

Abstract
Small molecules continue to dominate drug discovery because of their ease of use, lower cost of manufacturing and access to intracellular targets. However, despite these advantages, small molecules are more likely to fail in clinical trials compared to biologicals, and their development remains limited to a small subset of disease-relevant “druggable” targets. Targeted protein degradation has recently emerged as a novel pharmacological modality that promises to overcome small molecule limitations whilst retaining their key advantages. Here, we use a Strengths-Weaknesses-Opportunities-Threats (SWOT) framework to critically assess the current status of this rapidly evolving field. We expect that degrader molecules are only the beginning of a range of novel targeting modalities that hijack existing endogenous cellular machineries to chemically redirect biological targets and pathways. Therefore, this piece may offer a roadmap for enhancing development of both degraders and related modalities.
Keywords: targeted protein degradation, small molecule drug discovery, degrader molecules, undruggable targets
Overview of small molecule drug discovery
If measured by the number of US Food and Drug Administration (FDA) approvals, 2018 was a remarkable year. A record-setting 59 new drugs were approved, including the first small interfering RNA-based drug [1], [2]. This continued the trend of approvals for novel pharmacological modalities, such as the first gene therapy [3] and the first cell therapies [4] approved in the previous year. Moreover, biologies approved in 2018 constituted almost 30% of the total number of approved drugs, close to 40% of the pipeline, and a steadily increasing market share [5]. The recent release of new drug approvals for 2019 aligns well with what we’ve seen in the two years prior [6]. Taken together, these trends could be interpreted as a signal that the dominance of small molecule drugs (see Glossary) is being challenged by biological modalities.
Small molecule-based drugs have been the mainstay of pharmacopeia for millennia. These types of drugs have several advantages that continue to make them indispensable as therapeutic agents. For example, small molecules drugs are cell permeable, making them ideal for engaging intracellular targets. Small molecule drugs are characterized by low molecular weights (usually under 600 Da) and synthetically tractable structures that can be optimized for oral bioavailability and metabolic stability. Low molecular weights facilitate their ability to cross the blood-brain barrier (BBB), and access target space that currently eludes biological modalities. Additionally, small molecule drugs are cheaper to manufacture, store and regulate, which usually makes them more affordable for health care systems and patients. However, these agents have important limitations such as: (1) off-target toxicity and side-effects given that, despite efforts to improve selectivity, most small molecules bind multiple targets; and (2) restricted target scope, as the number of proteins known to contain well-defined small molecule binding pockets remains limited (for a general comparison between small molecule drugs and biologies, see [7]). Moreover, small molecules are more likely to fail the drug approval process than biologicals, most commonly due to lack of efficacy [6]. Overall, this suggests that small molecule drug discovery and development would benefit from intentional and well-orchestrated disruptive innovations that would retain key advantages of small molecule drugs whilst mitigating the liabilities.
Traditionally, the majority of small molecule drugs act via reversible binding to their target. While the compound is bound to the target, it exerts its effect by modulating the target’s biological activity in a defined way that is of benefit in a specific disease context. For example, many small molecule drugs on the market are inhibitors of enzymatic activity, antagonists/agonists of G-protein coupled receptors (GPCRs), or inhibitors of nuclear receptors (NRs). Once the drug dissociates from the target, the effect is lost until the next binding event occurs. Recognizing this as a potential problem, several alternative modalities that extend pharmacological duration have recently emerged. One such modality is irreversible covalent targeting, whereby a small molecule forms a covalent bond with the target resulting in permanent occupancy and more durable effects [8] [9]. Covalent inhibitors have seen a resurgence of clinical and drug development relevance, driven primarily by the recent approvals of several covalent kinase inhibitors, such as afatinib, ibrutinib and osimertinib, for the treatment of cancer [10] [11]. Another recent modality that results in a more permanent perturbation is targeted protein degradation, where a small molecule binds both the target and an E3 ubiquitin ligase, an enzyme that mediates the last step in the ubiquitination process, thus hijacking the cellular ubiquitin-proteasome pathway [12]. The outcome of this mode of action is a polyubiquitinated target that undergoes proteasomal degradation (Fig. 1). Here, we will offer a brief introduction to targeted protein degradation and analyze some of the key issues surrounding this emerging topic (for those interested in a more comprehensive review of the area, please check a recent review [13]). We hope that our assessments will enhance future development of the field.
Fig. 1. Schematic representation of mechanism of action of small molecule degraders.
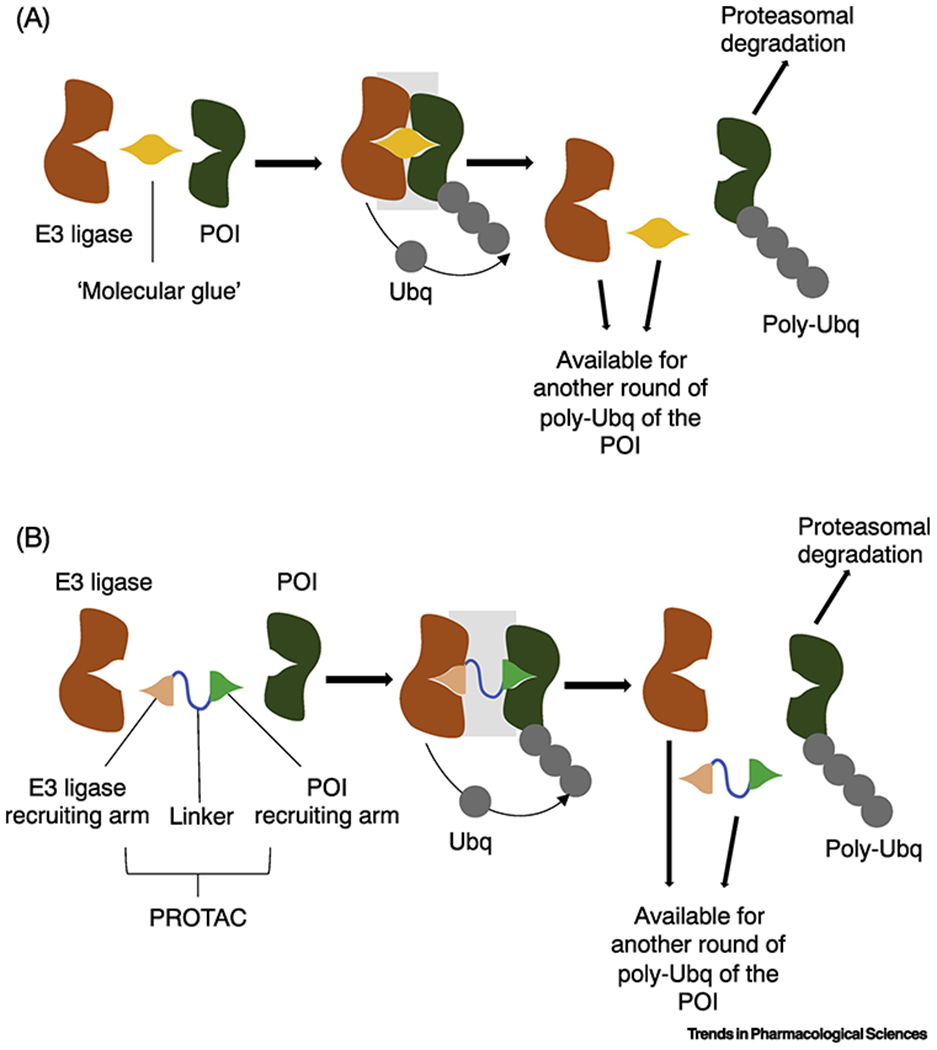
(a) “molecular glue” compounds are low-molecular weight degrader molecules that “glue” together the target protein of interest (POI) and an E3 ligase. In this instance, the “molecular glue” compound serves as a scaffold that induces protein-protein interactions that results in a productive, ubiquitination-competent ternary complex.
