

Abstract
Hepatocellular carcinomas (HCCs) are adapted to survive extreme genomic stress conditions imposed by hyperactive DNA replication and genotoxic drug treatment. The underlying mechanisms remain unclear, but may involve intensified DNA damage response/repair programs. Here, we investigate a new role of nucleostemin (NS) in allowing HCC to survive its own malignancy, as NS was previously shown to promote liver regeneration via a damage repair mechanism. We first established that a higher NS transcript level correlates with high HCC grades and poor prognostic signatures, and is an independent predictor of shorter overall and progression-free survival specifically for HCC and kidney cancer but not for others. Immunostaining confirmed that NS is most abundantly expressed in high-grade and metastatic HCCs. Genome-wide analyses revealed that NS is co-enriched with MYC target and homologous recombination (HR) repair genes in human HCC samples and functionally intersects with those involved in replication stress response and HR repair in yeasts. In support, NS-high HCCs are more reliant on the replicative/oxidative stress response pathways, whereas NS-low HCCs depend more on the mTOR pathway. Perturbation studies showed NS function in protecting human HCC cells from replication- and drug-induced DNA damage. Notably, NS depletion in HCC cells increases the amounts of physical DNA damage and cytosolic dsDNA, leading to a reactive increase of cytokines and PD-L1. This study shows that NS provides an essential mechanism for HCC to adapt to high genomic stress for oncogenic maintenance and propagation. NS deficiency sensitizes HCC cells to chemotherapy but also triggers tumor immune responses.
Keywords: genomic stability, liver tumor, replication stress, self-renewal, stem cells
INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth most common cancer and third leading cause of cancer-related death worldwide1. It accounts for up to half of all cancers in Southeast Asia and has become the fastest growing lethal cancer in the US. HCC may be treated by surgical resection, local ablation or transplantation at the early stage (~⅓) but becomes incurable at the advanced stage (~⅔) due to high incidence of relapse and metastasis as well as resistance to radiation and drug therapies.
Drug resistance poses a major obstacle for HCC treatment. Conventional chemotherapies have been proven ineffective individually or in combination. Current approved therapies for advanced HCC include the first-line sorafenib and lenvatinib [FDA-approved on 8/16/2018] and the second-line regorafenib, cabozantinib, nivolumab, and pembrolizumab. Sorafenib alone improves patients’ survival by 3 months2, 3. Nivolumab is moderately effective but only for selected patients (19%)4, reflecting the difficulty in developing targeted therapies for tumors with multiple oncogenic drivers, such as HCC. We reason that regardless of their many etiologies and mutations, the progression of HCCs would inevitably involve hyperactive cell division, leading to replication stress. Thus, determining the tumor-addicted mechanism(s) that enables HCCs to adapt to and evade replication stress-induced catastrophe (a.k.a. Achilles heel) may provide a first step toward finding a universal target to mitigate the mortality of advanced HCCs. Recent studies have indicated that genome maintenance may be beneficial for tumor progression at the late stage5–7. So far, these findings were primarily made in breast and ovarian cancers.
One protein that could help HCC cope with excess replication stress is nucleostemin (NS), a GTP-binding nucleolar protein first discovered in neural and embryonic stem cells8–10 and later found at high levels in other types of stem cells and cancers8, 11. Our initial interest in NS was driven by its role in stem cell self-renewal. A series of our work reveal that the primary activity of NS in stem cells is to protect the integrity of replicating genome9, 12–14. Recent studies have independently shown that the ability of cancer stem cells to respond to DNA damage is more intensified as compared to non-stem cancer cells, resulting in enhanced DNA repair and G2-M checkpoint activation, which may contribute to tumor resistance to replication stress and chemo/radiation therapies15. We speculate that NS may serve an essential mechanism for HCC to survive extreme genomic stress conditions. The role of NS in liver pathophysiology has been previously indicated by: (1) NS expression is upregulated in hepatic precursor cells and regenerating adult hepatocytes12, 16, (2) NS depletion sensitizes MHCC97 cells to UV and serum starvation-induced apoptosis17, and (3) high NS expression correlates with poor survival18 and sorafenib resistance in HCC patients19. Our current data show that NS correlates with tumor grade and uniquely predicts both overall survival (OS) and progression-free survival (PFS) in HCC patients. In addition, NS protects HCCs from drug-induced DNA damage, and NS-high HCCs are more addicted to stress response pathways than NS-low HCCs. Notably, NS depletion increases the amount of physical DNA damage and cytoplasmic accumulation of fragmented dsDNA, triggering innate immune responses in HCC cells. These results raise NS as a new HCC outcome predictor and a potential therapeutic target for sensitizing HCCs to conventional chemotherapy as well as immunotherapy.
MATERIALS AND METHODS
Animal care and procedures
Animals were housed by the Program for Animal Resources at the TAMHSC-Houston campus and handled in accordance with the principles described by the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the IACUC.
TCGA and Gene Set Enrichment Assay (GSEA) analyses
Processed RNA-seq gene expression data for HCC were acquired through the GDC data portal (https://portal.gdc.cancer.gov/). Other data were acquired through cBioPortal. Sample size determination followed the same TCGA recommendations described previously20. For GSEA, HCC RNA-seq data were run against the HALLMARK and Canonical gene sets in the Molecular Signatures Database (MsigDB). Spearman correlation coefficients between each of the genes and NS were determined by using pre-ranked gene set enrichment analysis. To compare NS expression between subtypes of cancers, normality was tested by the Kolmogorov-Smirnov test. For data not normally distributed, variances between groups were sufficiently similar to use non-normal statistical tests (a rank-sum test for two groups or a Kruskal-Wallis H test for multiple groups) to determine the statistical significance. For survival analysis, patients were stratified into high-NS (highest 33% of samples) and low-NS (bottom 66% of samples).
Molecular and chemical intervention of HCC cells
Hep3B and SNU449 cells were purchased from ATCC (Hep3B: Cat# HB-8064, RRID:CVCL_0326; SNU449: Cat#CRL-2234, RRID:CVCL_0454). HuH7 cells were obtained from David Moore (Baylor College of Medicine). All three cell lines were confirmed for their identity by Short Tandem Repeat (STR) fingerprinting. Mycoplasma was tested on 5/25/18. Cells were passaged every 2–4 days up to 10 passages after thawing. For RNAi knockdown, cells were treated with oligofectamine-complexed siRNA duplex (50nM) for 24 hours. For drug treatment, cells were treated with 5-FU (10μg/ml), oxaliplatin (OXP, 10μM), doxorubicin (DRB, 5μM), or olaparib (200μM).
Supplemental Data
More details of the above and other methods.
RESULTS
High NS expression correlates with high tumor grades and poor prognosis and survival in HCC patients
To establish the clinical relevance of NS in HCC, we first compared its total transcript levels between human HCC (n=369) and adjacent nontumor tissues (ANT, n=64). Based on the TCGA HCC cohorts, there is a significant increase of NS in HCC compared to ANT (Fig.1A). From a pan-cancer perspective, HCC is ranked among the top tertile in NS expression (Fig.S1A). Comparing against various clinicopathological parameters, NS expression shows a significant increase in WHO grade 3 tumors (G3, poorly differentiated) compared to G1 (well differentiated) and G2 (moderately differentiated) (p<0.001) (Fig.1B1). Despite the low case number of undifferentiated HCCs (G4) (n=12), it still shows the highest level of NS expression (Fig.1B2). HCCs with vascular macroinvasion (n=14) express more NS than do those with none or microinvasion (Fig.1C). We also found that NS expression in HCC correlates positively with serum α-fetoprotein (AFP), fibrosis score, or viral hepatitis, but not with clinical stages (Fig.S1B–S1E). To determine whether the NS level is prognostic of the clinical outcome of HCC patients, we compared it in different molecular categories of HCC21, and found that NS is expressed: (1) more in the S2 (aggressive, Myc/Akt-activated) than in the S1 (aggressive, TGF-β-activated) or S3 (differentiated) subtype based on the Hoshida’s classification22, (2) more in the robust cluster 2 (C2, advanced tumors with more vascular invasion and extrahepatic metastasis) than in the C1 subtype based on the hepatoblastoma classification23, and (3) more in the A (proliferation/unfavorable) than in the B subclass by the NCI proliferation classification24 (Fig.1D). Finally, univariate log-rank analyses demonstrated that patients with NS-high HCCs (n=182, G1/G2/G3/G4=20/76/75/11, Stage 1/2/3/4=80/50/42/3, age 16–85) show shorter overall survival (OS) or progression-free survival (PFS), compared to those with NS-low HCCs (n=181, G1/G2/G3/G4=35/101/44/1, Stage 1/2/3/4=35/101/44/1, age 17–90) (Fig.1E). Multivariate analyses by the Cox proportional hazard model demonstrated that NS expression level is a predictor of OS and PFS, independently of tumor grade and clinical stage (Fig.1F). Pan-cancer studies showed that out of 33 cancer types, NS is prognostic of either OS or PFS in eleven cancer types, and is prognostic of both OS and PFS in only three (i.e., kidney renal clear cell carcinoma (KIRC), HCC (LIHC), and pancreatic adenocarcinoma (PAAD)) by univariate analyses (Fig.S2A, red highlight). Of the eleven cancer types, NS is independently prognostic of both OS and PFS in only two (KIRC and HCC) by multivariate analyses (Fig.S2B).
