

Abstract
The tumour suppressor breast cancer type 1 susceptibility protein (BRCA1) promotes DNA double-strand break (DSB) repair by homologous recombination and protects DNA replication forks from attrition. BRCA1 partners with BRCA1-associated RING domain protein 1 (BARD1) and other tumour suppressor proteins to mediate the initial nucleolytic resection of DNA lesions and the recruitment and regulation of the recombinase RAD51. The discovery of the opposing functions of BRCA1 and the p53-binding protein 1 (53BP1)-associated complex in DNA resection sheds light on how BRCA1 influences the choice of homologous recombination over non-homologous end joining and potentially other mutagenic pathways of DSB repair. Understanding the functional crosstalk between BRCA1–BARD1 and its cofactors and antagonists will illuminate the molecular basis of cancers that arise from a deficiency or misregulation of chromosome damage repair and replication fork maintenance. Such knowledge will also be valuable for understanding acquired tumour resistance to poly(ADP-ribose) polymerase (PARP) inhibitors and other therapeutics and for the development of new treatments. In this Review, we discuss recent advances in elucidating the mechanisms by which BRCA1–BARD1 functions in DNA repair, replication fork maintenance and tumour suppression, and its therapeutic relevance.
The integrity of our genome is continually challenged by environmental agents such as high-energy radiation and mutagenic chemicals and also by reactive intermediates of cellular metabolism, such as free radicals and aldehydes1–3. Furthermore, DNA replication is replete with perils, including obstruction of the DNA polymerase ensemble by secondary DNA structures and by transcription-associated R-loops, which could lead to replication fork stalling or collapse. Failure to remove DNA lesions or to restart stalled replication forks can cause mutations or catastrophic genome rearrangements, leading to cell transformation and disease, in particular neurological disorders and cancer4–8.
Of the myriad of continually occurring DNA lesions, the DNA double-strand break (DSB), which is formed through direct chemical assault or from the collapse of stalled replication forks, poses the greatest threat to genomic stability. Several conserved, mechanistically distinct pathways of DSB repair have evolved to repair DSBs, including homologous recombination (HR), non-homologous end joining (NHEJ), alternative end joining and single-strand annealing9,10 (BOX 1). NHEJ, alternative end joining and single-strand annealing often entail deletion or insertion of several nucleotides and can also give rise to chromosome translocations. By contrast, HR is the most accurate DSB repair mechanism and is capable of faithfully restoring the original configuration of the broken DNA molecule. HR is also the default mechanism for replication fork repair11–15. However, since the HR machinery prefers to engage the sister chromatid over the homologous chromosome as a template for DSB repair, it is most active in S phase and G2 phase of the cell cycle, when sister chromatids become available16,17 (BOX 1).
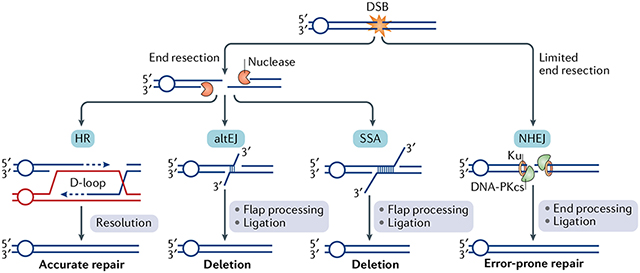
Box 1 |. Conservative and error-prone pathways of double-strand break repair.
Four conserved pathways of DNA double-strand break (DSB) repair have been characterized. In non-homologous end joining (NHEJ), the DNA ends are engaged by a complex of proteins including the Ku70-Ku80 (Ku) complex and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which further recruit other proteins to conduct nucleolytic trimming of the DNA ends, followed by DNA gap filling and ligation210 (see the figure). NHEJ is a highly efficient DsB repair pathway, but it generates DNA products that typically harbour the deletion or insertion of a few nucleotides.
The ends of a DsB can undergo nucleolytic degradation, in a process known as end resection. DNA end resection typically channels the DsB into the homologous recombination (HR) repair pathway. end resection removes a few hundred or more bases of the 5′-terminated strand to yield a 3′ single-stranded DNA (ssDNA) tail, which serves as the template for the assembly of protomers of the recombinase RAD51 to form a helical nucleoprotein filament. The RAD51-ssDNA ensemble then conducts a search for DNA homology in either the sister chromatid (preferred partner) or the homologous chromosome and catalyses the formation of a displacement loop (D-loop; see the figure). DNA synthesis occurs within the D-loop, followed by the resolution of the extended structure through one of several pathways211 to form different types of DNA products.
Occasionally, the 3′ tail originating from end resection can undergo annealing through sequences with microhomology, or through regions of more extensive homology (for example, DNA repeats) to yield an intermediate with 3′ DNA flaps. In both cases (microhomology or extensive homology), the 3′ flaps are trimmed and, following further processing, the DNA joint is sealed by ligation. The microhomology-mediated DNA repair process is termed ‘alternative DNA end joining’ (altEJ), whereas joining via the hybridization of DNA repeats is referred to as ‘single-strand annealing’ (SSA). Both altEJ and SSA require distinct cadres of factors, yet both pathways result in deletions212 (see the figure).
DSB repair pathway choice is intimately linked with cell cycle progression. Specifically, NHEJ remains active throughout the cell cycle and becomes the predominant pathway in G1 phase. By contrast, HR is most active in S phase and G2 phase, as the ability of cells in G1 phase to conduct DNA end resection is greatly attenuated75,213. Given that altEJ and SSA also require DNA end resection, they are primarily operational in S phase and G2 phase. DNA repair pathway choice is intricate and entails the exclusion of the resection machinery from DNA ends and also the cell cycle-specific regulation of HR repair complex assembly and chromatin modifications9,10.
HR is the only DSB repair pathway capable of restoring the original DNA sequence of the damaged site and is therefore conservative in nature. The mechanisms of different DSB repair pathways have been reviewed recently212,214, whereas the cell cycle-specific regulation of DNA end resection is discussed in this article.
Studies of DSB repair mechanisms and pathway choice have garnered intense interest owing to their relevance for the maintenance of genome integrity and cancer origin. In this Review, we focus on two key factors of the HR pathway in humans: breast cancer type 1 susceptibility protein (BRCA1) and its obligatory partner BRCA1-associated RING domain protein 1 (BARD1). Mutations in BRCA1 lead not only to familial breast and ovarian cancers but are also the likely driver of a variety of sporadic cancers18–21. There is compelling evidence that BARD1 is also a tumour suppressor22–27. We first introduce the original studies that led to the identification of BRCA1 as a suppressor of familial breast cancer and how BARD1 was isolated as an interactor and partner of BRCA1. We discuss how the BRCA1-BARD1 heterodimer, through its E3 ubiquitin ligase activity and the ability to interact with DNA and DNA damage response factors, helps channel DSBs, including at damaged replication forks, into the HR pathway for repair. We also discuss recent studies that implicate BRCA1-BARD1 in multiple stages of the HR process and a novel function of this tumour suppressor complex in the protection of stalled replication forks against deleterious attrition by cellular nucleases.
The recent approval of poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) by the US Food and Drug Administration and the European Medicines Agency as cancer therapeutic agents adds to the arsenal of drugs to treat HR-deficient tumours28 and also accords credence to the applicability of synthetic lethality in future drug development efforts29–31. However, most patients undergoing PARPi therapy ultimately develop resistance to these drugs, and we discuss the known mechanisms of PARPi resistance and the prospects for developing new therapeutics that further exploit the DSB repair deficiency of HR-deficient tumours.
The activity and interactions of BRCA1
BRCA1 and BARD1 together fulfil complex roles in DNA repair, replication fork protection, transcription and tumour suppression. Mouse studies have established that the Brca1 and Bard1 genes are essential for survival, as embryonic lethality is observed between 6.5 and 8.5 embryonic days in single-knockout or double-knockout mice. Cells derived from the mutant embryos exhibit chromosome structural abnormalities and aneuploidy32–36. Importantly, mammary-specific ablation of either Brca1 or Bard1 in combination with p53 deficiency leads to a high frequency of basal-like breast carcinomas with a propensity for being triple-negative tumours (that is, lacking the oestrogen, progesterone and HER2 (also known as ERBB2) receptors). The striking similarities between the mammary carcinomas of single-knockout and double-knockout mice again emphasize that BRCA1 and BARD1 function together in tumour suppression37–40. It is interesting that although heterozygous Brca1+/− and Bard1+/− mice are not particularly tumour-prone, women with one mutant BRCA1 or BARD1 allele exhibit tumour predisposition. In this section, we discuss the studies that led to the identification of BRCA1 and BARD1 as tumour suppressors and the functional domains and biochemical characteristics of BRCA1-BARD1 that are germane for its role in the maintenance of genome stability.
The characterization of BRCA1 as a familial breast cancer gene.
The BRCA1 gene encodes a large protein of 1,863 amino acids41 with several functional domains42–44 (FIG. 1; see later). BRCA1 was discovered as an early-onset breast cancer susceptibility gene in afflicted families by linkage analysis45. Subsequently, BRCA1 mutations were also linked to familial ovarian cancers46,47. Among carriers of BRCA1 mutations, the estimated lifetime risk of breast cancer is more than 80%, and that of ovarian cancer is 40–65%. This represents a sevenfold risk increase for breast cancer and a more than 20-fold increase for ovarian cancer compared with individuals without mutations in BRCA1 (REF.48). Typically, tumorigenesis in carriers of BRCA1 mutations entails loss of heterozygosity and the elimination of the wild-type BRCA1 allele. Therefore, BRCA1 fits the definition of a classical tumour suppressor. Importantly, BRCA1-deficient breast tumours can develop also with no familial linkage49, and many of these cancers may be explained by epigenetic silencing of the wild-type allele through hypermethylation of the BRCA1 gene promoter50. A substantial fraction of breast tumours arising from BRCA1 deficiency are triple negative, representing one of the most aggressive forms of the disease51.
Fig. 1 |. The functional domains of BRCA1 and BARD1.