(b) PROTACs (proteolysis targeting chimeras) are degrader molecules that employ two distinct recruiting arms, one that binds the POI and the other that binds the E3 ligase. The two arms are connected via a linker that may also be involved in mediating productive ternary complex formation.
Both types of degrader molecules result in POI poly-ubiquitination, marking the POI for proteasomal degradation and removal from the cell.
Targeted protein degradation as a pharmacological modality
Targeted protein degradation is not an unprecedented mode of action given that thalidomide, a drug with a troubled history of birth defects and currently in use as a treatment for multiple myeloma and leprosy [14], acts by inducing targeted degradation of several transcription factors by recruiting them to an E3 ligase called cereblon (CRBN) [15]. In the case of thalidomide the exact mechanistic insight into how this drug exerts its effects emerged some six decades later after its initial use in humans [16]. These mechanistic insights have recently spurred the intense interest in rational development of degrader molecules.
Degrader molecules fall into two distinct categories. Thalidomide and related molecules are often referred to as “molecular glues”. These molecules use one chemical scaffold to simultaneously bind and bring into proximity (“glue”) the target protein of interest (POI) and an E3 ligase (Figure 1a). On the other hand, proteolysis targeting chimeras (PROTACs; Figure 1b) [17] use two distinct recruiting arms, one optimized to bind the POI and the other to bind the E3 ligase, connected via a linker. Chemical structures of representative molecules from each of the two categories are shown in Figure 2 to illustrate the basic design principles [18], [19]. At the time of writing, only a handful of small molecule degraders have been approved for clinical use (thalidomide and its derivatives), and several additional degraders that target NRs have recently entered clinical trials (ARV-110, which targets androgen receptor [20] and ARV-471 that targets estrogen receptor [21]).
Fig. 2. Chemical structures of representative degrader molecules: thalidomide, example of a “molecular glue” compound, ARV-825 and dBET-1, examples of PROTAC compounds.
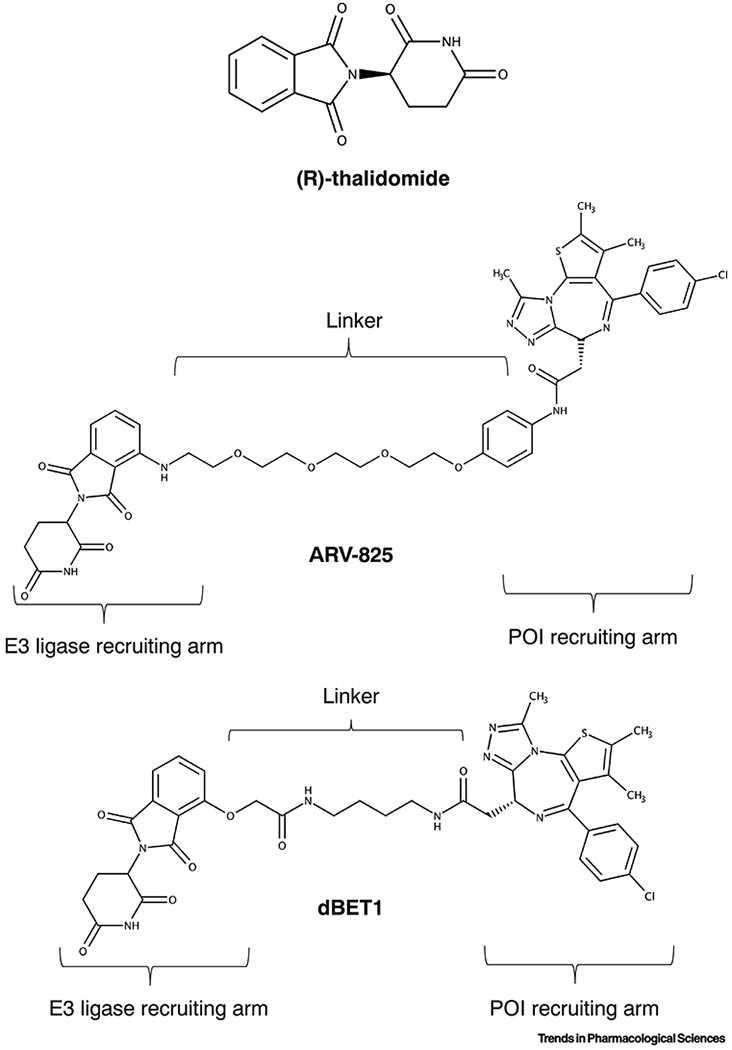
As clearly indicated, the two PROTAC molecules have an E3 recruiting arm (thalidomide-derived warhead optimized to bind E3 ligase cereblon (CRBN)), a protein-of-interest (POI) recruiting arm (JQ1 warhead targeted towards BET bromodomain containing proteins), and a variable linker.
Over the last five years, progress to rationally develop PROTAC degrader molecules has yielded compounds that induce degradation of a range of targets. In addition to targeting NRs, PROTAC-based degraders have been exploited to target kinases, including Bruton’s tyrosine kinase (BTK) [22] [23]. Bcr-Abl tyrosine kinase [24], anaplastic lymphoma kinase (ALK) [25] [26], receptor tyrosine kinases [27], and cyclin-dependent kinases like CDK4 and/or 6 [28]–[30] and CDK9 [31] [32], as well as epigenetic targets [18], [19], [33]–[35] (for an up-to date and exhaustive list of available PROTACs please refer to [36]). We have now reached the stage where new degraders, including those for targets previously considered to be difficult or even impossible to target, such as tau protein [37], are being disclosed, with interesting compounds being generated in both academia and industry.
SWOT-based strategic analysis
Given the growing enthusiasm for targeted protein degradation research, we will use this Opinion article as an opportunity to take a step back and highlight our views on strategic priorities for the next several years. To help us frame the discussion, we will employ a ‘Strengths-Weaknesses-Opportunities-Threats (SWOT)’ analysis, which is a widely used business development and strategic planning tool. In that context, the results of SWOT analyses are used to formulate key actionable points that, if implemented, will result in long-term business growth and competitor differentiation. Although scientific research may appear less predictable and more communal than business development, we think that strategic planning could help to expedite the translation of scientific discoveries into clinical solutions. This is especially relevant for an emerging area of research such as small molecule-mediated targeted protein degradation.