Figure 1.
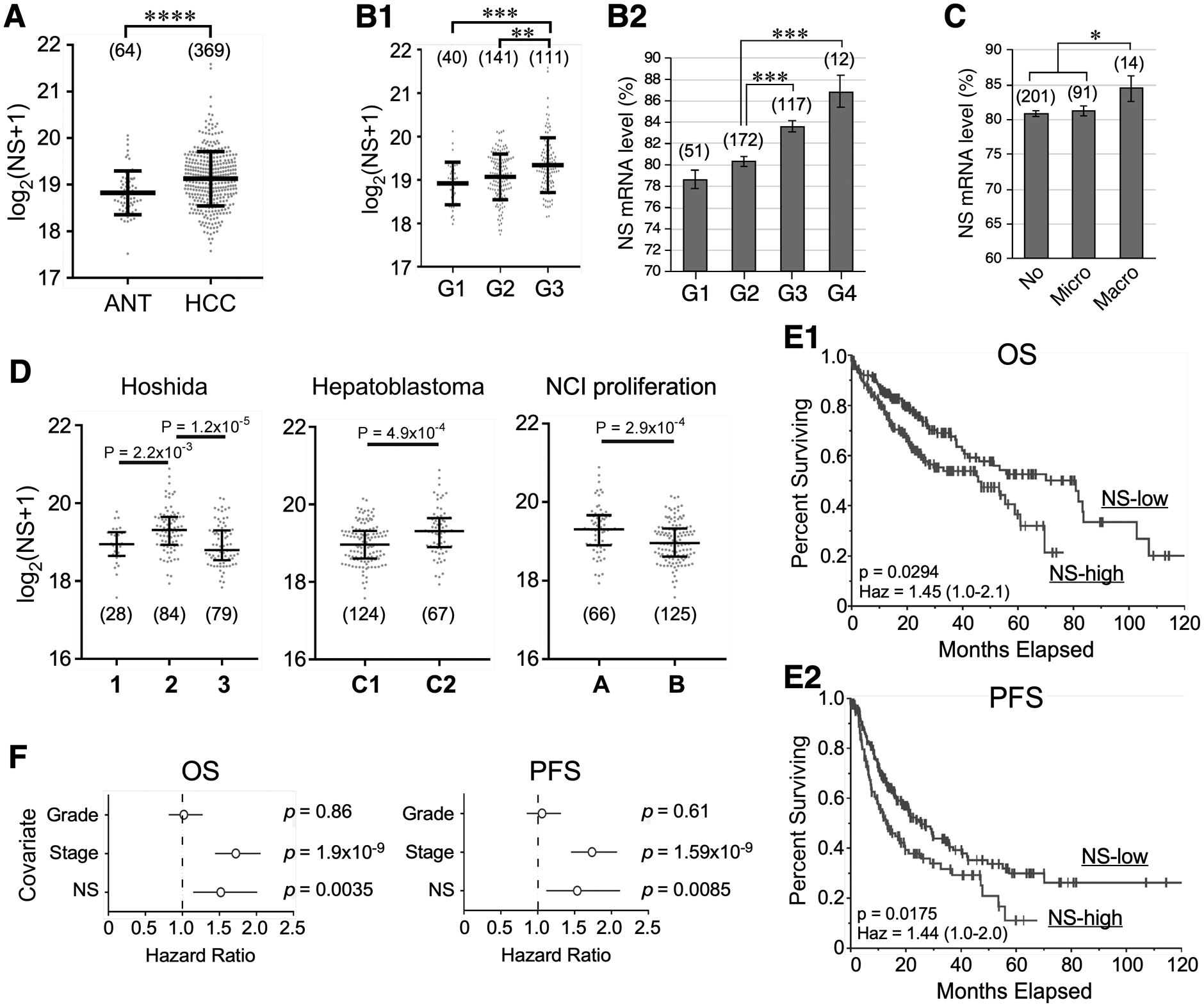
NS transcript level correlates with tumor grade and predicts survival outcome of hepatocellular carcinoma (HCC). TCGA analyses of RNA-seq-based NS expression in: (A) adjacent nontumor tissue (ANT) and HCC; (B) HCCs of different tumor grades (G) with or without grade 4 (G4); (C) HCCs of different vascular invasion; and (D) different molecular subtypes of HCCs. Statistical significance determined by the Wilcoxon Rank Sum test for two groups, by the Kruskal-Wallis H test followed by the Dunn’s Post Hoc test for ≥3 groups, and by t-test for B2 & C. Whiskers indicate median with interquartile distance. Numbers in parentheses show sample sizes. (E) Univariate analyses of overall survival (OS, E1) and progression-free survival (PFS, E2) of HCC patients with high and low NS expression by the Kaplan Meier plot (n=368). High and low NS are defined by the upper tertile and the remaining two-thirds, respectively. Significance determined by the log-rank (univariate) test. (F) Multivariate OS and PFS analyses of NS, tumor grade, and clinical stage in HCC patients by the Cox proportional hazards model. *, p <0.05; **, p <0.01; ***, p <0.001; ****, p <0.0001; Haz, hazard ratio.
NS upregulation during the malignant progression of HCC
To validate the relationship between NS expression and HCC progression at the protein level, we examined the NS staining pattern in human HCC samples (Biomax) (Fig.2A1). Our results showed that NS is expressed in much higher abundance in the nucleoli of distantly metastasized HCC cells (M1) compared to primary HCC, and more in high-grade HCCs compared to lower-grade HCCs, as determined by the NS+ tumor percentage in each grade (Fig.A2) as well as by the NS+ cell percentage and signal intensity of NS+ tumors (Fig.A3). Next, we used the diethylnitrosamine (DEN) mouse model to determine the timing of NS increase during HCC progression. DEN induces a continuous spectrum of preneoplastic/neoplastic lesions in the liver, including dysplastic nodules of increased nuclear pleomorphism and basophilic cytoplasm (8 m/o) and obvious carcinomas with higher nuclear-cytoplasmic ratios, hyperchromatic nuclei, and architectural distortion (14 m/o) (Fig.2B). Anti-NS (Ab2438) staining showed that the number of NS+ cells in dysplastic lesions is as low as that in normal livers but increases significantly in carcinomatous lesions. This dramatic increase of NS+ cells in carcinomatous lesions, which is also supported by qRT-PCR at the transcript level (Fig.2C), coincides with an increased number of Ki67+ mitotic cells on adjacent sections (Fig.2B3), suggesting a role of NS in mitotically active tumors.
Figure 2.