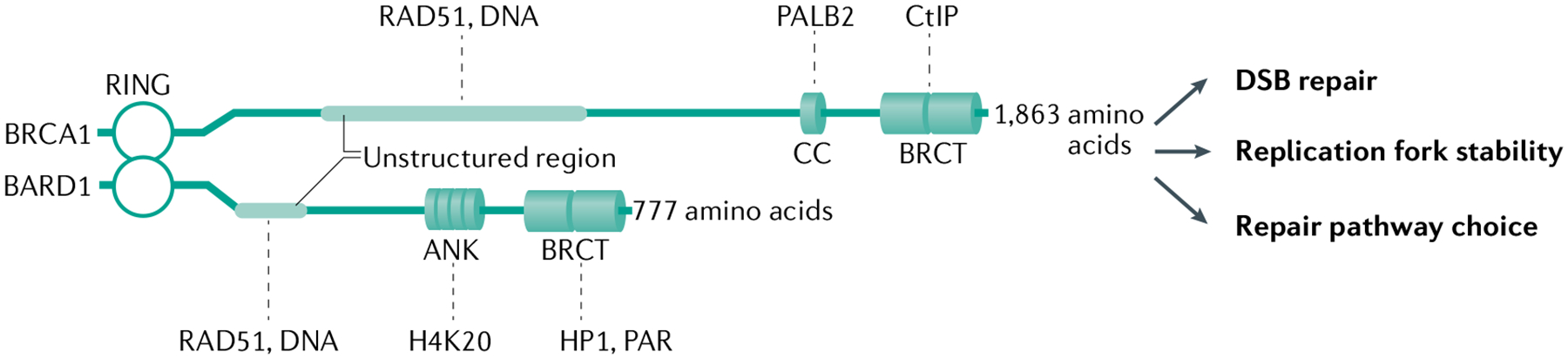
The RING (really interesting new gene) domain of breast cancer type 1 susceptibility protein (BRCA1) and BRCA1-associated RING domain 1 (BARD1) mediates their heterodimerization and is crucial for the E3 ubiquitin ligase activity of the BRCA1-BARD1 complex. Both proteins also harbour two BRCA1 C-terminal (BRCT) repeats arranged in tandem, which confer the ability to interact with various proteins involved in the DNA damage response, and unstructured regions that are involved in interactions with the recombinase RAD51 and with DNA. Unique features include a coiled-coil (CC) domain of BRCA1, which allows assembly of a higher-order complex with BRCA2-DSS1 through partner and localizer of BRCA2 (PALB2), and four ankyrin (ANK) repeats in BARD1, which serve to target the BRCA1-BARD1 complex to DNA lesions by interacting with unmethylated histone H4 Lys20 (H4K20), which is present in nucleosomes of newly replicated DNA75. The BRCT domain of BRCA1 interacts with CtBP-interacting protein (CtIP), which is involved in DNA end resection; the BRCT domain of BARD1 mediates its association with heterochromatin protein 1 (HP1), which is required for heterochromatin formation and maintenance, and with poly(ADP-ribose) (PAR), which is synthesized by poly(ADP-ribose) polymerase. PAR recognition is important for the recruitment of BRCA1-BARD1 to stalled replication forks and thus for maintaining the stability of these forks66. See TABLE 1 for a more complete list of protein interactors of this tumour suppressor complex. DSB, double-strand break.
BARD1 is the obligatory partner of BRCA1 and a tumour suppressor.
A screen based on yeast and mammalian two-hybrid analyses led to the isolation of BARD1, which has 777 amino acids and is the obligatory partner of BRCA1 (REF.52). Like BRCA1, BARD1 has a zinc-chelating RING (really interesting new gene) domain, and the two proteins assemble into a stable heterodimer through the association of their RING domains (FIG. 1). As discussed later, mutations that impair the BRCA1-BARD1 interaction, including those derived from tumours, result in deleterious proteolytic degradation of both proteins.
Compelling evidence that BARD1 is a tumour suppressor has been obtained from cancer association studies, which link BARD1 mutations to breast, ovarian and other tumour types22–26. For unclear reasons, BARD1 mutations are rarer than BRCA1 mutations in familial breast cancer and are generally associated with a lower breast cancer risk27. A number of single-nucleotide polymorphic variants of BARD1 result in differential expression of BARD1 isoforms, most notably the β-isoform, which lacks RING domain-encoding exons 2 and 3. RING-less BARD1 is thought to confer a dominant-negative effect on wild-type BARD1 and to contribute to the tumorigenesis of neuroblastoma and nephroblastoma53,54. Thus, not only is BARD1 a bona fide tumour suppressor but certain BARD1 isoforms appear to have an oncogenic effect as well55.
The domains of BRCA1 and BARD1 and their functions.
A solution NMR structure of the BRCA1-BARD1 RING dimer has been solved56, and a number of cancer-causing mutations in the BRCA1 RING domain have been described, which affect BRCA1-BARD1 heterodimer formation57–59. The C termini of BRCA1 and BARD1 each harbour two copies of the BRCA1 C-terminal (BRCT) repeat, which mediate interactions with different partner proteins. The BRCA1 BRCT repeats associate with the phosphorylated isoforms of interacting proteins60–64, whereas the BARD1 BRCT repeats specifically recognize poly(ADP-ribose) and other factors65–67 (FIG. 1). Substantial structural and functional information emphasizes the importance of the RING dimer and the BRCT domains for the tumour suppression activity of BRCA1-BARD1 (REFS68,69). The BRCA1 coiled-coil domain mediates complex formation with partner and localizer of BRCA2 (PALB2)70–72, itself a tumour suppressor73,74. BARD1 possesses four tandem ankyrin motifs that specifically recognize unmethylated histone H4 Lys20 (K20) (REF.75). Biochemical analyses have identified protein domains that mediate DNA binding and interaction with the recombinase RAD51 in both BRCA1 and BARD1 (REF.76) (FIG. 1).
In 2001, BRCA1 was shown to bind DNA77, thereby providing key evidence of a direct involvement of BRCA1-BARD1 in DNA repair and replication fork maintenance. Recently, a DNA-binding domain was found in BARD1 as well76 (FIG. 1). This study also revealed that both BRCA1-BARD1 and BARD1 bind a variety of DNA substrates, with the highest affinity being for the displacement loop (D-loop) structure, which is a DNA intermediate generated by RAD51 during HR, followed by the affinities for the replication fork structure, double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA)76.
The E3 ubiquitin ligase activity of BRCA1-BARD1.
Like many other RING domain proteins, BRCA1 and BARD1 can interact with E2 ubiquitin-conjugating enzymes and function as E3 ubiquitin ligases78,79. There is compelling evidence that optimal E3 ligase activity requires complex formation between the two tumour suppressors56,80–82. BRCA1-BARD1 interacts with a number of E2 enzymes to assemble K6-linked, K48-linked or K63-linked ubiquitin chains83,84. BRCA1-BARD1 is unique among E3 ubiquitin ligases in being capable of assembling K6-linked ubiquitin chains.
Initial studies in mouse breast and pancreatic tumour models involving the BRCA1-I26A mutant protein, which is impaired in ubiquitin ligase activity, concluded that ubiquitin conjugation by BRCA1-BARD1 is dispensable for the suppression of tumorigenesis69,85. Characterization of another E3 ligase-deficient BRCA1-BARD1 complex (harbouring the BARD1R99E mutation) revealed an intriguing pattern of cellular sensitivities to genotoxic agents86. Specifically, mutant cells are hypersensitive to inhibitors of topoisomerase I and topoisomerases II, to ionizing radiation and to the PARPi olaparib, but unlike BARD1-depleted cells, they are resistant to cisplatin (a DNA crosslinking agent), aphid-icolin (a DNA polymerase inhibitor) and hydroxyurea (another inhibitor of DNA replication). Moreover, the mutant cells show defects in DNA end resection, which is a crucial early step in the repair of DSBs by HR86. These observations suggest that the BRCA1-BARD1 E3 ligase activity is important for DNA repair but not for DNA replication fork maintenance.
Several physiological substrates of BRCA1-BARD1 have been identified (TABLE 1). Specifically, histone H2A (H2A) ubiquitylation may lead to the eviction of p53-binding protein 1 (53BP1), which is a reader of histone modifications and an antagonist of DNA end resection, to allow access to DNA ends to the HR machinery86 (see below). Mutations in BARD1 that impair H2A ubiquitylation, but do not destabilize the BRCA1-BARD1 heterodimer, have been identified in families afflicted with breast cancer87. There is evidence that BRCA1-BARD1-mediated H2A ubiquitylation is important for transcriptional repression of satellite repeats and that this function is relevant for tumour suppression88,89.
Table 1 |.
Interactors and antagonists of BRCA1-BARD1
| Direct interaction partners | Proteins | Refs |
|---|---|---|
| DNA repair machinery | PALB2 | 71,72 |
| RAD51 | 76,92 | |
| CtIP | 230 | |
| FANCJ | 61,231 | |
| MRE11-RAD50-NBS1 | 64 | |
| PIN1 | 129 | |
| FANCA | 232 | |
| DNA-PKcs | 233 | |
| MSH2 | 234 | |
| CSB | 235 | |
| DNA damage signalling | ATM | 236 |
| ATRIP | 237 | |
| ATR | 238,239 | |
| Histone H2A | 126 | |
| Histone H4 | 75 | |
| Merlin (NF2) | 240 | |
| HP1 | 67 | |
| ABRAXAS | 241,242 | |
| CDK-cyclin | 243,244 | |
| Protein phosphatase 1α | 245 | |
| TGFβ1 (SMAD3) | 246 | |
| Transcription (or R-loop metabolism) | RNA polymerase II | 247,248 |
| SETX | 156 | |
| CBP (p300) | 249,250 | |
| COBRA1 (NELFB) | 251 | |
| p53 | 249 | |
| Cell growth | BAP1 | 252 |
| RB | 253 | |
| Centromere regulation | KIAA0101 | 254 |
| OLA1 | 255 | |
| Chromosome segregation | Topoisomerase 2α | 256 |
| Chromatin remodelling | BRG1 | 257 |
| BRD7 | 258 | |
| HDAC1, HDAC2 | 259 | |
| Other | Acetyl-CoA carboxylase 1 | 260 |
| Importin-α subunit | 261 | |
| E3 ligase substratesa | Merlin (NF2) | 240 |
| Claspin | 262 | |
| Histone H2A | 126 | |
| Histone macroH2A | 263 | |
| RNA polymerase II | 264,265 | |
| Aurora kinase A, aurora kinase B | 53 | |
| Antagonists | 53BP1 | 100 |
| RIF1 | 99,122,215 | |
| Shieldin complex (REV7-SHLD1-SHLD2-SHLD3) | 187,188,216–219 | |
| CST complex (CTC1-STN1-TEN1) | 218,219 | |
| DNA polymerase-α-primase | 218 | |
| DYNLL1 | 222,223 | |
| USP48 | 266,267 |
BARD1, breast cancer type 1 susceptibility protein-associated RING domain 1; BRCA1, breast cancer type 1 susceptibility protein; CDK, cyclin-dependent kinase; CtIP, CtBP-interacting protein; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; TGFβ1, transforming growth factor-β1.
Other E3 ligase substrates have been identified but are not listed.
Considerable controversies remain about the involvement of the BRCA1-BARD1 E3 ligase activity in HR and in tumour suppression. Specifically, the BRCA1-I26A mutant protein that was tested in mouse models of tumorigenesis69 possesses residual capacity to interact with certain E2 enzymes and in ubiquitin conjugation when tested in vitro90. On the other hand, even though the BRCA1R99E mutation has been reported to impair HR and confer sensitivity to genotoxic agents86, another study found that the expression of this mutant allele in BARD1-depleted cells restores cellular resistance to PARPi75. Clearly, more work is needed to critically evaluate the BRCA1-BARD1 E3 ligase activity and its roles in tumour suppression in humans.
Protein interactions involving BRCA1-BARD1.
Numerous protein interactors of BRCA1-BARD1 have been described, including factors that function in HR, in transcription and in the avoidance of transcription-replication conflicts (TABLE 1). Some of the protein-protein interactions that are important for HR are mediated by the BRCT repeats of BRCA1 or BARD1. Complex formation with other factors that directly influence HR efficiency is dependent on the ankyrin repeats of BARD1, on the coiled-coil domain of BRCA1 and apparently on unstructured regions of both BRCA1 and BARD1 (FIG. 1). As we discuss in the following sections, the functional importance of some of the protein complexes involving BRCA1-BARD1 has been delineated.
