

Abstract
Understanding the evolutionary role of environmentally-induced phenotypic variation (i.e., plasticity) is an important issue in developmental evolution. A major physiological response to environmental change is cellular stress, which is counteracted by generic stress reactions detoxifying the cell. We describe a model, Stress-Induced Evolutionary Innovation (SIEI), whereby ancestral stress reactions and their corresponding pathways can be transformed into novel structural components of body plans, such as new cell types. We describe previous findings suggesting that the cell differentiation cascade of a cell type critical to pregnancy in humans, the decidual stromal cell, evolved from a cellular stress reaction. We hypothesize that the stress reaction in these cells was elicited ancestrally via inflammation caused by embryo attachment. We propose SIEI is a distinct form of plasticity-based evolutionary change leading to the origin of novel structures rather than adaptive transformation of pre-existing characters.
Keywords: stress response, evolutionary innovation, novelty, cell type origin, plasticity
Graphical Abstract
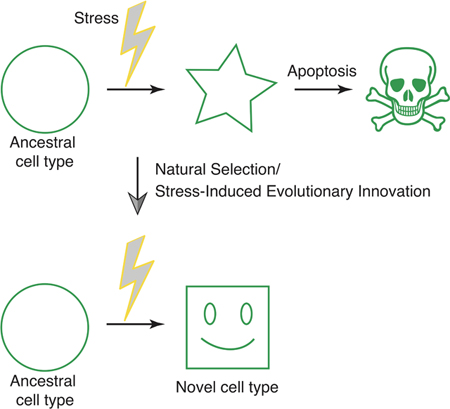
Cells frequently counteract environmental stress by conserved molecular mechanisms, leading to stress mitigation or apoptosis. Increasingly, studies on cellular stress responses intersect with cell type differentiation programs. We hypothesize that integration of these conserved pathways is a mechanism of stress-induced evolutionary innovation that is capable of generating novel cell types.
1. Cellular stress is counteracted by conserved molecular mechanisms
“It’s not stress that kills us; it is our reaction to it” (Hans Selye, 1956)
A “happy” cell is one that lives in relative harmony with its environment: nutritional input closely matches metabolic need, and the nature and rate of anabolic processes is balanced with the catabolic decay and loss of cellular components. When this equilibrium is disturbed, either by a lack of nutrients or toxin-induced malfunction of its components, the cell experiences stress and reacts with a stress response[1]. Cellular stress leads to the accumulation of unfolded proteins, DNA damage, and toxic side-products of metabolism, such as reactive oxygen species (ROS) and other radical-like, reactive nitrogen oxides. The stress response activates genes and pathways that minimize this damage by producing enzymes that neutralize ROS (e.g., catalase or super oxide dismutase [SOD]), degrade or refold misfolded proteins (e.g. ubiquitin proteasomes or chaperones), and repair DNA damage[2, 3]. If these responses fail, the cell undergoes senescence or commits suicide (e.g. apoptosis). The study of all these phenomena constitutes the field of cellular stress biology, which has been separated from “normal” developmental biology and physiology for decades. Increasingly, however, the boundary between these fields is blurred because mechanisms and agents common to the field of stress biology, such as apoptosis or ROS, have been found to play a role in “normal” biological processes like cell differentiation[4–8]. This overlap between stress biology and developmental biology is not coincidental. We argue that it constitutes the footprint of evolutionary processes that convert stress reactions into normal developmental and physiological processes, and in the course can give rise to evolutionary novelties such as new cell types.
Across all multicellular domains of life, ROS are produced by specialized enzymes of the cell, e.g. members of the family of NADPH oxidases NOX1 and NOX2, to accomplish particular tasks such as defense against microbes at the cell surface[9]. Nevertheless, it is now well established that ROS play an important part in the normal differentiation of mammalian cells, a phenomenon called ROS signaling. ROS signaling has been shown to function in the maintenance and proliferation of neuronal progenitor cells[6]. Similarly, the differentiation of mesenchymal stem cells in chondrocytes, osteocytes, and adipocytes is also regulated through the production of ROS[10]. In angiogenesis and cardiogenesis, NOX-derived ROS promote differentiation of blood vessels and cardiomyocytes, respectively[11, 12]. In Drosophila, ROS signaling precedes and is necessary for differentiation of hematopoietic progenitors[13], and also functions in proliferation of intestinal stem cells in the midgut[14]. In Xenopus, ROS activate the Wnt/β-catenin pathway during tail regeneration[15]. These observations, stemming from evolutionarily-distant clades, suggest that pathways functioning in the generation of ROS have been utilized as an evolutionary mechanism to induce cell differentiation.
In addition to these general considerations about the dual role of ROS signaling in normal physiology and under conditions of cellular stress, new results from research on the molecular mechanisms for the evolutionary origin of the decidual stromal cell type of eutherian mammals substantiate empirically the evolutionary significance of cellular stress responses[16]. Having encountered the significant role of stress response mechanisms in the evolution of cell differentiation, we formulated a model—Stress Induced Evolutionary Innovation (SIEI)—based on the role of stress pathways in the evolution of differentiation cascades that offers a new mechanism to account for the origin of novel cell types. After reviewing these recent results, we discuss the relationship of our model to previous analyses of the importance of plasticity in evolutionary change[17, 18] and its potential application to the origin of cell types in the eye that also have an affinity to stress derived mechanisms.