For the purposes of this Opinion, we will define strengths as specific advantages of degrader molecules over other targeting modalities. Degraders add to the translational (and basic research) toolbox an ability to directly ‘knockdown’ a POI. These protein knockdowns are comparatively rapid and reversible and therefore complementary to genetic strategies such as CRISPR/Cas9. As a result, targeted degradation is emerging as a valuable method for preclinical target validation [38], [39]. Beyond that, it is broadly believed that degraders offer advantages over other small molecule-based agents because:
degraders do not require tight binding to exert their effects, meaning that degrader development, even for POIs that lack well-defined small molecule binding pockets, could be feasible. This would open opportunities to target POIs that are currently considered to be “undruggable” [40]. Related to this is the fact that degraders don’t need to inhibit POIs function, they only need to bind the target, which, again, strongly suggests that targeted degradation is a strategy worth pursuing for targets that are currently considered to be beyond the reach of traditional small molecule modalities;
degrader molecules often possess improved selectivity profiles over inhibitors [22], [41]. To be effective they must mediate productive complex formation between the POI and an E3 ligase, thus forming a unique set of binding requirements and recognition elements that span the degrader molecule, POI and the ligase. This decreases the range of targets for which a single degrader molecule would lead to productive target ubiquitination. The rates of proteasomal processing could also be significantly different for ubiquitinated proteins. However, here caution needs to be taken as degrader molecules could still bind to a number of different targets without resulting in their degradation, while still causing a perturbation of their function (as discussed below under weaknesses);
degraders are likely to be more efficacious. Although the efficacy of PROTAC-based degrader molecules in the clinical setting is yet to be confirmed, preclinical work suggests that these molecules exert their effects at lower concentrations and for longer time periods [42]. Moreover, because degradation removes the entire POI, the effects are expected to be more profound and durable [13] [21]. This is especially important for multifunctional proteins that in addition to enzymatic activity play scaffolding or other roles. Here, while a traditional small molecule inhibitor affects the activity only, a degrader obliterates the full range of functions through protein removal.
Taken together, degrader molecules are proposed to exhibit some unique strengths over existing targeting modalities. In our view, the most far-reaching features are 1) possible access to novel targets and 2) the potential for improved efficacy. Currently approved degrader molecules (thalidomide and its derivatives) are examples of compounds that deplete transcriptions factors, previously considered “undruggable”. Although the two PROTAC-based degrader molecules in clinical trials (ARV-110 [20] and ARV-471 [21]) target “druggable” NRs (androgen receptor (ER) and estrogen receptor (AR), respectively), we anticipate that agents for difficult and/or currently impossible-to-drug targets will soon follow suit.
Despite the strengths we highlight, degrader molecules also have some weaknesses that should be acknowledged and discussed. One of the most often mentioned weakness of PROTAC-based degrader molecules is their non-compliance with Lipinski’s “Rule-of-5” (Ro5) [43]. This issue has recently been reviewed [44], [45], and we will not go into details here. Suffice to say, PROTACs in current clinical trials are orally bioavailable. However, as pointed out by Churcher [44], we may need to formulate new and/or additional rules to support development of this kind. In our view, other relevant issues are:
current lack of clear design rules and principles means that the field is still largely empirical. At the moment, for the targets with no known scaffolding functions, it is not possible to predict whether a degrader strategy will offer an efficacy advantage over inhibition. Additionally, it is difficult to predict which E3 ligase will result in a better performing PROTAC molecule, and as a result several different series may need to be developed and characterized (note that in vitro native mass spectrometry may help address this issue [46]). Generally, the PROTAC field has been somewhat slow to discuss and develop design principles optimized for enhancing their pharmacodynamic and pharmacokinetics parameters (e.g. considerations of rotatable bond count in long and flexible PROTAC linkers and its negative influence on cellular permeability and oral absorption), although a useful commentary of these issue has recently been published [47]. Overall, better understanding of structural, biochemical, kinetic and thermodynamic aspects of PROTAC design are needed to facilitate formulating useful guidelines and a growing number of efforts have been aimed in this direction [48]–[52];
the variety of E3 ligases currently used for PROTAC-mediated targeting is low. Despite the fact that the human genome encodes approximately 600 E3 ligases (not all mediating ubiquitination) [53], most efforts thus far have focused on CRBN and von Hippel-Lindau (VHL). Further research is needed to expand the pool of E3 ligases that can be hijacked by PROTAC and/or “glue” degraders. This requires the discovery of novel binders for the ligases, a task that has not been trivial thus far. Therefore, despite some recent progress [54], [55], we expect that this will remain a challenge for the field over the next few years;
“Hook effect” is a signature bell-shaped concentration dependence of activity for PROTAC degraders [56]. At high concentrations, PROTAC molecules can saturate binding sites on either the POI or the E3 ligase, without forming the required ternary complex. This results in decreased activity, and protection from induced degradation [57]. We expect that the “hook effect” will pose certain challenges to clinical studies and optimizing dosing regimens;
as mentioned above, degrader molecules offer an opportunity for enhanced selectivity. For example, several highly promiscuous kinase inhibitors displayed a significantly improved selectivity when developed into degraders [22], [41]. However, for PROTACs that use inhibitors as the POI recruiting arm, it is important to rule out the role of the intrinsic inhibitory activity to establish that the observed phenotype is driven by degradation of the intended target;
safety issues resulting from whole protein depletion. Although this is of course a key efficacy ‘strength’, removing the entire protein may also bring with it toxicities, which may be difficult to predict;
synthetic complexity of larger compounds. This slows down development and increases costs because each synthetic step needs optimization and each intermediate often needs purification. Also, pharmaceutical properties such as a stable solid form can be more difficult to achieve;
current lack of community-endorsed standards and guidelines for degrader validation. Although the number of disclosed molecules reported to act as selective degraders has increased exponentially, the extent to which these compounds have been characterized in terms of their POI and E3 ligase engagement, proteasome and ubiquitin dependence or their activity, cellular permeability, in-cell target selectivity, and phenotype dependence on degradation has varied. Recently, Collins et al. proposed a set of validation experiments that could form a useful framework for degrader characterization [58], which follows previous guidelines for target validation using chemical probes [59], [60]. Additionally, a recent commentary voiced similar suggestions [61], further highlighting the need for more guidelines in this area. We summarize main aspects of the degrader validation and characterization process in Figure 3. As pointed out by Collins et al., the workflow for degrader validation needs to include experimental evidence that the degrader engages with the target as well as the E3 ligase, and that this leads to formation of a productive ternary complex. Additionally, evidence of target degradation must be provided, as well as evidence that the decrease in target level is dependent on polyubiquitination and proteasome activity. Also important is to map the selectivity profile of a degrader molecule (preferably using quantitative proteomics methods), and provide cell-based evidence that the degrader engages the target, resulting in its depletion. Here, validation experiments should include appropriate control compounds (for example PROTAC-like molecules that do not bind to the E3 ligase due to small chemical modifications to the E3 ligase recruiting arm), and efforts should be made to establish that the observed phenotype is caused by POI degradation. In our view, Figure 3 offers an overview of basic expectations for degrader molecule validation if such molecules are to be used as cell-based research tools. For a degrader to advance into in vivo animal studies, further characterization of pharmacokinetic and pharmacodynamic properties should be a required step, as well as toxicology studies.