The role of BRCA1-BARD1 in HR
Early studies revealed HR defects in Brca1−/− mouse embryonic stem cells, manifested as impaired gene targeting and diminished repair of an induced, site-specific DSB91. The observation that BRCA1 interacts with the recombinase RAD51 and that they colocalize to ionizing radiation-induced nuclear foci92 provided the first evidence for the involvement of BRCA1 in HR repair. Furthermore, BRCA1 abrogation in mouse and human cells impairs DNA damage-induced RAD51 focus formation93,94. Likewise, ablation of the BARD1 gene in mouse and human cells also decreases the formation of damage-specific RAD51 foci and causes a defect in HR that is equivalent to the loss of BRCA1 (REFS36,76). These findings thus reveal an important function of BRCA1-BARD1 in RAD51 recruitment to DSBs, which is a crucial early event in HR. In this regard, BRCA1-BARD1 likely functions as part of a higher-order ‘HR mediator’ complex with two other tumour suppressors, BRCA2 and PALB2 (REFS4,95,96) (fig. 2; see later). In addition to RAD51 recruitment, there is evidence that BRCA1-BARD1 has a role in the nucleolytic resection of DSB ends (FIG. 2), as first proposed on the basis of the cell cycle-dependent association of BRCA1 with the MRE11-RAD50-NBS1 nuclease complex (MRE11 being the catalytic subunit) and with the end resection factor CtBP-interacting protein (CtIP)64,97.
Fig. 2 |. The roles of BRCA1-BARD1 in DSB repair by homologous recombination.
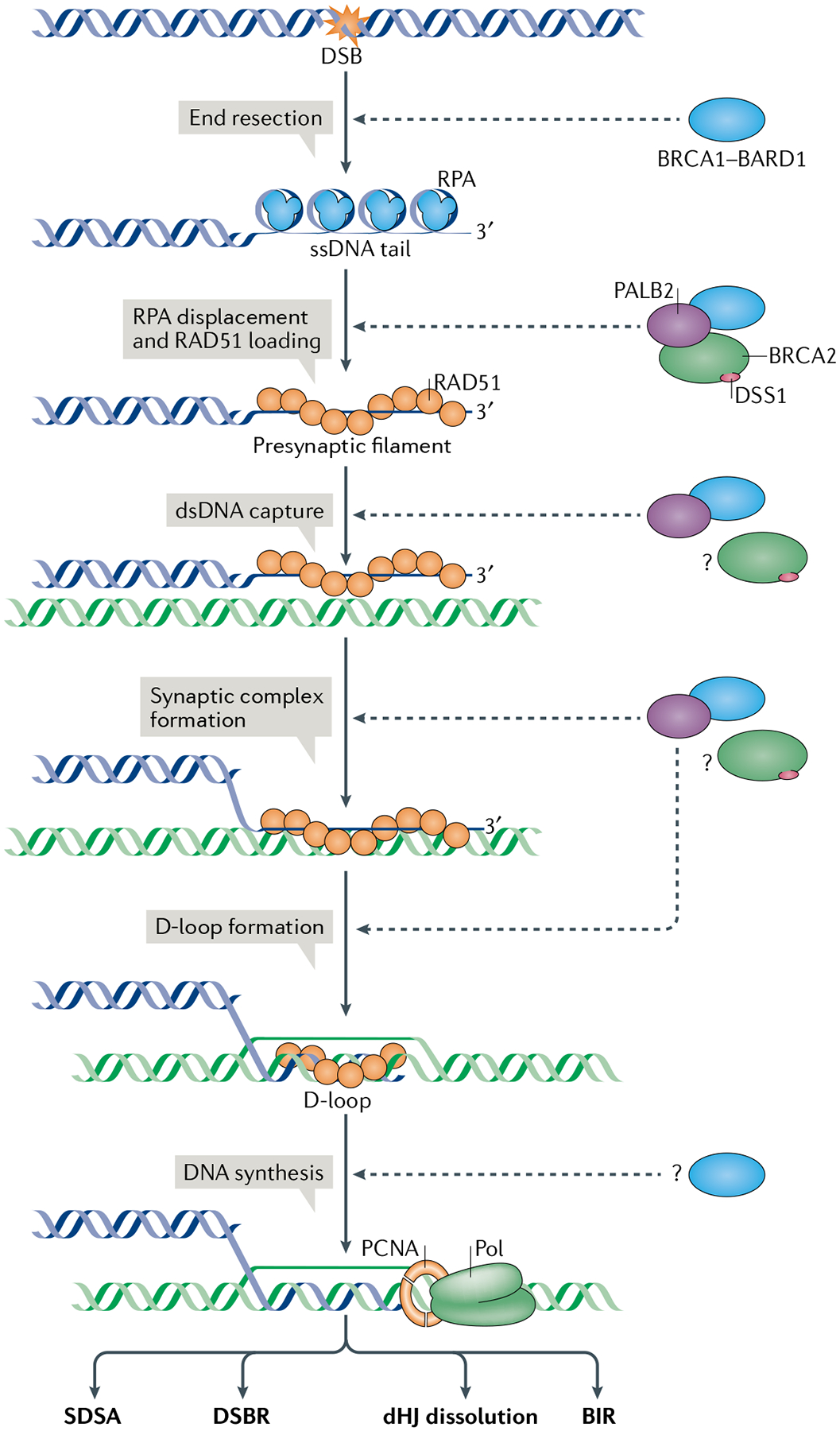
The single-stranded DNA (ssDNA) tail generated by end resection, which is aided by the breast cancer type 1 susceptibility protein (BRCA1)-BRCA1-associated RING domain 1 (BARD1) complex, is occupied by the abundant ssDNA-binding factor replication protein A (RPA), which must be replaced by the recombinase RAD51 for downstream repair steps to occur. The exchange of RPA with RAD51 on DNA is mediated by BRCA2-DSS1, with the involvement of BRCA1-BARD1 and partner and localizer of BRCA2 (PALB2). Importantly, BRCA1-BARD1 also enhances the ability of the RAD51-ssDNA nucleoprotein complex to capture the homologous duplex DNA and assemble the synaptic complex, which gives rise to the displacement loop (D-loop). Following repair DNA synthesis by DNA polymerase-δ (Pol) associated with PCNA, the extended D-loop structure can be resolved by one of four mechanistically distinct pathways — synthesis-dependent single strand annealing (SDSA), canonical double-strand break (DSB) repair (DSBR), double Holliday junction (dHJ) dissolution and break-induced DNA replication (BIR)11,95 — that yield distinct products. Homologous recombination steps that are facilitated by BRCA1-BARD1 or BRCA2-DSS1 are indicated by dashed arrows. Whether BRCA1-BARD1 and its homologous recombination partners also function in other steps of the repair reaction (for example, in repair DNA synthesis), remains to be determined (denoted by question marks). For simplicity, only one of the two DNA DSB ends is depicted. dsDNA, double-stranded DNA.
A central step in HR is the assembly of a RAD51 nucleoprotein filament on the 3′ ssDNA tail, which is capable of catalysing pairing with homologous DNA11,95,98 (FIG. 2). BRCA1 inactivation triggers a modest reduction in ssDNA levels compared with the abrogation of an essential resection factor such as CtIP99–102. Moreover, ablation of the BRCA1-CtIP complex affects resection efficiency only mildly102–104. Thus, it would appear that BRCA1-BARD1 is an accessory factor of DNA end resection, but is not a component of the core resection machinery105,106. Since resection can still occur in BRCA1-deficient cells, attenuated formation of RAD51 foci cannot be explained solely by lack of ssDNA formation. A more likely scenario is that BRCA1 not only influences the efficiency of DNA end resection but also helps recruit RAD51 to resected DNA ends (see later).
The antagonistic activities of BRCA1 and 53BP1 in DNA end resection.
Whereas nullizygous Brca1 mice die in utero32–35, mice carrying the Brca1 exon 11 deletion (Brca1Δ11/Δ11) are partially viable or show embryonic lethality later in embryogenesis37,107. BRCA1-deficient mice die embryonically due to the accumulation of endogenous DNA damage, which activates DNA damage signalling through the kinase ataxia telangiectasia mutated (ATM) (FIG. 3a). It was therefore anticipated that abolishing DNA damage sensors would increase tolerance and promote survival on the occurrence of DNA damage in these mice. This assumption was proved correct, as deletion of Atm or the gene encoding its effector checkpoint kinase 2 (Chk2) rendered Brca1Δ11/Δ11 mice viable108. Two research groups introduced the Brca1Δ11/Δ11 deletion into mice lacking the Trp53bp1 gene (orthologue of the gene encoding human 53BP1), which encodes a p53 interacting protein109,110 that functions as a signal transducer in the ATM-dependent DNA damage checkpoint111–113. Deletion of Trp53bp1 restored viability in Brca1Δ11/Δ11 mice114.
Fig. 3 |. Antagonistic roles of BRCA1-BARD1 and the 53BP1 complex.