2. Stress responses have shaped the evolution of the decidual stromal cell
The decidual stromal cell (DSC) is a cell type that evolved in the stem lineage of eutherian mammals[19] after the most recent common ancestor of marsupials and eutherian or “placental” mammals, and before the most recent common ancestor of eutherian mammals (Figure 1A). This inference is based on the fact that DSCs are only found in eutherian mammals and not in marsupials. Marsupials do have a cell type that is homologous to the DSC, namely the endometrial stromal fibroblast or ESF[20]. However, marsupial ESF cannot differentiate into a DSC[16]. In eutherians, and in humans specifically, the DSC differentiates from an ESF, which we label “neo-ESF” because it can differentiate into a DSC[21]. Figure 1B shows the cell type tree which implies that human ESF and DSC are sister cell types, as reflected in the similarity of their transcriptomes compared to other mesenchymal cell types[22].
Figure 1.

Evolutionary history of viviparity and the decidual stromal cell (DSC) in mammals. A) Large scale evolutionary history of mammals: there are three main groups (clades) of mammals. Monotremes are egg laying and likely diverged from the mammalian lineage before the evolution of viviparity, which is a shared derived characteristic of marsupials and eutherians. However, marsupials do not implant and do not have the DSC. In contrast, the ancestral situation in eutherian mammals is implantation, invasive (hemochorial) placentation, and the presence of the DSC, which is a maternal cell type essential for the accommodation of the embryo and maintenance of pregnancy. B) The distribution of endometrial stromal cells among therian mammals. Marsupials have an endometrial stromal fibroblast (ESF) that does not decidualize, which is referred to as “paleo-ESF”. In eutherians, the ESF can differentiate into a DSC and is therefore called “neo-ESF”.
2.1. Embryo implantation leads to cellular stress responses
The evolution of the DSC occurred in the stem lineage of eutherian mammals in parallel with the evolution of invasive placentation. Invasive placentation involves the embryo invading the lining of the uterus and, in its most severe form, destroying maternal blood vessels. This form of placentation is called “hemochorial” because the outer layer of the placenta, the chorion, stands in direct contact with maternal blood. Phylogenetic reconstruction suggests that hemochorial placentation was already present in the most recent common ancestor of eutherian mammals[19, 23, 24]. Hemochorial placentation has only been described in eutherian mammals; in marsupials and reptiles, placental invasion is comparatively mild and never leads to a hemochorial interface between the mother and the fetus[25].
It has long been known that embryo implantation in humans is a process that activates elements of the inflammatory response, and that this inflammation is beneficial for embryo implantation[26]. Later in pregnancy, the inflammatory processes are suppressed, and this enables extended gestational duration. More recently, it has been shown that embryo-attachment in the opossum (a marsupial) leads to a quasi-inflammatory process quite similar to that observed in eutherian mammals[27]. The difference is that inflammation in the opossum leads directly to birth, while in eutherians the inflammation is suppressed to allow an extended maintenance of the fetal-maternal interface.
Given this natural history, it is likely that DSC evolved in response to the inflammatory conditions caused by attachment and invasion of the embryo. In fact, the decidual cell is necessary for the maintenance of pregnancy in eutherians that have an invasive placenta[28], and also plays a role in controlling the inflammatory processes at the site of implantation[29]. Because embryo attachment leads to an inflammatory reaction in the maternal tissue and thereby creates a stressful environment for the endometrial stromal cells at the site of embryo attachment and implantation, the evolution of a stable fetal-maternal interface required the evolution of a new cell type, the DSC, that is capable of surviving in these circumstances and that specialized to foster a sustainable environment for the developing placenta.
2.2. Decidual stromal cell differentiation utilizes stress response pathways
Much is known about the transcription factors and signals involved in the differentiation of human DSCs[28, 30]. The most salient features are that differentiation depends on progesterone via nuclear progesterone receptor A (PGR-A) and the activation of the PKA pathway that produces cyclic adenosine monophosphate (cAMP). cAMP can be activated by prostaglandins[31], such as prostaglandin E2 (PGE2) via the prostaglandin receptors PTGER2 or PTGER4, as well as through human chorionic gonadotropin (hCG) or relaxin[28]. Comparative data suggest that PGE2 may have been the ancestral ligand activating the PKA pathway in these cells. hCG expression in the trophoblast is only found in humans and their close relatives. Other ligands also have been shown to also play a role, such a EGF, IHH, BMP, and WNT[30, 32, 33]. Downstream of the cAMP signal, the transcription factor FOXO1 is stabilized and retained in the nucleus rather than degraded. Nuclear FOXO1 then physically interacts with a number of other transcription factors, including PGR, CEBPB, and HOXA11[34, 35]. These transcription factor complexes then activate decidual effector genes—transcription factors that target effector and transcription factor genes like ZBTB16[36, 37]. Decidual marker genes in humans include prolactin (PRL), IGFBP1, and somatostatin.