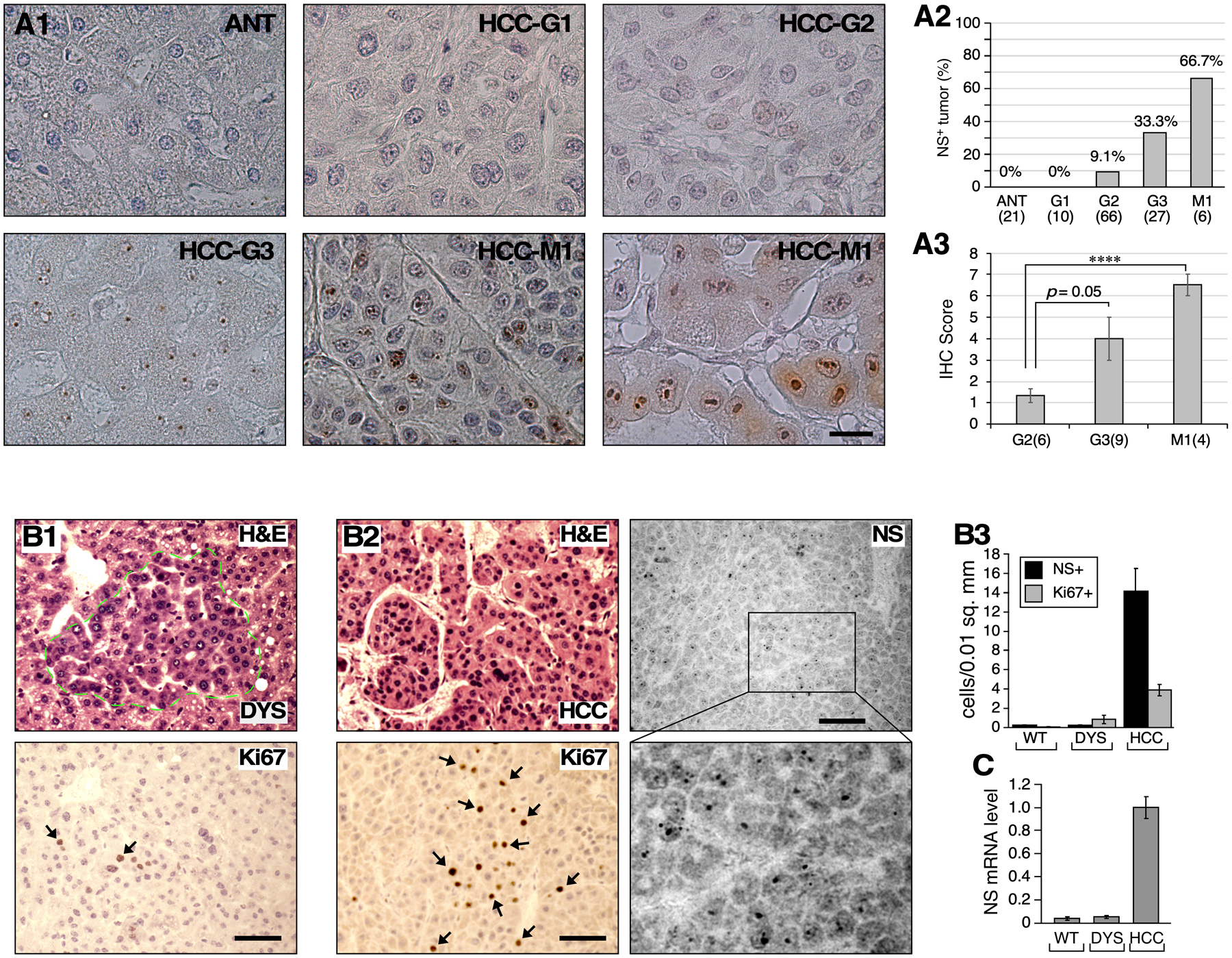
NS protein expression in high-grade HCC. (A1) Immunohistochemistry of NS (brown) with light hematoxylin counterstain (blue) in human metastasized HCCs (M1) compared to primary HCCs of different grades. (A2) Percentages of NS+ tumors in different HCC grade categories. (A3) IHC scores of NS for NS+ tumors of different grades. Numbers in parentheses indicate tumor case numbers. (B) DEN-induced mouse HCCs collected at 8 months old (m/o) (B1) and 14 m/o (B2) and determined for their pathologies (H&E), mitotic activities (Ki67), and NS+ cells. (B3) Bar graphs show quantification (mean ± sem) of NS+ and Ki67+ cells in liver normal tissue (WT), dysplastic nodules (DYS), and carcinoma (HCC). Y-axis indicates cells per 0.01mm2. (C) NS expression by quantitative RT-PCR. Scale bars, 20μm (A), 50μm (B).
NS is co-enriched with HR repair genes in HCC and functionally intersects with replication stress responsors in yeasts
To explore how NS might act mechanistically in HCC in a genome-wise manner, we applied the Gene Set Enrichment Analysis (GSEA) on RNA-seq data to identify pathway gene sets that are co-enriched with NS in human HCC samples. Of the 50 HALLMARK pathways analyzed (h.all.v5.1.symbols.gmt), ten show positive and three show negative co-enrichment with NS (p <0.05) (Fig.3A). NS co-enrichment with the MYC targets (Fig.3B) supports previous reports that NS is a c-Myc target25, 26. Of the ten positively co-enriched pathways, four are related to genomic damage/repair and cell cycle checkpoints. Canonical pathway analysis (c2.cp.v5.1.symbols.gmt) showed that HR repair is the only one of the many DNA repair (Fig.3C) and cell signaling pathways (Fig.S3A) that meets the threshold for significance in HCC samples. On an individual gene basis, NS is more co-enriched with HR pathway genes (NES>2.3, p<0.05) than with those involved in early DNA damage detection or processing (Fig.3D and S3B). While there is no significant co-enrichment of NS with the p53 pathway (Fig.S3A), there is a positive correlation between NS and wildtype p53 at the RNA expression level (Fig.S3C1), and NS is slightly more abundant in p53-mutant than in p53-wildtype HCCs (Fig.S3C2). To identify the genes with which NS might functionally intersect, we performed yeast genetic analyses to determine the genes whose knockout showed synthetic lethality with the knockout of Nug1 (the orthologue of NS in Saccharomyces cerevisiae)27. We found that Nug1KO shows synthetic lethality with the knockout of genes involved in the cellular response to replication stress (cdc45, cdc7, dbf4, esc2, cdc20) or DNA damage (rad1, rtt101) (Fig.3E). Somatic mutations in cancer genome reflect a combined outcome of the initial mutagenic exposure and a series of subsequent cell response and selection events. To probe the relationship between NS and the different genetic experiences of HCC, we calculated the correlation coefficients between NS expression and 22 mutational signatures of substitutions and indels in HCCs28, and found that the NS level in HCC correlates positively with Signatures #16 and #5 and negatively with Signature #17 (Fig.3F), which implies that NS may preferentially react to certain mutational events and their related pathways (see Discussion). Some of the correlations between NS and various gene or clinicopathological events show a low P and a low R value, suggesting that even though those correlations are likely to be true (hence a low p value), there may be other factors involved, which is not uncommon in the analysis of clinical samples, where multiple factors over a wide range of variation intersect with one another.
Figure 3.
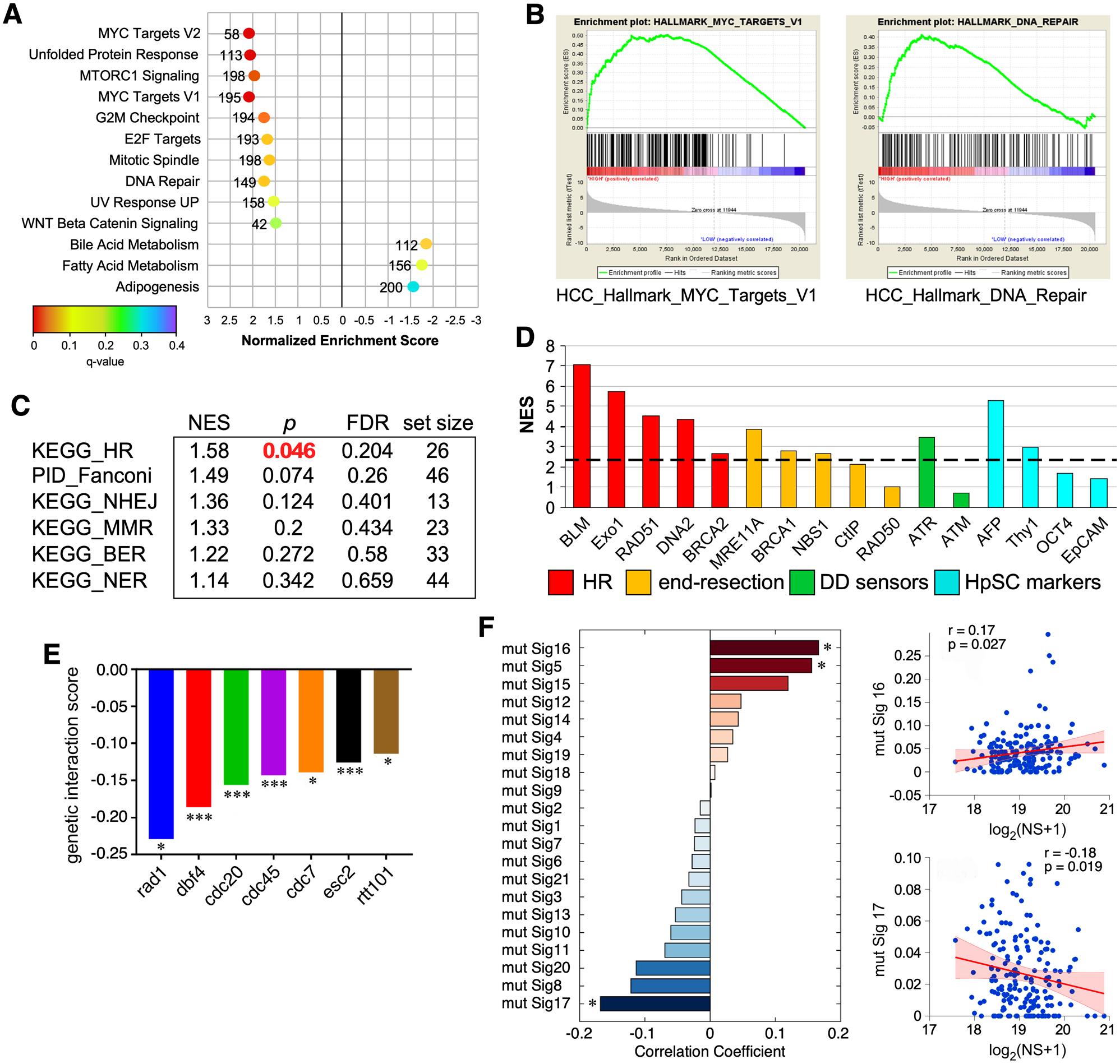
Genome-wide analysis of NS co-enrichment with pathways and mutational signatures in human HCC samples and its functional intersects in yeast. (A) Top-ranked HALLMARK pathways with positive (NES >1.5) or negative (NES <−1.5) co-enrichment with NS and p values <0.05 by GSEA. X-axis shows NES (−3 to 3); color scheme depicts FDR q values (0 to 0.4); numbers indicate pathway size. (B) Enrichment plots for the MYC Targets (left) and DNA Repair (right) pathways. (C) Canonical pathway analysis of NS co-enrichment with DNA repair sub-pathways in HCC. (D) NS co-enrichment with genes involved in HR repair (red), limited end resection (yellow), DNA damage sensing (green), and hepatic stem cell markers (HpSC, blue) in human HCC. Y-axis shows NES. Genes with NES values above the dashed line show significant correlation with NS, except for Thy1 (see Fig.S2B). (E) Genetic interaction network of Nug1 (NS orthologue) in yeast based on synthetic lethality. (F) Correlation between NS expression and mutational signatures in HCC. ES, enrichment scores; NES, normalized enrichment scores; FDR, false discovery rate.