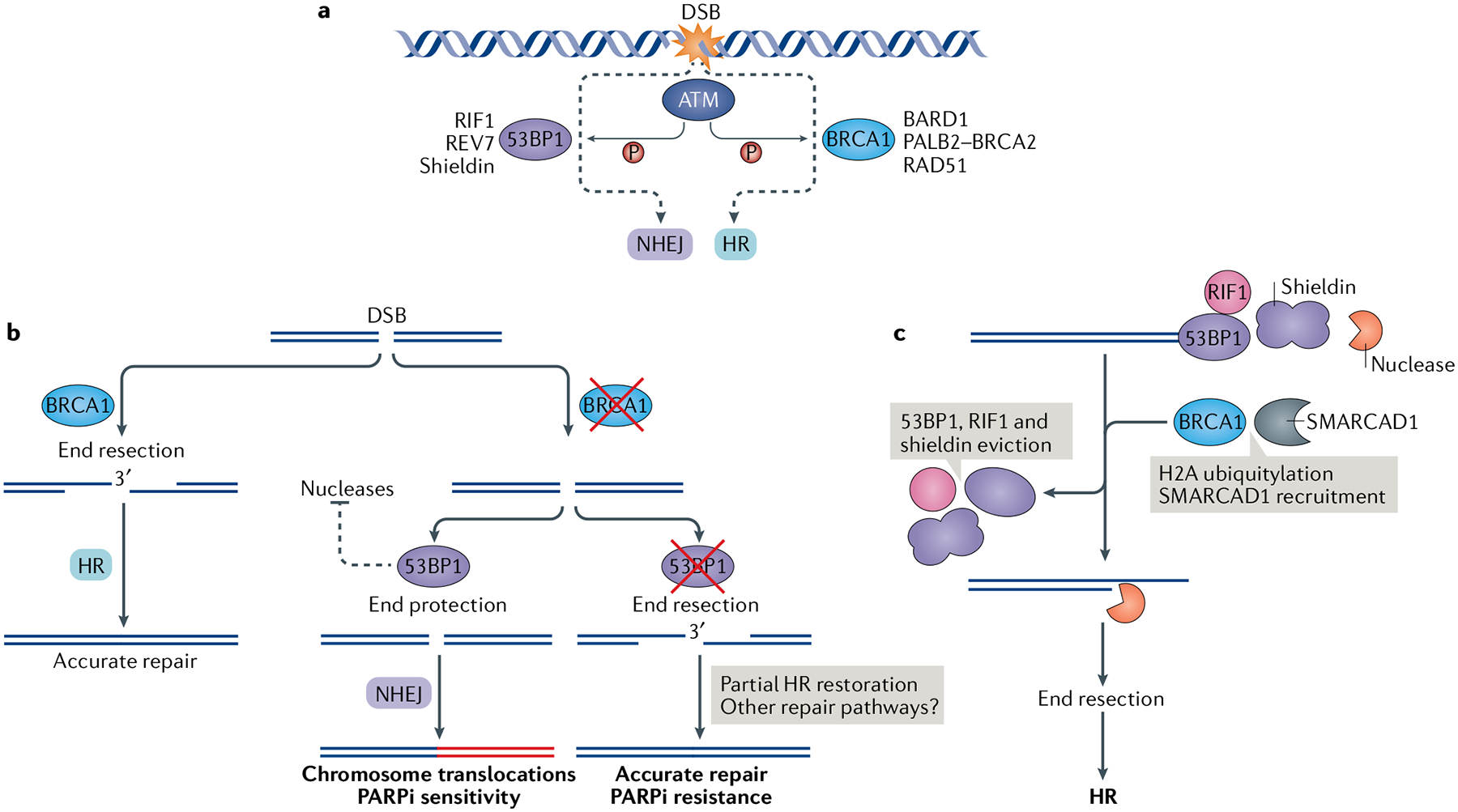
a | A DNA double-strand break (DSB) elicits ataxia telangiectasia mutated (ATM)-dependent checkpoint responses, which activate and recruit breast cancer type 1 susceptibility protein (BRCA1)-containing and/or p53-binding protein 1 (53BP1)-containing complexes to the DNA lesion. BRCA1 and 53BP1 compete and perform opposing functions at broken DNA ends by engaging either homologous recombination (HR) or non-homologous end joining (NHEJ) for DSB repair, respectively. b | BRCA1 promotes DNA end resection and HR repair (left). If BRCA1 is abrogated, DSB ends are protected from resection by 53BP1-containing complexes and are channelled into NHEJ for repair (right). This process can lead to deleterious chromosome translocations, which underline the sensitivity of BRCA1-deficient cells to poly(ADP-ribose) polymerase inhibitors (PARPi). If, in addition to BRCA1, 53BP1 is concomitantly abrogated, the DNA ends are accessible to resection nucleases, leading to partial HR restoration and PARPi resistance. c | 53BP1-containing complexes (53BP1—RIF1—shieldin) protect DNA ends from nucleolytic digestion by nucleases. This end protection is overcome by BRCA1-mediated histone H2A ubiquitylation and recruitment of the helicase SMARCAD1. The eviction of the 53BP1-containing complexes leads to DNA end resection and HR. BARD1, BRCA1-associated RING domain 1; PALB2, partner and localizer of BRCA2.
The protein 53BP1 interacts with epigenetically modified histones to nucleate the formation of a multisubunit complex, which includes the telomere-associated proteins RIF1, REV7 (also known as MAD2L2 or MAD2B), shieldin complex subunit 1 (SHLD1), SHLD2 and SHLD3 and prevents access of the resection machinery to DNA ends in G1 phase of the cell cycle (BOX 2). The removal of the 53BP1-associated protein complex from DNA ends occurs in S phase and G2 phase of the cell cycle and requires BRCA1-BARD1. Abrogation of 53BP1 renders BRCA1-mutant cells proficient in the repair of DNA damage induced by camptothecin, ionizing radiation and olaparib100. This discovery has provided the generally accepted model, in which 53BP1 ablation allows BRCA1-mutant cells to process DSBs for HR repair (FIG. 3b). Consequently, BRCA1 and 53BP1 double-mutant cells and tumours are resistant to PARPi such as olaparib, which induce the formation of DSBs that can be channelled into the HR pathway for repair (FIG. 3b). This has important clinical implications (discussed later). However, an important yet often overlooked aspect of these results is that 53BP1 abrogation in cells lacking BRCA1 does not restore HR and genome stability to wild-type levels76,100,114,115. For example, targeted DNA integration mediated by HR at some specific loci occurs at only 10–50% efficiency in Trp53bp1 depleted Brca1−/− mouse embryonic stem cells relative to the BRCA1-proficient counterparts115. Likewise, even though formation of RAD51 foci in response to ionizing radiation is upregulated in BRCA1-deficient and 53BP1-deficient cells relative to the BRCA1-deficient and 53BP1-proficient cells, it does not reach the wild-type level100,115. It is also important to note that 53BP1 deletion fails to suppress the meiotic defects of Brca1Δ11/Δ11 mice116,117. Notably, however, the DNA-damage sensitivity and tumour susceptibility caused by the RING-less Brca1Δ2 allele can be suppressed by 53BP1 ablation118,119. Emerging evidence suggests that a functional BRCA1 protein is in fact needed to mediate HR steps that occur downstream of DNA end resection in order to make possible the observed cellular resistance to genotoxic agents and partial restoration of HR in 53BP1-null cells120,121.
Box 2 |. Protein complexes that antagonize DNA end resection.
Multiple protein interactors help p53-binding protein 1 (53BP1) to restrict DNA end resection (TABLE 1). RIF1 inhibits DNA double-strand break (DsB) resection and homologous recombination repair similarly to 53BP1. Much like 53BP1 abrogation, the loss of RIF1 restores ionizing radiation-induced RAD51 focus formation and triggers resistance to poly(ADP-ribose) polymerase inhibitors (PARPi) in breast cancer type 1 susceptibility protein (BRCA1)-deficient cells99,122,215. REv7 (also known as MAD2L2 or MAD2B) is another component of the 53BP1-RIF1-associated complex that helps restrict DNA end resection; REV7 was isolated in a screen for factors whose inactivation leads to PARPi resistance in a BRCA1-deficient background101. Recent efforts by multiple groups have led to the identification of three new 53BP1-interacting factors — shieldin complex subunit 1 (SHLD1), SHLD2 and SHLD3 — that form the stable shieldin complex with REV7, thus named because it acts in conjunction with 53BP1-RIF1 to ‘shield’ DSB ends from the DNA end resection machinery187,188,216–219. Independently, characterization of the REV7 interactome in B lymphocyte extracts led to the isolation of shieldin220. Studies involving CRISPR-Cas9-based genetic screens for factors that, when disrupted in a BRCA1-deficient background, confer resistance to DNA damaging agents also identified shieldin188,217. Finally, SHLD2 was isolated as a REV7 interactor in BRCA1-defective cells that exhibit abnormal PARPi resistance187.
The CST complex (consisting of CTC1, STN1 and TEN1) functions in telomere replication to facilitate C-strand synthesis by recruiting the DNA polymerase-α (Polα)-primase complex221. CST inactivation leads to increased single-stranded DNA (ssDNA) at telomeres and at ionizing radiation-induced DSBs genome wide218. Importantly, CST interacts with shieldin, and its inactivation renders BRCA1-deficient cells resistant to olaparib. These results suggest that the 53BP1-RIF1-shieldin ensemble cooperates with CST and Polα to fill in resected DSBs, thereby adding a new dimension to the mechanism by which 53BP1 counteracts the formation of recombinogenic 3′ ssDNA overhangs218. A separate study isolated the CST complex as a resection antagonist and mediator of PARPi sensitivity in BRCA1-deficient cells219. It remains possible that the primary role of shieldin is to recruit and activate the protein ensemble comprising CST and Polα-primase.
In a loss-of-function CRISPR-Cas screen, dynein light chain 1 (DYNLL1) was found to be an antagonist of DNA end resection222. Like 53BP1 and its effectors, loss of DYNLL1 can suppress the DNA end resection and homologous recombination defects of BRCA1-mutant cells. Accordingly, DYNLL1 deletion results in resistance of BRCA1-deficient cells to platinum-based drugs and PARPi. DYNLL1 associates in cell extracts with components of the DNA end resection machinery, including the MRE11-RAD50-NBS1 complex, the helicase Bloom syndrome protein and the nuclease DNA2. Importantly, DYNLL1 interacts directly with MRE11 and attenuates its nuclease activity222. In addition to directly inhibiting the DNA end resection machinery, DYNLL1 promotes the oligomerization of 53BP1 (REF.223), which is a prerequisite for the association of 53BP1 with DSB-harbouring chromatin224–226.
Roles of BRCA1 and the 53BP1-associated complex in DSB repair pathway choice.
The discovery of how 53BP1 may antagonize BRCA1-dependent DSB resection prompted certain predictions about the interplay between BRCA1 and 53BP1 in engaging HR or NHEJ, respectively, for DSB repair (BOX 2; FIG. 3a). The most important prediction is that, by counteracting DNA end resection activity, 53BP1 helps channel DSBs into repair by NHEJ. Conversely, by promoting end resection, BRCA1 favours HR repair. According to this model, competition between BRCA1 and 53BP1 at DSBs is the defining factor in DSB repair pathway selection (FIG. 3b). This model is supported by extensive experimental evidence99,100,122. For example, whereas wild-type cells are able to assemble BRCA1 foci only in S phase and G2 phase of the cell cycle, such foci form in G1 phase on 53BP1 ablation99.
Immunostaining and high-resolution microscopy123 have revealed progressive, BRCA1-dependent exclusion of 53BP1 from DNA damage sites during S phase. Other studies have begun to provide mechanistic insights into how BRCA1-BARD1, through its E3 ubiquitin ligase activity and the ATPase-helicase SMARCAD1, contribute to the eviction of 53BP1, and presumably of its associated RIF1-shieldin complex, from the vicinity of DSB ends to facilitate DNA end resection and HR repair86. It has been known for quite some time that SMARCAD1 and its yeast orthologue Fun30 have a crucial role in the resection of DSB ends124,125. A recent study has provided tantalizing evidence that BRCA1-BARD1, through its ability to ubiquitylate three lysine residues of H2A (K125, K127 and K129)126, contributes to the recruitment of SMARCAD1 through its ubiquitin-binding CUE domains86. Specifically, introduction of the E3 ligase-inactivating BARD1-R99E mutant protein into the BRCA1-BARD1 complex leads to impairment of DNA end resection and, as mentioned earlier, causes hypersensitivity to genotoxic agents such as olaparib. Moreover, the expression of a ubiquitin-H2A fusion protein in cells lacking BARD1 helps to restore DNA end resection resistance to genotoxic agents in a SMARCAD1-dependent manner86. Thus, the E3 ligase activity of BRCA1-BARD1 likely contributes to SMARCAD1-dependent 53BP1 eviction from DSBs and helps initiate DNA end resection through H2A ubiquitylation. It remains to be determined whether BRCA1-BARD1 also participates in the ubiquitylation of other factors important for the removal of 53BP1, RIF1 and shieldin from DSB ends and for the end resection process itself (FIG. 3c).
Role of BRCA1-BARD1 in RAD51-mediated DNA strand invasion.