We believe that clarity on standards and working guidelines will help not only researchers developing and using targeted degraders, but also those evaluating them (editors, peer reviewers, grant panels and readers). Overall, in our view, the lack of design rules and standards for validation are the two most significant current weaknesses of the field. Addressing these gaps will create important opportunities for future growth.
Fig. 3. Degrader molecules validation criteria.
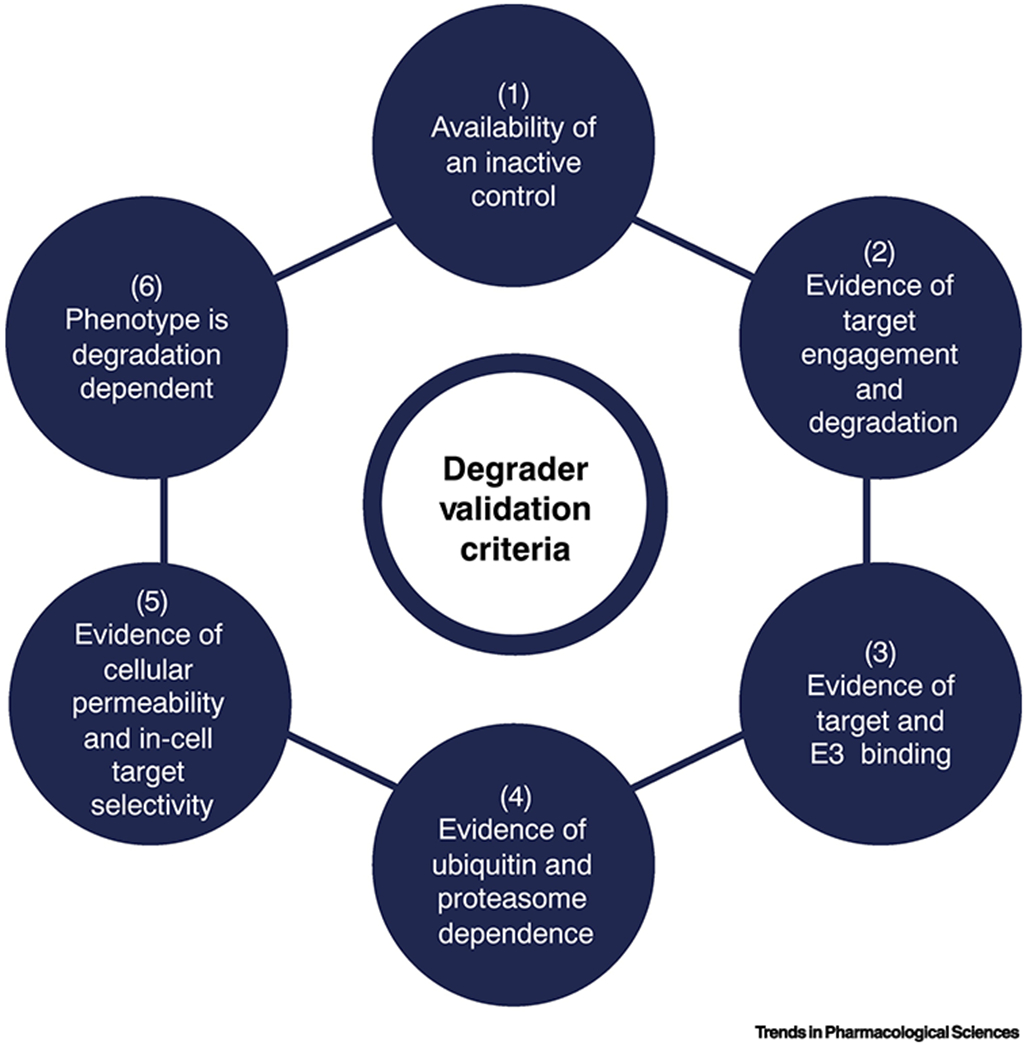
A set of criteria that can help formalize the process of degrader characterization and validation. The criteria can be useful for structuring laboratory workflows, as well as for evaluating newly reported degraders. The criteria presented here are based on Collins et al. [58]. Overall, in addition to control compounds that should be used in parallel throughout the validation process (1), the workflow for degrader validation needs to include experimental evidence of: (2) target engagement and degradation; (3) target and E3 ligase binding; (4) polyubiquitination and proteasome dependence of the process; (5) cellular permeability and in-cell target selectivity; and (6) phenotype dependence on degradation.
In the classical SWOT analysis Opportunities and Threats are defined based on a consideration of external factors that affect a given business enterprise. Here, we will step away from this viewpoint, as some opportunities and threats are, to some extent, internal to the field. Some of the most immediate opportunities that we see stem from the need to define rules and predictive models to accelerate the discovery of novel targets, E3 ligases and chemical matter. In addition to this, three other opportunities that we currently see as being of enormous value are:
the use of degrader molecules as research tools. Small molecule degraders, both “glues” and PROTACs, are emerging as powerful research tools for precise perturbation of the proteome. They represent protein knock-down reagents that achieve selective POI removal on timescales that are not accessible to genetic strategies (often as rapidly as 30 minutes to hours). They also offer an opportunity to readily assess dose-dependence which can facilitate target validation studies. Although the degrader modality has been hailed as a breakthrough in drug discovery and development, and potential advantages discussed from a more pharmacological perspective, we believe that using degraders as tools for basic research still remains an untapped opportunity. For example, a recent study described PROTACs that induce selective degradation of CDK4 and CDK6 that now offers, for the first time, an opportunity to study distinct activities of these two closely related cyclin-dependent kinases involved in regulation of the cell cycle [28], [29]. In order to realize their full potential, we would like to encourage those developing degraders to start thinking beyond therapeutic modalities and seize the opportunity to use these molecules as unique tools for probing biology. These efforts should go hand in hand with strengthening the ‘degrader community’ (see threats for more on this) and developing standards for degrader validation and utility (for more see weaknesses);
expanding the biological target space of current degraders. To date, the field has been dominated by PROTACs targeting hormone receptors, kinases and bromodomains, and immunomodulatory molecular glues that target transcription factors. As the modality gathers pace, there will no doubt be greater variety in the pharmacologically relevant degradable proteome in the future.
using lessons learned from targeted protein degradation to inform the development of additional modalities. Broadly speaking, degrader molecules represent a subset of small molecule dimerizers, compounds that enable chemically induced dimerization (CID) of biomolecules that have not evolved to form dimers [62], [63]. In some cases, this induced dimerization has an activity-based functional outcome, such as ubiquitination of a POI induced by the degrader molecules. One can imagine developing related systems that would recruit enzymes to POIs that control post-translational modifications beyond ubiquitination, as shown recently using phosphatases [64]. Small molecules may also be used to redirect proteins to different regions of the cell. For instance, ceapin was shown to induce the association of ATF6α in the ER with ABCD3 in the peroxisome, hence acting as an inter-organelle tether [65]. As such, we are optimistic that we will see a new wave of activity-hijacking CIDs in the near future. As the current leading edge of that wave, degrader molecules are paving the way and we have an opportunity to use what we learn through the development of degraders to help define rules, guidelines, use cases, and the community.
