the reference set but not in the directly genotyped target sample (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html).
Following imputation, into the genotype data, we carried out sensitivity analyses using different filtering thresholds for the imputed data to ascertain whether different info thresholds were associated with varying degrees of inflation in quantile-quantile (QQ) plots. QQ plots were generated from GEMMA mixed model analysis and compared to see if inflation was related to the threshold. applied However, no relationship was seen between inflation factors and stringency for thresholds used in filtering of imputed data (data not shown), suggesting that the vast majority of data were high quality and a threshold of 0.3 was therefore considered adequate, and consistent with previous GWAS.
We therefore opted to use an info score threshold of 0.3 for quality control. Furthermore, we also applied a threshold for type 2 SNPs (genotyped SNPs that are also in the imputation panel, and are also imputed, allowing an examination of correlation between genotyped and imputed data), requiring a minimum r2 of 0.60.
DDS and DCC
Consistent with filtering of the UGR dataset, we used an imputation info threshold of 0.3 for filtering (this was used across both cohorts, as imputation had been carried out across these cohorts) and a type 2 SNP correlation threshold of 0.6. GWAS analysis was carried out for each cohort separately, and a minimum MAF threshold of 0.5% was applied to each cohort for analysis. A total of 24,419,014 and 24,423,923 variants were analyzed for DCC, and DDS respectively.
AADM
The AADM dataset is a combination of multiple ethno-linguistic groups, where imputation has been carried out separately. We did not have access to individual genotype data, or cohorts for analysis, but had results for summary statistics for analysis across all cohorts. We therefore applied an info threshold filter of 0.3 consistent with the other datasets. As these data include multiple cohorts that have been imputed separately, a minimum threshold of 0.3 across all cohorts was applied for each variant. Data regarding correlation for type 2 SNPs were not available for this cohort; therefore, this filter could not be applied. GWAS analysis was carried out only for SNPs with a MAF threshold above 0.5%, consistent with other cohorts. A total of 19,580,546 variants were included in final analysis.
Merging UGR Sequence and Genotyping Data
As the Uganda Genome Resource included genotyped and sequenced indviduals, we merged imputed genotype and sequence data to create a single pooled dataset for analysis. We created a pooled dataset for analysis, rather than meta-analyzing separately, as cryptic relatedness and family structure existed across the genotyped and sequence data, which would make data correlated, and not independent. As such, mixed model analysis, explicitly modeling this relatedness would be likely to provide more accurate results.
Following a merger of imputed genotype and sequence data, we assessed and removed any systematic differences between imputed genotype data and sequence data. We did this by carrying out principal component analysis on these data to examine whether there was separation by data mode (imputed genotype data and sequenced data) among 343 individuals who had been genotyped and sequenced in duplicate. We noted clear separation of data points of genotype imputed and sequence data on PCA. We evaluated different thresholds of concordance between sequence and imputed genotype data for these 343 samples, filtering out SNPs that showed a concordance < 0.80 and < 0.90. We found that a minimum concordance threshold of 0.90 was required to abolish systematic effects observed between genotype array and sequence data on PCA. Following exclusion of 904,283 variants (2.3% of all variants) that showed < 90% concordance in genotypes between the sequence and imputed genotype data (for 343 samples that had been genotyped and sequence), PCAs did not show any systematic differences between imputed genotype and sequence data. We inspected the first ten PCs to ensure that systematic differences did not represent an important axis of variation in the genetic data. Following filtering, a total of 39,312,112 autosomal markers in the joint set of 6,407 samples were taken forward for analyses. For GWAS association analyses, we only included a subset of variants (n = 20,594,556) that met an MAF threshold of at least 0.5%.
Curation and Transformation of Phenotype data
Transformation of traits was carried out uniformly for each cohort to make effect sizes comparable across cohorts, allowing meta-analyses of summary results. A list of phenotypes used for analysis can be found in Tables S1.2 and S1.3. For association analysis, inverse normal transformation was carried out on residuals after regressing on age, age2 and sex. For HbA1c alone, regression was carried out on age, age2, sex & month of sample collection as an indicator variable, to allow for seasonal trends in HbA1C that have been described previously (Tseng et al., 2005).
Phenotypic trait information available across cohorts were variable, as shown in Table S1.3. For each trait, all cohorts with relevant data on phenotype were included in the meta-analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of population structure and admixture
Principal Component Analysis
To examine population structure in the Ugandan ethno-linguistic groups, we carried out principal component analyses (PCA) among 4,778 genotyped individuals (UGWAS). PCA was carried out in the Ugandan dataset among unrelated individuals, projecting onto others, as well as for unrelated individuals in a global context, including individuals from the 1000 Genomes Project,(Abecasis et al., 2012) the African Genome Variation Project (Gurdasani et al., 2015) and the Human Origins dataset (Table S1.4) (Patterson et al., 2012). For these analyses, only markers with a minor allele frequency (MAF) of above 1% were included, and LD pruning was carried out to an r2 threshold of 0.2 using PLINK (Purcell et al., 2007). A summary of these datasets is provided in Table S1.4.
FineSTUCTURE Analysis
In order to take advantage of the linkage disequilibrium structure in the cohort, and the dense genotyping on the Illumina Omni 2.5M array, we carried out fineSTRUCTURE analysis (Lawson et al., 2012), which provides detailed information about structure among populations without loss of information due to LD pruning by identifying shared haplotypes among individual along the chromosome (with ChromoPainter), and estimating a co-ancestry matrix. This has previously been shown to produce more detailed information about population structure (Lawson et al., 2012). Only unrelated individuals were included in these analyses as relatedness itself can contribute to structure. A total of 1,899 unrelated individuals were identified by serially removing related individuals, until all individuals had a pairwise IBD of less than 0.10. Haplotype information was extracted for these individuals from phased data from the entire cohort, as phasing was likely to be more accurate due to relatedness within the cohort. Recombination files were generated from the Hapmap build 37 recombination map available for each chromosome.
To input parameters into fineSTRUCTURE, for the analysis of unrelated individuals from UGWAS, we first estimated the mutation rate and effective population size, with a subset of 10 individuals (one in every 200 individuals sampled) across all chromosomes. These parameters were then input into ChromoPainter, and a co-ancestry matrix was calculated for all individuals, with haplotypes of each individual sequentially considered as recipients, and haplotypes of all other individuals in the cohort considered donors (using the -a option). Hence, we were able to estimate the average number of chromosome chunks and chunk lengths that could be considered as donated to each individual from every other individual. Apart from quality filtering, no other filtering (LD pruning/MAF thresholds) was carried out, with a view to maximizing information within the co-ancestry matrix. The co-ancestry matrix was used to generate trees of ethno-linguistic groups based on sharing of ancestry. Furthermore, we generated principal components from the co-ancestry matrix to study the relationships between these ethno-linguistic groups.
First pass analyses showed several outliers belonging to the Baganda and Barundi ethno-linguistic groups on all principal components obtained (data not shown). On closer examination, these samples were noted to have much higher co-ancestry sharing with another sample in the cohort, and high heterozygosity, suggesting these were pairs of samples, with one contaminating another. We excluded 6 samples, and reran fineSTRUCTURE analysis, as this element of contamination was predominating many principal components. FineSTRUCTURE inferred PCs in our second pass analysis did not show the clines observed due to the few contaminated samples, so this was considered as our primary analysis.
In addition to PCA analysis, we also inferred tree structure among the populations using the co-ancestry matrix generated by fineSTRUCTURE. We also ran unsupervised fineSTRUCTURE analysis without ethno-linguistic clusters, to infer population clusters from within the data, and assess the correlation of these inferred clusters with ethno-linguistic group, historical geographical structure, admixture, and current geographical coordinates (using GPS coordinates).
Correlation of genetic structure with geographical structure
Next, we compared principal components to current GPS coordinates to identify if there was genetic correlation with current spatial structure in the cohort. We carried out Procrustes analysis on combinations of principal components and rotated this matrix and re-scaled to best fit with the transformed GPS coordinates for individuals.
In order to assess correlation between principal components and historical geographical structure prior to migration, we carried out Procrustes analysis using geographical coordinates based on the average coordinate of the center of the region individuals are likely to have migrated from, as identified by their ethno-linguistic group, based on the map in Figure 1C. We considered that the migrant populations Basoga, Bakiga, Banyarwanda, Baganda, Barundi, Banyankole, Bafumbira migrated from the historical Soga, Kiga, Rwanda, Buganda, Urundi, Nkole, and Kisoro districts, respectively (Figure 1C) (Richards, 1954). The latitude and longitude for each ethno-linguistic groups was assigned as the center of each district as on the map (Figure 1C; Table S2.1). The same coordinate was used for all individuals belonging to a given ethno-linguistic group, as historical geographical origins of individuals were not available. Additionally, admixture was not considered in this assignment. We note that admixture and migration among co-located migrant populations are likely to distort and dilute the association between genetic structure and historical geographical origins, producing conservative results.
Analyses of population admixture
We used several approaches to examine admixture among Ugandan populations, including unstructured ADMIXTURE analysis, in the Ugandan and in a global context. Additionally, we also formally confirmed the presence of historical Eurasian and hunter-gatherer admixture among several populations using different approaches, including admixture linkage disequilibrium based approaches (MALDER) (Pickrell et al., 2014; Loh et al., 2013) f3, double conditioned site frequency spectrum analysis (Yang et al., 2012), analysis of Neanderthal ancestry (Sankararaman et al., 2014) and analysis of MT and Y chromosome haplotypes.
ADMIXTURE Analysis
Clustering of genetic data from the Ugandan discovery cohort (UGWAS) was carried out using ADMIXTURE in the context of the global dataset, including data from the Human Origins array (Table S1.4) (Patterson et al., 2012). Analysis was carried out specifiying K = 2 to 20 clusters. ADMIXTURE analyses were repeated 20 times using a seed derived from the time of analysis, and results were combined using the LargeKGreedy algorithm in CLUMPP with 1000 repeats (Jakobsson and Rosenberg, 2007). LD pruning to an r2 of 0.2 was carried out prior to analysis, and known regions of long range LD were removed, as previously described (Price et al., 2008).
F3 Tests
We formally assessed admixture in the Ugandan populations among unrelated individuals from the genotyped dataset (UGWAS) using the f3 test. In order to examine Eurasian ancestry in AGVP populations, we tested a model with admixture between populations related to European/Middle Eastern populations and YRI by using these as reference populations testing the tree (European/Middle eastern population, YRI; X), X being each of the Ugandan ethno-linguistic groups. Here, Eurasian ancestry/gene flow refers to ancient gene flow from an ancestral population that is closely related to populations currently living in Western Europe. However, as it is diffi-cult to identify the precise source of this ancestry, which may be the result of multiple population movements—including through Europe, the Middle-East, or from other parts of Africa, we shall henceforth broadly refer to this as ‘Eurasian gene flow/ancestry’. f3 tests are robust to complex ancestry in the admixing populations, ascertainment bias and the choice of reference populations, as has been described previously (Patterson et al., 2012). The test statistic is negative if X has complex history and admixture from populations related to the reference populations, as this topology that would lead to a negative term in the f3 parameter. We note that results from these tests would be subject to the outgroup case, where the reference population is an outgroup to the true mixing population.
Linkage disequilibrium based tests for admixture
In order to confirm the presence of admixture, and date this, we used admixture-LD based approaches (Pickrell et al., 2014). This approach is based on the relationship between admixture-LD, time since admixture and the difference in allelic frequency between SNPs in mixing populations. It leverages the fact that admixture LD between 2 SNPs weighted by difference in allelic frequency between two mixing populations decays exponentially as a function of time since admixture. The amplitude of the curve allows estimation of admixture proportions. We applied two methods that use similar principles of admixture LD decay for inferring admixture: MALDER (Pickrell et al., 2014; Loh et al., 2013) and GLOBETROTTER (Hellenthal et al., 2014).
Admixture inference with MALDER
We assessed multiple admixture events and identified populations most similar to ancestral mixing populations for Ugandan populations, using methods described previously (Pickrell et al., 2014). For these analyses, we estimated curves from a minimum distance of 0.5cM. We estimated the lower bound and upper bounds of the number of generations since admixture for each event, by assessing the rate of decay of each exponential curve.
In addition to identifying multiple admixture events and most likely source populations using MALDER (Pickrell et al., 2014; Loh et al., 2013), we assessed the probability of each Eurasian and HG-like admixture event by using a process similar to that described by Pickrell et al. (2014) as described previously (Gurdasani et al., 2015). We recapitulate these methods here.
We identified combination of source populations that were associated with the highest amplitude as the most likely representatives of ancestry within the target population (if Z > 3). Where the highest amplitude of admixture LD in a target population was produced by the combination of Eurasian and African reference populations, we compared the highest amplitude with the highest amplitude produced when both reference populations had < 1% Eurasian admixture, as reported previously (Gurdasani et al., 2015). We calculated this as follows:
ZEUR represents the statistical difference between the highest amplitude and the highest amplitude when both populations have < 1% Eurasian ancestry. Similarly, we estimated the probability of HG admixture when the highest amplitude included either a Khoe-San, Hadza or rf-HG (Pygmy) population, as follows:
where the proportion of HG admixture was estimated from ADMIXTURE analysis as the sum of Khoe-San and Pygmy like ancestry.