As discussed already, early studies showed that BRCA1 co-immunoprecipitates with RAD51 from mammalian cell extracts92. Additionally, BRCA1 binds DNA and is required for the assembly of DNA damage-induced RAD51 nuclear foci76,77,86,127,128, suggesting a direct role of BRCA1-BARD1 in the enhancement or regulation of the recombinase activity of RAD51. Recent studies using highly purified BRCA1-BARD1 complex and RAD51 directly support this idea. Specifically, both BRCA1 and BARD1 were shown to bind DNA and interact directly with RAD51 (REF.76). The interaction of BRCA1-BARD1 with RAD51 is species specific, because yeast Rad51 and Escherichia coli RecA (the bacterial orthologue of RAD51) fail to associate with BRCA1-BARD1. In DNA-binding assays, both BRCA1-BARD1 and the DNA-binding domain of BARD1 show the highest affinity for the D-loop structure — the DNA structure formed by RAD51-catalysed pairing of homologous DNA molecules (FIG. 2). Importantly, in a reconstituted in vitro system of D-loop formation, BRCA1-BARD1 strongly enhanced the RAD51-mediated reaction but had no effect on either yeast Rad51 or E. coli RecA76.
As a prerequisite for D-loop formation, the RAD51-ssDNA complex must capture a duplex DNA molecule, conduct a search for DNA homology and align the recombining ssDNA and dsDNA in homologous registry. The three-stranded aligned nucleoprotein intermediate is referred to as the ‘synaptic complex’76,95 (FIG. 2). Biochemical and single-molecule biophysical analyses have provided evidence that BRCA1-BARD1 works in conjunction with RAD51 to assemble the synaptic complex76.
The functional relevance of the BRCA1-BARD1-RAD51 complex was established by isolation of a BARD1 mutant (BARD1-AAE (F133A, D135A and A136E)) that compromises the ability of BARD1 and BRCA1-BARD1 to associate with RAD51 but that has no effect on DNA binding by these protein species76. Importantly, the BRCA1-BARD1-AAE mutant complex is greatly impaired in its ability to enhance RAD51-mediated synaptic complex assembly and D-loop formation in vitro. Cells that are devoid of endogenous BARD1 but express the BARD1-AAE mutant exhibit a deficiency in DSB repair by HR and hypersensitivity to mitomycin C and olaparib. However, these mutant cells are still competent in the assembly of DNA damage-induced RAD51 foci. Taken together, the results from this study provide evidence that BRCA1-BARD1 functions in synergy with RAD51 in synaptic complex assembly and D-loop formation76.
Postulated role of BRCA1-BARD1 in the assembly of the RAD51 repair complex.
One of the strongest phenotypes associated with BRCA1 deficiency is impaired assembly of DNA damage-induced RAD51 foci76,93. That BRCA1-BARD1 likely has an important role in the assembly of the RAD51 repair complex to initiate HR relies on the following findings: BRCA1 and BARD1 physically interact with RAD51 (REFS76,92); mutations in BARD1 that affect its interaction with RAD51 cause a defect in the RAD51-dependent protection of stalled DNA replication forks against nucleolytic attrition129; BRCA1 interacts with PALB2, which functions to recruit the RAD51 loader BRCA2 to DNA lesions for assembling RAD51 repair complexes, and mutations that abrogate the interaction between BRCA1 and PALB2 cause strong HR defects71,72; and the suppression of HR defects associated with BRCA1 deficiency by 53BP1 depletion is far from complete100,115.
In its postulated role in RAD51-ssDNA complex assembly, BRCA1-BARD1 likely functions as part of a larger protein ensemble that includes PALB2 and BRCA2-DSS1 (REFS95,96) (FIG. 2). BRCA2, by binding both DNA and RAD51, nucleates the assembly of RAD51 filaments on ssDNA occupied by replication protein A (RPA), which is an abundant factor with high affinity for ssDNA130–132 (FIG. 2). The exchange of RPA with RAD51 on ssDNA is enhanced by the BRCA2-associated factor DSS1, which is a small, highly acidic protein that weakens the grip of RPA on ssDNA130. Thus, BRCA2-DSS1 is a ‘mediator’ of HR11,130,133,134. In summary, aside from their roles in the recruitment of BRCA2-DSS1 to sites of DNA damage95,96, BRCA1-BARD1 and PALB2 likely also enhance the HR-mediator activity of BRCA2-DSS195,96. In the future, it will be important to isolate protein complexes that include these factors and to test them for HR-mediator activity to reveal the mechanism of function of BRCA1-BARD1 and PALB2 in RAD51 repair complex assembly.
Functions of BRCA1-BARD1 in DNA replication
In addition to its well-documented DSB repair function, BRCA1-BARD1 has crucial roles in the repair and restart of stalled and damaged DNA replication forks and in their protection from nucleolytic attack and attrition (FIG. 4). As we discuss in the following sections, the involvement of BRCA1-BARD1 in the preservation of replication fork integrity is complex and likely also involves bypass of potentially pathogenic secondary DNA structures — R-loops (which arise following transcription perturbation) — and G-quadruplexes.
Fig. 4 |. Roles of the Brca1 complex in protecting regressed replication forks and resolving r-loops.
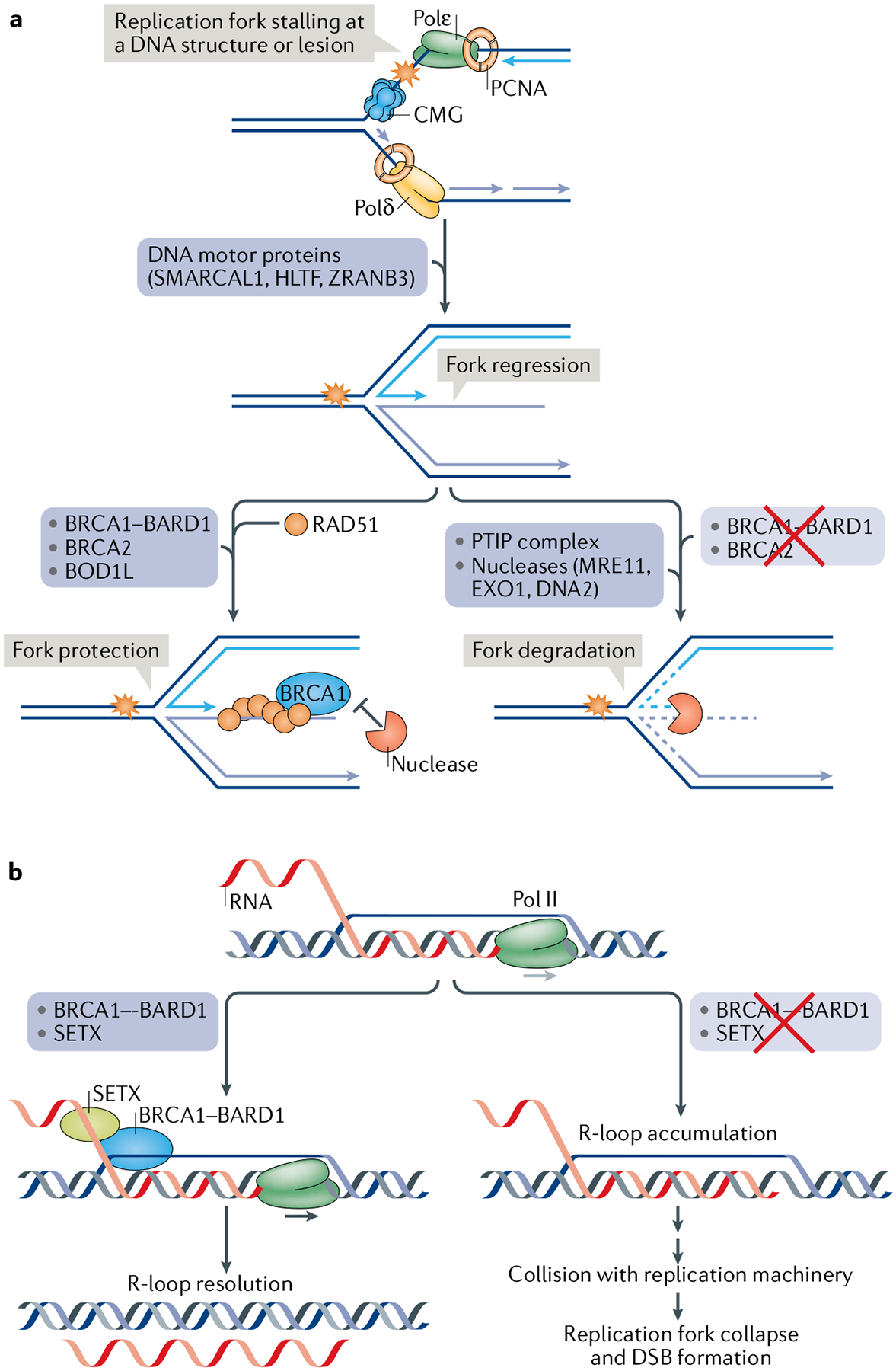
a | During DNA replication, when the DNA polymerase ensemble comprising DNA polymerase-ε (Polε), PCNA and the CDC45-MCM-GINS (CMG) helicase complex on the leading strand and Polδ-PCNA on the lagging strand is impeded by a DNA structure or lesion, one of the nucleic acid motor proteins SMARCAL1, HLTF or ZRANB3 can catalyse the reversal of the replication fork, a process often referred to as ‘replication fork regression’, to generate a four-armed DNA structure that harbours a free DNA end. Formation of the regressed fork allows a replicative mechanism of lesion bypass15,227. However, the free DNA end is prone to attack by MRE11 and other nucleases in conjunction with the PTIP complex148, unless RAD51 is deposited on the regressed fork. The stability of the regressed fork is also dependent on breast cancer type 1 susceptibility protein (BRCA1)-BRCA1-associated RING domain 1 (BARD1), BRCA2 and biorientation of chromosomes in cell division protein 1-like 1 (BOD1L), which are postulated to stabilize the RAD51-DNA interaction to prevent nucleolytic attrition139,140,146. Other pathways of replication fork repair and restart have been identified, and readers are referred to several comprehensive reviews on this topic227–229. b | Stalling of RNA polymerase II (Pol II) or delay in mRNA processing can lead to accumulation of R-loops (RNA-DNA hybrids). An R-loop can stall the DNA replication machinery, thereby causing fork collapse and the formation of a DNA double-strand break (DSB). BRCA1-BARD1 and the putative RNA-DNA helicase senataxin (SETX) facilitate R-loop resolution155.
The multifaceted role of BRCA1-BARD1 in replication fork repair and protection.
The first indication of a role of BRCA1 in DNA replication came from a study on the response of BRCA1-mutant cells to hydroxyurea, which causes replication stress by inhibiting ribonucleotide reductase, leading to depletion of nucleotides needed for DNA synthesis135. In hydroxyurea-treated cells, BRCA1 colocalizes with RAD51 at S phase-specific foci containing the DNA polymerase accessory factor PCNA, which likely correspond to stalled replication forks135. Later studies showed colocalization of BRCA1 with other factors that act on stalled replication forks, including the MRE11-RAD50-NBS1 nuclease complex and the helicase Bloom syndrome protein136.