Overall, degrader molecules are one of the newest additions to drug discovery and development pipelines, with numerous exciting opportunities. In particular, the ability to tackle difficult targets for which the development of traditional inhibitors is impossible is a notable prospect for the area. Here, we chose to comment on opportunities that have not been remarked upon previously, namely the use of degraders as research tools and as a founding modality of a broader category of activity-hijacking CIDs. We hope that this will encourage others across areas and sectors to join and expand these efforts, and realize some of the broader possibilities.
However, there are definitely potential threats that we should acknowledge. The entire field is currently keeping a close eye on the two PROTAC-based degraders in clinical trials, as their outcome will shape current interest in this therapeutic modality. In addition to this uncertainty, we would like to highlight several factors that we feel may pose serious limitations on how far and how fast degrader molecules advance.
incomplete knowledge of biology. This includes not only incomplete knowledge of the ubiquitin-proteasome system but also the superficial characterization of how degrader molecules function – these deficiencies will hinder progress. Currently, the majority of targeted degradation research is centered on progressing to the clinic as rapidly as possible. This prioritizes different aspects of degrader development that do not necessarily include a deeper mechanistic investigation of molecular pharmacology. Therefore, to mitigate this threat we would like to encourage the broader research community to participate in targeted protein degradation, both as users and developers. Moreover, we are convinced that the use of degrader molecules as research tools will yield basic biological insights inaccessible to other strategies;
lack of a cohesive and inclusive community. There is a great deal of work to be done to develop and foster the adoption of standards and policies for reporting, data sharing, publishing and review, and to advocate for the field within academic, funding and regulatory agencies. Moreover, evidence suggests that disciplinary fragmentation limits scientific progress [66], further highlighting the need for cohesion and collaboration. Therefore, we invite comments on our opinions expressed here in any format in the hope that it may help nucleate the community. Additionally, we are heartened by the rise in grass roots (nonprofit) scientific conferences in this area, as we view this as an effective way to build and strengthen the community;
naivety. In the early days of therapeutic RNAi research, there was a belief that the modality would enable all targets to yield to therapeutic intervention. This led to some false hope and was something of a distraction to the field. Over time, it was realized that certain targets are not well-suited to the RNAi modality, while others, such as those in the liver, are a great fit. Our hope is that the hype surrounding PROTACs in particular does not lead to unrealistic expectations.
The main take-home messages of the SWOT analysis discussed here are summarized in Figure 4, Key Figure. Collectively, degrader molecules have unique strengths that set them apart from other targeted therapies and research tools. They also serve as an inspiration for additional innovative targeting modalities under the umbrella of activity-hijacking CIDs. However, we should acknowledge and embrace the challenges in the area and come together as a community to address them.
Fig. 4, Key Figure. Summary of the SWOT analysis of targeted protein degradation.
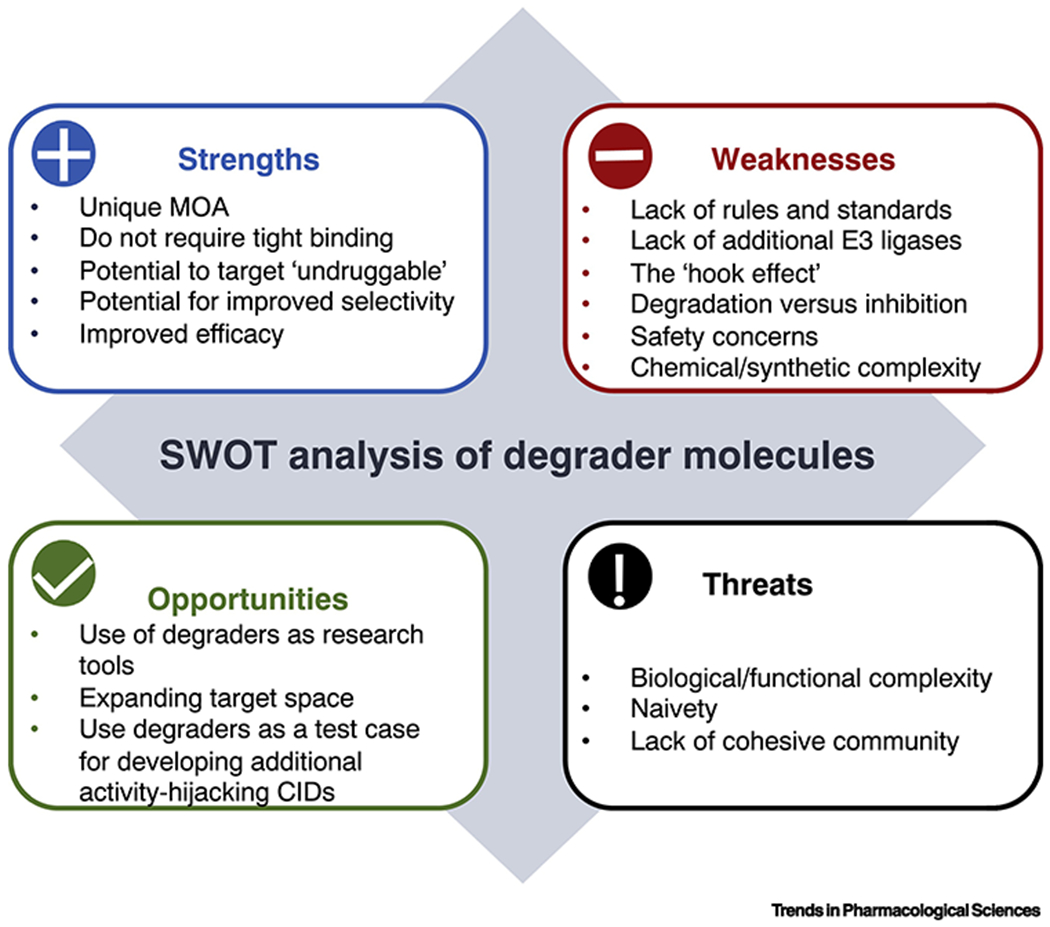
Here we summarize the different strengths, weaknesses, opportunities and threats in the field in an attempt to help in a strategic development of the field. Key Strengths of targeted protein degradation as pharmacological and research tool modality are: (1) their unique mechanism of action (MOA) as they induce degradation of the target, therefore removing not only its activity (which is the feature inhibitors affect) but scaffolding or other roles the target may have; (2) potential to target proteins currently considered to be difficult to address using small molecules given that degrader molecules does not require tight binding or the presence of high-affinity binding pockets on the target, and (3) potential for improved selectivity and efficacy. On the other hand, major Weaknesses of degrader molecules are: (1) lack of rules and standards to guide their development and optimization, as well as sufficient numbers of E3 ligases that can be employed for degrader development; (2) concentration dependence that results in decreased activity at high concentrations called “hook effect”, which may render dosing difficult and raise concerns regarding the safety; (3) potential for chemical/synthetic complexity, something that may become increasingly important as attempts to decouple inhibitory activity from degradation activity become more prominent. In terms of Opportunities, the major ones are: (1) use of degrader molecules as research tools to better understand biological function and disease physiology, as well as potentially expand the target space; and (2) use of degraders as a test case for developing additional modalities that use similar activity-hijacking mode of action (activity-hijacking chemical inducers of dimerization (CIDs). In terms of threats, the most significant ones are: (1) biological and functional complexity of the biology coupled with potentially inflated expectations; and (2) lack of cohesive community to drive standard and technology development in a more organized and open manner. Taken together this roadmap identifies clear opportunities to explore and points to potential pitfalls to avoid in order to accelerate future growth in this area.