For some populations, admixture events with the highest amplitude included both a hunter-gatherer (Khoe-San, Mbuti Pygmy or Hadza) and Eurasian population. For these we calculated the separate probability of HG and Eurasian admixture using the Z scores described above. The source of admixture was considered to be the population with a Z score of above 2. If both ZHG and ZEUR were >2, this was considered a dual admixture event where a HG-like population had admixed with a Eurasian-like population. When only one of these events were high probability, and the other one was low probability, this was considered an admixture event with a single source. We also note that MALDER can only indicate the source population most similar to the ancestral mixing population among a set of modern populations provided. However, gene flow may arise from an ancestral population with allele frequencies correlated to the source population inferred by MALDER.
We note that, as described by Pickrell et al. (2014) – Supplementary Material 1.2.3 (https://www.pnas.org/content/pnas/suppl/2014/01/29/1313787111.DCSupplemental/sapp.pdf), MALDER is biased toward identifying source populations that are more drifted, even if they contribute little proportionally to ancestry. This is because the drift parameter predominates over the proportion weight in this instance, favoring the most drifted population as the source population (this will have greatest amplitude in these scenarios).
Admixture inference with GLOBETROTTER
We also carried out GLOBETROTTER (Hellenthal et al., 2014) analysis to assess potential sources and dating of admixture among Ugandan populations. We included all individuals for all nine ethno-linguistic groups, except Baganda, where 200 individuals were randomly subsampled to make this analysis computationally tractable. We also examined admixture within Jola and LWK to assess specificity of events to Ugandan populations, and within East Africa. Consistent with previous applications of GLOBETROTTER (Tambets et al., 2018; Hudjashov et al., 2017), we conducted our analyses in two ways: 1) including all possible source populations within the Human-origins and 1000 Genomes combined dataset, including regional east African populations (‘all population’ analysis; and 2) including only a subset of donor populations representative of certain types of ancestry: YRI representing Bantu ancestry, TSI and CHB representing Eurasian ancestry; Dinka representing east African Nilo-Saharan ancestry, Hadza, and rfHG (Mbuti Pygmy) representing east-African hunter-gatherer ancestry; and Juhoan_North representing Khoe-San ancestry. We refer to this analysis as ‘limited population’ analysis. The purpose of the limited population analysis was to help identify ancestral sources representative of certain types of ancestry, where the least admixed representative populations of these groups were used. While the ‘all population’ analysis is the most informative with regards to admixture events, inferences drawn regarding ancestral components that mixed may be limited due to the admixed nature of donor populations. Hence, we analyzed the data using these two approaches to better understand the ancestral populations representing source populations inferred as contributing to modern Ugandans.
For each of the analyses described above (‘all population’ and ‘limited population’), a first run of fineSTRUCTURE was used to estimate a global mutation rate and effective population size. This was carried out on a subset of the dataset by randomly subsampling 1 in 10 individuals, for efficiency. The global mutation rate and effective population size estimated through ten iterations then input into a second run, to estimate the length of chunks copied from each donor population haplotype. The merged Ugandan, AGVP genotype African data, 1000 Genomes Project and Human origins dataset was used for these analysis (Table S1.4).
For the ‘all population analysis’, a total of 87 populations were considered donors and 12 Ugandan ethno-linguistic groups, LWK and Jola were considered recipients. As mentioned, LWK was included to assess ancestral components and admixture specific to Uganda, and Jola, a West African population was included to assess events specific to East Africa. For computational tractability, we randomly subsampled 25 individuals from all populations (and included all individuals when < 25 individuals were in a population group. All donor populations were also allowed to be recipients in the algorithm, in line with the suggested mode of analysis for GLOBETROTTER. However, Ugandan populations, LWK and Jola were only considered recipients to avoid loss of power, in line with guidance for running GLOBETROTTER to identify admixture.
For the ‘limited population’ analysis, only 7 donor populations were considered, as described above. The output across all chromosomes was combined with ChromoCombine, following which GLOBETROTTER was run to assess admixture among recipient populations, allowing for > 2 donor populations, multiple events and dates of admixture. We identified the source population and admixture events based on the ‘best guess’ event inferred by GLOBETROTTER: these included three types of events – 1) oneway admixture involving a single event with two source populations; 2) multi-way admixture involving a single time point of admixture but with multiple source populations; and 3) multiple-dates events which involved admixture at different time points with two source populations at each point. In order to better understand the ancestral components represented by source populations, we also extracted these source components from GLOBETROTTER output based on principal component analyses. For multiple events, bootstrapping could not be carried out to resolve confidence intervals (as this capability is not currently present in GLOBETROTTER); hence we have presented CIs only for MALDER analysis.
Delineation of Eurasian ancestry in Ugandans using the double conditioned site frequency spectrum
The results from f3, fineSTRUCTURE and MALDER may suggest gene flow, or shared ancestry with a population with Eurasian affinity. However, this does not confirm that this ancestry originated out of Africa, as an alternate hypothesis of gene flow from a population in East Africa with ancient substructure with Eurasians would also lead to similar results in these tests. In other words, statistically significant results in f3, and MALDER may result from allele sharing or gene flow from an ancestral population within Africa with allele frequencies correlated with modern European populations.
Two possible models: Ancient structure and recent admixture
In order to differentiate deep ancient structure in Ugandans with shared history with European populations from more recent shared ancestry due to gene flow, we used a method that has been previously used to examine affinity observed between European and Neanderthal genomes, and study the interrelationship between modern humans within Africa, out of Africa and Neanderthals (Yang et al., 2012). This method utilizes a double conditioned site frequency spectrum (dcsfs), where the site frequency spectrum (sfs) in the population being examined is conditioned on alleles being derived in a random haplotype in one population, and ancestral in a random haplotype in another population (Yang et al., 2012).
We now apply this method to consider two models (see Figures S5A and S5B): the ancient structure model within Africa, and the recent gene-flow model. The ancient structure model postulates that there were two or more deeply structured populations of hominins within Africa, with limited gene flow among them. The ancestors of modern day Eurasians originated from the same structured subpopulation from which modern day East Africans (in this case, Ugandans) arose. As a result of this, modern day Ugandans share a more recent common ancestor with modern day Eurasians, as compared with other modern day populations within Africa (e.g., West Africans) (Figure S5B). This would explain the correlation of allele frequencies observed between Ugandans and modern day Europeans on D statistics. We further hypothesize that following the out-of-Africa migration that gave rise to modern Europeans and Asians, the gene flow between ancestors of modern Africans that share a more recent common ancestor with Europeans, and ancestors of other modern Africans would have made these modern African populations more similar to each other, than either is to Europeans.
We also consider a second model: the recent gene flow model, which postulates that Ugandans arose from an African population that did not have more recent common ancestry with Eurasians compared with other African populations, and shared alleles observed with Europeans in D stastistics are a direct consequence of recent gene flow from Eurasian populations due to back migration into Africa (Figure S5A).
The double conditioned site frequency spectrum (dcsfs)
To examine these hypotheses, we calculate a site frequency spectrum (sfs) among Ugandans conditioned on alleles being derived in a random European haplotype, and ancestral in a random West African haplotype. Such a double-conditioned sfs is expected to be approximately uniform in the scenario of ancient structure, as has been shown previously (Yang et al., 2012). In the event of recent gene flow, we would expect to see an excess of derived allele sharing with modern day Europeans due to the more recent shared common ancestry, producing an L shaped rather than flat curve. The exact shape of the curve is likely to be determined by the amount of admixture. We also carried out simulations to confirm this, and assess goodness of fit to observed data. It must be noted, however, that our observed derived sfs is based on low coverage data. Although we use methods to minimize bias associated with lower coverage, it is likely that the derived sfs may be biased toward less enrichment of rarer alleles. This would be likely to bias our inferences against the hypothesis of recent gene flow, as the sfs would appear more uniform.
Assessment of the observed sfs among Ugandans
In order to assess the two hypotheses, we first examined the shape of the observed dcsfs among Ugandans. We calculated a double conditioned site frequency spectrum (dcsfs) among 100 randomly sampled Ugandans. In order to condition the sfs, we first sampled one YRI and one French sample from the Simons Genome Diversity Project (SGDP) (https://www.simonsfoundation.org/simons-genome-diversity-project/). As these data were unphased, we randomly sampled, one allele at each site for the YRI and French sample each. We used the human ancestral reference (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_ancestor_GRCh37_e59.tar.bz2) provided by the 1000 Genomes Project to assign alleles as ancestral or derived. We only used sites with high confidence (at least two alignments supporting the ancestral allele). We identified sites where the allele was derived in the French sample, and ancestral in the YRI sample. We note that although YRI has been shown to have small amounts of Eurasian ancestry, this would be unlikely to bias results, as this would only reduce the number of sites contributing to the dcsfs (ancestral in YRI and derived in the French sample), potentially reducing power slightly (Prüfer et al., 2014).
We then calculated the sfs for these sites in the Uganda sample. In order to minimize bias in calculation of sfs in our low coverage data, we used ANGSD (Korneliussen et al., 2014), which uses a probabilistic estimation of sfs, using genotype likelihoods (GLs) at sites. This method directly estimates the sfs from sequencing data by first computing site allele frequency (SAF) likelihood for each site. In order to assess bias due to low coverage, we also re-calculated sfs in the sample of 100 Ugandans, including only sites with a minimum depth of 8 by specifying the -setMinDepthInd 8 filter in ANGSD. This removes sites with a read depth < 8 for each individual sample, so that sfs is only analyzed among samples with adequate coverage at each site. We used the GATK model to estimate genotype likelihoods within ANGSD. We did not find any differences n the sfs between low coverage sequence, and sequence data limited to high coverage regions, suggesting that the sfs estimated by ANGSD from low coverage data was unbiased. In both analyses, dcsfs appeared non-linear, with enrichment of derived alleles consistent with a recent admixture model, as we show subsequently. We present all results based on analysis of low coverage data in subsequent analyses. In order to assess whether the observed dcsfs was a better fit with the recent admixture model in comparison with the ancient structure model, we carried out simulations of different models, as we describe below.
Simulation of dcsfs for ancient structure and recent admixture models
Following estimation of sfs using ANGSD, we carried out several simulations to identify which population genetics model was the best fit to the observed data. We used the coalescent simulator ms(Hudson, 2002) for simulation. The two models used in simulation are shown in Figures S5A and S5B. The various parameters used in simulation are shown in Table S3.3.
We used the coalescent simulator ms to simulate models of recent admixture and ancient structure. We assumed an effective population size of N = 10,000 for European populations, and N = 20,000 for African populations, and a generation time of 25 years per generation (Yang et al., 2012). We simulated 3 populations, using both these models. We refer to these as West Africans, East Africans and Europeans, given the current context. These represent two African populations, one of which may share a more recent common ancestor with Europeans (‘East Africans’) compared with another African population (‘West Africans’) in the ancient structure model. For the gene flow model, these represent African popualtions symmetrically related to Europeans, with a recent common ancestor ranging between 280–800 generations ago (Schiffels and Durbin, 2014). We generated 10,000 replicates of each model. In each replicate of both models, the simulated sample consisted of one European chromosome, one West African chromosome, and 200 East African haplotypes.
In the recent admixture model, tN was set to 4,500 generations ago (112.5 kya) (Yang et al., 2012). Several values were assigned to f(0.03, 0.05, 0.07, 0.10, 0.15). tGF was varied between 10–100 generations, consistent with results from MALDER. The parameter ranges for the simulations for both models are shown in Table S3.5. In the ancient structure model, T was varied between 4,600 generations and 5,000 ago, in steps of 200 generations. The intensity of ancient migration m was set to 4Nm = {0..20}. For both models, the population split time between YRI and the east-Africans (tH) was varied between 280 to 800 generations ago, consistent with our MSMC analyses, and previous reports of divergence between YRI and LWK (Schiffels and Durbin, 2014).
For each model, a bottleneck reducing the effective size of the non-African populations by a factor of 100 (b) for 100 generations was set to (tb) 2000 generations (50 kya) (Yang et al., 2012). We also considered ongoing symmetric gene flow between YRI and the East African population with rates 4Nmt = {1,5,10}. We simulated 100 fold population growth in Africans and non-Africans occurring 300–820 generations ago. (20 generations before tH in each case).
We also simulated a scenario of both ancient structure and admixture, and assessed differences in dcsfs with different proportions of gene flow.
Given that parameters for models were approximates, and the true demographic history of several events in these populations are unknown, we simulated a range of values for each parameter, examining the impact on varying these parameters on the dcsfs. For the recent admixture model, we varied tH, tGF, f, mt and tG, varying each of these one at a time, keeping all other parameters constant. To assess variability in each parameter, we chose near mid-range values for the remaining parameters. The standard values chosen while assessing variability for each parameter were tH = 0.01, 4Nmt = 5, tGF = 0.001, f = 0.05, and tG = 0.0105. We also visually assessed the fit of each model to our observed dcsfs among 100 Ugandans. Curves were smoothed using the smooth.spline function in R, checking visually, that the fit to individual values was good in each case.
The following is the command line for implementing the recent admixture model using ms:
Next, we simulated ancient structure, and examined the effects of varying various parameters, while keeping others fixed on the observed dcsfs in simulations. We varied th, mt, m, and T, one at a time, keeping all other parameters constant. The fixed parameters chosen were in the mid-range of all evaluated values. We used th = 0.02, 4Nmt = 5, 4Nm = 4 and T = 0.12 as the fixed parameters. The command line implemented in ms was as follows:
We also simulated a model of ancient structure with recent admixture. As we did not see any variation in simulation sfs by varying different parameters, except with variation of proportion of gene flow in the recent admixture model, we fixed the remaining parameters (th = 0.01, 4Nmt = 5, 4Nm = 5, t = 0.12, tGF = 0.001 and tG = 0.0105). We varied the proportion of gene flow per generation (f) from 5%–15%.