In contrast, stimulating opossum ESFs with the same signaling molecules (i.e., progesterone and PGE2) leads to a mixed response[16]. Many of the same transcription factor genes are upregulated at the transcriptional level, as observed in decidual cells, and FOXO1 protein is stabilized and retained in the nucleus, similar to the observed response in human DSC differentiation. However, none of the highly-induced effector genes of the human DSC are expressed in opossum paleo-ESFs after stimulation with these molecules. Instead, intracellular ROS are produced and genes related to ROS scavenging, detoxification, and decomposition of H2O2 (e.g., catalase, GPX3, GPX4, SOD1, and SOD3) are expressed as part of the cellular stress response. A model of the regulatory network induced in paleo-ESFs was reported in Erkenbrack et al.[16] and is summarized in Figure 2A. In brief, progesterone prevents the degradation of FOXO1 protein and leads to its accumulation in the cytoplasm. PGE2 activates the PKA pathway via the PTGER4 receptor, thereby increasing intracellular cAMP. This, in turn, leads to the expression of NOX4 that produces intracellular ROS, activating the oxidative stress response. FOXO1 and FOXO3 contribute to the protective function of the stress response. Figure 2B depicts a plausible reconstruction of the ancestral generic oxidative stress response. The derived stress response seen in the opossum consists of two additional pathways: (1) the production of ROS stimulated by PGE2, which in itself is not a stressor, and (2) the inhibition of FOXO1 degradation by progesterone. The latter effect could facilitate a more efficient induction of anti-oxidant gene expression because FOXO1 ancestrally activates these gene cassettes and translocates to and accumulates in the nucleus.
Figure 2.

Regulation of FOXO1 protein activation. A) Regulatory network revealed in paleo-ESF of the opossum Monodelphis domestica (for evidence, see Erkenbrack, et al., 2018). Progesterone leads to the inhibition of degradation of FOXO1 protein, which leads to its accumulation in the cytoplasm. Presence of ROS and cAMP, as well as PGE2 through the activation of PGE2 receptor 4 (PTGER4 aka EP4) stimulation, inhibits the nuclear export of FOXO1 and thus retention of FOXO1 in the nucleus, where it positively influences the transcription of anti-oxidant enzymes like catalase and gluthathione peroxidase. In this model we assume that PGE2 acts through PKA and cAMP to induce the NADPH oxidase 4 (NOX4) to produce the cytoplasmic ROS, which is parsimonious but not yet directly shown. B) Ancestral activation of FOXO1 in response to ROS, through activation of ERK1/2 signaling found in many other cells. The network in the opossum ESF, as shown in A), is likely derived from this generic oxidative stress pathway by two evolutionary events. First, the evolution of NOX4 activation by PGE2, though this also could be a generic pathway because PGE2 has been shown to activate NOX4 through PTGER4 in liver cells[63]. Second, the inhibition of FOXO1 degradation by progesterone through its receptor isoform A, PGR-A. In this way, the activation of FOXO1 through the generic oxidative stress reaction becomes internalized, i.e., activated through physiological signals rather than stressors.
The similarities between the human DSC and the opossum paleo-ESF are the activation of PKA, the production of ROS[38], and the activation of FOXO1 protein as well as the upregulation of many “decidual” transcription factor genes. Intriguingly, human DSCs also produce ROS during decidualization, which plays a positive role in their differentiation[38]. The notable differences between human DSC and opossum paleo-ESF are that the latter do not express decidual marker genes like PRL and IGFBP1, but rather activate a cellular stress response. The generic cellular stress response has two branches: one that activates genes that counteract the damaging effects of stress, like anti-oxidant enzymes and chaperones, and another that readies components of the apoptotic pathway in the event that the protective pathway fails. In human DSC, components of the stress response are inhibited, in particular signaling pathways that activate apoptosis such as JNK and P38. In the human endometrium, the phosphorylated (active) form of JNK is not expressed[39] because of the action of a specific phosphatase, DUSP1/MEK1[40]. Thus, in the human DSC, the generic stress response is modified so that the likelihood of apoptosis is lower than it is in ESF.
Another surprising feature is that the signals that activate the cellular stress response (i.e., progesterone and PGE2) are not themselves stressors. Instead, PGE2 seems to “simulate” oxidative stress by activating NOX4, leading to some level of intracellular ROS, which in turn activates a protective oxidative stress response. These features of the regulatory network seem to indicate that we are dealing with an “internalized” stress response that is activated by physiological rather than pathological signals, but which nevertheless is evolutionarily derived from, and thus homologous to, a reactive stress response.