Concluding remarks
Targeted protein degradation is an exciting therapeutic modality that addresses the key reason for attrition in clinical trials – lack of efficacy in Phase II. In theory, a degrader will deliver enhanced efficacy and more closely mimic genetic methods of whole protein depletion. The field is developing at a remarkable pace and there has been considerable investment in biotech and pharma companies to further exploit targeted protein degradation [67].
Our SWOT analysis (Figure 4) highlights not only the advantages and challenges, but also the considerable opportunities facing targeted protein degradation research. We expect that degrader molecules represent the tip of the iceberg for a range of novel targeting modalities that are based on using synthetically tractable molecules as CIDs and hijackers of existing endogenous cellular machineries towards neo-substrates (see Outstanding Questions).
Outstanding Questions Box.
What are the validation standards that each newly reported degrader molecule (regardless whether it is monovalent or bivalent) needs to satisfy?
How does one effectively decouple degradation effects from inhibition?
Which targets should be prioritized for degradation vs. inhibition (or other modes of functional modulation)?
How can we more effectively discover additional monovalent (“glue”) degraders?
Which E3 ligases should be pursued for degrader development?
What happens to the interactome and/or proteome after a mature protein is removed from it?
Do degrader molecules represent a tip of the iceberg and a shift from inhibitor-based mentality toward a binder-based mentality in drug discovery and development?
Overall, we think that one of the first steps the community would need to take is towards defining validation standards. In our view, the standards are urgently needed to prevent disclosures and wide-spread use of suboptimal degrader molecules that may lead to inaccurate scientific record and conclusions, not unlike what has been happening with other research tools, like chemical probes [68] and antibodies [69].
Some additional outstanding questions that will challenge the researchers in this area in years to come revolve around developing new methods and platforms to streamline the process of discovering new “glue” degraders and new ligands for E3 ligases. Additional technological development is also needed in order to quantify and investigate changes, including dynamic ones, to the interactome and/or proteome upon degrader treatment. The field will also need to develop a strategy that will help distinguish targets that are best addressed via traditional inhibition from those that are best degraded. Here, rules to stratify targets as well as match the target with the ligase and with the most suitable degrader molecule will require a significant amount of future work.
Broadly speaking, we consider degraders as examples of a current shift from inhibitor-based targeting strategies towards binder-based approaches [70]. There are reasons to believe that many small molecules in current use affect the levels of their targets, and we strongly recommend that every small molecule characterization/validation workflow includes assessment of its effects on protein levels [71]. Small molecule ligands should also be investigated for their effects on protein location as this may play a key role in their molecular mode of action [72].
Taken together, we expect that degrader molecules are only the beginning of a range of novel targeting modalities that hijack existing endogenous cellular machineries to chemically redirect biological targets and pathways. Therefore, we hope that the roadmap for enhancing development of both degraders and additional modalities we present here will be a valuable contribution to these efforts.
Highlights.
Degrader molecules, small molecules that induce targeted degradation of a protein of interest, are of increasing interest in drug development and discovery.
What makes degrader molecules attractive is that they offer several advantages over both small molecule inhibitors and biologies.
Examples of degrader molecules that have been approved for human treatment are immunomodulatory drugs (IMiDs), while additional degrader molecules are undergoing clinical trials.
The Strengths-Weakness-Opportunities-Threats (SWOT) analysis was used as a framework to identify key components that need to be in place to ensure accelerated future growth and prepare for some of the challenges.
In our view, the field represent just the beginning of a new wave of small molecule based modalities that act via hijacking specific elements of endogenous cellular machineries.
Acknowledgement
M.K. is supported by the Linde Program in Cancer Chemical Biology and the NCI T32 CA236754. M.K. also acknowledges attendees of the 1st Proteome-Targeted Drug Discovery Summit 2019 for very helpful discussion.
Disclaimer Statement
M. K. is a paid consulting editor at Life Science Editors.
L. H. J. receives funding from Deerfield.
GLOSSARY
- “Hook effect”:
phenomenon observed for PROTAC degrader molecules (and previously documented for antibodies) where the effects decrease with increasing concentration of the degrader past a certain peak point. This is due to higher concentrations of PROTACs favoring binary complex formation, rather than ternary resulting in “hook”-shaped activity vs. concentration curve
- Lipinski’s “Rule-of-5” (Ro5):
an empirical set of rules which states that poor absorption or permeation is more likely when a compound contains >5 hydrogen bond donors, >10 hydrogen bond acceptors, a LogP >5 or when the molecular weight is >500 (Lipinski et al)
- Small molecule drug:
a low molecular weight compound (often less than 600 Daltons) that is approved for the treatment of human disease
- Strengths-Weaknesses-Opportunities-Threats (SWOT) analysis:
a tool that dissects SWOT characteristics of an entity to identify areas of strategic priority
- Targeted protein degradation:
a novel therapeutic modality that uses small molecules to hijack endogenous protein degradation machinery towards neo-substrates. In this way, small molecules induce degradation of a protein target of interest
- Ubiquitin-proteasome pathway:
a cellular process involving an enzymatic cascade consisting of three enzymes E1, E2 and E3 that work together to transfer a small protein called ubiquitin onto a substrate protein. The result of this process is a ubiquitinated substrate. A substrate that becomes poly-ubiquitinated is recognized by the proteasome, the intracellular proteolytic machinery, and degraded. This process regulates homeostasis of many cellular proteins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Milliard A, “2018 FDA drug approvals,” Nat. Rev. Drug Discov, vol. 18, no. 2, pp. 85–89, Feb. 2019, doi: 10.1038/d41573-019-00014-x. [DOI] [PubMed] [Google Scholar]
- [2].de la Torre BG Albericio F, “The Pharmaceutical Industry in 2018. An Analysis of FDA Drug Approvals from the Perspective of Molecules,” Molecules, vol. 24, no. 4, p. 809, Feb. 2019, doi: 10.3390/molecules24040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Trapani I and Auricchio A, “Seeing the Light after 25 Years of Retinal Gene Therapy,” Trends Mol. Med, vol. 24, no. 8, pp. 669–681, Aug. 2018, doi: 10.1016/j.molmed.2018.06.006. [DOI] [PubMed] [Google Scholar]