The command line used to generate these simulations was as follows:
Assessment of fit of simulated models to observed data
In addition to visual assessment of fit of simulated data to the observed dcsfs among Ugandans, we assessed the relative statistical fit of various models by examining differences in squared errors between each model and observed data. For this, we calculated error terms for each model (models 1 and 2) in relation to the observed data for each point (smoothed). We then calculated the sum of differences in squared errors across all points as follows:
, where i2 is the squared error term for model m at point i. We then calculated a Z score for S being different from 0 as follows:
, where
Here, σ is the standard deviation of differences in squared error terms between models. The Z score here is indicative of whether one model shows a better fit to observed data compared to another. A positive Z score would indicate that model 1 is better than model 2 as the mean difference in squared error terms is positive. We calculated 2 sided p values for the Z score. We used a p value threshold of < 0.005 to define statistical significance (corrected for 10 tests).
Neanderthal Ancestry among Ugandans
To better understand the source of Eurasian-like ancestry in modern Ugandans, we examined whether Neanderthal ancestry could be detected in these populations. The presence of Neanderthal ancestry in Uganda would suggest that at least some of the Eurasian-like ancestry entered Uganda though back-to-Africa migrations, as one would not expect to otherwise observe Neanderthal ancestry within Africans. Given the likely small proportion of Neanderthal ancestry among Ugandans, in the event of ancient Eurasian admixture, we used a Conditional Random Field (CRF) to identify potential sites of admixture among 100 randomly selected Ugandans. The CRF model developed by Sankararaman et al. (2014) identifies Neanderthal ancestry among samples using the following three features of variation at a given site: the model prioritises 1) sites at which a panel of sub-Saharan-African individuals (YRI, in this case) carry the ancestral allele and in which the sequenced Neanderthal and the test haplotype carry the derived allele, 2) genomic segments in which the divergence of the test haplotype to the sequenced Neanderthal is low, whereas the divergence to a panel of sub-Saharan-African individuals (YRI, in this case) is high; and 3) segments that have a length consistent with what is expected from Neanderthal-to-modern-human gene flow approximately 2,000 generations ago, corresponding to a size of about 0.05 cM = (100 cM per Morgan)/(2,000 generations). Although the CRF model was originally trained for detecting Neanderthal ancestry among non-Africans, it is possible that the model may function reasonably well in inferring high confidence Neanderthal ancestral regions in the genome, if stringent probability thresholds are used, as opposed to marginal probabilities which may not be as accurate (in correspondence with Sankararaman S). We therefore evaluated the model in simulated data prior to running this on Ugandan sequence data.
Simulation analysis
We simulated European ancestry in an African population among 50 haplotypes for chromosome 10, at different proportions and different time points. We used the method used previously by Price et al. (2009) In brief, we simulated 50 admixed haplotypes (chromosome 10 only) from the 198 Esan (ESN) and 198 CEU haplotypes. To construct each admixed genome, we randomly sampled an ESN and CEU haplotype to simulate admixture. Sampling was carried out without replacement, so each admixed genome had unique ESN and CEU ancestral haplotypes. To construct an admixed genome, we began at the first marker on chromosome 10 and sampled CEU ancestry with probability p and ESN ancestry with probability 1-p. We simulated recombination with a probability of 1-e−λg, where λ is the number of generations in the past when admixture occurred, and g is the genetic distance in Morgans between sites. At each recombination event, we resampled CEU probability p and ESN ancestry with probability 1-p. For each individual, we chose a value of p by sampling from a beta distribution with mean p and standard deviation σ. We simulated admixture 10 and 100 generations ago (in keeping with results from MALDER which inferred two events of gene flow into Ugandans, at approximately these time points), with p = 0.10, and σ = 0.02. Pairs of haploid admixed individuals were merged to form 25 diploid admixed individuals. We then ran CRF to identify segments of Neanderthal ancestry among these individuals on chromosome 10. As we simulated European ancestry among Africans, true segments of Neanderthal ancestry were not known in the simulated data. However, as Neanderthal segments of ancestry were likely to lie within European ancestral segments, we examined overlap of inferred Neanderthal segments with European simulated segments (see Table S3.7). Additionally, a map of Neanderthal ancestry among Europeans and Asians in the 1000 Genomes project has been published (Sankararaman et al., 2014). We compared the regions of inferred Neanderthal ancestry to this map, as most regions would be expected to lie within these segments (see Table S3.7). We also carried out permutation analysis (1000 permutations), permuting random segments of the genome of the same length as inferred segments to calculate the statistical significance of overlap with European segments in our simulated data, and overlap with current maps of Neanderthal ancestry among Eurasians (see Table S3.7). CRF inference of Neanderthal ancestry was carried out using default parameters, and 100 YRI individuals, and 1 Altai Neanderthal as reference populations. Sites with > 0.90 probability of Neanderthal ancestry were inferred as Neanderthal.
Direct assessment of Neanderthal ancestry among Ugandans
Following validation of this approach in simulated data, we used CRF to identify segments of Neanderthal ancestry in real sequence data in a random sample of 100 Ugandans on chromosome 10. We identified regions of Eurasian ancestry in the same samples using fineSTRUCTURE (Lawson et al., 2012) on low coverage sequence data for the same samples. We first estimated parameters for fineSTRUCTURE on this sample set using the -i 20 -in -im -ip flags in ChromoPainter. This was carried across 425 chunks of the genome, and parameters were calculated using ChromoCombine. We next ran ChromoPainter with parameters estimated, using 216 YRI, 198 CEU and 206 CHB donor haplotypes. The Ugandan haplotypes were recipient haplotypes and were painted based on these donor haplotypes. The Hapmap recombination map was used to provide information regarding recombination rates/bp. Ancestry was inferred on haplotypes, and then combined across two haplotypes for each individual to make this comparable with CRF output, which provides diploid ancestral inference.
Distribution of introgressed Neanderthal segments
We assessed the distribution of inferred Neandethal ancestry among Ugandans with respect to Eurasian ancestral segements in these genomes, as well as known maps of Neanderthal ancestry among Eurasians. Eurasian ancestry was inferred as the sum of CEU and CHB ancestry > 0.90 within a haplotype.
Background selection in inferred tracts of Neanderthal ancestry
To help provide additional evidence for accurate inference of Neanderthal ancestry among Ugandans, we assessed background selection in regions of inferred Neanderthal ancestry. Previous work has suggested that tracts of Neanderthal ancestry are depleted in functionally important regions in the genome, and have suggested that collectively, regions which carry Neanderthal ancestry in modern humans are less likely to be under purifying selection (Sankararaman et al., 2014). We examined this using the B statistic (McVicker et al., 2009), which is likely to be lower in regions of purifying selection. We hypothesized that true regions of Neanderthal ancestry would have significantly higher B statistics, as compared to random regions of the genome, as has been shown before (Sankararaman et al., 2014). We compared the B score distribution across inferred regions of Neanderthal ancestry with an empirical distribution of B statistics generated by 1000 permutations where we sampled an equivalent number of sites, with the same segment length per site across the genome.
Examination of admixture using uniparental marker
To further examine admixture in the Ugandan population, we examined possible signatures of Eurasian admixture among uniparental markers. Since uniparental markers (mitochondrial DNA and the Y chromosome in males) do not undergo recombination from generation to generation, examining these provides an alternative strategy to identify Eurasian admixture. It must be noted that absence of haplotypes from ancestral admixing populations do not necessarily suggest the absence of admixture, as drift or purifying selection can eliminate such haplotypes, given enough time (Serre et al., 2004).
Mitochondrial DNA analysis
We reconstructed the mitochondrial genomes of 1,978 UG2G and 2,535 1000 Genomes phase III samples using a majority/consensus rule caller, i.e., calling the most frequent base at each site. We did not consider insertions or deletions and required a minimum depth of coverage (DP) of 5 and a minimum base quality of 30. We generated a gVCF file for each of the genomes and predicted their mitochondrial haplogroups using Haplogrep (Kloss-Brandstätter et al., 2011) which relies on PhyloTree build 17 (van Oven and Kayser, 2009).
To reconstruct the evolutionary history of UG2G and 1000 Genomes mitochondrial genomes, we aligned the UG2G and 1000 Genomes mitochondrial genomes (n = 4,513) using Mafft (v7.222) and reconstructed the phylogenetic tree using the BioNJ method (Gascuel, 1997) implemented in Seaview v4.5.4 (Gouy et al., 2010); distances were calculated with the Jukes-Cantor model. Similar results were obtained with a maximum likelihood tree reconstruction approach (results not shown)
Y-haplogroups in the Uganda genome resource
We examined Y chromosomal haplogroups to assess possible admixture within the Ugandan cohort. The prediction of Y haplogroups is harder than the prediction of mitochondrial haplogroups because the sequencing coverage for the mitochondrial genome is much higher than for the nuclear genome. With low coverage sequencing data, probabilistic methods like YFitter (https://arxiv.org/abs/1407.7988) are particularly appropriate. Instead of calling variants, YFitter analyses genotype likelihoods, and it does so for a set of 439 marker sites that discriminate the known Y haplogroups (Karafet et al., 2008). YFitter selects the haplogroup that best fits the data and also provides estimates of uncertainty.
We obtained YFitter predictions for 829 UG2G and 1,244 1000 Genomes Project males. To assess the reliability of the haplogroup assignments we built a phylogenetic tree using the 439 sites used by YFitter. To call these sites we required a minimum depth of 1 and a minimum base quality of 30. We reconstructed the phylogenetic tree with different methods (neighbor joining and maximum likelihood) consistently obtaining topologies consistent with YFitter haplogroup predictions, suggesting these topologies are robust.
Deconvolution of admixture in Uganda using ancient populations
In order to understand complex admixture within Ugandan populations and identify source populations most closely representing ancestry in Uganda, we examined a combination of modern and ancient African and non-African data.
Curation of ancient and modern genomic data
We merged data including several ancient East African and South African genomes (Skoglund et al., 2017) with Eurasian ancient genomes (Lazaridis et al., 2016) and the Human origins array (Lazaridis et al., 2016). In order to maximize power, we included sequence data from 1,978 Ugandans, and sequence data for Dinka (Mallick et al., 2016) extracted across the sites enriched for in the 1240K capture (Skoglund et al., 2017). In order to minimize ascertainment bias, we used Ugandan sequence data on 2,100 individuals called and refined across the Ugandan and 1000 Genomes Project phase 3 (Auton et al., 2015) and AGVP panel (Gurdasani et al., 2015). We subsequently removed all related individuals (IBD > 0.10), and included a final set of 1,154 Ugandans (893 Baganda, 130 Banyarwanda, 27 Banyankole, 26 Barundi, 42 Rwandese Ugandans, 19 Bakiga, 7 Batanzania, 7 Basoga, and 3 Batooro). We only included transversions (n = 228,656) in our analyses to minimize bias due to ancient DNA damage.
Three distinct streams of ancestry in Ugandan populations
We first used qpwave (ADMIXTOOLS) (Patterson et al., 2012) to estimate the number of distinct streams of ancestry in modern Ugandans. For these analyses, we used 19 populations as global outgroups, as previously outlined (Skoglund et al., 2017), including Mbuti, Dinka, Mende, South_Africa_2000BP, Tanzania_Luxmanda_3100BP, Ethiopia_4500BP (Mota), Levant_Neolithic, Anatolia_Neolithic, Iran_Neolithic, Denisova, WHG, Ust_Ishim, Georgian, Iranian, Greek, Punjabi, Orcadian, Ami, and Mixe. In this context, Ethiopia_4500BP/Mota represents East African hunter-gatherer ancestry (Gallego Llorente et al., 2015), the Mbuti represent central African rainforest hunter-gatherer ancestry, and Tanzania_ Luxmanda_3100BP represents an early pastoralist lineage from eastern Africa (Skoglund et al., 2017). Dinka represents modern Nilotic speakers in East Africa. Consistent with previous approaches, we rejected a model if p < 0.01 for the rank of a given matrix (Skoglund et al., 2017). If the rank of a matrix was not rejected (p > 0.01), we considered the number of streams of ancestry as rank+1.
We then successively removed ethnolinguistic groups to identify whether distinct streams could be localized to specific ethno-linguistic groups. Given the identified clade structure in fineSTRUCTURE (Figure 1), we removed populations in the order of clades identified.
Delineation of source populations of complex admixture in Uganda
In order to further understand the sources of ancestry among these populations, we used qpAdm (Patterson et al., 2012). QpAdm tests whether the ancestral components in a given target population can be explained by ancestral components contributed by pre-specified source populations, and then estimates the ancestral components contributed by source-like populations. QpAdm first tests whether inclusion of a target population to a set of source populations adds an additional stream of ancestry (increases rank of f4 statistics matrix by one). If the ancestry in the target population is fully represented among source populations, one would expect the number of streams inferred to remain constant, even with addition of the target population, as these streams are already represented in the source populations. Following this, it uses a matrix f4 statistics calculated of the form (target; sourcen; outgroup1, outgroupm) to infer admixture proportions, where n source populations are included along with the target population on the left, and m outgroup populations are included on the right. Negatively inferred proportions suggest that the model is incorrect.
In order to examine the sources of ancestry among Ugandan populations, we used the approach described previously by Skoglund et al. (2017) first examining single source admixture, dual source admixture, and three sources of admixture, when single and dual admixture models did not fit for a given target populations. We moved outgroup populations from the right to the left population set in turn to assess whether these fit as source populations.