3. Stress-Induced Evolutionary Innovation (SIEI) generates novel cell types
The comparison of human DSC differentiation and the response of the sister cell type in the opossum—the paleo-ESF—suggests two homologies: a) the inflammatory reaction induced by embryo attachment in the opossum and the implantation reaction in the endometrium of eutherian mammals[27]; and, b) the stress reaction of the opossum ESF and the differentiation pathway of the human DSC[16]. It is likely that the evolution of implantation and decidualization are not only historically associated but in fact causally intertwined. The stress reaction of the opossum ESF is likely caused by attachment-induced inflammation, and therefore the evolution of the DSC in eutherian mammals was a key step in transforming the inflammatory attachment reaction into the sustainable implantation[27, 41].
3.1. SIEI is the evolution of an initial protective response into a phenotype that compensates for a stressor
Overall, we have a monophyletic group of animals (i.e., a clade)—the eutherian mammals—in which two signals (progesterone and PGE2) lead to the differentiation of a cell type, the DSC. The DSC, in turn, interacts with the fetus to establish a sustainable fetal-maternal interface. In contrast, we have an outgroup species, the opossum, in which there is a homologous cell type—the opossum ESF—that reacts to the same signals by deploying a cellular stress response. The homology of the gene regulatory network activated in opossum ESF and human DSC by progesterone and PGE2 implies that the differentiation pathway of the DSC evolved from an ancestral stress response. In eutherians, the stress response was transformed into a novel cellular phenotype that compensates the perturbation caused by the invading embryo. It is important to notice both the different nature of the phenotypes induced in these two cells (stress response versus decidualization), and that the stimuli that cause the deployment of the stress cascade in opossum (progesterone and prostaglandins) are not in themselves stressors.
The generic cellular stress reaction activates genes that protect against the harmful effects of stress like ROS. Examples in the opossum ESF are the upregulation of catalase and SOD to break down H2O2, the activation of the NRF2 signaling pathway that leads to the production of ROS-detoxifying enzymes like glutathione peroxidases (e.g., GPX3/4), and the fact that activated FOXO1 protein protects against apoptosis[16]. In contrast, the phenotype of the DSC is both enabling and limiting trophoblast invasion, as well as modifying the reaction of the innate immune system to the perturbation caused by the embryo[42, 43]. Therefore, the function of the DSC is not only to protect the cell from damage, like the stress reaction seen in opossum, but also to compensate for the effects of the stressor: embryo attachment, inflammation, and invasion. These observations lead to a model where the initial, environmentally-induced reaction that is protective later evolves into a phenotype that compensates for the initial stressor of implantation by suppressing the inflammatory process.
This transition from protection to compensation is an important feature of the SIEI model when compared to other models for the role of plasticity in evolution, such as the classical Baldwin effect[44, 45], the “plasticity first model” (PFH) of Levis and Pfennig[46], and the genetic accommodation model of West-Eberhard[17]. In these models, the initial plastic response prompted by a novel environment involves an overproduction of variation that is random with respect to organismal fitness[47]. A subset of this variation is then accommodated within an organism’s homeostatic capacities as a phenotype that facilitates an adaptive fit with the environment and hence survival. Over time, if the same plastic response is evoked by similar environmental circumstances, then the phenotype can be heritable stabilized through the emergence of genetic variance that accommodates it. The implication is that the plastic reponse produces an approximation of the adaptive phenotype itself (Figure 3). There is a transition from temporary compensation to permanent compensation. In the SIEI model, the initially plastic reaction is a generic, protective stress response that becomes the target of natural selection to produce a novel compensatory phenotype later—a transition from temporaroy protection to permanent compensation (Figure 4).
Figure 3.

The plasticity first hypothesis (PFH) of adaptive evolution, modified from Figure 1 of Levis and Pfennig (2016). A) On the top is a population with different genotypes, indicated by different colors, and similar phenotypes (shown here in dashed shapes). An environmental change elicits a plastic response in the phenotypes as shown here in dashed shapes. These phenotypes depend on the genotype of the individual. Genotypes differ in their plastic responses, and hence natural selection will select for the genotypes that have the most adaptive phenotype. The most adaptive phenotype is then further refined by natural selection, but, importantly, this phenotype is still dependent on the environment. Finally, the now refined phenotype is genetically assimilated, meaning that the environmental trigger is no longer necessary for its development, indicated here by replacing the dashed lines with solid lines. B) A greatly simplified schema of the structure of PFH. Plastic changes are indicated by horizontal arrow and evolution by the vertical arrow. Note that in this model the derived adaptive phenotype is more or less the immediate product of some genotype. In other words, the PFH is a model where the plastic reaction itself is producing the adaptive phenotype. The result is a transformation of the phenotype, not the origin of a novel part of the body, as in the SIEI model.
Figure 4.

The model of Stress-Induced Evolutionary Innovation includes three steps. A) A stressor induces a generic stress response that protects the affected cells against the consequences of the stressor, such as oxidative stress. B) If the stress becomes frequent or predictable, then the stress reaction becomes “anticipatory protection,” meaning that it is triggered by physiological signals before the stressor arrives. Note that the phenotype remains largely similar to that of the generic stress response; only the signals that elicit the response change. This is the situation in the opossum ESF, where progesterone and prostaglandin together trigger the expression of anti-oxidant genes. One can speculate that the induction of the stress pathway by inflammatory cytokines could be an anticipatory response. C) Once the stress reaction is under the control of physiological rather than pathological signals, the phenotype of the affected cells can become the target of natural selection. If the stressor is routinely manifested in the life cycle of the animal, then the phenotype can evolve towards compensation. An example of this is the fact that DSCs specifically downregulate the apoptotic branch of the oxidative stress reaction[40], leading to a compensated cellular phenotype that is, to a degree, autonomous from the original stressor.