NS-high and NS-low HCCs show preferential dependency on different pathways
To address whether NS-high and NS-low HCCs display preferential addiction to selective cellular pathways, we analyzed the ACHILLES genome-wide pooled CRISPR data to determine the relative vulnerabilities of NS-high versus NS-low HCC cells to CRISPR knockout of 17,760 genes29. Using relative gene depletion/enrichment determined by CERES30, we calculated the Spearman correlation coefficient between the NS expression level and changes in cell abundance following individual gene knockout. GSEA analyses showed that pathways with negative NES values represent those to which NS-high HCCs are preferentially addicted compared to NS-low HCCs. Hallmark pathway studies showed that NS-high HCC cells are more sensitive to gene knockout in the oxidative phosphorylation, ROS, Myc, adipogenesis, Notch, apoptosis, and E2F pathways, whereas NS-low HCC cells are more sensitive to gene knockout in the PI3K-AKT-mTOR, angiogenesis, and mitotic spindle pathways (Fig. 4A, p<0.05). To explore the idea that NS may be used to predict the drug sensitivity of HCC, we analyzed the drug screening data from the COSMIC (Catalogue of Somatic Mutations In Cancer) and CTRPv2 (Cancer Therapeutics Response Portal) databases to determine the relative vulnerabilities of NS-high versus NS-low HCC cell lines to the COSMIC and CTRP lists of inhibitors. The relationships between NS expression levels and changes in cell abundance following individual inhibitor treatment were analyzed by their relative sensitivity (Sen), with positive values indicating synthetic lethality with NS-low cells and negative values indicating synthetic lethality with NS-high cells. Those with absolute Sen values >1.4 and p values <0.05 are listed in Fig.4B, along with their FDR values. Calculations for select inhibitors (underlined) were plotted in Fig.4C. Notably, NS-high cells show multiple hits with inhibitors of Aurora kinase B (AURAB), and NS-low cells show multiple hits with inhibitors of proteins in the mTOR/PI3K pathway across different datasets. To experimentally validate the exploratory analysis of drug sensitivity, we measured the survival of two HCC cell lines (SNU449 and HuH7) in response to two mTOR inhibitors (OSI-027, omipalisib) and two AURKB inhibitors (baraserib, ZM-447439) by MTT assay. qRT-PCR confirmed that SNU449 cells express more NS compared to HuH7 cells (Fig.S7B1). MTT results demonstrated that SNU449 cells are less sensitive to mTOR inhibitors and more sensitive to AURKB inhibitors compared to HuH7 cells (Fig.4D). Together, these results indicate that NS-high HCC cells are more addicted to oxidative and/or replicative stress response pathways and are more sensitive to AURKB inhibitors compared to NS-low HCC cells. Conversely, NS-low HCC cells are more addicted to the PI3K-AKT-mTOR pathway and are more sensitive to mTOR inhibitors.
Figure 4.
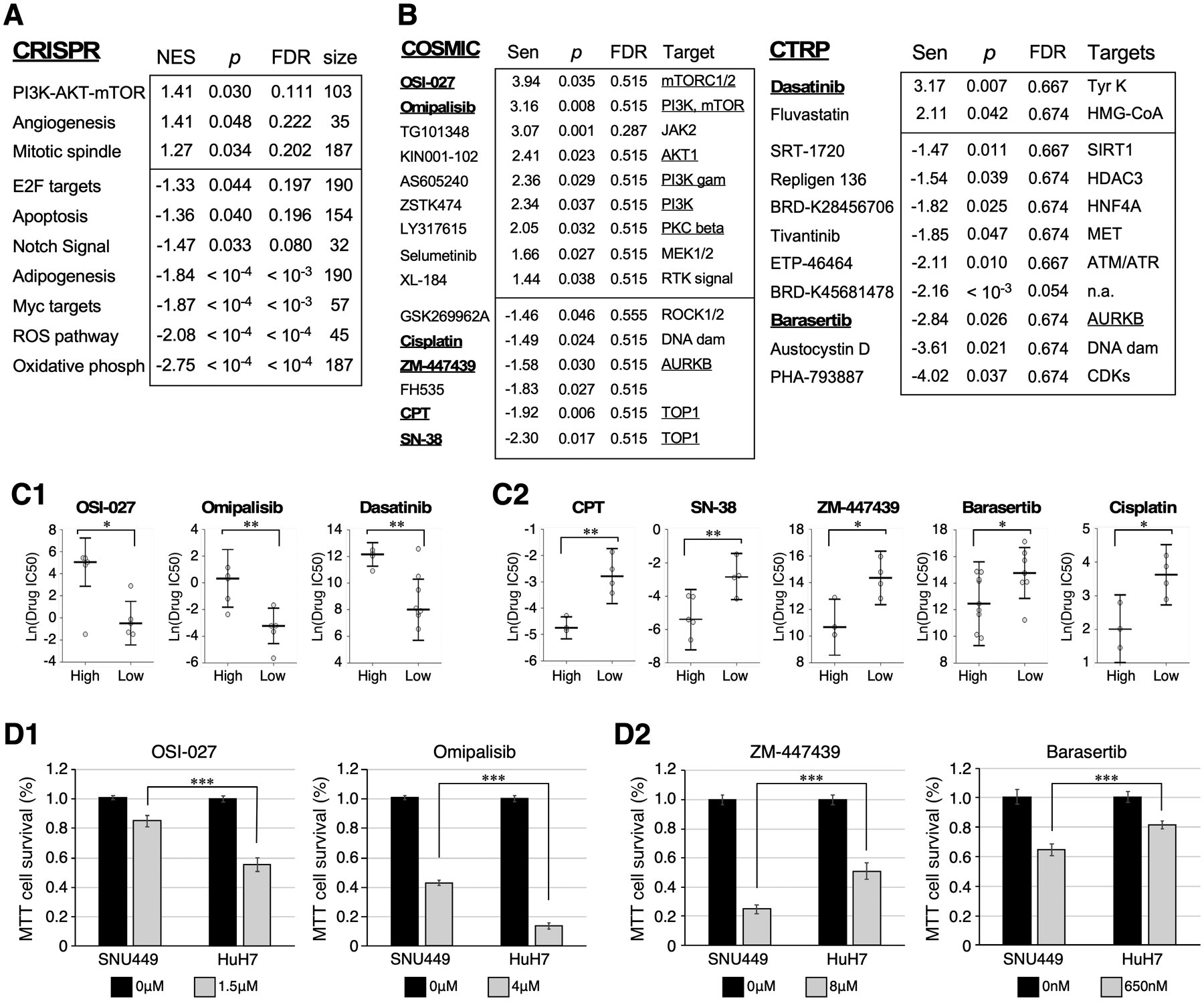
NS-high and NS-low HCC cells show preferential reliance on different classes of gene knockout and pathway inhibitors. (A) Top-ranked HALLMARK Pathways whose knockout shows synthetic lethality in NS-low (NES>0) or NS-high (NES<0) HCC cells, with p values <0.05 by GSEA. (B) Tables list top-ranked inhibitors from the COSMIC (left) or CTRP (right) panels showing significant synthetic lethality with NS-high (Sen <−1.4) or NS-low (Sen >1.4) HCC cells, with p values <0.05. (C) Relative viability of NS-high and NS-low HCC cells following treatment of specific inhibitors underlined in 4B. (D) MTT-based cell survival assays of SNU449 (NS-high) and HuH7 (NS-low) cells treated with mTOR inhibitors (D1, OSI-027, omipalisib) and AURKB inhibitors (D2, ZM-447439, Baraserib). Y-axis shows percentages of surviving cells following drug treatment compared to non-treated controls. Data consist of four biological repeats with three technical triplicates.