We examined a subset of outgroup populations as source populations; these included Mende, Mbuti, Dinka, Ethiopia_4500BP (Mota), Tanzania_Luxmanda_3000BP, South_Africa_2000BP, Anatolia_N, Levant_N, Iran_N and Orcadian.
Sensitivity analyses to identify appropriate source and outgroup populations
Given the recent identification of basal admixture in West African Bantu populations (Skoglund et al., 2017), we first evaluated whether these would provide appropriate source populations to represent bantu-like ancestry in East Africans. We first evaluated the presence of basal ancestry in Ugandans relative to Mende, and Yoruba.
Asymmetry of East and West African populations with ancient South Africans
In order to evaluate possible basal ancestry within Uganda, we carried out F4 tests of the form (chimp, South_Africa_2000BP; Mende/YRI, X), where X is a Ugandan population. We find asymmetry in test statistics f4(chimp, South_Africa_2000BP; Mende, X) and f4(chimp, South_Africa_2000BP; Yoruba, X) (Table S3.10), suggesting possibly higher levels of basal ancestry in Mende and Yoruba relative to Uganda. Another explanation for this asymmetry may be low levels of Hadza-like admixture in Uganda (Hadza is thought to be related to Khoe-San populations in South Africa). F4 statistics of the form (chimp, South_Africa_2000BP; Mota, X) did not show any asymmetry and were consistent with ancient South Africans being an outgroup to Mota and Ugandans, suggesting either no basal ancestry among these populations, or similar proportions of this ancestry.
Subsequent sensitivity analyses suggested that Mende provided poor representation of Bantu-like ancestry in Uganda. Given the possibly lower basal ancestry observed in West African populations, we considered inclusion of an East African ancient Bantu-like source population to represent bantu-like ancestry in modern Ugandans. Tanzania_Pemba_700BP is an ancient East African sample represented most closely by Bantu ancestry in West Africans, as previously reported (Skoglund et al., 2017). However, our tests suggest that this sample is symmetrical to Ugandans with respect to ancient South Africans, potentially making this more appropriate as a source population for East African Bantu ancestry. We therefore included 11 source populations in our analyses of admixture in Ugandans. We therefore tested 11 single sources of admixture, (11, 2) = 55 dual admixture models, and (11,3) = 165 three-way models of admixture for each of the nine ethno-linguistic groups.
Rf-HGs may have admixture from a Uganda-like East African population
QpWave and qpAdm analyses assume there is no post-admixture gene flow between left and right populations. Although rf-HG (Mbuti) and Dinka have generally been considered unadmixed, and has been previously used as a right sided population to assess admixture in modern East African populations, we formally assessed admixture in Mbuti rf-HGs and Dinka. We therefore carried out ALDER analysis to assess whether Ugandan population related gene flow was observed in East African right sided populations (Mbuti and Dinka). We found evidence suggestive of Uganda-like ancestry in Mbuti rf-HGs (Baganda as reference, Z = 18.4). We therefore carried out two sets of sensitivity analyses with qpAdm, including and excluding Mbuti as a right sided population (although this was assessed as a left sided source population for all target populations). Although results were broadly similar, populations most representative of ancestry in Ugandans were found to be different in some cases; we therefore present both sets of results, with Mbuti excluded from right sided populations as the primary set of results.
Inference of Demographic history from High Coverage Genome Sequences
We explored the demographic history of the Ugandan population in relation to other African and global populations. In order to study this, we used the multiple sequentially Markovian coalescent model (MSMC2) (Schiffels and Durbin, 2014) to estimate the population size history of the Ugandans using a high coverage (30x) trio sequenced from the Baganda population. The trio was sequenced with paired end sequencing on the Hiseq 2000 platform. Aligment was carried out to the 1000Genomes_hs37d5 reference with bwa aln. Duplicates were marked with Picard, following which re-aligment around indels was carried out with GATK. SNPs were called, and mask files were generated for each sample using samtools with the command:
samtools mpileup −q 20 −Q 20 −C 50 −u −r < chrx > −f < ref.fasta > < sample.bam > | bcftools call −c −V indels |./msmc-tools/bamCaller.py < mean_coverage > sample_mask_chrx.bed.gz j
Input files were generated using scripts provided in the MSMC2 tutorial, the mask files generated with the above command, and additional mappability mask files downloaded from https://oc.gnz.mpg.de/owncloud/index.php/s/RNQAkHcNiXZz2fd. These masks include all regions across the genome for each chromosome where reads from short read sequence data can be uniquely mapped.
1000 Genomes high coverage sequence data available were also processed in the same way as described above for a CEU trio, one high coverage LWK sample, a YRI trio and samples from GWD, ESN and MSL populations (Table S1.6). We used PCR-free samples, where available. Mapped bams were downloaded from the 1000 Genomes home page, and processed in the same way as the Ugandan samples for consistency.
For samples that belonged to trios, trio based phasing was carried out, as implemented in msmc-tools. Reference based statistical phasing was carried out for unrelated samples that did not belong to trios. SHAPEIT2 r790 was used for phasing of these samples using a merged reference panel combining 1000 Genomes Project phase3, AGVP populations, and the Uganda GWAS dataset (see Method Details). For phasing, only sites within the reference panel were included. These phased sites were then merged back into the original calls using run_shapeit.sh from msmc-tools, leaving non-phased sites as ambigiously phased, as described in the MSMC2 tutorial (https://github.com/stschiff/msmc-tools).
As only two LWK haplotypes were available in the high coverage 1000 Genomes sequence data, we also examined high coverage whole-genome sequences generated by Complete Genomics (Drmanac et al., 2010) with a larger sample of LWK haplotypes (Table S1.7a). We also analyzed data from corresponding Europeans (CEU) and Africans (YRI) from these data for comparability with results from the 1000 Genomes sequence data (Table S1.7b). Complete Genomics data were called using msmc-tools ./ cgCaller.py, calling the consensus sequence from the masterVarBeta file. YRI and CEU samples were part of trios, and were phased as such, while LWK were phased using reference data, as described previously.
We estimated the effective population size over time of all popualtions using MSMC2, as well as split times between Uganda and other populations by estimation of the cross-coalescence rate (CCR) with MSMC2. We implemented MSMC2 on 4 haplotypes from every population, except for LWK, ESN, MSL and GWD from the 1000 Genomes dataset, where only 2 haplotypes were available for analysis for each population. For all initial analyses, we specified 32 time segments -p 1*2+25*1+1*2+1*3. We excluded ambiguous sites from analyses for estimation of cross-coalescence rates. Inclusion or exclusion of ambiguously phased sites did not appear to impact estimation of effective population sizes. Here, we present all results estimated with exclusion of ambiguously phased sites, as recommended. We used a generation time of 30 years and a rate of 1.25 × 10−8 mutations per nucleotide per generation for estimation of coalescence rates. We also conducted sensitivity analyses to assess the impact of different modes of phasing on split times estimated by MSMC2, assessing more recent population growth my finer-scale parametrisation of segments, using the option -p 27*1+1*2+1*3 with MSMC2, allowing parameters to be different in the leftmost 27 time segments (of 32 time segments in total) (Table S1.7c) and (−p 30*1+1*20), examining population history and finer scale.
To examine split times between Ugandans and other populations we used MSMC2 estimation of cross-coalesence rates and considered splits to have occurred when gene flow between the populations dropped to below 50%. We examined cross-coalescence between Uganda and other African populations in the 1000 Genomes high coverage data, and the Complete Genomics dataset (Tables S1.6 and S1.7).
Previous studies have that suggested that phasing inaccuracies can lead to split times being biased and appearing more recent in comparison with experimentally phased data on samples (Song et al., 2017). We, therefore also examined the robustness of dating of cross-coalescence to errors in statistical phasing. In order to examine the impact of reference based phasing on results, we reanalyzed the Uganda-YRI CCR, using reference panel guided phasing for Uganda, and YRI from both the 1000 Genomes Project data and Complete Genomics data. We used trio phasing as the gold standard in this context, and compared results to results obtained with trio phased data.
Sharing of f2 variants and estimation of dates of shared variants between Uganda and other populations
To better understand recent population history among Ugandan populations, and between Ugandan populations and others, we examined f2 variation in our sequence data combined with the AGVP and the 1000 Genomes Project phase 3 dataset. Curation of this merged dataset is detailed in Method Details. The number of individuals in each population group is provided in Table S1.5. F2 variants are variants that occur only two times in a dataset, in two different individuals. Examining such rare variants can provide important information about recent population history as well as population demography, recent bottlenecks, ancient splits, and relationships between populations. As dating of haplotypes shared within and between populations would provide important insights into split times among populations, we sought to date haplotypes around f2 variation as has been described previously (Mathieson and McVean, 2014).
To explore population relationships, we first examined sharing of f2 variants among populations. Our large sample of WGS allowed us to examine very rare variants, and hence more recent population history among these populations. Given the differences in numbers of samples from each population, inferences about f2 variant sharing are likely to be biased, with f2 variants from large populations likely to be rarer than f2 variants in a smaller number of individuals. We examined f2 variants in a set of combined sequences including UG2G, AGVP and 1000 Genomes Phase III sequence (N = 3,895) (Table S1.5). Although we ascertained f2 variants across the entire sample set of 3,895 individuals, we subsequently subsampled 40 haplotypes from each population 100 times and calculated the mean number of shared f2 variants. These were then normalized by the total number of f2 variants existing in each population (Figure S7).
We further explored these f2 variants by defining the extent (length) of haplotypes around these, and estimating most likely dates in generations of each haplotype using a maximum likelihood approach described by Mathieson and McVean (2014). We removed low complexity regions of sequence as defined by the 1000 Genomes Project, as described previously (Auton et al., 2015). We defined the extent of haplotypes by scanning along the genome on both sides until homozygote inconsistencies were observed between individuals. We used the HapMap recombination map to estimate haplotype length. We used an estimate of power of 0.60 for singleton discovery for these data, and a mutation rate of 1.2e-08 for our analyses.
We observed a total of 12,477,686 f2 variants in our dataset belonging to 9,875,361 f2 haplotypes. Given our ascertainment of f2 variants in a sample size comprising largely Ugandans, we expect f2 variation within Ugandans to be more recent than within other populations; therefore, we decided only to focus on the relationship of f2 variation between Ugandan and other populations, as this is likely to be relatively unbiased. We compared the relationship of Baganda to other Ugandan and 1000 Genomes Project and AGVP populations by examining the dating of shared f2 variants between Baganda and other population groups (Figures S7B and S7C).
We first examined sharing of f2 variation between European and African populations. We observe old sharing of f2 variation between African and European populations (median f2 sharing between YRI and Europe ~51,000 ya), (Figure S7B) consistent with previous reports (Mathieson and McVean, 2014), and with known divergence times between these populations. Compared with other African populations, f2 sharing between Baganda and European populations was noted to be more recent (median f2 sharing = 19,500 ya). We hypothesized that this might be due to greater Eurasian admixture in Uganda, compared with YRI. However, this might also reflect ascertainment of f2 variants in a large sample of Ugandans, resulting in these being more recent. However, we found that median shared f2 dating between LWK-Europe was more recent than between YRI-Europe, with sharing between Ethiopian populations and Europe being even more recent (in spite of the small sample size of these populations) (Figure S7B), strongly suggesting that recent dating was a consequence of Eurasian gene flow. This is consistent with possible gene flow from Europe into Uganda as a result of back migration.
Analysis of Mutational Spectrum in UGR
Comparison of diversity with other low coverage WGS resources
We compared the variants (SNPs and Indels) discovered with UG2G with discovery within global low coverage sequencing datasets, including the 1000 Genomes Project Phase 3, sequence data on 320 individuals from the African Genome Variation Project, and the UK10K cohorts. It must be noted that average coverage for the 1000 Genomes Project and the UK10K cohorts was higher than for UG2G (average coverage 7x, 6x and 4x, respectivelyu). We also compared the variants discovered in UG2G with those in the GnoMAD database. To better characterize individual level variation, we examined the number of variants per individual within each of these datasets to examine diversity at individual level. In order to examine the spectrum of variation, and compare this with other resources, we compared variation in a random sample of 379 unrelated Ugandans with an equal number of European individuals (n = 379) from the 1000 Genomes Project, which has more comparable depth of coverage. For this comparison, we excluded target exonic regions sequenced to higher depth in the 1000 Genomes project, for consistency.
In order to compare diversity among African populations, we examined heterozygosity among different Ugandan populations in the context of AGVP.
To assess the influence of sample size of the resource on discovery of variants, we performed subsampling of individuals in incremental steps (see Figure S8). This provides a direct observation of the variant discovery in large sampling projects, and provides useful information for future large-scale sequencing endeavours in African populations. We also compared gains in discovery as a function of sample size between UG2G and the UK10K ALSPAC data. For homogeneity in the analysis, for analysis within UG2G, we analyzed the Baganda population only, for which we sequenced 1,549 individuals. We picked randomly a combination of individuals for each sample size and calculated the number of variants in each combination. Then we averaged the number of variants in intervals of 10 additional samples to reduce the effect of chance on sampling. We carried out similar analyses with the same sample sizes in UK10K ALSPAC data.
Functional Variation in UG2G
In order to understand the relative distribution of functional variants in UG2G, we examined the spectrum of these variants in this cohort, and compared putatively functional variants in UG2G with other European cohorts.