The second unexpected feature of our findings in the opossum ESF is that we can induce a stress reaction with physiological stimuli (progesterone and PGE2). It is unlikely that these signals are stressors in this context because they do not act as stressors for other cells in the body. For the opossum ESF, it is more plausible that the co-activation of its progesterone receptor and PTGER4 carries information that embryo attachment and oxidative stress could occur soon. Elevated progesterone levels indicate that ovulation has happened, and thus fertilized eggs are likely coming down the oviduct. The presence of PGE2 may indicate that copulation has occurred, which generically leads to transient post-copulatory inflammation in the female reproductive tract[48]. This latter suggestion is speculative because the regulation of PGE2 production in the opossum uterus has not been elucidated. Regardless, the stress reaction in opossum is triggered by intra-organismal signals, hormones, and paracrine factors rather than by an actual stressor. Interestingly, production of ROS in human keratinocytes has been shown to stimulate production of PGE2[49]. Thus, it likely represents an anticipatory activation of the stress network (i.e., an internalized stress reaction), rather than a trigger by an acute stressor. This internalization of the molecular trigger (i.e., the environmental stimulus that induces the phenotype) of the stress reaction in the SIEI model is very closely aligned with the models developed from the classical notion of the Baldwin effect (sensu Simpson 1953) or genetic assimilation[50].
Another feature of the SIEI model is that the internalized stress response network becomes a target of natural selection. Only a subset of cells reacts to the internal signal and deploys a stress response. This set of cells in individualized functionally and therefore can be a phenotypic target of natural selection because genetic variation that specifically affects these cells can be adaptive and subsequently fixed in the population. SIEI not only leads to an adaptive phenotype through the transformation of a stress response, but also to a new part of the organism—in this case, a novel cell type—that has its own developmental identity and differentiation pathway, derived from the original stress response. In the standard genetic assimilation and accomodations models (Figure 3), such as PFH[46], the adaptive phenotype is a quantitative transformation of an existing trait, such as the frequency of a life-history polyphenism (e.g. herbivory versus carnivory in spadefoot toad larvae)[51]. In the SIEI model, a new morphological or behavioral trait results (novelty or innovation sensu Love[52]); the eutherian DSC is not a quantitative transformation of the gene expression variation already present in the opossum ESF.
We can summarize the above considerations with a visual model (Figure 3). Initially, some environmental change leads to a stressed state in a subset of cells in the body (Figure 3A). When this stressor becomes a regular feature of the life cycle of the species, such as in reproduction or seasonal changes, natural selection will favor the deployment of an anticipatory stress reaction, one that is triggered by physiological signals so that these cells are poised for action when the actual stressor arrives (Figure 3B). Anticipatory mechanisms are widespread among organisms and seem to be adaptively favored. The most obvious example is the mobilization of nutrients from the root system of deciduous trees before the arrival of spring and the growth of leaves[53]. Another potential example is spontaneous decidualization, which is associated with menstruation. In most eutherians, decidualization depends on a signal from the embryo, but in great apes and old world monkeys (Catarrhini), as well as a few other mammals, the endometrium decidualizes even without the embryo, likely as an anticipatory mechanism because of the highly invasive nature of the placenta in these species[54].
Once the trigger of the stress reaction is internalized, as it is in opossum, the entire process can become the target of natural selection to produce a phenotype that addresses the stressor. Over time, the stress reaction becomes transformed into a phenotype that not only protects but also engages the stressor itself (Figure 3C). In the marsupials the stress reaction became the trigger for parturition and in eutherians it was transformed to become the implantation process. This eventually leads to a situation that fully compensates for the influence of the stressor so that it becomes a normal part of the life cycle of the species (i.e., it is no longer a stressor). In the eutherians the result is a novel cell type, specialized to deal with a specific functional challenge (the stressor), and an organism where the original stressor has become a part of the normal physiology of the species, i.e. decidualization and implantation.
3.2. The SIEI model applies to cell types
To our knowledge a mechanistic link between the cellular stress response and the evolution of novel cell types or organs has only been proposed in the context of eye evolution by Oakley and Speiser[55]. They argue: “One logical hypothesis about cellular environments and origins [of cell types] is that light creates a stressful environment for cells, such that the origins of new expression patterns of light-interacting genes may often be responses to light-induced stress.”
Specifically, Oakley and Speiser highlight that opsin genes, coding for the main light-sensitive pigment in photoreceptor cells, are phylogenetically related to melatonin receptors[56, 57]. Melatonin degrades in light and likely had an ancestral role as an anti-oxidant[56, 58]. Similarly, some lens proteins are related to and derived from stress enzymes. For example, the αB-crystallins also are known as small heat shock proteins that protect mitochondria from oxidative damage due to light[59, 60]. The authors suggest that proteins that now interact with light in eye cells originally were expressed in light-exposed cells as part of their stress response[55]. By extension, one can argue that cell types dedicated to visual functions may arise from the stress response to light-induced stress.