NS depletion predisposes HCC cells to replication-dependent DNA damage
Co-enrichment and synthetic lethality data indicate the NS function in reducing genomic stress-induced damage in HCC. To confirm this idea, we tested the effect of NS depletion in predisposing human HCC (HuH7 and Hep3B) cells to DNA damage. We first established the efficiencies of two independent RNAi constructs (siNS and siNS2, which target different NS mRNA regions) in decreasing NS protein by 76% and 58% in HuH7 cells and by 90% and 80% in Hep3B cells, respectively, as compared to control RNAi (siScr) (Fig.5A). Colony formation assays showed that siNS reduces the clonogenic survival of HuH7 cells by ~45% compared to siScr (Fig.5B). NS-knockdown (NSKD) by either siNS or siNS-2 increases the percentages of γH2AX+ cells in HuH7 and Hep3B cells (Fig.5C, S4A). Propidium iodide-labeled cell cycle analysis showed that NS depletion by siNS decreases the percentage of G1 cells and increases the percentages of S, G2, and > G2 cells in Hep3B cells compared to control knockdown by siScr, indicating that NS loss results in cell cycle blockage at the late S and G2 phase (Fig.5D). To demonstrate that NS deficiency causes physical breakage of genomic DNAs in a replication-dependent manner, Comet assays were performed to show that the amounts of damaged DNA, as measured by the increased tail moment, are indeed increased by NSKD in actively dividing HuH7 cells grown in 10% serum-containing medium but not in G0-arrest cells grown in 0.1% serum-containing medium for 5 days (Fig.5E). To determine if NS constitutes an essential mechanism for HCCs to survive genotoxic stress and hence contributes to their drug resistant property, HuH7 cells were treated with siScr or siNS, and then treated with doxorubicin (DRB), 5-FU, oxaliplatin (OXP), or olaparib. Clonogenic survival assays showed that siNS-treated HuH7 cells display a significantly reduced survival compared to siScr-treated cells in response to all the drugs (Fig.5F). As siNS alone decreases cell survival, we normalized the drug response of siScr and siNS-treated HuH7 cells to their respective no-drug controls and showed that NSKD is synthetically lethal when combined with 5-FU (reducing IC50 from 17μg/ml to 2.5μg/ml) or OXP (reducing IC50 from 14μM to 4μM) but not with DRB or olaparib (Fig.S4B).
Figure 5.
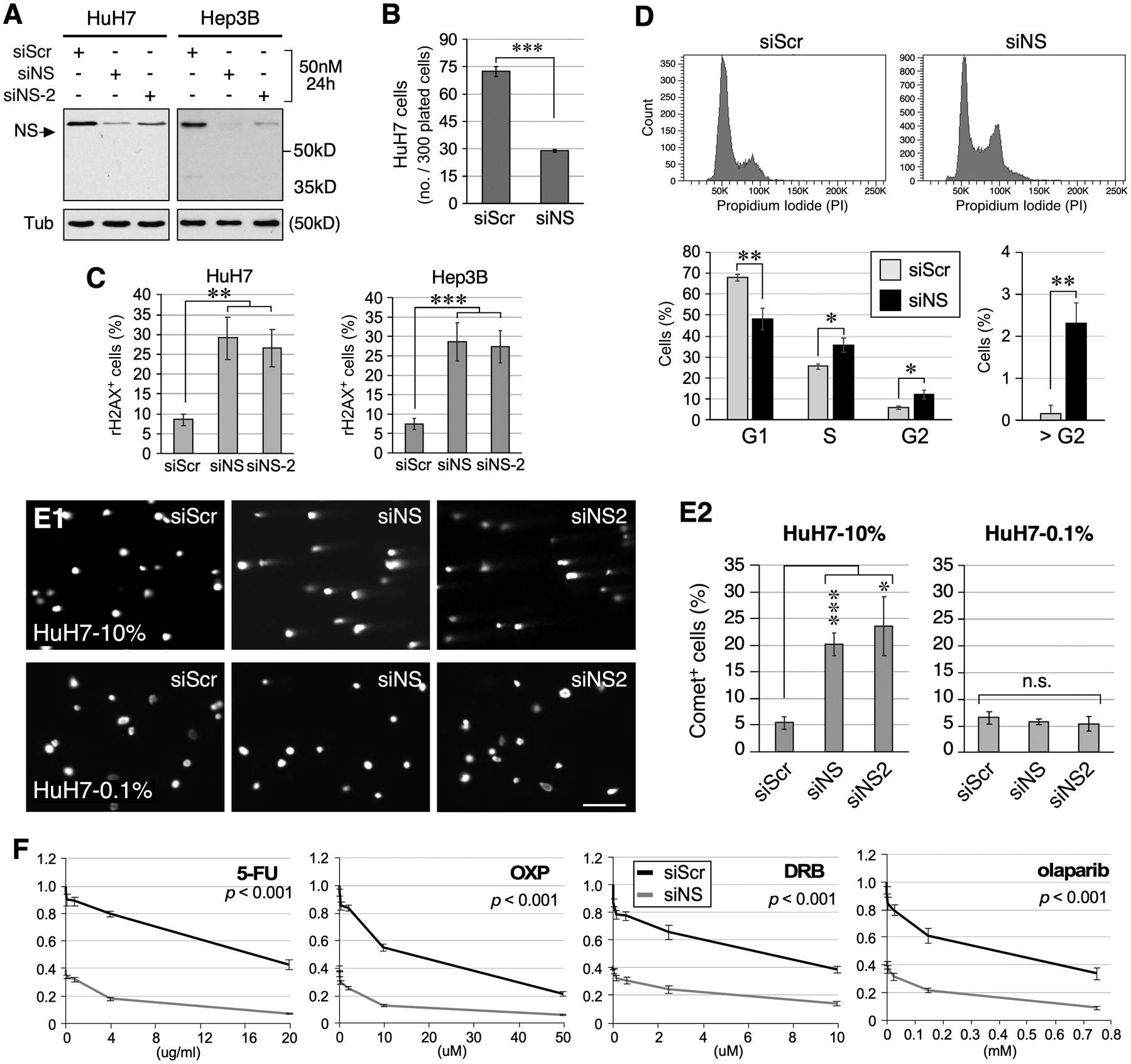
Loss of NS predisposes HCC cells to replication-dependent DNA damage. (A) Western blots of RNAi-treated HuH7 and Hep3B cells. Tub, α-tubulin. (B) Clonogenic survival of RNAi-treated HuH7 cells. (C) Quantitative analyses of γH2AX+ HuH7 and Hep3B cell percentages following RNAi treatment. (D) Cell cycle analysis of Hep3B cells treated with siScr or siNS. Top and bottom panels show representative profiles and statistically analyzed results, respectively. (E) Images and analyses of DNA tail moments of RNAi-treated HuH7 cells under the normal growing (10% FBS) or G0-arrested (0.1% FBS) condition by Comet assay. (F) Dose-dependent curves of clonogenic survival of siScr (black) and siNS (grey) treated HuH7 cells in response to 5-FU, oxaliplatin (OXP), doxorubicin (DRB), and olaparib treatment. Y-axis shows the relative survival compared to the no-drug, siScr-treated group. Significance calculated by two-way ANOVA.