The aforementioned studies were performed in the presence of hydroxyurea and thus in conditions of replication stress. The question remains whether BRCA1 has a role in DNA replication in physiological settings. Early on, analysis of mouse embryonic fibro-blasts derived from Brca1Δ11/Δ11 mice revealed spontaneous complex chromosomal aberrations in the form of radial chromosomes37, which are a molecular signature of misrepaired replication-associated DNA damage. Importantly, BRCA1 haploinsufficiency is associated with defects in replication fork restart137, even though HR-mediated DSB repair capacity remains unaffected. This suggests BRCA1 has a primary function in DNA replication, for which the normal level of fully functional BRCA1 is required; alternatively, mutant BRCA1 might exert a dominant-negative effect on the wild-type protein66. Replication stress stemming from a defect in the timely resolution of stalled replication forks and the resulting genomic instability are hallmarks of BRCA1-deficient cells and tumours96,138.
In addition to its role in the stabilization, repair and restart of stalled DNA replication forks, BRCA1 is also very important for the protection of stressed forks from nucleolytic degradation by MRE11 (REFS139,140) and other nucleases141–145. The newly identified biorientation of chromosomes in cell division protein 1-like 1 (BOD1L) appears to function with BRCA1 and BRCA2 to protect stressed replication forks from the destructive activity of the nuclease DNA2 (REF.146) (FIG. 4a).
Several members of the SNF2 family of helicases, namely SMARCAL1, ZRANB3 and HLTF, remodel stalled replication forks in a process termed ‘fork regression’ into a branched structure that harbours a single DNA end, which becomes prone to degradation by the nuclease MRE11 when BRCA1 is absent147 (FIG. 4a). This is mediated by PTIP, a component of the histone methyltransferase complex MLL3 (KMT2C)-MLL4 (KMT2B) and an effector of 53BP1 in DSB end protection, which recruits MRE11 to stalled replication forks in BRCA1-deficient cells to trigger fork attrition148. Whether the recruitment of the helicase-like proteins SMARCAL1, ZRANB3 and HLTF is also PTIP dependent has not been determined.
Another mechanism of fork protection is BRCA1 independent and instead relies on CtIP149. Unlike in BRCA1-deficient cells, in which stalled forks are degraded by MRE11, CtIP deficiency renders replication forks susceptible to DNA2. Thus, loss of CtIP in BRCA1-deficient cells further increases the vulnerability of replication forks to nucleolytic attrition.
The precise mechanisms by which BRCA1 protects and restarts stalled replication forks are unknown, but RAD51 loading at the ssDNA that accumulates at the stalled fork or at the free end of a regressed fork is likely required (FIG. 4a). In Xenopus laevis egg extracts, Rad51 associates with replication intermediates in a Brca1-dependent manner to protect and restart stalled forks150,151. A recent study has implicated the proline isomerase PIN1 in BRCA1-dependent replication fork protection against the destructive activity of MRE11. Specifically, PIN1 converts the BRCA1-BARD1 complex into a form that has higher affinity for RAD51. This PIN1 function requires cyclin-dependent kinase 1 (CDK1)-mediated or CDK2-mediated BRCA1 phosphorylation129.
Replication fork collapse and misrepair are major sources of mutagenesis and chromosomal rearrangements and are likely relevant to tumorigenesis. However, whether the activity of BRCA1-BARD1 at replication forks contributes to its tumour suppressor function is unclear. Better mechanistic understanding of how BRCA1-BARD1 functions at replication forks versus at DSBs and the development of separation-of-function mutants that affect DNA repair or replication fork maintenance will be necessary to answer this question.
BRCA1 facilitates the progression of replication through specific obstructions.
During DNA replication, the DNA polymerase ensemble may stall at genomic sites that are difficult to replicate. One such replication barrier is the R-loop, an RNA-DNA hybrid with an appended displaced ssDNA strand. R-loops accumulate in sites where strong DNA secondary structures, such as the G-quadruplexes (see later), are formed on perturbation of transcription or of transcription-coupled processes such as mRNA splicing152,153. Head-on collisions between the DNA replication machinery and RNA polymerase-associated R-loops lead to replication fork collapse and DSB formation154. BRCA1 interacts with senataxin (SETX), which is a putative RNA-DNA hybrid helicase and partner of RNA polymerase II involved in transcription termination and R-loop resolution155 (FIG. 4b). BRCA1 and SETX resolve conflicts between transcription and replication at transcription termination sites156. Consistent with this function, BRCA1-deficient tumours show high mutation rates in transcription termination regions. However, the manner by which BRCA1-BARD1 regulates the activity of SETX in R-loop resolution remains to be determined. SETX may also function to resolve RNA-DNA hybrids at DSB sites157 created as a result of de novo transcription at the DNA lesions158–161. This activity of SETX is believed to suppress DSB-induced chromosome translocations162.
G-quadruplex DNA structures can form at G-rich sequences when the DNA becomes single stranded during DNA replication or transcription163. Such G-quadruplex structures present a strong impediment to replication fork progression. Cells and tumours deficient in BRCA1 are highly sensitive to chemicals that interact with and stabilize the G-quadruplex structure164,165. These observations are consistent with the premise that BRCA1-BARD1 promotes the restart of replication forks arrested at G-quadruplex sites.
By-products of cellular metabolism (for example, acetaldehyde) can cause interstrand crosslinks (ICLs), which interfere with DNA replication. BRCA1-deficient cells and tumours are exquisitely sensitive to ICL-inducing agents, which underlies their vulnerability to certain chemotherapeutic drugs such as platinum salts and mitomycin C. Abrogation of detoxification pathways results in accumulation of endogenous acetaldehyde and replication stress in BRCA1-deficient cells166,167. These observations highlight the role of BRCA1 in the resolution of ICL lesions during DNA replication through the Fanconi anaemia pathway of DNA damage response168–171. Diseases resembling Fanconi anaemia were identified in two individuals homozygous for BRCA1 mutations172,173, and consequently the BRCA1 gene was classified as the Fanconi anaemia gene FANCS.
The clinical impact of BRCA1 mutations
The deficiency of BRCA1-mutant cells and tumours in DNA repair and in replication fork restart and protection renders them prone to oncogenic genome alterations (FIG. 3b). Importantly, BRCA1 deficiency is also a vulnerability that can be therapeutically targeted28,29,174,175 with chemical compounds (such as platinum salts, DNA crosslinking agents and PARPi) that increase the load of DNA damage or generate replication stress176,177. It is unfortunate that the mutator phenotype of HR-deficient tumours can give rise to drug resistance through secondary mutations that either allow the expression of functional BRCA1 or inactivate or downregulate HR repressors such as 53BP1. Overall, tumour cells possess surprising plasticity in circumventing the effects of drugs. We now discuss studies that are beginning to elucidate the underlying basis of innate and acquired drug resistance in animal models and in clinical settings.
Targeting BRCA1-deficient tumours and the emergence of PARPi resistance.
BRCA1-deficient tumours are hypersensitive to drugs that exacerbate their intrinsic DNA replication defects178. DNA crosslinking and alkylating agents, such as platinum salts, mitomycin C, cyclophosphamide and melphalan, impair replication fork progression by inducing ICLs, whereas topoisomerase I and topoisomerase II inhibitors such as camptothecin, irinotecan and etoposide covalently trap these enzymes on the DNA and generate a strong DNA replication block. More recently, inhibitors of PARP1 and PARP2 (PARPi) were shown to ‘trap’ these proteins on DNA to interfere with DNA replication. PARPi such as olaparib are generally well tolerated by normal cells and tissues but potently induce the demise of BRCA1-deficient cells and tumours28. PARPi have been approved by the US Food and Drug Administration and the European Medicines Agency as therapeutic agents for advanced-stage or recurrent ovarian179 and breast180,181 cancers with germline BRCA1 or BRCA2 mutations. In addition, some of the inhibitors mentioned are used in the clinic for maintenance treatment of patients with ovarian cancer who have successfully completed platinum-based chemotherapy182,183.
It is important to note that mutations in other HR genes also increase the vulnerability of cells to PARPi; this is known as the BRCAness concept178,184. For example, patients with prostate cancer with mutations in BRCA2, PALB2, FANCA, RAD51 or CHK2 are responsive to olaparib178,185. As such, PARPi may be efficacious for a much wider variety of tumours than those specifically lacking BRCA1 function.
The early discovery that 53BP1 inhibition in BRCA1-deficient cells leads to partial HR reactivation and resistance to PARPi100 was subsequently corroborated by in vivo data. In a mouse Brca1-mutant cancer model, tumours that are sensitive to olaparib acquire resistance following prolonged exposure to the drug. Silencing of 53BP1 expression is a major cause of drug resistance and tumour regrowth186. This discovery suggests that the expression level of 53BP1 could be a predictor of the response of BRCA1-deficient tumours to PARPi. Consistent with this premise, analysis of two large cohorts of patients with breast cancer (the Yale study and the Helsinki study)115 revealed an association of low 53BP1 expression with poor prognosis. Moreover, early-stage triple-negative breast tumours with reduced 53BP1 levels exhibited a higher risk of metastasis115. In congruence with these observations, REV7 was found to be downregulated in a significant fraction of triple-negative human breast carcinomas101. Moreover, the REV7-interacting protein and shieldin complex component SHLD2 is absent in the BRCA1-mutated breast cancer-derived HCC1937 cell line, which shows abnormal resistance to PARPi treatment187.
Studies of tumour xenografts derived from patients with triple-negative breast cancer with germline BRCA1 mutations have led to the characterization of specific 53BP1 (also known as TP53BP1) and REV7 (also known as MAD2L2) mutations that likely give rise to PARPi resistance in tumours. In addition, loss of SHLD1 or SHLD2 expression has been observed to cause PARPi resistance188. These studies were conducted with tumour xenografts derived from patients who had received prior therapy and, as such, it was not possible to determine whether the secondary mutations and gene silencing events arose spontaneously (innate resistance) or during treatment (acquired resistance). To answer this question, patients who receive PARPi as monotherapy at the onset of treatment will need to be examined. Such efforts would also shed light on whether the expression status of the 53BP1, RIF1 and shieldin axis might serve as a reliable biomarker for predicting drug response. BRCA1-deficient cells and tumours that have become olaparib resistant through abrogation of 53BP1 or shieldin components remain susceptible to ionizing radiation188,189 and to chemotherapeutic treatments, including G-quadruplex ligands164,165, chlorambucil190 and cisplatin117,188,191. In a recent clinical trial, patients with BRCA1-mutated or BRCA2-mutated, PARPi-resistant ovarian cancers were found to respond favourably to platinum-based therapies192, suggesting that the level of HR restoration in these tumours is insufficient for the timely removal of drug-induced DNA damage117. In addition, abrogation of 53BP1-dependent NHEJ may be dispensable for the repair of olaparib-induced DNA damage, but not for that induced by other cancer therapeutics117.
PARPi resistance mechanisms that are independent of HR restoration have been reported in Brca1-deficient mouse cells and tumours. In Brca1−/−Trp53−/− mouse mammary tumours, olaparib resistance, as well as resistance to doxorubicin and docetaxel, is mediated by activation of the P-glycoprotein drug efflux transporter193,194. Studies in vitro or using patient-derived tumour xenografts have identified inhibition of the poly(ADP-ribose)-degrading enzyme poly(ADP-ribose) glycohydrolase195 and abrogation of NEDD8 (REF.196) as additional mechanisms of resistance in the context of BRCA1-deficiency. PTIP abrogation in BRCA-deficient tumours leads to restoration of replication fork stability by preventing access ofMRE11 to stalled forks148 (FIG. 4a). This, in turn, causes resistance not only to PARPi but also to cisplatin. Of note, PTIP interacts with artemis, a nuclease that trims DNA ends and promotes NHEJ197. Thus, PTIP abrogation also leads to partial HR restoration and inhibition of PARPi sensitivity198.