Considering how the SIEI model might apply in the context of the origin of novel cell types in the eye suggests a strategy for testing its applicability more widely as a mode of evolution. In particular, organ systems that are subjected to specific forms of stress that induce responses with characteristic physiological signatures, such as gene expression that neutralizes ROS, and exhibit an overlap between stress biology and developmental biology, such as ROS signaling, would be plausible candidates for the SIEI model. Sufficient comparative data will be necessary to evaluate any specific hypotheses, and in many cases this will not be available. However, comparative physiological data on a variety of candidate systems, such as endothelial cells and shear stress due to blood flow in the cardiovasculature or kidney glomerulus podocytes and chemical stress due to continuous blood filtration, are likely to point in the direction of where further experimentation would make it possible to confirm whether the SIEI model applies to the origin of other cell types. Overall, the first line of testing the SIEI model is to see whether there are significant homologies between differentiation cascades and cell type relevant stress pathways.
4. Comparing SIEI to other models of plasticity in evolution
Our proposed model of SIEI is related to other models that have been advanced to understand the role of phenotypic plasticity in evolution. Models about the role of plasticity come in different flavors, including the Baldwin effect and genetic assimilation[50, 61, 62], genetic accommodation[17], and the “plasticity first hypothesis”[46, 51]. The Baldwin effect is named after the American psychologist James Mark Baldwin who proposed a cluster of ideas about how behavior influences the course of evolution[44]. His model was a complex (and not altogether transparent) argument that included “intelligent” choices made by organisms, selection for those individuals who make the best choices (from the vantage point of fitness), imitation by other members of the population, phenotypic modifications caused by the changed behavior, and also the genetic fixation of those changes. The modern notion and the term Baldwin effect derives from G. G. Simpson[45], who reduced the Baldwin effect to the genetic fixation of plastic phenotypic changes, which is identical to the notion of “genetic assimilation” as proposed by Waddington[50, 61].
The most recent instantiation of a plasticity-based model of adaptation is the Plasticity First Hypothesis (PFH); we consider it an exemplary representative of models describing the role of phenotypic plasticity in evolution (after[46]). According to this model (Figure 3A), the initial effect of an environmental change is to elicit a plastic response in the phenotype of individuals in the population. This response is genetically heterogeneous; individuals with different genotypes will show different phenotypic responses or different degrees of plasticity (i.e., an overproduction of variation that is random with respect to organismal fitness that is accommodated within an organism’s homeostatic capacities). Because not all of this variation will be equally adaptive, natural selection will eliminate genotypes exhibiting less adaptive phenotypes in the new environment, assuming these conditions continue to be present. Next, there is a phase in which the environmentally-induced phenotype is further refined by natural selection. Finally, the induced phenotype will be “assimilated” so that the development of this phenotype no longer needs the environmental stimulus; it becomes part of the “genetic heritage” of the species. There is good experimental evidence that this has occurred in nature, in particular for amphibian larvae[51].
There are two salient features of this model, which can be seen as a more precise articulation of the Baldwin effect (Figure 3B). First, the environmental change directly induces at least an approximation of the adaptive phenotype in some individuals of the population. Hence, the role of plasticity is to (more or less) directly transform the ancestral plastic character into a more adaptive trait that becomes genetically fixed by genetic assimilation. Second, the model explains the transformation of a character state of an organism (i.e., a quantitative transformation of an existing trait), rather than the origin of a novel character[52]. Both of these features distinguish PFH from SIEI. The main result of an SIEI process is a novel body part, such as a novel cell type, that can evolve an adaptive phenotype that addresses a specific functional challenge—for example, from protection to compensation in mammalian pregnancy. The main result of a PFH process is the stabilization and refinement or elaboration of an adaptive, plastic character state.
Importantly, there are also similarities between PFH and SIEI. Both include environmentally-induced changes in the traits of organisms, whether that be a stress response (SIEI) or a plastic phenotypic change (PFH). And both models postulate processes that make the induced state independent of the original stimulus: genetic assimilation of the derived plastic phenotype (PFH) and an “internalized” or normalized stress response (SIEI).
Given these similarities, it is not surprising that there are examples that fall somewhat between the two models, such as the evolution of congenital calluses in ostriches and camels. In these cases, the derived character (the callus) is induced by the environment, like in the PFH, but it also leads to the individuation of new parts of the organism—the individual calluses at specific locations of the body—like the origin of a novel body part in SIEI. Thus, PFH and SIEI are not competing models but explain different kinds of evolutionary processes that are sometimes distinct and sometimes combined over evolutionary time. PFH is primarily concerned with adaptive transformations, whereas SIEI is primarily concerned with the origin of novel cell types and body parts.