NS overexpression protects HCC cells from drug-induced DNA damage
Since the NSKD effect in sensitizing HCC cells to anti-mitotic drugs may be counteracted by the decreased proliferation of NS-depleted cells, we created three NS overexpression (NSOE) HuH7 cell lines by stable transfection of EF1α promoter-driven, HA-tagged human NS (Fig.S5). Western blots showed that the protein levels of recombinant NS in NSOE #2, #26, and #28 lines are ~30–50% of the endogenous NS amount (Fig.6A). To determine if NSOE protects HCC cells from drug-induced damage, we tested the sensitivities of NSOE HuH7 cells to DRB, 5-FU, OXP, and olaparib. Anti-γH2AX staining showed that NSOE effectively reduces the number of γH2AX+ cells in response to DRB, 5-FU, OXP, and olaparib treatment in all three NSOE lines (Fig.6B and S6). The only exception is olaparib-treated #26 cells. This exception may be caused by a combination of the lower genotoxic effect of olaparib (~30%) compared to the other three drugs (~60–70%) and the lower amount of recombinant NS expressed by #26 compared to #2 and #28. Immunostaining confirmed that the baseline mitotic activities of all three NSOE HuH7 lines, as determined by their Ki67+ cell percentages, are comparable to that of wildtype HuH7 cells (Fig.6C).
Figure 6.
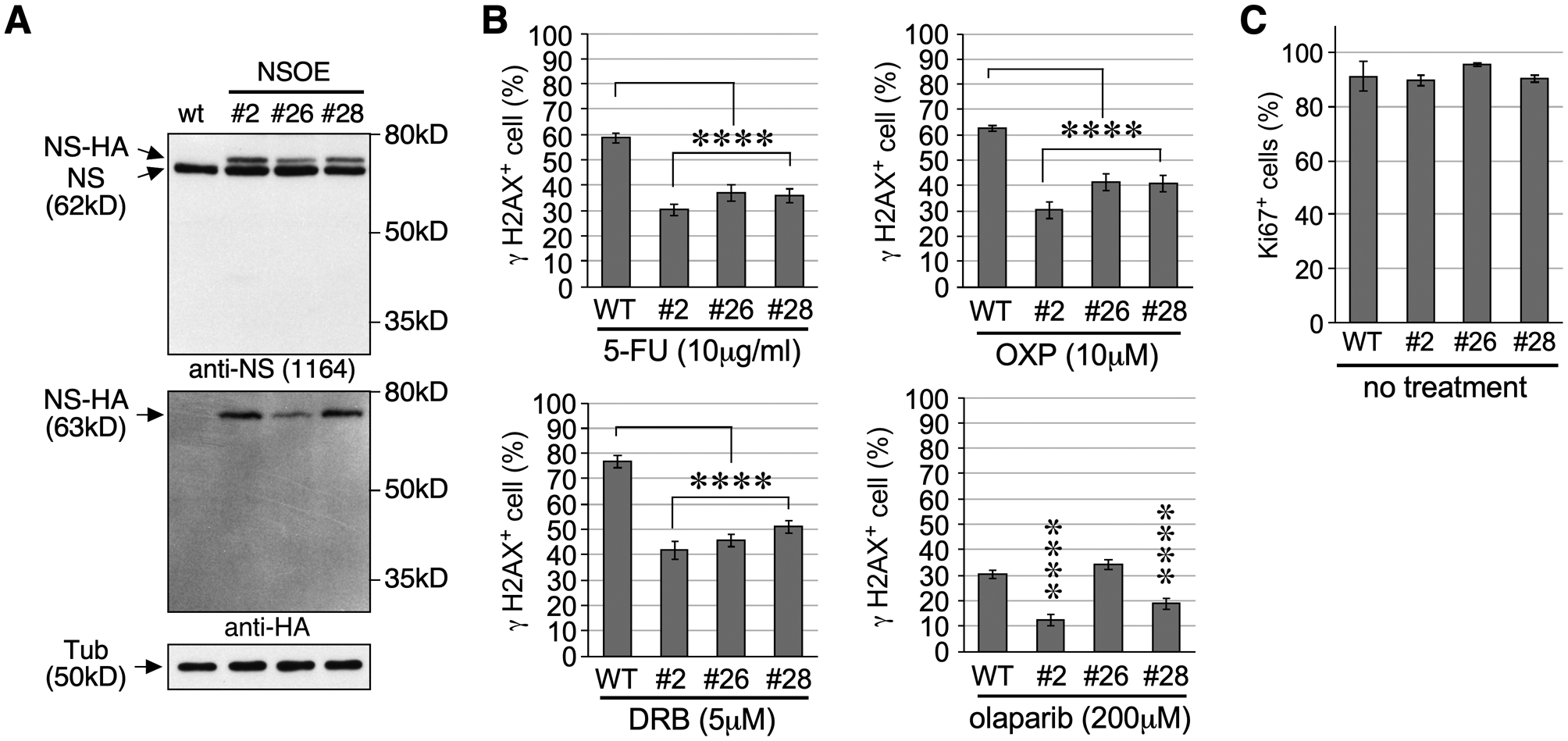
NS overexpression protects HuH7 cells from drug-induced DNA damage. (A) Western blots of parental (WT) HuH7 cells and three NSOE lines overexpressing hemagglutinin (HA)-tagged human NS (NS-HA) from an EF1α promoter. Tub, α-tubulin. (B) DNA damage response of WT and NSOE cells to 5-FU, OXP, DRB, or olaparib. Y-axis shows γH2AX+ cell percentages. (C) Baseline mitotic activities of WT and NSOE cells determined by Ki67+ cell percentages. Bars represent means (± SEM); asterisks, see Fig.1.
NS depletion increases cytosolic double-stranded DNA (dsDNA) and upregulates PD-L1 and cytokine expression in HCC cells
Excess fragmented nucleic acids in the nucleus might be transported into the cytoplasm and trigger innate immune response. To address this possibility, we first used Comet assays to show that NSKD by either siNS or siNS2 also increases physical breakage on genomic DNAs in Hep3B and SNU449 cells (Fig.7A), as seen in HuH7 cells (Fig.5D). Anti-dsDNA (clone AE-2) staining demonstrated that NSKD increases the percentage of cytosolic dsDNA+ cells to 30.3% by siNS and 31.1% by siNS2 in Hep3B cells and to 26.6% by siNS and 24.7% by siNS2 in SNU449 cells, compared to siScr-treated Hep3B (3.3%) and SNU449 (1.0%) cells or no RNAi-treated Hep3B (0.8%) and SNU449 (1.8%) cells (Fig.7B). Excess cytosolic dsDNA can be sensed by cyclic GMP-AMP synthase (cGAS), which then activates the STING pathway to turn on the expression of type I interferons, cytokines, and PD-L131–33. We speculated that NSKD-induced cytosolic dsDNA may trigger tumor innate immune response that contributes to its therapeutic effectiveness. To explore this possibility, we first used qRT-PCR to establish the baseline expression of NS and tumor immune response genes in three HCC cell lines (Fig.S7A), as well as their NSKD efficiencies by siNS and siNS2 (Fig.S7B). The NSKD effect on the expression of STING and its downstream cytokines and PD-L1 were then measured in two or three of the HCC cell lines that express them at high enough levels to be reliably detected by qRT-PCR (with Ct values ≤ 28). Our results showed that NSKD by either siNS or siNS2 increases the expression of CXCL2, CCL5, and CXCL10 in Hep3B and/or HuH7 cells but decreases their expression in SNU449 cells (Fig.7C1). NSKD also increases the expression of STING in HuH7 cells but decreases its expression in SNU449 cells (Fig.S7C). Notably, NSKD increases the expression of PD-L1 in all three HCC cells (Fig.7C2). To validate that NSKD induces the expression and secretion of cytokine from HCC cells at the protein level, we measured the amounts of CXCL10 in the culture medium of RNAi-treated Hep3B and HuH7 cells by ELISA. Our results confirmed that both siNS-treated or siNS2-treated Hep3B and HuH7 cells secret more CXCL10 compared to siScr-treated cells (Fig.7D). To validate that NSKD induces PD-L1 protein expression in HCC cells, we determined the percentages of PD-L1+ cells in RNAi-treated SNU449 cells by anti-PD-L1 FACS analysis. Our results showed that NSKD by either siNS or siNS2 increases the percentage of PD-L1+ SNU449 cells compared to control-KD by siScr (Fig.7E). Together, these results show that NS depletion increases genomic damage and cytoplasmic dsDNAs, thereby triggering tumor immune responses, including increased PD-L1 expression and cell-dependent responses of cytokines.
Figure 7.