In order to understand the burden of these mutations among individuals in our cohort, we assessed the spectrum of the annotations given in the Human Gene Mutation Database (HGMD) (Stenson et al., 2003) in relation to our sequence data (Figure 3). We specifically studied the burden of the most deleterious variants according to the HGMD annotations, namely the DM (disease-causing mutations), counting the number of DM alleles per individual. We also examined the frequency of ClinVar mutations of clinical significance (clinical significance = 5). As DMs are likely to be rare, they may be underestimated in the Ugandan cohort due to low-coverage sequencing leading to under-calling of rare variation. We assessed this by comparing estimated DMs in three high coverage sequence (30x) samples belonging to a Ugandan trio, with the DM calls for the same samples from the low coverage sequence data. The sensitivity of detection of DMs in these samples was 94% and the specificity is 91%. Additionally, the mean number of DMs detected by high and low coverage data in these three samples were very similar (33 and 34, respectively), thus validating our DM discovery.
We also validated our DM discovery by using ANGSD, a method that accounts As an account for genotype calling biases in low coverage (Korneliussen et al., 2014). The approach implemented in ANGSD has been shown to produce accurate site frequency spectra even in low coverage data (Han et al., 2014). Our results comparing ANGSD calls from low coverage data to our standard calls with Unified Genotyper (UG) produced highly comparable results (median of 28.4 and 29DMs/individual in ANGSD and UG called data respectively), providing further validation, and suggesting that results from comparisons are likely to be accurate and closely approximate the true distribution of DMs among individuals in the UG2G cohort.
We sought to evaluate the clinical relevance of the DM annotations in the context of the UG2G resource. We closely examined these DMs that were common in our data, to assess the effect of these on relevant hematological and cardiometabolic traits. DMs are considered to be primarily mutations that cause severe disease phenotypes or monogenic disorders; therefore, one would expect them to be very rare in a given population as a result of purifying selection. There are several reasons we might find DMs to be common in a given population cohort; 1. The mutations truly cause monogenic disease, but confer protection against a competing disease, and are therefore under positive or balancing selection; 2. The mutations are not functionally relevant, and are incidental findings that have been erroneously associated with a given phenotype; 3. The mutations are in LD with the true causal mutation in European populations and a proxy for this, but not in our East African cohort; 4. The true penetrance of these mutations is much lower than previously thought; or 5. The mutations have different phenotypic effects among different populations due to differences in epigenetic or epistatic factors. Interrogation of these mutations in an independent cohort of different ancestry allows us to identify DMs that may need further exploration to better understand their effects and disease penetrance among different population groups.
Due to limitated availability of phenotypic data, we were only able to assess the impact of DMs associated with cardiometabolic diseases with those specific phenotypes. This may limit inferences relating to potential impact of these mutations on other phenotypes. We focused on 38 DMs that were common (> 5%) in the UG2G cohort but rare or absent (< 1%) in the UK10K cohort (Table S4.3).
The Uganda Genome Resource as an Imputation Reference Panel
In order to assess improvement in imputation accuracy when using UG2G as a reference panel, we compared three panels: the 1000 Genomes Phase III dataset, the 1000 Genomes Phase III dataset merged with the African Genome Variation Project sequences from 320 individuals, and a combination of the 1000 Genomes Phase III dataset, the AGVP sequences and the UG2G data from 1,071 unrelated individuals from the GPC. The generation of the UG2G+AGVP panel has been outlined in Method Details.
For imputation, we used Omni 2.5M genotype data available for UGWAS and AGVP populations, as the target set. We measured the accuracy of imputation using the leave one out method in IMPUTE2 and calculating the correlation (r2) between the imputed and original genotype calls. This method systematically leaves out each SNP from the target data treating this as missing, and then imputes the marker from the reference data. Accuracy of imputation at each of these sites is then determined by calculating the correlation between imputed genotype calls and the original genotype data in the target set.
Heritability of traits in the General Population Cohort
We examined the heritability of traits within the General Population Cohort using the genotype data within the Uganda genome resource. In addition to assessing the narrow sense heritability across multiple traits, we were also able to examine the contribution of shared environment to the phenotypic variance, using novel methodology, and show that not accounting for this can lead to marked overestimation in estimates (Heckerman et al., 2016). We recapitulate our methods here.
Statistical model
The linear mixed model (LMM) is now routinely used to estimate narrow sense heritability. Unfortunately, LMM estimates of heritability can be inflated when environmental correlation is not explicitly modeled. To help avoid inflated estimates, we can use an LMM with two random effects—one based on genetic markers and one based on environmental factors. In order to assess narrow sense heritability, we used a mixed model approach in FaST-LMM using similar methodology to previous studies (Zaitlen et al., 2013). Given the unique pedigree structure in the cohort, we were able to phase the haplotypes for 4,778 individuals (UGWAS) included in the analysis very accurately and generate very accurate estimates of IBD. We have previously shown that haplotype phasing in this cohort using methods that leverage relatedness such as SHAPEIT2 are very accurate, even when the pedigree structure is not explicitly input into the algorithm (O’Connell et al., 2014). We further improved on accuracy by including the known complex pedigree structure into SHAPEIT2, using duo-HMM to correct any phasing errors. For this, we excluded pedigrees where the age differences did not match pedigree structure (parent was reported to be aged > 60 when the child was born, parent-offpsring pairs with an age difference < 12). We ran KING (http://people.virginia.edu/wc9c/KING/) to check the pedigrees, and further excluded any sibling pairs where it was unclear whether these were full or half siblings and parent-offspring pairs where the inferred parent seemed incorrect. These produced a highly accurate set of pedigrees for phasing. MERLIN (Abecasis et al., 2002) was used for error correction before using duo-HMM in SHAPEIT2 for phasing. For the remaining individuals who were excluded, haplotypes inferred from phasing the entire cohort as unrelated individuals were used, and merged with the haplotypes inferred using duo-HMM. Using these combined phased haplotypes, we calculated an IBD matrix, using methods that have been outlined previously (Price et al., 2011). This IBD matrix was used in the mixed model to provide accurate estimates of narrow sense heritability.
Previous studies examining heritability and genetic associations in large cohorts have tended to regard related individuals as having uncorrelated environment that contributes to phenotypic variance. This assumption is very unlikely to be true, and previous work has suggested that not accounting for this shared environment can lead to overestimation of heritability (Zaitlen et al., 2013). Here, we modeled environmental correlation using spatial distances and assessed the impact of this on heritability and GWAS estimates. These methods are described in detail elsewhere (Heckerman et al., 2016). We recapitulate these methods here:
For the environmental random effect, we constructed a radial basis function kernel, where the entry for a pair of individuals was the exponential of the negative scaled distance between the two individuals. The scaling parameter as well as the weights of the two random effects were determined by maximizing the restricted likelihood of the data. As the impact of various types of environmental clustering on phenotypic variance and estimates of heritability was unclear, we fit a number of models outlined:
-
1In the first model, we only estimated the contribution of the IBD matrix to phenotype variance, and considered environmental effects as independent, as studies have done previously.
Var(Y) is the phenotype variance, while and are the genetic and environmental components of variance, respectively. IBD represents the IBD matrix, while I is an identity matrix representing uncorrelated environmental components. Narrow sense heritability is calculated as follows:
-
2Given the relatedness among individuals and the clustered nature of the cohort into villages, and households, the above model is an unlikely representation of the truth, as environment among individuals living in the same household/village is likely to be more correlated than those living further apart. Modeling environment as unrelated, in this case could potentially overestimate the genetic contribution to heritability. We modeled the effect of correlated environment as follows:
Here, GPS represents the distance matrix derived from GPS coordinates, and represents residual variance. Here, heritability was calculated as:
and the contribution of shared environment to phenotypic variance was calculated as:
All parameters were estimated using maximum likelihood estimation. Standard errors for heritability for models 1 and 2 were calculated using a bootstrapping approach allowing comparison with published estimates for heritability in studies that have used a similar methodology.
In addition to the above two models, we also assessed gene-environment interaction for all phenotypes by fitting a model with the IBD matrix, distance matrix, in addition to a GXE term, as follows:
where i2 is the proportion of phenotypic variance explained by the interaction component.
For the variance estimates, we tested the null hypothesis that the variance component was equal to zero. To test , we performed a permutation test by permuting the distance matrix, by randomly shuffling identifiers of individuals. A p value for , was similarly determined by permuting entries of the KGXE matrix. In both cases, we performed 10,000 permutations. We carried out a comparison of our heritability estimates with those from Zaitlen et al. (2013) that had been obtained using very similar methods. We also re-calculated heritabilities using GCTA for consistency, using the same method they outlined in the paper (Zaitlen et al., 2013) and found that using this for estimation of heritability in the Ugandan cohort did not materially alter comparisons of estimates with the Icelandic study (data not shown). The methods implemented in Fast-LMM are described in detail in Heckerman et al. (2016) and the relevant code is available at: https://github.com/MicrosoftGenomics/FaST-LMM.
Comparison of heritabilities with European cohorts
We compared heritability estimated using our method with heritability estimated using similar methods in an Icelandic population (Zaitlen et al., 2013). We also compared our estimates with a pedigree based study from Pilia et al. (2006) in a Sardinian population which reported heritability on a large number of complex traits.
We note that the shared environment model of heritability estimated in the Sardinian study only modeled shared environment within pedigrees, and is therefore not directly comparable to estimation within the Ugandan cohort, where geographical distances were modeled; therefore, estimates from the Sardinian study may be more biased than the estimates from the Ugandan data.
We additionally evaluated whether the differences observed between European and Ugandan populations with regard to heritability could arise due to reduced bias in heritability estimation due to better correction of environmental sharing in the Ugandan cohort. In order to examine this, we compared uncorrected heritabilities in Uganda (heritability estimates calculated assuming un-correlated environment among indivduals) with those estimated by Pilia et al. (2006), Zaitlen et al. (2013), and Kang et al. (2010). Here, we used uncorrected estimates from the basic model in Pilia et al. (2006) not accounting for shared environment for consistency of comparison. The estimates in Zaitlen et al. (2013) are adjusted for geographical region, but not for any additional environmental sharing. We also compared with estimates of pseudoheritability estimated by Kang et al. (2010) using the EMMAX model. We note that pseudoheritability estimates calculated by EMMAX based on the IBS matrix are not actually estimates of narrow sense heritability, as outlined in Zaitlen et al. (2013) As these estimates do not utilize the thresholded IBS matrix, which provides similar estimates to the IBD matrix, these would provide estimates of heritability intermediate between GWAS heritability and narrow sense heritability (Zaitlen and Kraft, 2012); therefore, these are likely to be underestimates of narrow sense heritability. We, however, include these for completeness.
Genome-wide association study of 34 traits
Meta-analysis across cohorts to maximize discovery
To discover loci associated with traits, we carried out a meta-analysis of association statistics across four cohorts: the Ugandan Genome Resource (n = 6,400), the Durban Diabetes Study (DDS) (n = 1,165), the Diabetes Case control study (n = 1,542), and the AADM study (n = 5,231). Details regarding studies are below.
Mixed model analysis
Uganda Genome Resource.
In order to identify loci associated with traits within the Uganda Genome Resource, we used a linear mixed model (LMM) approach to account for relatedness (including cryptic relatedness) and population structure.
Given the influence of environmental correlation on estimation of heritability, we first evaluated whether modeling environmental correlation influenced beta estimates and p values obtained in GWAS of the 34 traits, and independent signals observed above the threshold for statistical significance. We conducted these analyses using the Fast-LMM model discussed before. We did not identify any additional loci at the genome-wide significance threshold using the 2 kernel (IBD and GPS coordinates) versus the 1 kernel (IBD matrix only) model (Table S5.5) and found no systematic difference in p values between the two models (kruskall wallis p = 0.46), suggesting that although modeling environmental correlation did alter heritability estimates, GWAS results were not altered significantly. We, therefore opted to use a simple LMM approach with uncorrelated environment for GWAS.
For this, we used the exact linear mixed model approach implemented in GEMMA v24 for analysis of pooled data from 6,407 individuals in the Uganda Genome Resource. We evaluated different approaches for generation of the kinship matrix to control type I error in analysis. It has been shown that inclusion of causal SNPs in the kinship matrix can lead to overly conservative results for these SNPs, and reduction in power for GWAS discovery. In order to maximize discovery, we used the leave one chromosome out (LOCO) approach for analysis (Listgarten et al., 2012; Yang et al., 2014). In this approach each chromosome is excluded from generation of the kinship matrix in turn, for association analysis for markers along that chromosome. This ensures that causal SNPs at a locus on a given chromosome are not used for generation of the kinship matrix used in analysis of that specific chromosome. Therefore, we generated 22 kinship matrices for analysis, each excluding the chromosome being analyzed using the given matrix.
For computational efficiency, and to avoid correlation effects due to LD, we LD pruned the data prior to calculation of the GRM matrix for each LOCO analysis. We carried out sensitivity analyses using different r2 thresholds for pruning, to examine whether type I error was appropriately controlled on examining genome inflation factors from QQ plots. We finally used all markers with an MAF > 1%, pruned to an r2 threshold of 0.5, using PLINK (Purcell et al., 2007) with the flags–maf 0.01 and–indep-pairwise 100 10 0.5, where 0.01 is the minimum MAF threshold of 1% and 0.5 is the r2 threshold within each 100 marker window sliding by a step size of 10 markers during each iteration. All genome inflation factors for traits were noted to be below 1.05 using this approach.
We also included a covariate to indicate whether data originated from imputed genotyped individuals or sequenced individuals to allow for any systematic differences between data (although earlier PCA suggested no systematic effects in filtered data). A MAF threshold of 0.5% was applied in GEMMA analysis.