5. Conclusions and outlook
The role of phenotypic plasticity in organismal evolution has puzzled biologists since the beginnings of evolutionary thought. It is now clear that phenotypic plasticity is a general biological property of most (if not all) aspects of an organism. It is also clear that evolution proceeds through a complicated interaction between natural selection and developmental processes subject to environmental plasticity. The best supported model of this kind—PFH—explains how initially plastic modifications of the phenotype can become genetically fixed adaptations[51]. In this paper, we have described a model that involves another potential mode of interaction between plasticity and natural selection. The SIEI model includes the hypothesis that a stress response, exhibited by a group of cells in an ancestral lineage can collectively, becomes the target of natural selection and thereby lead both to a transformation of the stress response into an adaptive phenotype and the genetic individuation of this group of cells into a novel cell type. There is circumstantial evidence that the main light-interacting cell types of eyes also evolved from an ancestral response to light induced stress[55], and a clear strategy for testing further applications of the model by comparatively scrutinizing recurring patterns of stresss in different organ systems. This model has the potential to explain the widespread correspondence between molecular mechanisms that play a role in the cellular stress response and those involved in cell differentiation. This fact may be the footprint of a wide spread mode of evolution that transforms cellular stress reactions into developmental differentiation pathways.
Acknowledgements
This work was funded by John Templeton Foundation Grant #54860 awarded to G.P.W. and A.C.L., as well as a Revson Foundation Senior Biomedical Fellowship to E.M.E. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number U54CA209992. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- [1].Ray PD, Huang BW, Tsuji Y, Cell Signal. 2012, 24, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kultz D, Annu. Rev. Physiol. 2005, 67, 225. [DOI] [PubMed] [Google Scholar]
- [3].Flick K, Kaiser P, Semin. Cell Dev. Biol. 2012, 23, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cliff TS, Dalton S, Curr. Opin. Genet. Dev. 2017, 46, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schieber M, Chandel NS, Curr. Biol. 2014, 24, R453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borquez DA, Urrutia PJ, Wilson C, van Zundert B, Nunez MT, Gonzalez-Billault C, J Neurochem. 2016, 137, 506. [DOI] [PubMed] [Google Scholar]
- [7].Li J, Dong S, Stem Cells Int. 2016, 2016, 2470351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fuchs Y, Steller H, Cell. 2011, 147, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lambeth JD, Neish AS, Annu. Rev. Pathol. 2014, 9, 119. [DOI] [PubMed] [Google Scholar]
- [10].Atashi F, Modarressi A, Pepper MS, Stem Cells Dev. 2015, 24, 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD, Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, Steger K, Krause KH, Jaconi ME, Mol. Biol. Cell. 2006, 17, 3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Owusu-Ansah E, Banerjee U, Nature. 2009, 461, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hochmuth CE, Biteau B, Bohmann D, Jasper H, Cell Stem Cell. 2011, 8, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Love NR, Chen Y, Ishibashi S, Kritsiligkou P, Lea R, Koh Y, Gallop JL, Dorey K, Amaya E, Nat. Cell Biol. 2013, 15, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Erkenbrack EM, Maziarz JD, Griffith OW, Liang C, Chavan AR, Nnamani MC, Wagner GP, PLoS Biol. 2018, 16, e2005594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].West-Eberhard MJ, Developmental Plasticity and Evolution, Oxford University Press, Oxford: 2003. [Google Scholar]
- [18].Badyaev A, in Variation: A Central Concept in Biology, (Eds: Hallgrímsson B, Hall BK), Academic Press, San Diego, CA: 2005, 277. [Google Scholar]
- [19].Mess A, Carter AM, J. Exp. Zool. Part B (Mol. Dev. Evol.). 2006, 306B, 140. [DOI] [PubMed] [Google Scholar]
- [20].Kin K, Maziarz J, Wagner GP, Biol. Reprod. 2014, 90, 111. [DOI] [PubMed] [Google Scholar]
- [21].Kin K, Maziarz J, Chavan AR, Kamat M, Vasudevan S, Birt A, Emera D, Lynch VJ, Ott TL, Pavlicev M, Wagner GP, Genome Biol. Evol. 2016, 8, 2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kin K, Nnamani MC, Lynch VJ, Michaelides E, Wagner GP, Cell Reports. 2015, 10, 1398. [DOI] [PubMed] [Google Scholar]
- [23].Wildman DE, Chen C, Erez O, Grossman LI, Goodman M, Romero R, Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Elliot MG, Crespi BJ, J. Evol. Biol. 2008, 21, 1763. [DOI] [PubMed] [Google Scholar]