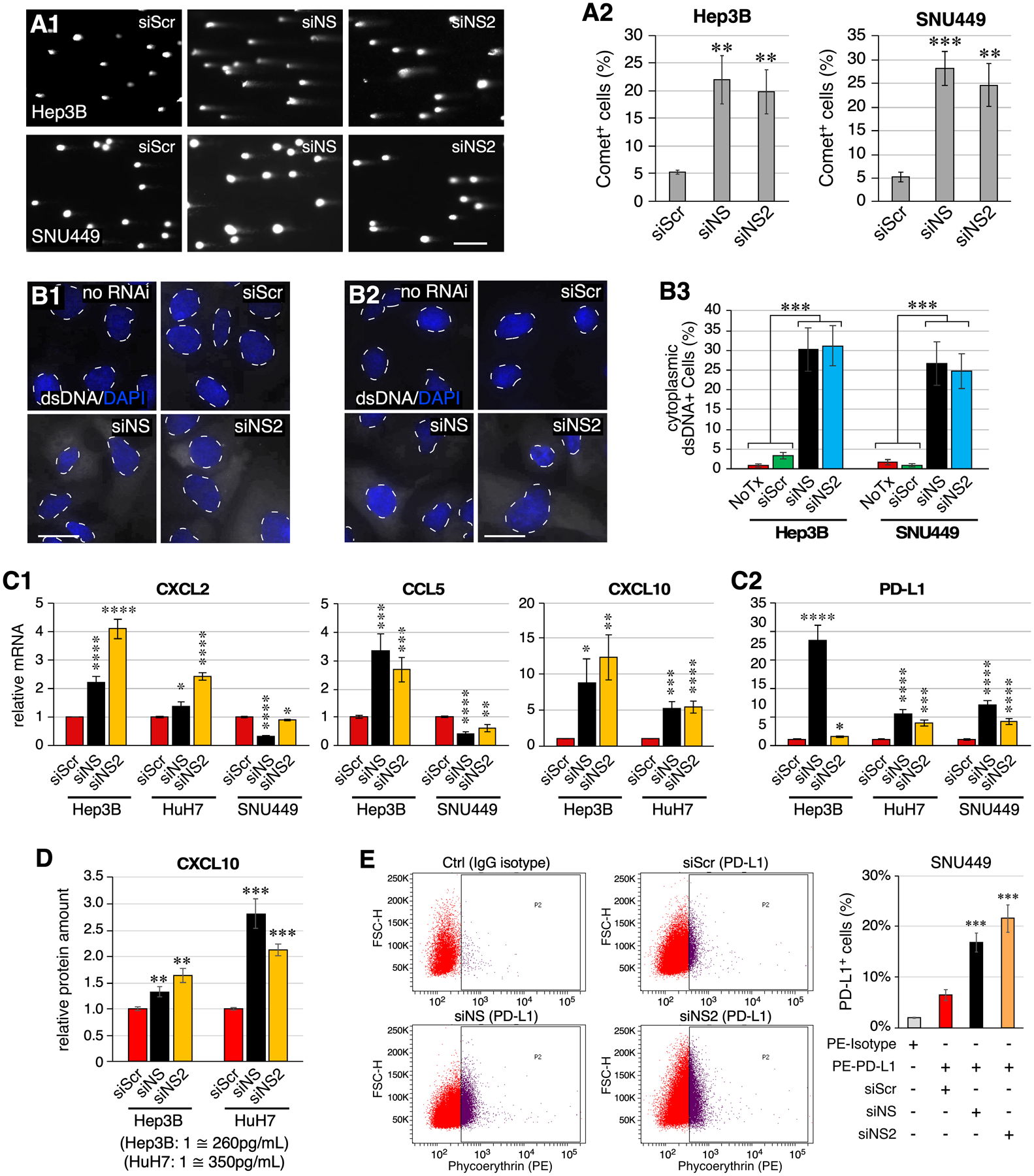
NS depletion leads to increased cytosolic double-stranded DNAs and triggers innate immune response in HCC cells. (A) Images and analyses of DNA tail moments of RNAi-treated Hep3B and SNU449 cells by Comet assay. (B) Images of dsDNA staining in no-RNAi and RNAi-treated Hep3B cells (B1) and SNU449 cells (B2). Signals of dsDNA and DAPI are shown in white and blue colors, respectively. Nuclear-cytoplasmic borders are demarcated by dashed lines. Statistical analyses are shown in (B3). (C) Expression levels of CXCL2, CCL5, CXCL10, and PD-L1 in RNAi-treated Hep3B, HuH7, and/or SNU449 cells by qRT-PCR. Levels were referenced to Rplp0 in each sample and compared to their respective siScr controls. (D) ELISA analysis of CXCL10 in the medium collected from RNAi-treated Hep3B and HuH7 cell cultures. Data consist of four biological repeats with two technical replicates. (E) FACS analysis of PD-L1+ cell percentage of RNAi-treated SNU449 cells labeled with PE-conjugated anti-PD-L1 antibody. Untreated SNU449 cells labeled with PE-conjugated IgG isotype antibody were used as the gating control. Scale bars, 200μm in (A) and 20μm in (B). The NSKD responses of CCL5 in HuH7 cells, CXCL10 in SNU449 cells, and STING in Hep3B cells were not included because of their low baseline expression levels in those cell lines, limiting the reliability of qRT-PCR results.
DISCUSSION
NS as a new outcome predictor of HCC
The clinical relevance of NS in HCC is revealed by the following evidence. First, HCC expresses more NS compared to adjacent nontumor liver tissue, which is confirmed by immunostaining showing that high-grade and metastatic HCCs express more NS than do low-grade HCCs. Second, high NS expression correlates with tumor factors that predict early recurrence, i.e., tumor differentiation/grade and vascular invasion. Third, high NS is associated with the most aggressive subclass of HCC with poor prognosis based on three different molecular classifications (Fig.1D). Finally, multivariate analyses demonstrate that high NS is an independent predictor of poor OS and PFS in HCC patients. Among the 33 cancer types analyzed, HCC is one of the only two cancer types (along with KIRC) whose NS expression shows a significant correlation with both OS and PFS outcomes by multivariate analyses. The NS ability to predict survival outcome is not related to its basal expression level, as KIRC is the lowest NS-expressing cancer type and testicular germ cell tumor, being the highest NS-expressing cancer type, shows no survival prediction by NS. For those that show no survival prediction by NS, it remains possible that some subclasses of them may still do. Longitudinal studies of the DEN model indicate that NS is more involved in the late progression than early initiation of HCC.
NS allows HCC to adapt to high genomic stress conditions
We have used several genome-wide analysis tools to broadly explore the mechanism of NS in HCC, all of which converge on cell response to genomic stress. GSEA shows that NS is co-expressed with two oncogenic networks, Myc and E2F, as well as those involved in DNA damage repair, G2M checkpoint, or mitotic spindle checkpoint in human HCC samples. The Myc gene, although seldom mutated in any cancer, is quite commonly overexpressed in HCC34, 35 and is a dominant oncogene that confers oncogene addiction in HCC36, 37. NS has been previously shown to be a Myc target in rats and mice25, 26. We reason that NS may be an effector underpinning the Myc oncogene addiction of HCC. To date, the mechanistic connection between E2F and NS regulation has not yet been shown experimentally and may warrant further investigation. Of all the DNA repair sub-pathways, NS is most significantly co-expressed with the HR repair components in HCC. A yeast genetic screen supports that NS functionally intersects with most HR repair genes and with pathways that respond to replication stress and DNA damage. NS involvement in genomic stress-induced damage is supported by data showing that NSKD increases DNA damage and NSOE protects against DNA damage triggered by DNA-damage drugs in HCC cells. As the G2/M checkpoint control is often disabled during the tumorigenic process, cancer cells experiencing genomic stress-induced damage and G2 slippage may enter mitosis prematurely, resulting in mitotic failure and, ultimately, senescence and death. To avoid those detrimental outcomes, HCCs may rely on an intensified NS activity to minimize replication stress-induced damage.
HCCs facing hyperactive mitosis are addicted to NS for survival
Genomic instability has long been regarded as a contributing factor to tumor initiation and progression. Yet excess genomic defects may also subject cancer cells to mitotic failure and cell death, revealing a dual role of genome maintenance mechanisms in preventing tumor initiation at the early stage and allowing tumor addiction at the late stage. It has been shown that low rates of chromosome mis-segregation promote tumorigenesis, whereas high rates prevent tumor growth38. Similarly, DNA damage response and repair pathways may contribute to cancer resistance to radiation and chemotherapies39, 40. Recent studies are now converging on the idea that the survival of malignant tumors may be indispensably reliant on a heightened DNA damage response pathway. Our data raise NS as a novel tumor adaptation mechanism critical for advanced HCC to survive high genomic stress conditions. Given the diverse tumor-initiating mutation events in HCCs, it has been difficult to develop a universally effective strategy by targeting a single hepatocarcinogenic pathway for most HCCs. Our findings suggest an attractive solution by targeting the NS pathway as the “Achilles heel” of malignant HCC. Notably, other than its effect on tumors, NS is also known to play critical roles in normal biology. Therefore, a non-discriminative NS depletion therapy may incur unwanted effects on organ development or tissue homeostasis. To avoid these potential side effects, one may need to incorporate additional strategies, such as tumor-specific delivery methods or combination therapies to create tumor-unique synthetic lethality – a concept explored in Fig. 4.