DDS and DCC
Analyses for the DDS and DCC datasets were carried out in exactly the same way as described for the UGR dataset. LOCO analysis was used for each chromosome, and 22 GRM matrices were generated for each dataset. Analyses were carried out separately for DDS and DCC in GEMMA using an MAF threshold of 0.5% for each cohort. We confirmed that genome inflation factors were < 1.05 for all traits.
AADM
For AADM, analyses of all markers were carried out using EPACTS (Efficient and Parallelizable Association Container Toolbox) pipeline (https://genome.sph.umich.edu/wiki/EPACTS), which includes an implementation of EMMAX (Kang et al., 2010), which is an approximate linear mixed model approach similar to GEMMA. While a LOCO approach was not used in these analyses, we note that this would only make results more conservative for a given locus, and would not generate increased type I error. We carried out filtering for info score (0.3) following analysis, in this case, as we only had access to summary statistics. On examining QQ plots, we confirmed that genome inflation factors were < 1.05 for all traits.
Meta-analysis Methods
In order to maximize power for discovery, we carried out meta-analysis of results across all four cohorts (UGR, DDS, DCC and AADM), subject to availability of phenotypic data for given traits (Table S1.3). Given genomic diversity, admixture, and geographical distribution of studies, we used a union set of all SNPs (rather than the intersection), to maximize discovery, and allow for heterogeneity of effect, as well as to examine population-specificity and reproducibility of associations. Rather than exclude associated variants with heterogeneity in effect observed, we explored the underlying factors contributing to this heterogeneity of effect. While we do expect for heterogeneity in effect to arise as a result of artifactual associations in some cases, we opted to use this approach to allow for real heterogeneity in effect across populations; we discuss this in more detail subsequently, and describe implications of heterogeneity in meta-analyses of GWAS among diverse African populations.
Consistent with previous literature, we used the Han-Eskin random effects meta-analysis approach implemented in METASOFT (RE2) (Han and Eskin, 2011). This approach corrects for the overly conservative standard random effects meta-analysis approach by correctly assuming no heterogeneity of effect sizes if the null hypothesis is true (i.e., all betas are zero).
We find this approach gives highly comparable results to MANTRA meta-analysis (Morris, 2011), a bayesian approach used commonly for trans-ethnic meta-analyses, but is more computationally tractable, and easily interpretable with the output including a frequentist p value for combined effect. This also provides insight into heterogeneity across studies, which we examine in greater detail subsequently. We observed a strong correlation of 0.80, p < 2.2e-16, between log10(BF) from MANTRA analysis and −log10(pval) from METASOFT analysis (for variants with support from single studies the p value from GEMMA analysis within the study was used instead of the RE2 p value). For LDL METASOFT analyses, we observed that of 546 variants with p values < 5e-09 in METASOFT analysis, 97.4% (532) exceeded a log10(BF) threshold of 7 in MANTRA analysis, suggesting high concordance between results, and validating the approach used here.
Derivation of a genome-wide significance threshold for GWAS in African populations
Genome-wide association studies examining common variation across the genome for association with complex traits typically use a significance threshold of p < 5.0×10−08, with more stringent thresholds suggested for examination of rare variants (Xu et al., 2014). The 5×10−08 threshold has been derived from the total number of effective common variant (MAF ≥ 0.05) tests in European populations and has been based on HapMap data. When studying populations of African descent, a new statistical significance level needs to be defined, as lower levels of linkage disequilibrium between common variants may necessitate a more stringent threshold in Africans compared to Europeans.
Many methods exist which exploit the correlation structure, either haplotypic or genotypic, between variants to estimate the effective number of independent tests, and then use standard techniques for independent tests (Sidak or Bonferroni correction for multiple testing) to calculate an appropriate significance threshold. Some methods use the eigenvalues of the correlation matrix, since their absolute values correspond to the amount of the overall variance accounted for by the corresponding principal component (see for instance Gao et al., 2008). However, for large datasets of SNPs, it is not feasible to calculate the eigenvectors, and instead techniques have been developed which rely solely on the coefficients (see for instance Chen and Liu, 2011 and Moskvina and Schmidt, 2008). In Chen and Liu (2011) the correlation coefficients are used directly to estimate the effective number of tests, while in Moskvina and Schmidt (2008) the joint distributions of the event that the markers are not deemed significant are found based upon the correlation coefficients. We implemented 4 of these methods: SimpleM (Gao et al., 2008), Chen and Liu Method (Chen and Liu, 2011), Keffective (Moskvina and Schmidt, 2008), and Cheverud-Nyholt (Table S6.1) (Nyholt, 2004). The Keffective method produced the most robust results, which we present here. It uses the pairwise haplotypic Pearson’s correlation coefficients between SNPs to estimate the statistical independence between each SNP and those which preceded it, and sums them to estimate the total number of independent tests (Table S6.1).
Three populations from the 1000 Genomes Project (sequence data, phase 1 integrated public data release) were used; Luhya in Webuye, Kenya (LWK) and Yoruba in Ibadan, Nigeria (YRI), to estimate the significance thresholds for African data, and Utah residents (CEPH) with Northern and Western European ancestry CEU dataset as a European comparison. Standard quality control steps were performed on all autosomes after excluding indels.
Definition of distinct loci
Based on our derivation of a new threshold for statistical significance in African popualtions, we applied a statistical significance threshold of 5.0×10−9 to define statistical significance at a given locus in Han-Eskin meta-analysis. MANTRA-meta-analysis (Morris, 2011) was carried out for fine mapping across loci identified to be statistically significant. We calculated 99% credible intervals, and credible sets, as has been discussed previously (Morris, 2011).
We defined a significant locus based on the peak SNP with the lowest p value in a given region. A significant locus was defined as a 500MB region flanking a peak SNP on either side (total 1MB region). If there were SNPs outside this region that were statistically significant, these were defined as separate associated loci, once again identifying the variant with the lowest p value in the region, and defining a 500MB region around it on either side. We note that this definition is arbitrary, and in regions of high LD, or regions with strong association signals, statistically significant variation can extend across several MB. Therefore, where loci were adjacent to each other, we considered the hypothesis, that these loci represented one locus primarily, with ‘satellite’ loci representing the same peak signal. In order to understand whether these adjacent loci represented the same causal signal, we carried out joint conditional analyses to examine whether joint conditional analysis abolished the association at the ‘satellite’ locus. Following joint analyses, we reported ‘distinct’ loci as those that were associated with traits independently from surrounding regions. We found that for almost all adjacent loci, these satellite loci represented the same peak signal; we therefore collapsed these into single ‘distinct’ signals.
Conditional analyses and conditional meta-analyses
We carried out joint and conditional analyses to identify distinct association signals; these analyses were carried out for two scenarios:
To identify whether two or more adjacent loci (defined based on distance) represented a single locus, or multiple distinct loci.
To examine whether a peak variant at a known locus (previously associated with the given trait) was distinct from previously associated variants at that locus.
In both these scenarios, we carried out joint and conditional analyses either on the most significant SNP in the region (in scenario 1), or on all previously known SNPs identified to be associated with the trait (in scenario 2). Joint conditional analyses were carried out in GEMMA separately for each cohort, and these conditional estimates were then meta-analyzed using the Han-Eskin method implemented in METASOFT. As we did not have access to individual level data for the AADM cohort, or accurate LD reference data, we could not carry out conditional analysis for AADM. As a result, whether a locus was distinct was determined by a comparison of the conditional meta-analyzed p value from random-effects meta-analysis, to the original p value from meta-analysis across all cohorts excluding AADM. Association signals were considered distinct if the conditional meta-analytic p value < 5×10−09, or if in joint analyses with all other SNPs, the given SNP emerged most statistically significant in joint conditional analysis.
Previously known trait-associated SNPs within a given locus were extracted from the NHGRI catalog (MacArthur et al., 2017), from large consortium meta-analyses for given traits, and from a literature search.
Analyses of Transferability
Analyses across populations of differing ancestry and from different geographical regions across Africa allows an examination of transferability of association signals across regions. Understanding transferability of association signals has implications for the design and analysis of medical genetic studies in Africa.
For statistically significant association signals observed, we define transferability as the presence of nominally significant p values (p < 0.05) in at least two or more studies.
We note that the lack of transferability of association signals across diverse cohorts or populations does not always indicate artifactual signals. This can arise from differences in statistical power to observe association, including from differences in demographic structure of cohorts, measurement error in phenotypes, allele frequency differences, differential LD of sampled variants with the causal SNP(s), differences in accuracy of imputation across different populations and sample size and real differences in effect size due to gene-environmental interactions.
We examined statistical heterogeneity of effect as one of the factors affecting transferability of association signals across cohorts. In order to assess heterogeneity, we used the Cochran’s Q statistic, as output by METASOFT. We assessed this genome-wide, applying a stringent threshold of 5×10−09, equivalent to the genome wide association threshold for statistical significance. We note that this statistic is likely to be highly conservative, and that statistically significant heterogeneity is unlikely to be due to chance and is suggestive of real differences in effect size (as a result of either artifactual or biological factors). These differences are unlikely to be due to differences in allele frequency or sample size (which would affect the SE). We also evaluated the consistency of this statistic with the null (using QQ plots), and examined whether there were regions of high heterogeneity in associations with traits across the genome, and whether these were in regions of known associations with given traits. We confirmed that heterogeneity statistics did not show any inflation relative to the null (lambda < 1.05 for all traits).
Classification of discovered Loci
We defined distinct loci based on conditional analysis and distance metrics as defined in previous sections. Among identified independent loci, we define different categories of association signal as follows:
-
Novel locus (NL):
A locus that has not been previously associated with the given trait, or any biologically similar and correlated traits in previous GWAS, or in the literature.
-
Novel locus – known for related trait (NL-KRT):
A locus that has not been previously associated with the given trait, but has been associated with biologically similar or correlated traits in previous GWAS, or in the literature.
-
Known locus:
A locus that has been previously associated with the trait in a previous GWAS or in the literature. These loci can be divided into the following sub-categories:-
Known locus – known SNP (KS):The peak associated SNP at the locus has been previously identified as associated with the trait of interest in a previous GWAS or in the literature.
-
Known locus – unknown SNP:The given locus has been associated with the trait in a previous GWAS or in the literature, but the specific SNP is not known to be associated with the given trait. This can be further divided into two subcategories:
- Known locus – distinct association (KL-DA): On joint and conditional analysis the peak associated SNP is distinct from previously known SNPs associated at this locus.
- Known locus- non-distinct association (KL-NDA): On joint and conditional analysis, the peak associated SNP is not distinct from previously known SNPs associated at this locus.
-
Assessment of allelic heterogeneity
We assessed allelic heterogeneity at this locus by examining whether multiple causal variants were present in joint and conditional analysis within the Ugandan cohort. We carried out joint and conditional analysis by conditioning SNPs within a 1MB region (500KB flanks) around the peak SNP, and examining if another distinct signal below p < 5e-09 was observed following conditioning. If this was the case, we continued iteratively, conditioning on the two distinct SNPs identified, and so on. At each stage, prior to the conditioning step, we carried out joint analysis of the distinct SNPs identified, and dropped any SNPs with a p < 5e-09 in joint analysis.
Fine-Mapping at the HBA1/HBA2 Locus
In order to fine-map identified associations with serum total bilirubin and HbA1c locus, we considered that peak associations identified at this locus may be tagging a known common alpha thalassemia variant observed in African populations. This thalassemia variant has been thought to have risen to high frequencies in Africa due to protection conferred against severe malaria (Mockenhaupt et al., 2004), and has previously been associated with several hematological markers in cohorts including individuals of African-American ancestry (Chen et al., 2013). This deletion was not called in the 1000 Genomes Phase 3 project data, but was present in a previous release of the 1000 Genomes Project Phase 1 Project data with an MAF = 22% among Africans. In order to assess whether associations identified at this locus were being driven by the α−3.7 thalassemia deletion, we re-imputed data within this region with the 1000 Genomes Phase I imputation panel, and re-analyzed data using the same methods across all cohorts where phenotype data on bilirubin and HbA1c were available. We carried out joint conditional analysis between the α−3.7 thalassemia deletion, and the peak SNP identified in the region within our analysis in the data that did not include the deletion to identify the primary driver of the association signal within the region.
DATA AND CODE AVAILABILITY
Summary GWAS and allele frequency data are publicly available at https://www.ebi.ac.uk/gwas/downloads/summary-statistics. The combined UG2G+AGV imputation panel is available for imputation from the Haplotype Reference Consortium: http://www.haplotype-reference-consortium.org/participating-cohorts. All individual level data, phenotype, genotype and sequence data are available under managed access to researchers. Requests for access to the phenotypic data will be granted for all research consistent with the consent provided by participants. This would include any research in the context of health and disease, that does not involve identifying the participants in any way. The UMIC committees are responsible for curation, storage, and sharing of phenotypic and genetic data under managed access. The array and low and high depth sequence data have been deposited at the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/, accession numbers EGAS00001001558/EGAD00010000965, EGAS00001000545/EGAD00001001639 and EGAS00001000545/EGAD00001005346 respectively). Requests for access to data may be directed to segun.fatumo@mrcuganda.org. While data cannot be released on public databases as this would conflict with the study protocol and participant consent under which data were collected, we aim to facilitate data access for all bona fide researchers. Applications are reviewed by an independent data access committee (DAC) and access is granted if the request is consistent with the consent provided by participants within two weeks of submission. The data producers may be consulted by the DAC to evaluate potential ethical conflicts. Requestors also sign an agreement which governs the terms on which access to data is granted.
Supplementary Material
KEY RESOURCES TABLE
Highlights.