- [25].Wagner GP, Ed. Comparative Placentation-Mammals, Vol. 2, Academic Press, 2018. [Google Scholar]
- [26].Mor G, Cardenas I, Abrahams V, Guller S, Ann. N. Y. Acad. Sci. 2011, 1221, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Griffith OW, Chavan AR, Protopapas S, Maziarz J, Romero R, Wagner GP, Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gellersen B, Brosens JJ, Endocr. Rev. 2014, 35, 851. [DOI] [PubMed] [Google Scholar]
- [29].Chavan AR, Griffith OW, Stadtmauer D, Maziarz J, Pavlicev M, Fishman R, Koren L, Romero R, Wagner GP, Biorxiv. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wetendorf M, DeMayo FJ, Mol. Cell Endocrinol. 2012, 357, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Frank GR, Brar AK, Cedars MI, Handwerger S, Endocrinology. 1994, 134, 258. [DOI] [PubMed] [Google Scholar]
- [32].Li Q, Kannan A, Das A, Demayo FJ, Hornsby PJ, Young SL, Taylor RN, Bagchi MK, Bagchi IC, Endocrinology. 2013, 154, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Filant J, Spencer TE, Biol. Reprod. 2013, 88, 93. [DOI] [PubMed] [Google Scholar]
- [34].Lynch VJ, Brayer K, Gellersen B, Wagner GP, PLoS One. 2009, 4, e6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lynch VJ, May G, Wagner GP, Nature. 2011, 480, 383. [DOI] [PubMed] [Google Scholar]
- [36].Kommagani R, Szwarc MM, Vasquez YM, Peavey MC, Mazur EC, Gibbons WE, Lanz RB, DeMayo FJ, Lydon JP, PLoS Genet. 2016, 12, e1005937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Szwarc MM, Hai L, Gibbons WE, Peavey MC, White LD, Mo Q, Lonard DM, Kommagani R, Lanz RB, DeMayo FJ, Lydon JP, Biol. Reprod. 2018, 98, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Al-Sabbagh M, Fusi L, Higham J, Lee Y, Lei K, Hanyaloglu AC, Lam EW, Christian M, Brosens JJ, Endocrinology. 2011, 152, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kizilay G, Cakmak H, Yen CF, Atabekoglu C, Arici A, Kayisli UA, Histochem. Cell Biol. 2008, 130, 761. [DOI] [PubMed] [Google Scholar]
- [40].Leitao B, Jones MC, Fusi L, Higham J, Lee Y, Takano M, Goto T, Christian M, Lam EW, Brosens JJ, FASEB J. 2010, 24, 1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chavan AR, Griffith OW, Wagner GP, Curr. Opin. Genet. Dev. 2017, 47, 24. [DOI] [PubMed] [Google Scholar]
- [42].Nancy P, Siewiera J, Rizzuto G, Tagliani E, Osokine I, Manandhar P, Dolgalev I, Clementi C, Tsirigos A, Erlebacher A, J. Clin. Invest. 2018, 128, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nancy P, Tagliani E, Tay CS, Asp P, Levy DE, Erlebacher A, Science. 2012, 336, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Baldwin JM, American Naturalist. 1896, 30, 441. [Google Scholar]
- [45].Simpson GG, Evolution. 1953, 7, 110. [Google Scholar]
- [46].Levis NA, Pfennig DW, Trends Ecol. Evol. 2016, 31, 563. [DOI] [PubMed] [Google Scholar]
- [47].Badyaev AV, Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schjenken JE, Robertson SA, Adv. Exp. Med. Biol. 2015, 868, 127. [DOI] [PubMed] [Google Scholar]
- [49].Valencia A, Kochevar IE, J. Invest. Dermatol. 2008, 128, 214. [DOI] [PubMed] [Google Scholar]
- [50].Waddington CH, Nature. 1959, 183, 1654. [DOI] [PubMed] [Google Scholar]
- [51].Levis NA, Isdaner AJ, Pfennig DW, Nat. Ecol. Evol. 2018, 2, 1289. [DOI] [PubMed] [Google Scholar]
- [52].Love AC, Theory Biosci. 2006, 124, 317. [DOI] [PubMed] [Google Scholar]
- [53].Taiz L, Zeiger E, Møller IM, Murphy A, Plant Physiology and Development, Sinauer Associates, Sunderland, MA: 2015. [Google Scholar]
- [54].Emera D, Romero R, Wagner G, Bioessays. 2012, 34, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Oakley TH, Speiser DI, Annu. Rev. Ecol. Evol. S. 2015, 46, 237. [Google Scholar]
- [56].Feuda R, Hamilton SC, McInerney JO, Pisani D, Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 18868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Plachetzki DC, Fong CR, Oakley TH, Proc. Biol. Sci. 2010, 277, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tan DX, Hardeland R, Manchester LC, Paredes SD, Korkmaz A, Sainz RM, Mayo JC, Fuentes-Broto L, Reiter RJ, Biol. Rev. Camb. Philos. Soc. 2010, 85, 607. [DOI] [PubMed] [Google Scholar]
- [59].Klemenz R, Frohli E, Steiger RH, Schafer R, Aoyama A, Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].McGreal RS, Kantorow WL, Chauss DC, Wei J, Brennan LA, Kantorow M, Biochim. Biophys. Acta. 2012, 1820, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Waddington CH, Nature. 1942, 150, 563. [Google Scholar]
- [62].Crispo E, Evolution. 2007, 61, 2469. [DOI] [PubMed] [Google Scholar]
- [63].Sancho P, Martin-Sanz P, Fabregat I, Free. Radic. Biol. Med. 2011, 51, 1789. [DOI] [PubMed] [Google Scholar]