The heterogeneity of NS expression in HCC also highlights the idea that different HCC subtypes rely on different pathways for survival. CRISPR/inhibitor screens reveal that the survival of NS-high HCC is more dependent on genes involved in cellular response to oxidative and replication stress, whereas NS-low HCC is more reliant on the PI3K-AKT-mTOR pathway, which has been shown to promote tumor survival by inhibiting MYC-induced apoptosis41. We also found that certain mutational signatures are distinctively clustered in NS-high (#16) or NS-low (#17) HCCs. Signature #16 is characterized by T>C mutations at the ATA, ATG, and ATT sequences, is present exclusively in HCCs, and shows the strongest transcriptional strand bias of all signatures, which may reflect the event of bulky DNA helix-distorting adducts28. The different genetic constitution of NS-high and NS-low HCCs may on one hand capture their different mutational experience during development, and on the other hand dictate their preferential addictions thereafter. Furthermore, subpopulations of NS-heterogeneous cells with distinct pathway reliance may exist within a single tumor, providing a mechanism for HCC to escape drug toxicity initially, which may later evolve into a stable drug resistance pathways. Thus, using combination therapies to target multiple addiction pathways may be necessary.
NS depletion increases genomic damage, triggering tumor immune response
Genomic instability may also trigger tumor inflammatory responses via the cGAS-cGAMP-STING pathway that senses excess cytosolic dsDNAs33. The possibility of NSKD in triggering innate immune responses was investigated in three HCC cell lines, which may bear interesting implications for HCC treatment given the recent FDA approval of immunotherapies (nivolumab, and pembrolizumab) as 2nd-line therapies for unresectable HCC. Immunotherapies work by disabling the PD-1-mediated immune checkpoint, thereby unleashing the anti-tumor activity of tumor-infiltrating lymphocytes (TILs). Following NSKD, all three HCC lines show elevated expression of PD-L1 but opposite cytokine responses (up in Hep3B/HuH7 cells and down in SNU449 cells). Given that PD-L1 is a ligand of PD-1 that subverts the immunosurveillant function of TILs, our findings suggest that NS depletion may unleash both tumor innate immune and checkpoint response in some HCCs, thereby sensitizing them to anti-PD-1 immunotherapy.
Supplementary Material
Implications Statement:
Hepatocellular carcinoma (HCC) employs a novel, nucleostemin-mediated adaptive mechanism to survive high genomic stress conditions, a deficiency of which sensitizes HCC cells to chemotherapy but also triggers tumor immune responses.
ACKNOWLEDGEMENTS
We thank the Cellular & Molecular Morphology Core of the Texas Medical Center Digestive Diseases Center (NIDDK-P30-DK056338), Pamela Parsons, and Kay Pham for help with immunohistochemistry.
This work is supported by NCI-PHS grants R03 CA201988 and R21AG052006 to R.Y.L. Tsai, CPRIT Individual Investigator Research Awards RP200081 to R.Y.L. Tsai, G. Komen PDF17483544 to D.J. McGrail, and NIH grant P30 DK56338 to the Texas Medical Center Digestive Diseases Center to M.J. Finegold.
Footnotes
Disclosure: The authors declare no potential conflicts of interest financially, professionally, or personally relevant to this manuscript.
REFERENCES
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365: 1118–1127. [DOI] [PubMed] [Google Scholar]
- 2.Chan SL, Yeo W. Development of systemic therapy for hepatocellular carcinoma at 2013: Updates and insights. World J Gastroenterol. 2014;20: 3135–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Germano D, Daniele B. Systemic therapy of hepatocellular carcinoma: Current status and future perspectives. World J Gastroenterol. 2014;20: 3087–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kudo M. Immune Checkpoint Blockade in Hepatocellular Carcinoma: 2017 Update. Liver Cancer. 2016;6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361: 123–134. [DOI] [PubMed] [Google Scholar]
- 6.Graeser M, McCarthy A, Lord CJ, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16: 6159–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin T, Lin TC, McGrail DJ, et al. Nucleostemin reveals a dichotomous nature of genome maintenance in mammary tumor progression. Oncogene. 2019;38: 3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai RY, McKay RD. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 2002;16: 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng L, Lin T, Peng G, et al. Nucleostemin deletion reveals an essential mechanism that maintains the genomic stability of stem and progenitor cells. Proc Natl Acad Sci U S A. 2013;110: 11415–11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai RY. Turning a new page on nucleostemin and self-renewal. J Cell Sci. 2014;127: 3885–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin T, Meng L, Li Y, Tsai RY. Tumor-initiating function of nucleostemin-enriched mammary tumor cells. Cancer Res. 2010;70: 9444–9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin T, Ibrahim W, Peng C-Y, Finegold MJ, Tsai RY. A novel role of nucleostemin in maintaining the genome integrity of dividing hepatocytes during mouse liver development and regeneration. Hepatology. 2013;58: 2176–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin T, Meng L, Wu LJ, Pederson T, Tsai RY. Nucleostemin and GNL3L exercise distinct functions in genome protection and ribosome synthesis, respectively. J Cell Sci. 2014;127: 2302–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai RY. Balancing self-renewal against genome preservation in stem cells: How do they manage to have the cake and eat it too? Cell Mol Life Sci. 2016;73: 1803–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carruthers RD, Ahmed SU, Ramachandran S, et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018;78: 5060–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shugo H, Ooshio T, Naito M, et al. Nucleostemin in injury-induced liver regeneration. Stem Cells Dev. 2012;21: 3044–3054. [DOI] [PubMed] [Google Scholar]
- 17.Yuan F, Cheng Q, Li G, Tong T. Nucleostemin Knockdown Sensitizes Hepatocellular Carcinoma Cells to Ultraviolet and Serum Starvation-Induced Apoptosis. PLoS One. 2015;10: e0141678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu B, Hua L, Ni W, et al. Nucleostemin/GNL3 promotes nucleolar polyubiquitylation of p27(kip1) to drive hepatocellular carcinoma progression. Cancer Lett. 2017;388: 220–229. [DOI] [PubMed] [Google Scholar]
- 19.Hua L, Hu B, Yan D, et al. Upregulated expression of Nucleostemin/GNL3 is associated with poor prognosis and Sorafenib Resistance in Hepatocellular Carcinoma. Pathol Res Pract. 2017;213: 688–697. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173: 400–416.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erstad DJ, Fuchs BC, Tanabe KK. Molecular signatures in hepatocellular carcinoma: A step toward rationally designed cancer therapy. Cancer. 2018;124: 3084–3104. [DOI] [PubMed] [Google Scholar]
- 22.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69: 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairo S, Armengol C, De Reynies A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14: 471–484. [DOI] [PubMed] [Google Scholar]
- 24.Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40: 667–676. [DOI] [PubMed] [Google Scholar]
- 25.O’Connell BC, Cheung AF, Simkevich CP, et al. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem. 2003;278: 12563–12573. [DOI] [PubMed] [Google Scholar]
- 26.Zwolinska AK, Heagle Whiting A, Beekman C, Sedivy JM, Marine JC. Suppression of Myc oncogenic activity by nucleostemin haploinsufficiency. Oncogene. 2012;31: 3311–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costanzo M, VanderSluis B, Koch EN, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353: aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a Cancer Dependency Map. Cell. 2017;170: 564–576.e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49: 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36: 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14: 19–26. [DOI] [PubMed] [Google Scholar]
- 33.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215: 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18: 3004–3016. [DOI] [PubMed] [Google Scholar]
- 35.Kawate S, Fukusato T, Ohwada S, Watanuki A, Morishita Y. Amplification of c-myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology. 1999;57: 157–163. [DOI] [PubMed] [Google Scholar]
- 36.Beer S, Zetterberg A, Ihrie RA, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2: e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shachaf CM, Kopelman AM, Arvanitis C, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431: 1112–1117. [DOI] [PubMed] [Google Scholar]
- 38.Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A. 2013;110: E4134–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim CH, Park SJ, Lee SH. A targeted inhibition of DNA-dependent protein kinase sensitizes breast cancer cells following ionizing radiation. J Pharmacol Exp Ther. 2002;303: 753–759. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda S, Urata Y, Fujiwara T. Ataxia-telangiectasia mutated and the Mre11-Rad50-NBS1 complex: promising targets for radiosensitization. Acta Med Okayama. 2012;66: 83–92. [DOI] [PubMed] [Google Scholar]
- 41.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385: 544–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.