The Uganda Genome Resource comprises genetic and phenotypic data on 6,400 individuals
Ugandans show geographically correlated genetic substructure and complex admixture
The Uganda sequence panel substantially improves imputation in African populations
The Uganda Genome Resource enables novel discovery of loci associated with traits
ACKNOWLEDGMENTS
We acknowledge the H3Africa Bioinformatics Network (H3ABioNet) Node, National Biotechnology Development Agency (NABDA), and the Federal Ministry of Science and Technology (FMST) Abuja, Nigeria for funding S.F. for his postdoctoral research. D.N.C. wishes to acknowledge the financial support of Qiagen through a License Agreement with Cardiff University. We also acknowledge the 1000 Genomes Project, UK10K, Simon’s Foundation Genome Diversity Project, and African Genome Variation Project (AGVP) for providing data resources that were used to contextualize the UG2G data. The GATK3 program was made available through the generosity of Medical and Population Genetics program at the Broad Institute. The research was partially supported by the NIHR Leicester Biomedical Research Centre. L.V.W. holds a GSK/British Lung Foundation Chair in Respiratory Research. This work was funded by the Wellcome Trust, The Wellcome Sanger Institute (WT098051), the U.K. Medical Research Council (G0901213-92157, G0801566, and MR/K013491/1), and the Medical Research Council/Uganda Virus Research Institute Uganda Research Unit on AIDS core funding. This work was funded in part by IAVI with the generous support of the United States Agency for International Development (USAID) and other donors. The full list of IAVI donors is available at https://www.iavi.org. The contents of this manuscript are the responsibility of IAVI and co-authors and do not necessarily reflect the views of USAID or the U.S. Government. D.G. is funded by a UKRI HDR-UK Innovation Fellowship (reference MR/S003711/1). We thank the African Partnership for Chronic Disease Research (APCDR) for providing a network to support this study as well as a repository for deposition of curated data. We thank all participants who contributed to this study. We also acknowledge the NIH Research Cambridge Biomedical Research Centre. The authors wish to acknowledge the use of the Uganda Medical Informatics Centre (UMIC) compute cluster. Computational support from UMIC was made possible through funding from the Medical Research Council (MC_EX_MR/L016273/1). We acknowledge the Sanger core pipeline teams for their help with sequencing and mapping the whole genome sequence data. The authors acknowledge, with thanks, the participants in the AADM project, their families, and their physicians. The study was supported in part by the Intramural Research Program of the NIH in the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is supported by the National Human Genome Research Institute (NHGRI), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the Center for Information Technology, and the Office of the Director at the NIH (1ZIAHG200362). The research by N.S. is supported by the Wellcome Trust (WT098051 and WT091310), the EU FP7 (EPIGENESYS 257082 and BLUEPRINT HEALTH-F5-2011-282510), and the NIH Research Blood and Transplant Research Unit (NIHR BTRU) in Donor Health and Genomics at the University of Cambridge in partnership with NHS Blood and Transplant (NHSBT). D.G. was funded by the MRC (MR/S003711/1). A.J.M. was funded by the Well-come Trust (WT106289). S.F. received salary support from NIH grant U01MH115485 and the Makerere University-Uganda Virus Research Institute Centre of Excellence for Infection and Immunity Research and Training (MUII). MUII is supported through the DELTAS Africa Initiative (grant 107743). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS), Alliance for Accelerating Excellence in Science in Africa (AESA), and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107743) and the U.K. government. We acknowledge the H3Africa Bioinformatics Network (H3ABioNet) Node at the Center for Genomics Research and Innovation (CGRI), National Biotechnology Development Agency (NABDA), Abuja, Nigeria for supporting S.F. H3ABioNet is supported by the National Institutes of Health Common Fund (National Human Genome Research Institute) under grant number U41HG006941 We acknowledge use of summary data from the Global Lipids Genomics Consortium (GLGC) (Willer et al., 2013). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health, or NHSBT.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.10.004.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abadie JM, and Koelsch AA (2008). Performance of the Roche second generation hemoglobin A1c immunoassay in the presence of HB-S or HB-C traits. Ann. Clin. Lab. Sci 38, 31–36. [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, and Cardon LR (2002). Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet 30, 97–101. [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Hand-saker RE, Kang HM, Marth GT, and McVean GA; 1000 Genomes Project Consortium (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, Kozlitina J, Stender S, Wood GC, et al. (2018). A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med 378, 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeyemo AA, Tekola-Ayele F, Doumatey AP, Bentley AR, Chen G, Huang H, Zhou J, Shriner D, Fasanmade O, Okafor G, et al. (2015). Evaluation of Genome Wide Association Study Associated Type 2 Diabetes Susceptibility Loci in Sub Saharan Africans. Front. Genet 6, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, and Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altunkan S, Ilman N, Kayatürk N, and Altunkan E (2007). Validation of the Omron M6 (HEM-7001-E) upper-arm blood pressure measuring device according to the International Protocol in adults and obese adults. Blood Press. Monit 12, 219–225. [DOI] [PubMed] [Google Scholar]
- Altunkan S, Iliman N, and Altunkan E (2008). Validation of the Omron M6 (HEM-7001-E) upper arm blood pressure measuring device according to the International Protocol in elderly patients. Blood Press. Monit 13, 117–122. [DOI] [PubMed] [Google Scholar]
- Amorim CEG, Gao Z, Baker Z, Diesel JF, Simons YB, Haque IS, Pickrell J, and Przeworski M (2017). The population genetics of human disease: The case of recessive, lethal mutations. PLoS Genet 13, e1006915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiki G, Murphy G, Nakiyingi-Miiro J, Seeley J, Nsubuga RN, Karabarinde A, Waswa L, Biraro S, Kasamba I, Pomilla C, et al. ; GPC team (2013). The general population cohort in rural south-western Uganda: a platform for communicable and non-communicable disease studies. Int. J. Epidemiol 42, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer PL, Johnsen JM, Johnson AD, Logsdon BA, Lange LA, Nalls MA, Zhang G, Franceschini N, Fox K, Lange EM, et al. (2012). Imputation of exome sequence variants into population-based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am. J. Hum. Genet 91, 794–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR; 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmanov AA, and Beauchamp GK (2007). Taste receptor genes. Annu. Rev. Nutr 27, 389–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot A, Dupuy AM, Badiou S, Bargnoux AS, and Cristol JP (2012). Evaluation of three turbidimetric assays for automated determination of hemoglobin A1c. Clin. Lab 58, 1171–1177. [PubMed] [Google Scholar]
- Bergmeyer HU, Hørder M, and Rej R (1986a). International Federation of Clinical Chemistry (IFCC) Scientific Committee, Analytical Section: approved recommendation (1985) on IFCC methods for the measurement of catalytic concentration of enzymes. Part 2. IFCC method for aspartate aminotransferase (L-aspartate: 2-oxoglutarate aminotransferase, EC 2.6.1.1). J. Clin. Chem. Clin. Biochem 24, 497–510. [PubMed] [Google Scholar]
- Bergmeyer HU, Hørder M, and Rej R (1986b). International Federation of Clinical Chemistry (IFCC) Scientific Committee, Analytical Section: approved recommendation (1985) on IFCC methods for the measurement of catalytic concentration of enzymes. Part 3. IFCC method for alanine aminotransferase (L-alanine: 2-oxoglutarate aminotransferase, EC 2.6.1.2). J. Clin. Chem. Clin. Biochem 24, 481–495. [PubMed] [Google Scholar]
- Browning SR, and Browning BL (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MC, and Tishkoff SA (2010). The evolution of human genetic and phenotypic variation in Africa. Curr. Biol 20, R166–R173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, et al. ; Alcohol Genome-wide Association (AlcGen) Consortium; Diabetes Genetics Replication and Meta-analyses (DIAGRAM+) Study; Genetic Investigation of Anthropometric Traits (GIANT) Consortium; Global Lipids Genetics Consortium; Genetics of Liver Disease (GOLD) Consortium; International Consortium for Blood Pressure (ICBP-GWAS); Meta-analyses of Glucose and Insulin-Related Traits Consortium (MAGIC) (2011). Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet 43, 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EA, Tomov ML, Suhr ST, Luo J, Olmsted ZT, Paluh JL, and Cibelli J (2015). Derivation of Ethnically Diverse Human Induced Pluripotent Stem Cell Lines. Sci. Rep 5, 15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, and Liu Q (2011). A new approach to account for the correlations among single nucleotide polymorphisms in genome: wide association studies. Hum. Hered 72, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Tang H, Qayyum R, Schick UM, Nalls MA, Handsaker R, Li J, Lu Y, Yanek LR, Keating B, et al. ; BioBank Japan Project; CHARGE Consortium (2013). Genome-wide association analysis of red blood cell traits in African Americans: the COGENT Network. Hum. Mol. Genet 22, 2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JP, and Morris AP (2016). Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur. J. Hum. Genet 24, 1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Filippo C, Bostoen K, Stoneking M, and Pakendorf B (2012). Bringing together linguistic and genetic evidence to test the Bantu expansion. Proc. Biol. Sci 279, 3256–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Balick D, Li H, Adzhubei I, Sunyaev S, and Reich D (2015). No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet 47, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. (2010). Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81. [DOI] [PubMed] [Google Scholar]
- Durbin R (2014). Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 30, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECCLS (1989a). Determination of the catalytic activity concentration in serum of L-alanine aminotransferase. Klin Chem Mitt 20, 204–211. [Google Scholar]
- ECCLS (1989b). Determination of the catalytic activity concentration in serum of L-aspartate aminotransferase. Klin Chem Mitt 20, 198–204. [Google Scholar]
- Eriksson A, and Manica A (2014). The doubly conditioned frequency spectrum does not distinguish between ancient population structure and hybridization. Mol. Biol. Evol 31, 1618–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Kelly DE, Beltrame MH, Hansen MEB, Mallick S, Ranciaro A, Hirbo J, Thompson S, Beggs W, Nyambo T, et al. (2019). African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol 20, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. ; ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JK (2007). Evaluation of HbA1c on the Roche COBAS Integra 800 closed tube system. Clin. Biochem 40, 822–827. [DOI] [PubMed] [Google Scholar]
- Gallego Llorente M, Jones ER, Eriksson A, Siska V, Arthur KW, Arthur JW, Curtis MC, Stock JT, Coltorti M, Pieruccini P, et al. (2015). Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science 350, 820–822. [DOI] [PubMed] [Google Scholar]
- Gao X, Starmer J, and Martin ER (2008). A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol 32, 361–369. [DOI] [PubMed] [Google Scholar]
- Gascuel O (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol 14, 685–695. [DOI] [PubMed] [Google Scholar]
- Genome of the Netherlands, C. (2014). Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet 46, 818–825. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, and Gascuel O (2010). SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol 27, 221–224. [DOI] [PubMed] [Google Scholar]
- Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, Karthikeyan S, Iles L, Pollard MO, Choudhury A, et al. (2015). The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, and Di Rienzo A (2002). Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet 70, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, and Eskin E (2011). Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet 88, 586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han E, Sinsheimer JS, and Novembre J (2014). Characterizing bias in population genetic inferences from low-coverage sequencing data. Mol. Biol. Evol 31, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CM, and Davis OS (2014). From observational to dynamic genetics. Front. Genet 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckerman D, Gurdasani D, Kadie C, Pomilla C, Carstensen T, Martin H, Ekoru K, Nsubuga RN, Ssenyomo G, Kamali A, et al. (2016). Linear mixed model for heritability estimation that explicitly addresses environmental variation. Proc. Natl. Acad. Sci. USA 113, 7377–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW (2012). Resistance to malaria in humans: the impact of strong, recent selection. Malar. J 11, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, and Myers S (2014). A genetic atlas of human admixture history. Science 343, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn BM, Botigué LR, Gravel S, Wang W, Brisbin A, Byrnes JK, Fadhlaoui-Zid K, Zalloua PA, Moreno-Estrada A, Bertranpetit J, et al. (2012). Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet 8, e1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn BM, Botigué LR, Peischl S, Dupanloup I, Lipatov M, Maples BK, Martin AR, Musharoff S, Cann H, Snyder MP, et al. (2016). Distance from sub-Saharan Africa predicts mutational load in diverse human genomes. Proc. Natl. Acad. Sci. USA 113, E440–E449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman WH, and Cohen RM (2012). Racial and ethnic differences in the relationship between HbA1c and blood glucose: implications for the diagnosis of diabetes. J. Clin. Endocrinol. Metab 97, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird TR, Young EH, Pirie FJ, Riha J, Esterhuizen TM, O’Leary B, McCarthy MI, Sandhu MS, and Motala AA (2016). Study profile: the Durban Diabetes Study (DDS): a platform for chronic disease research. Glob. Health Epidemiol. Genom 1, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, and Abecasis GR (2012). Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet 44, 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudjashov G, Karafet TM, Lawson DJ, Downey S, Savina O, Sudoyo H, Lansing JS, Hammer MF, and Cox MP (2017). Complex Patterns of Admixture across the Indonesian Archipelago. Mol. Biol. Evol 34, 2439–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR (2002). Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18, 337–338. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, and Rosenberg NA (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. [DOI] [PubMed] [Google Scholar]
- Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, and Kamatani N (2010). Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet 42, 210–215. [DOI] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, and Eskin E (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, and Hammer MF (2008). New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 18, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson EK, Kwiatkowski DP, and Sabeti PC (2014). Natural selection and infectious disease in human populations. Nat. Rev. Genet 15, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AI, Kerfoot SM, Heit B, Liu L, Andonegui G, Ruffell B, Johnson P, and Kubes P (2004). Role of CD44 and hyaluronan in neutrophil recruitment. J. Immunol 173, 7594–7601. [DOI] [PubMed] [Google Scholar]