- [4].Dyer O, “FDA panel recommends approval of first gene therapy in US,” BMJ, p. j3443, Jul. 2017, doi: 10.1136/bmj.j3443. [DOI] [PubMed] [Google Scholar]
- [5].Smietana K, Siatkowski M, Moller M, “Trends in clinical success rates,” Nat. Rev. Drug Discov, vol. 15, no. 6, pp. 379–380, Jun. 2016, doi: 10.1038/nrd.2016.85. [DOI] [PubMed] [Google Scholar]
- [6].Mullard A, “2019 FDA drug approvals,” Nat. Rev. Drug Discov, vol. 19, no. 2, pp. 79–84, Feb. 2020, doi: 10.1038/d41573-020-00001-7. [DOI] [PubMed] [Google Scholar]
- [7].Samanen J, “Similarities and differences in the discovery and use of biopharmaceuticals and small-molecule chemotherapeutics,” in Introduction to Biological and Small Molecule Drug Research and Development, Elsevier, 2013, pp. 161–203. [Google Scholar]
- [8].Zhang T, Hatcher JM, Teng M, Gray NS, and Kostic M, “Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation,” Cell Chem. Biol, vol. 26, no. 11, pp. 1486–1500, Nov. 2019, doi: 10.1016/j.chembiol.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baillie TA, “Targeted Covalent Inhibitors for Drug Design,” Angew. Chem. Int. Ed, vol. 55, no. 43, pp. 13408–13421, Oct. 2016, doi: 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- [10].Singh J, Petter RC, Baillie TA, and Whitty A, “The resurgence of covalent drugs,” Nat. Rev. Drug Discov, vol. 10, no. 4, pp. 307–317, Apr. 2011, doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- [11].Zhao Z and Bourne PE, “Progress with covalent small-molecule kinase inhibitors,” Drug Discov. Today, vol. 23, no. 3, pp. 727–735, Mar. 2018, doi: 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- [12].Kleiger G and Mayor T, “Perilous journey: a tour of the ubiquitin-proteasome system,” Trends Cell Biol, vol. 24, no. 6, pp. 352–359, Jun. 2014, doi: 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schapira M, Calabrese MF, Bullock AN, and Crews CM, “Targeted protein degradation: expanding the toolbox,” Nat. Rev. Drug Discov, vol. 18, no. 12, pp. 949–963, Dec. 2019, doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- [14].Brynner R and Stephens TD, Dark remedy: the impact of Thalidomide and its revival as a vital medicine. New York: Basic Books, 2001. [Google Scholar]
- [15].Ito etal T., “Identification of a Primary Target of Thalidomide Teratogenicity,” Science, vol. 327, no. 5971, pp. 1345–1350, Mar. 2010, doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- [16].Donovan KA et al. , “Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome,” eLife, vol. 7, Aug. 2018, doi: 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ, “Protacs: Chimeric molecules that target proteins to the Skpl-Cullin-F box complex for ubiquitination and degradation,” Proc. Natl. Acad. Sci, vol. 98, no. 15, pp. 8554–8559, Jul. 2001, doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lu et ah J, “Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4,” Chem. Biol, vol. 22, no. 6, pp. 755–763, Jun. 2015, doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Winter etal GE, “Phthalimide conjugation as a strategy for in vivo target protein degradation,” Science, vol. 348, no. 6241, pp. 1376–1381, Jun. 2015, doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Neklesa T et al. , “An oral androgen receptor PROTAC degrader for prostate cancer.,” J. Clin. Oncol, vol. 36, no. 6_suppl, pp. 381–381, Feb. 2018, doi: 10.1200/JC0.2018.36.6_suppl.381. [DOI] [Google Scholar]
- [21].Flanagan J et al. , “Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer,” in Poster Session Abstracts, 2019, pp. P5-04-18–P5-04-18, doi: 10.1158/1538-7445.SABCS18-P5-04-18. [DOI] [Google Scholar]
- [22].Huang H-T et al. , “A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader,” Cell Chem. Biol, vol. 25, no. 1, pp. 88–99.e6, Jan. 2018, doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sun Y et al. , “PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies,” Cell Res, vol. 28, no. 7, pp. 779–781, Jul. 2018, doi: 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lai AC et al. , “Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL,” Angew. Chem. Int. Ed, vol. 55, no. 2, pp. 807–810, Jan. 2016, doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Powell CE et al. , “Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK),” J. Med. Chem, vol. 61, no. 9, pp. 4249–4255, May 2018, doi: 10.1021/acs.jmedchem.7b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang C et al. , “Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK),” Eur. J. Med. Chem, vol. 151, pp. 304–314, May 2018, doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Burslem GM et al. , “The Advantages of Targeted Protein Degradation Over Inhibition: AnRTK Case Study,” Cell Chem. Biol, vol. 25, no. 1, pp. 67–77.e3, Jan. 2018, doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brand M et al. , “Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML,” Cell Chem. Biol, vol. 26, no. 2, pp. 300–306.e9, Feb. 2019, doi: 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang B et al. , “Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6,” Angew. Chem. Int. Ed, vol. 58, no. 19, pp. 6321–6326, May 2019, doi: 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Su S et al. , “Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders,” J. Med. Chem, vol. 62, no. 16, pp. 7575–7582, Aug. 2019, doi: 10.1021/acs.jmedchem.9b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Robb CM et al. , “Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC),” Chem. Commun, vol. 53, no. 54, pp. 7577–7580, 2017, doi: 10.1039/C7CC03879H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Olson CM et al. , “Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation,” Nat. Chem. Biol, vol. 14, no. 2, pp. 163–170, Feb. 2018, doi: 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zengerle M, Chan K-H, and Ciulli A, “Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4,” ACS Chem. Biol, vol. 10, no. 8, pp. 1770–1777, Aug. 2015, doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hsu JH-R et al. , “EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex,” Cell Chem. Biol, vol. 27, no. 1, pp. 41–46.e17, Jan. 2020, doi: 10.1016/j.chembiol.2019.11.004. [DOI] [PubMed] [Google Scholar]
- [35].Potjewyd F et al. , “Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader,” Cell Chem. Biol, vol. 27, no. 1, pp. 47–56.e15, Jan. 2020, doi: 10.1016/j.chembiol.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun X et al. , “PROTACs: great opportunities for academia and industry,” Signal Transduct. Target. Ther, vol. 4, no. 1, Dec. 2019, doi: 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Silva MC et al. , “Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models,” eLife, vol. 8, Mar. 2019, doi: 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chessum NEA et al. , “Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766),” J. Med. Chem, vol. 61, no. 3, pp. 918–933, Feb. 2018, doi: 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yesbolatova A, Tominari Y, and Kanemaki MT, “Ligand-induced genetic degradation as atool for target validation,” Drug Discov. Today Technol, vol. 31, pp. 91–98, Apr. 2019, doi: 10.1016/j.ddtec.2018.11.001. [DOI] [PubMed] [Google Scholar]