BRCA1 promoter methylation versus gene mutation in tumour response to therapy.
Tumours are classified as BRCA1 deficient on the basis of several criteria, including the presence of germline BRCA1 mutations, spontaneously acquired deleterious mutations causing intragenic BRCA1 deletions or epigenetic silencing of BRCA1 through promoter methylation. Germline BRCA1 mutations are the most reliable marker for BRCA1 deficiency in predicting tumour response to PARPi or platinum-based therapy. Most cases of cisplatin resistance in these tumours are attributed to secondary mutations that restore the BRCA1 open reading frame and the expression of a functional protein199,200. A consequence of these secondary mutations is that mutant cells regain the ability to eliminate ICLs or DSBs induced by cisplatin or PARPi.
A common feature of sporadic triple-negative breast tumours is BRCA1 promoter methylation and silencing, which is a good predictor of PARPi sensitivity183. Consequently, treatment-induced loss of BRCA1 promoter methylation could restore normal BRCA1 expression levels or trigger a hypomorphic phenotype (low levels of BRCA1 mRNA and protein expression) associated with PARPi resistance201. A study of tumour xenografts derived from patients with triple-negative breast cancer identified cases in which BRCA1 promoter methylation was retained yet, surprisingly, BRCA1 mRNA and protein were still produced and caused resistance to olaparib and cisplatin treatments202. Next-generation sequencing revealed that genomic rearrangements had placed the BRCA1 gene under the transcriptional control of a heterologous promoter. This is a unique example of the genomic plasticity of BRCA1-deficient tumour adaptation to treatment, which, unfortunately, allows tumour regrowth.
Recently, a panel of tumour xenografts assembled from patients with triple-negative and metastatic breast cancers and from patients with metastatic ovarian cancer carrying germline BRCA1 mutations was assessed for BRCA1 expression and response to therapy191. PARPi resistance emerged in a subset of these tumours even though germline BRCA1 mutations and the loss of the second allele remained detectable by sequence analysis. BRCA1 mRNA and protein analyses revealed the expression of a hypomorphic isoform in these tumours191. The same study identified a mutation in SLFN11 in a BRCA1-mutated patient-derived tumour xenograft that likely gave rise to PARPi resistance. Studies have shown that SLFN11, which encodes the putative DNA and RNA helicase Schlafen family member 11, is needed for PARPi sensitivity irrespective of BRCA status203. A recent CRISPR-Cas9 screen in human cell lines identified multiple gene deletions that sensitize cells to PARPi treatment in the presence or absence of functional BRCA1, similarly to the SLFN11 deletion204. It will be interesting to determine whether mutations in these genes are present in BRCA1-mutated tumours from human patients and whether they can be used as biomarkers for PARPi response regardless of BRCA1 inactivation (TABLE 2).
Table 2 |.
Mechanisms of resistance to PARP inhibitors in BRCA1-deficient cells and tumours
| Mode of resistance | Mechanism | Refs |
|---|---|---|
| Restoration of BRCA1 function | Secondary mutations in BRCA1 | 199,200 |
| Promoter demethylation | 201,202 | |
| Promoter switch | 202 | |
| Partial restoration of homologous recombination | Loss of 53BP1, RIF1, REV7, PTIP, DYNLL1 or the shieldin complex (BOX 2) |
99–101,115, 122,186–189,197, 198,215,222 |
| Restoration of replication fork stability | Loss of PTIP, SMARCAL1, ZRANB3 or HLTF | 147,148 |
| Restoration of PARP signalling | Loss of poly(ADP-ribose) glycohydrolase (PARG) | 195 |
| Other | Loss of SLFN11 (putative DNA/RNA helicase) | 191,203 |
BRCA1, breast cancer type 1 susceptibility protein; PARP, poly(ADP-ribose) polymerase.
Conclusion and future perspective
In this Review, we have analysed the multifaceted role of BRCA1 in the initial and later stages of the HR process, which is crucial for the timely elimination of DSBs and the repair and restart of damaged DNA replication forks. BRCA1, acting in concert with its obligatory partner BARD1, clearly fulfils an important function in DNA end resection, by overcoming the restriction imposed by 53BP1 and its associated complex. In addition, emerging evidence has implicated BRCA1-BARD1 in the RAD51-dependent assembly of the synaptic complex in D-loop formation. As we have discussed at length, BRCA1-BARD1 also very likely helps mediate the exchange of RPA with RAD51 on ssDNA. BRCA1-BARD1 may act alone (in DNA end resection) or likely functions with PALB2 (in synaptic complex assembly) and with PALB2-BRCA2-DSS1 (in the RAD51 for RPA exchange). Importantly, recent evidence has revealed a role for BRCA1-BARD1 in the protection of stalled replication forks against deleterious attrition by cellular nucleases in a manner that is independent of its canonical DSB repair function.
During DNA end resection, BRCA1-BARD1 not only help overcome the various restrictive factors to allow access to the resection machinery but may also influence the activity of the resection machinery75,205,206 (unpublished results from the P.S. laboratory). Moving forward, the major challenge in fully understanding the roles of BRCA1-BARD1 in HR and in replication fork protection will be to develop reconstituted systems consisting of purified factors to interrogate the functions of BRCA1-BARD1 and test mutants that are impaired in either DNA binding or protein-protein interactions. Likewise, it will be important to ascertain whether the E3 ligase activity of BRCA1-BARD1 modifies proteins that are relevant for the execution or regulation of HR repair and replication fork protection.
On the clinical front, there is an urgent need to delineate different mechanisms that cause innate and acquired resistance of BRCA-deficient and HR-deficient tumours to drugs such as platinum-based compounds and PARPi. In this regard, the recent discovery of the HR inhibitory axis comprising the 53BP1-RIF1-shieldin and CST complexes, the mutational inactivation or silencing of which can help restore HR proficiency to HR mutant cells, can be expected to lead to refined understanding of these processes. Intense efforts are being expended on the development of new chemical inhibitors of DNA repair and of DNA damage checkpoint pathways for clinical applications. There can be little doubt that some of these inhibitors will ultimately make their way into the clinic as cancer prophylactics and/or for the treatment of incalcitrant tumours207–209.
R-loops.
Three-stranded nucleic acid structures that arise during transcription, consisting of an RNA-DNA hybrid formed by annealing of the nascent transcript with its DNA template. The non-template DNA is displaced as single-stranded DNA.
Synthetic lethality.
Induction of cell death (lethality) by simultaneously inactivating two different biological pathways or genes, which normally do not affect cell viability inactivated individually.
Ionizing radiation-induced nuclear foci.
Subnuclear domains into which factors needed for DNA damage signalling and repair concentrate on exposure to ionizing radiation. Their formation and resolution reflect the robustness of the cellular response to ionizing radiation.
G-quadruplexes.
Non-canonical DNA (or RNA) structures consisting of stacks of two or more guanine quartets, each stabilized by a monovalent cation. G-quadruplexes form spontaneously on guanine-rich single-stranded DNA during DNA replication and transcription.
Radial chromosomes.
Fused chromosomes that arise from aberrant repair of DNA double-strand breaks or stalled replication forks, or from incomplete resolution of repair intermediates.
Fanconi anaemia.
A multigenic disorder characterized by bone marrow failure and cancer predisposition, owing to an inability to properly process DNA interstrand crosslinks and other DNA lesions.
Acknowledgements
The studies in the laboratories of the authors were supported by Cancer Research UK, the UK Medical Research Council, the University of Oxford and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 722729 (M.T.), and by Cancer Prevention and Research Institute of Texas (CPRIT) REI award RR180029, a Grey Foundation Team Science award and US National Institutes of Health research grant awards R35 CA241801, RO1 CA168635 and RO1 ES007061 (to P.S.). P.S. is a CPRIT Scholar of Cancer Research and the Robert A. Welch Distinguished Chair in Chemistry (AQ-0012). The authors are grateful to Y. Kwon for help with artwork and to W. Zhao and J. Daley for providing valuable feedback on the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Molecular Cell Biology thanks Joanna Morris and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Saini N & Gordenin DA Somatic mutation load and spectra: a record of DNA damage and repair in healthy human cells. Env. Mol. Mutagen 59, 672–686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tubbs A & Nussenzweig A Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeb LA & Harris CC Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res. 68, 6863–6872 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prakash R, Zhang Y, Feng W & Jasin M Homologous recombination and human health:the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol 7, a016600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konstantinopoulos PA, Ceccaldi R, Shapiro GI & D’Andrea AD Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5, 1137–1154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKinnon PJ Genome integrity and disease prevention in the nervous system. Genes. Dev 31, 1180–1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alt FW & Schwer B DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 71, 158–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorthi A & Bishop AJR Ewing sarcoma fusion oncogene: at the crossroads of transcription and DNA damage response. Mol. Cell Oncol 5, e1465014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daley JM, Niu H, Miller AS & Sung P Biochemical mechanism of DSB end resection and its regulation. DNA Repair 32, 66–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma P & Greenberg RA Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.San Filippo J, Sung P & Klein H Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem 77, 229–257 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Sollier J & Cimprich KA Breaking bad: R-loops and genome integrity. Trends Cell Biol. 25, 514–522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Techer H, Koundrioukoff S, Nicolas A & Debatisse M The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet 18, 535–550 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Feng W & Jasin M Homologous recombination and replication fork protection: BRCA2 and more! Cold Spring Harb. Symp. Quant. Biol 82, 329–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickman K & Smogorzewska A Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol 218, 1096–1107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ira G et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A & Jackson SP CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorodetska I, Kozeretska I & Dubrovska A BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J. Cancer 10, 2109–2127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavanagh H & Rogers KM The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered. Cancer Clin. Pract 13, 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noh JM et al. Associations between BRCA mutations in high-risk breast cancer patients and familial cancers other than breast or ovary. J. Breast Cancer 15, 283–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mersch J et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121, 269–275 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghimenti C et al. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer 33, 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Karppinen SM, Heikkinen K, Rapakko K & Winqvist R Mutation screening of the BARD1 gene: evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J. Med. Genet 41, e114 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Brakeleer S et al. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat 31, E1175–E1185 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Rudd MF et al. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res. 16, 693–701 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esteban-Jurado C et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med 17, 131–142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suszynska M et al. BARD1 is a low/moderate breast cancer risk gene: evidence based on an association study of the central European p.Q564X recurrent mutation. Cancers 11, 740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lord CJ & Ashworth A PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashworth A & Lord CJ Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol 15, 564–576 (2018). [DOI] [PubMed] [Google Scholar]
- 30.D’Andrea AD Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71, 172–176 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Annunziato S, Barazas M, Rottenberg S & Jonkers J Genetic dissection of cancer development, therapy response, and resistance in mouse models of breast cancer. Cold Spring Harb. Symp. Quant. Biol 81, 141–150 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Ludwig T, Chapman DL, Papaioannou VE & Efstratiadis A Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 11, 1226–1241 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Hakem R et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85, 1009–1023 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Liu CY, Flesken-Nikitin A, Li S, Zeng Y & Lee WH Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev. 10, 1835–1843 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Gowen LC, Johnson BL, Latour AM, Sulik KK & Koller BH Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet 12, 191–194 (1996). [DOI] [PubMed] [Google Scholar]
- 36.McCarthy EE, Celebi JT, Baer R & Ludwig T Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol. Cell Biol 23, 5056–5063 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet 22, 37–43 (1999). [DOI] [PubMed] [Google Scholar]
- 38.McCarthy A et al. A mouse model of basal-like breast carcinoma with metaplastic elements. J. Pathol 211, 389–398 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Liu X et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl Acad. Sci. USA 104, 12111–12116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shakya R et al. The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl Acad. Sci. USA 105, 7040–7045 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miki Y et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Koonin EV, Altschul SF & Bork P BRCA1 protein products. Functional motifs. Nat. Genet 13, 266–268 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Castilla LH et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat. Genet 8, 387–391 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Friedman LS et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet 8, 399–404 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Hall JM et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250, 1684–1689 (1990). [DOI] [PubMed] [Google Scholar]; This study maps the first genomic region linked to inherited breast cancer.