- Kloss-Brandstätter A, Pacher D, Schönherr S, Weissensteiner H, Binna R, Specht G, and Kronenberg F (2011). HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum. Mutat 32, 25–32. [DOI] [PubMed] [Google Scholar]
- Kleinberger JW, Copeland KC, Gandica RG, Haymond MW, Levitsky LL, Linder B, Shuldiner AR, Tollefsen S, White NH, and Pollin TI (2018). Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet Med 20, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, and Nielsen R (2014). ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanktree MB, Elbers CC, Li Y, Zhang G, Duan Q, Karczewski KJ, Guo Y, Tragante V, North KE, Cushman M, et al. (2015). Genetic meta-analysis of 15,901 African Americans identifies variation in EXOC3L1 is associated with HDL concentration. J. Lipid Res 56, 1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DJ, Hellenthal G, Myers S, and Falush D (2012). Inference of population structure using dense haplotype data. PLoS Genet 8, e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, et al. (2016). Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J, Lippert C, Kadie CM, Davidson RI, Eskin E, and Heckerman D (2012). Improved linear mixed models for genome-wide association studies. Nat. Methods 9, 525–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little RR, Rohlfing CL, Hanson S, Connolly S, Higgins T, Weykamp CW, D’Costa M, Luzzi V, Owen WE, and Roberts WL (2008). Effects of hemoglobin (Hb) E and HbD traits on measurements of glycated Hb (HbA1c) by 23 methods. Clin. Chem 54, 1277–1282. [DOI] [PubMed] [Google Scholar]
- Liu X, Ong RT, Pillai EN, Elzein AM, Small KS, Clark TG, Kwiatkowski DP, and Teo YY (2013). Detecting and characterizing genomic signatures of positive selection in global populations. Am. J. Hum. Genet 92, 866–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, and Berger B (2013). Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193, 1233–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finu-cane H, Schoenherr S, Forer L, McCarthy S, Abecasis GR, et al. (2016). Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet 48, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE (2014). The impact of population demography and selection on the genetic architecture of complex traits. PLoS Genet 10, e1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, et al. (2008). Proportionally more deleterious genetic variation in European than in African populations. Nature 451, 994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 45 (D1), D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher D, Smeeth L, and Sekajugo J (2010). Health transition in Africa: practical policy proposals for primary care. Bull World Health Organ 88, 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, et al. (2016). The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, and McVean G (2014). Demography and the age of rare variants. PLoS Genet 10, e1004528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. ; Haplotype Reference Consortium (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, Gordon D, Davis C, and Green P (2009). Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet 5, e1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, et al. (2003). Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. USA 100, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockenhaupt FP, Ehrhardt S, Gellert S, Otchwemah RN, Dietz E, Anemana SD, and Bienzle U (2004). Alpha(+)-thalassemia protects African children from severe malaria. Blood 104, 2003–2006. [DOI] [PubMed] [Google Scholar]
- Morris AP (2011). Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol 35, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvina V, and Schmidt KM (2008). On multiple-testing correction in genome-wide association studies. Genet. Epidemiol 32, 567–573. [DOI] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. (2010). From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalwoga A, Maher D, Todd J, Karabarinde A, Biraro S, and Grosskurth H (2010). Nutritional status of children living in a community with high HIV prevalence in rural Uganda: a cross-sectional population-based survey. Trop. Med. Int. Health 15, 414–422. [DOI] [PubMed] [Google Scholar]
- Nyholt DR (2004). A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet 74, 765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, Traglia M, Huang J, Huffman JE, Rudan I, et al. (2014). A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 10, e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Siddle KJ, Laval G, Quach H, Harmant C, Becker N, Froment A, Régnault B, Lemée L, Gravel S, et al. (2014). The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nat. Commun 5, 3163. [DOI] [PubMed] [Google Scholar]
- Patin E, Lopez M, Grollemund R, Verdu P, Harmant C, Quach H, Laval G, Perry GH, Barreiro LB, Froment A, et al. (2017). Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science 356, 543–546. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, and Reich D (2006). Population structure and eigenanalysis. PLoS Genet 2, e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, and Reich D (2012). Ancient admixture in human history. Genetics 192, 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peprah E, Xu H, Tekola-Ayele F, and Royal CD (2015). Genome-wide association studies in Africans and African Americans: expanding the framework of the genomics of human traits and disease. Public Health Genomics 18, 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, and Reich D (2014). Toward a new history and geography of human genes informed by ancient DNA. Trends Genet 30, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Patterson N, Loh PR, Lipson M, Berger B, Stoneking M, Pakendorf B, and Reich D (2014). Ancient west Eurasian ancestry in southern and eastern Africa. Proc. Natl. Acad. Sci. USA 111, 2632–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilia G, Chen WM, Scuteri A, Orrú M, Albai G, Dei M, Lai S, Usala G, Lai M, Loi P, et al. (2006). Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet 2, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, and Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Price AL, Weale ME, Patterson N, Myers SR, Need AC, Shianna KV, Ge D, Rotter JI, Torres E, Taylor KD, et al. (2008). Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet 83, 132–135, author reply 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, Beaty TH, Mathias R, Reich D, and Myers S (2009). Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet 5, e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Helgason A, Thorleifsson G, McCarroll SA, Kong A, and Stefansson K (2011). Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet 7, e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulit SL, Voight BF, and de Bakker PI (2010). Multiethnic genetic association studies improve power for locus discovery. PLoS ONE 5, e12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, and Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AI (1954). Economic development and tribal change: a study of immigrant labour in Buganda (Heffer and Sons) [Google Scholar]
- Richards M, Macaulay V, Hickey E, Vega E, Sykes B, Guida V, Rengo C, Sellitto D, Cruciani F, Kivisild T, et al. (2000). Tracing European founder lineages in the Near Eastern mtDNA pool. Am. J. Hum. Genet 67, 1251–1276. [PMC free article] [PubMed] [Google Scholar]
- Rotimi CN, Dunston GM, Berg K, Akinsete O, Amoah A, Owusu S, Acheampong J, Boateng K, Oli J, Okafor G, et al. (2001). In search of susceptibility genes for type 2 diabetes in West Africa: the design and results of the first phase of the AADM study. Ann. Epidemiol 11, 51–58. [DOI] [PubMed] [Google Scholar]
- Rotimi CN, Chen G, Adeyemo AA, Furbert-Harris P, Parish-Gause D, Zhou J, Berg K, Adegoke O, Amoah A, Owusu S, et al. ; Africa America Diabetes Mellitus (AADM) Study (2004). A genome-wide search for type 2 diabetes susceptibility genes in West Africans: the Africa America Diabetes Mellitus (AADM) Study. Diabetes 53, 838–841. [DOI] [PubMed] [Google Scholar]
- Rotimi C, Abayomi A, Abimiku A, Adabayeri VM, Adebamowo C, Adebiyi E, Ademola AD, Adeyemo A, Adu D, Affolabi D, et al. ; H3Africa Consortium (2014). Research capacity. Enabling the genomic revolution in Africa. Science 344, 1346–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, and Reich D (2014). The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf SL, Molokie RE, Nouraie M, Sable CA, Luchtman-Jones L, Ensing GJ, Campbell AD, Rana SR, Niu XM, Machado RF, et al. (2014). Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr. Respir. Rev 15, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeldt LB, Soi S, Lambert C, Ko WY, Coulibaly A, Ranciaro A, Thompson S, Hirbo J, Beggs W, Ibrahim M, et al. (2019). Genomic evidence for shared common ancestry of East African hunting-gathering popula tions and insights into local adaptation. Proc. Natl. Acad. Sci. USA Published online February 19, 2019 10.1073/pnas.1817678116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherag A, Dina C, Hinney A, Vatin V, Scherag S, Vogel CI, Müller TD, Grallert H, Wichmann HE, Balkau B, et al. (2010). Two new Loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and german study groups. PLoS Genet 6, e1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S, and Durbin R (2014). Inferring human population size and separation history from multiple genome sequences. Nat. Genet 46, 919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Langaney A, Chech M, Teschler-Nicola M, Paunovic M, Mennecier P, Hofreiter M, Possnert G, and Pääbo S (2004). No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol 2, E57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Thompson JC, Prendergast ME, Mittnik A, Sirak K, Hajdinjak M, Salie T, Rohland N, Mallick S, Peltzer A, et al. (2017). Reconstructing Prehistoric African Population Structure. Cell 171, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Achilli A, Semino O, Davies W, Macaulay V, Bandelt HJ, Torroni A, and Richards MB (2010). The archaeogenetics of Europe. Curr. Biol 20, R174–R183. [DOI] [PubMed] [Google Scholar]
- Song S, Sliwerska E, Emery S, and Kidd JM (2017). Modeling human population separation history using physically phased genomes. Genetics 205, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, and Cooper DN (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat 21, 577–581. [DOI] [PubMed] [Google Scholar]
- Sugiuchi H, Uji Y, Okabe H, Irie T, Uekama K, Kayahara N, and Miyauchi K (1995). Direct measurement of high-density lipoprotein cholesterol in serum with polyethylene glycol-modified enzymes and sulfated alpha-cyclodextrin. Clin. Chem 41, 717–723. [PubMed] [Google Scholar]
- Sugiuchi H, Irie T, Uji Y, Ueno T, Chaen T, Uekama K, and Okabe H (1998). Homogeneous assay for measuring low-density lipoprotein cholesterol in serum with triblock copolymer and alpha-cyclodextrin sulfate. Clin. Chem 44, 522–531. [PubMed] [Google Scholar]
- Tambets K, Yunusbayev B, Hudjashov G, Ilumäe AM, Rootsi S, Honkola T, Vesakoski O, Atkinson Q, Skoglund P, Kushniarevich A, et al. (2018). Genes reveal traces of common recent demographic history for most of the Uralic-speaking populations. Genome Biol 19, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. (2009). The genetic structure and history of Africans and African Americans. Science 324, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topouchian JA, El Assaad MA, Orobinskaia LV, El Feghali RN, and Asmar RG (2006). Validation of two automatic devices for self-measurement of blood pressure according to the International Protocol of the European Society of Hypertension: the Omron M6 (HEM-7001-E) and the Omron R7 (HEM 637-IT). Blood Press. Monit 11, 165–171. [DOI] [PubMed] [Google Scholar]
- Tseng CL, Brimacombe M, Xie M, Rajan M, Wang H, Kolassa J, Crystal S, Chen TC, Pogach L, and Safford M (2005). Seasonal patterns in monthly hemoglobin A1c values. Am. J. Epidemiol 161, 565–574. [DOI] [PubMed] [Google Scholar]
- van Oven M, and Kayser M (2009). Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat 30, E386–E394. [DOI] [PubMed] [Google Scholar]
- Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JR, Xu C, Futema M, Lawson D, et al. ; UK10K Consortium (2015). The UK10K project identifies rare variants in health and disease. Nature 526, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Teder P, Judd NP, Noble PW, and Doerschuk CM (2002). CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am. J. Pathol 161, 2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. ; Global Lipids Genetics Consortium (2013). Discovery and refinement of loci associated with lipid levels. Nat. Genet 45, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2010). STEPS Surveillance Manual https://www.who.int/chp/steps/manual/en/index.html.
- Xu C, Tachmazidou I, Walter K, Ciampi A, Zeggini E, and Greenwood CM; UK10K Consortium (2014). Estimating genome-wide significance for whole-genome sequencing studies. Genet. Epidemiol 38, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, and Tyler-Smith C; 1000 Genomes Project Consortium (2012). Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet 91, 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MA, Malaspinas AS, Durand EY, and Slatkin M (2012). Ancient structure in Africa unlikelytoexplain Neanderthal and non-African genetic similarity. Mol. Biol. Evol 29, 2987–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zaitlen NA, Goddard ME, Visscher PM, and Price AL (2014). Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet 46, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, Vollenweider P, Stirnadel H, Johnson T, et al. (2008). Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am. J. Hum. Genet 83, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitlen N, and Kraft P (2012). Heritability in the genome-wide association era. Hum. Genet 131, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S, and Price AL (2013). Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 9, e1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. (2016). Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol 34, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, and Stephens M (2012). Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, and Salit M (2014). Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary GWAS and allele frequency data are publicly available at https://www.ebi.ac.uk/gwas/downloads/summary-statistics. The combined UG2G+AGV imputation panel is available for imputation from the Haplotype Reference Consortium: http://www.haplotype-reference-consortium.org/participating-cohorts. All individual level data, phenotype, genotype and sequence data are available under managed access to researchers. Requests for access to the phenotypic data will be granted for all research consistent with the consent provided by participants. This would include any research in the context of health and disease, that does not involve identifying the participants in any way. The UMIC committees are responsible for curation, storage, and sharing of phenotypic and genetic data under managed access. The array and low and high depth sequence data have been deposited at the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/, accession numbers EGAS00001001558/EGAD00010000965, EGAS00001000545/EGAD00001001639 and EGAS00001000545/EGAD00001005346 respectively). Requests for access to data may be directed to segun.fatumo@mrcuganda.org. While data cannot be released on public databases as this would conflict with the study protocol and participant consent under which data were collected, we aim to facilitate data access for all bona fide researchers. Applications are reviewed by an independent data access committee (DAC) and access is granted if the request is consistent with the consent provided by participants within two weeks of submission. The data producers may be consulted by the DAC to evaluate potential ethical conflicts. Requestors also sign an agreement which governs the terms on which access to data is granted.