- [40].Oprea TI et al. , “Unexplored therapeutic opportunities in the human genome,” Nat. Rev. Drug Discov, vol. 17, no. 5, pp. 317–332, May 2018, doi: 10.1038/nrd.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bondeson DP et al. , “Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead,” Cell Chem. Biol, vol. 25, no. 1, pp. 78–87.e5, Jan. 2018, doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Raina K et al. , “PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer,” Proc. Natl. Acad. Sci, vol. 113, no. 26, pp. 7124–7129, Jun. 2016, doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings,” Adv. Drug Deliv. Rev, vol. 23, no. 1–3, pp. 3–25, Jan. 1997, doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- [44].Churcher I, “Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?,” J. Med. Chem, vol. 61, no. 2, pp. 444–452, Jan. 2018, doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- [45].Edmondson SD, Yang B, and Fallan C, “Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges,” Bioorg. Med. Chem. Lett, vol. 29, no. 13, pp. 1555–1564, Jul. 2019, doi: 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- [46].Beveridge R, Kessler D, Rumpel K, Ettmayer P, Meinhart A, and Clausen T, “Native mass spectrometry can effectively predict PROTAC efficacy,” bioRxiv, Nov. 2019, doi: 10.1101/851980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Maple HJ, Clayden N, Baron A, Stacey C, and Felix R, “Developing degraders: principles and perspectives on design and chemical space,” MedChemComm, vol. 10, no. 10, pp. 1755–1764, 2019, doi: 10.1039/C9MD00272C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gadd MS et al. , “Structural basis of PROTAC cooperative recognition for selective protein degradation,” Nat. Chem. Biol, vol. 13, no. 5, pp. 514–521, May 2017, doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roy MJ et al. , “SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate,” ACS Chem. Biol, vol. 14, no. 3, pp. 361–368, Mar. 2019, doi: 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nowak RP et al. , “Plasticity in binding confers selectivity in ligand-induced protein degradation,” Nat. Chem. Biol, vol. 14, no. 7, pp. 706–714, Jul. 2018, doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Riching KM et al. , “Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action,” ACS Chem. Biol, vol. 13, no. 9, pp. 2758–2770, Sep. 2018, doi: 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- [52].Fisher SL and Phillips AJ, “Targeted protein degradation and the enzymology of degraders,” Curr. Opin. Chem. Biol, vol. 44, pp. 47–55, Jun. 2018, doi: 10.1016/j.cbpa.2018.05.004. [DOI] [PubMed] [Google Scholar]
- [53].Bett JS, “Proteostasis regulation by the ubiquitin system,” Essays Biochem, vol. 60, no. 2, pp. 143–151, Oct. 2016, doi: 10.1042/EBC20160001. [DOI] [PubMed] [Google Scholar]
- [54].Spradlin JN et al. , “Harnessing the anti-cancer natural product nimbolide for targeted protein degradation,” Nat. Chem. Biol, vol. 15, no. 7, pp. 747–755, Jul. 2019, doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, and Cravatt BF, “Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16,” Nat. Chem. Biol, vol. 15, no. 7, pp. 737–746, 2019, doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tate J and Ward G, “Interferences in immunoassay,” Clin. Biochem. Rev, vol. 25, no. 2, pp. 105–120, May 2004. [PMC free article] [PubMed] [Google Scholar]
- [57].Pettersson M and Crews CM, “PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future,” Drug Discow Today Technol, vol. 31, pp. 15–27, Apr. 2019, doi: 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Collins I, Wang H, Caldwell JJ, and Chopra R, “Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway,” Biochem. J, vol. 474, no. 7, pp. 1127–1147, Apr. 2017, doi: 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bunnage ME, Chekler ELP, and Jones LH, “Target validation using chemical probes,” Nat. Chem. Biol, vol. 9, no. 4, pp. 195–199, Apr. 2013, doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- [60].Arrowsmith CH et al. , “The promise and peril of chemical probes,” Nat. Chem. Biol, vol. 11, no. 8, pp. 536–541, Aug. 2015, doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jaeger MG and Winter GE, “Expanding the Degradable Proteome: Designing PROTACs by the Book,” Cell Chem. Biol, vol. 27, no. 1, pp. 14–16, Jan. 2020, doi: 10.1016/j.chembiol.2019.12.009. [DOI] [PubMed] [Google Scholar]
- [62].Fegan A, White B, Carlson JCT, and Wagner CR, “Chemically Controlled Protein Assembly: Techniques and Applications,” Chem. Rev, vol. 110, no. 6, pp. 3315–3336, Jun. 2010, doi: 10.1021/cr8002888. [DOI] [PubMed] [Google Scholar]
- [63].Choi J, Chen J, Schreiber SL, and Clardy J, “Structure of the FKBP12-Rapamycin Complex Interacting with Binding Domain of Human FRAP,” Science, vol. 273, no. 5272, pp. 239–242, Jul. 1996, doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- [64].Yamazoe S et al. , “Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study,” J. Med. Chem, Jan. 2020, doi: 10.1021/acs.jmedchem.9b01167. [DOI] [PubMed] [Google Scholar]
- [65].Torres SE et al. , “Ceapins block the unfolded protein response sensor ATF6a by inducing a neomorphic inter-organelle tether,” eLife, vol. 8, May 2019, doi: 10.7554/eLife.46595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Balietti S, Mas M, and Helbing D, “On Disciplinary Fragmentation and Scientific Progress,” PLOS ONE, vol. 10, no. 3, p. e0118747, Mar. 2015, doi: 10.1371/journal.pone.0118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Scudellari M, “Protein-slaying drugs could be the next blockbuster therapies,” Nature, vol. 567, no. 7748, pp. 298–300, Mar. 2019, doi: 10.1038/d41586-019-00879-3. [DOI] [PubMed] [Google Scholar]
- [68].Blagg J and Workman P, “Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology,” Cancer Cell, vol. 32, no. 1, pp. 9–25, 10 2017, doi: 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bordeaux J et al. , “Antibody validation,” BioTechniques, vol. 48, no. 3, pp. 197–209, Mar. 2010, doi: 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schreiber SL, “A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines,” Isr. J. Chem, vol. 59, no. 1–2, pp. 52–59, Feb. 2019, doi: 10.1002/ijch.201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Field SD, Arkin J, Li J, and Jones LH, “Selective Downregulation of JAK2 and JAK3 by an ATP-Competitive pan-JAK Inhibitor,” ACS Chem. Biol, vol. 12, no. 5, pp. 1183–1187, May 2017, doi: 10.1021/acschembio.7b00116. [DOI] [PubMed] [Google Scholar]
- [72].Targett-Adams P et al. , “Small Molecules Targeting Hepatitis C Virus-Encoded NS5A Cause Subcellular Redistribution of Their Target: Insights into Compound Modes of Action,” J. Virol, vol. 85, no. 13, pp. 6353–6368, Jul. 2011, doi: 10.1128/JVI.00215-11. [DOI] [PMC free article] [PubMed] [Google Scholar]