- 46.Steichen-Gersdorf E et al. Familial site-specific ovarian cancer is linked to BRCA1 on 17q12–21. Am. J. Hum. Genet 55, 870–875 (1994). [PMC free article] [PubMed] [Google Scholar]
- 47.Narod SA et al. Familial breast-ovarian cancer locus on chromosome 17q12-q23. Lancet 338, 82–83 (1991). [DOI] [PubMed] [Google Scholar]
- 48.King M-C, Marks J & Mandell J Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Futreal PA et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science 266, 120–122 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Esteller M et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl Cancer Inst 92, 564–569 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Foulkes WD, Smith IE & Reis-Filho JS Triple-negative breast cancer. N. Engl. J. Med 363, 1938–1948 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Wu LC et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet 14, 430–440 (1996). [DOI] [PubMed] [Google Scholar]; This study isolates the BRCA1 partner BARD1.
- 53.Bosse KR et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 72, 2068–2078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu W et al. BARD1 gene polymorphisms confer nephroblastoma susceptibility. EBioMedicine 16, 101–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cimmino F, Formicola D & Capasso M Dualistic role of BARD1 in cancer. Genes 8, 375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brzovic PS, Rajagopal P, Hoyt DW, King MC & Klevit RE Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol 8, 833–837 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Hashizume R et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem 276, 14537–14540 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Densham RM & Morris JR The BRCA1 ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus 8, 116–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brzovic PS, Meza J, King MC & Klevit RE The cancer-predisposing mutation C61G disrupts homodimer formation in the NH2-terminal BRCA1 RING finger domain. J. Biol. Chem 273, 7795–7799 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Yu X, Wu LC, Bowcock AM, Aronheim A & Baer R The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem 273, 25388–25392 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Yu X, Chini CC, He M, Mer G & Chen J The BRCT domain is a phospho-protein binding domain. Science 302, 639–642 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Manke IA, Lowery DM, Nguyen A & Yaffe MB BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302, 636–639 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Yu X, Fu S, Lai M, Baer R & Chen J BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 20, 1721–1726 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen L, Nievera CJ, Lee AY & Wu X Cell cycle-dependent complex formation of BRCA1.CtIP. MRN is important for DNA double-strand break repair. J. Biol. Chem 283, 7713–7720 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Li M & Yu X Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23, 693–704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Billing D et al. The BRCT domains of the BRCA1 and BARD1 tumor suppressors differentially regulate homology-directed repair and stalled fork protection. Mol. Cell 72, 127–139 e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu W et al. Interaction of BARD1 and HP1 Is required for BRCA1 retention at sites of DNA damage. Cancer Res. 75, 1311–1321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drost R et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20, 797–809 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Shakya R et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia B et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 22, 719–729 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Sy SM, Huen MS & Chen J PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl Acad. Sci. USA 106, 7155–7160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang F et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol 19, 524–529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reid S et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet 39, 162–164 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Rahman N et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet 39, 165–167 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakamura K et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol 21, 311–318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao W et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550, 360–365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; BRCA1-BARD1 is shown to synergize with RAD51 in the formation of D-loops, which are a crucial DNA intermediate in DSB repair by homologous recombination.
- 77.Paull TT, Cortez D, Bowers B, Elledge SJ & Gellert M Direct DNA binding by Brca1. Proc. Natl Acad. Sci. USA 98, 6086–6091 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a DNA-binding activity in BRCA1, thereby providing evidence for a direct involvement.
- 78.Ruffner H, Joazeiro CA, Hemmati D, Hunter T & Verma IM Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl Acad. Sci. USA 98, 5134–5139 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lorick KL et al. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl Acad. Sci. USA 96, 11364–11369 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baer R & Ludwig T The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev 12, 86–91 (2002). [DOI] [PubMed] [Google Scholar]
- 81.Mallery DL, Vandenberg CJ & Hiom K Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 21, 6755–6762 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia Y, Pao GM, Chen HW, Verma IM & Hunter T Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem 278, 5255–5263 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Christensen DE, Brzovic PS & Klevit RE E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol 14, 941–948 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Wu-Baer F, Lagrazon K, Yuan W & Baer R The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem 278, 34743–34746 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Reid LJ et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl Acad. Sci. USA 105, 20876–20881 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Densham RM et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol 23, 647–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stewart MD et al. BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc. Natl Acad. Sci. USA 115, 1316–1321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu Q et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nat. 477, 179–184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu Q et al. Heterochromatin-encoded satellite RNAs induce breast cancer. Mol. Cell 70, 842–853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stewart MD et al. Tuning BRCA1 and BARD1 activity to investigate RING ubiquitin ligase mechanisms. Protein Sci. 26, 475–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moynahan ME, Chiu JW, Koller BH & Jasin M Brca1 controls homology-directed DNA repair. Mol. Cell 4, 511–518 (1999). [DOI] [PubMed] [Google Scholar]
- 92.Scully R et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88, 265–275 (1997). [DOI] [PubMed] [Google Scholar]; This study reveals interactions between BRCA1 and the recombinase RAD51 and documents a DNA repair phenotype of BRCA1-deficient cells.
- 93.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR & Bishop DK The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem 275, 23899–23903 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Huber LJ et al. Impaired DNA damage response in cells expressing an exon 11-deleted murine BRCA1 variant that localizes to nuclear foci. Mol. Cell. Biol 21, 4005–4015 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao W, Wiese C, Kwon Y, Hromas R & Sung P The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu. Rev. Biochem 88, 221–245 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen CC, Feng W, Lim PX, Kass EM & Jasin M Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu. Rev. Cancer Biol 2, 313–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhong Q et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 285, 747–750 (1999). [DOI] [PubMed] [Google Scholar]
- 98.Symington LS Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Escribano-Diaz C et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Bunting SF et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu G et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cruz-Garcia A, Lopez-Saavedra A & Huertas P BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9, 451–459 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Reczek CR, Szabolcs M, Stark JM, Ludwig T & Baer R The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol 201, 693–707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Polato F et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med 211, 1027–1036 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kakarougkas A et al. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 41, 10298–10311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alagoz M et al. SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res. 43, 7931–7944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu XL et al. Genetic interactions between tumor suppressors BRCA1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet 28, 266–271 (2001). [DOI] [PubMed] [Google Scholar]
- 108.Cao L et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 25, 2167–2177 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iwabuchi K, Bartel PL, Li B, Marraccino R & Fields S Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl Acad. Sci. USA 91, 6098–6102 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Iwabuchi K et al. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem 273, 26061–26068 (1998). [DOI] [PubMed] [Google Scholar]
- 111.Schultz LB, Chehab NH, Malikzay A & Halazonetis TD p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol 151, 1381–1390 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.DiTullio RA Jr. et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol 4, 998–1002 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Wang B, Matsuoka S, Carpenter PB & Elledge SJ 53BP1, a mediator of the DNA damage checkpoint. Science 298, 1435–1438 (2002). [DOI] [PubMed] [Google Scholar]
- 114.Cao L et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35, 534–541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bouwman P et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol 17, 688–695 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Bouwman et al. (2010) and Bunting et al. (2010) demonstrate the antagonism between BRCA1 and 53BP1 in the DNA damage response.
- 116.Li M et al. 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO Rep. 17, 1532–1541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bunting SF et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 46, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Y et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Invest 126, 3145–3157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Drost R et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Invest 126, 2903–2918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nacson J et al. BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Rep. 24, 3513–3527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zong D et al. BRCA1 haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Mol. Cell 73, 1267–1281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chapman JR et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chapman JR, Sossick AJ, Boulton SJ & Jackson SP BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci 125, 3529–3534 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen X et al. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489, 576–580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Costelloe T et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581–584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kalb R, Mallery DL, Larkin C, Huang JT & Hiom K BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 8, 999–1005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Simons AM et al. BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res. 66, 2012–2018 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Martin RW et al. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 67, 9658–9665 (2007). [DOI] [PubMed] [Google Scholar]
- 129.Daza-Martin M et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 571, 521–527 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Zhao W et al. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol. Cell 59, 176–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jensen RB, Carreira A & Kowalczykowski SC Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.San Filippo J et al. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J. Biol. Chem 281, 11649–11657 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sung P Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem 272, 28194–28197 (1997). [DOI] [PubMed] [Google Scholar]
- 134.Zelensky A, Kanaar R & Wyman C Mediators of homologous DNA pairing. Cold Spring Harb. Perspect. Biol 6, a016451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scully R et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90, 425–435 (1997). [DOI] [PubMed] [Google Scholar]
- 136.Wang Y et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14, 927–939 (2000). [PMC free article] [PubMed] [Google Scholar]
- 137.Pathania S et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun 5, 5496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 139.Schlacher K et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schlacher K, Wu H & Jasin M A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals the role of BRCA1 in DNA replication fork protection and documents its relationship with RAD51 and other DNA damage repair factors in fork protection.
- 141.Wang AT et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol. Cell 59, 478–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lemacon D et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thangavel S et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Iannascoli C, Palermo V, Murfuni I, Franchitto A & Pichierri P The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 43, 9788–9803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hashimoto Y, Ray Chaudhuri A, Lopes M & Costanzo V Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol 17, 1305–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Higgs MR et al. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell 59, 462–477 (2015). [DOI] [PubMed] [Google Scholar]
- 147.Taglialatela A et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chaudhuri AR et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Przetocka S et al. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, 568–582 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Hashimoto Y, Puddu F & Costanzo V RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol 19, 17–24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kolinjivadi AM et al. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67, 867–881(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Aguilera A & Garcia-Muse T R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Santos-Pereira JM & Aguilera A R loops:new modulators of genome dynamics and function. Nat. Rev. Genet 16, 583–597 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Hamperl S & Cimprich KA Conflict resolution in the genome: how transcription and replication make it work. Cell 167, 1455–1467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hill SJ et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 28, 1957–1975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hatchi E et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 57, 636–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that BRCA1 acts with the putative RNA-DNA helicase senataxin to prevent the accumulation of R-loops at transcription pause sites.
- 157.Cohen S et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun 9, 533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li L et al. DEAD box 1 facilitates removal of RNA and homologous recombination at DNA double-strand breaks. Mol. Cell Biol 36, 2794–2810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Britton S et al. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 42, 9047–9062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ohle C et al. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 167, 1001–1013 (2016). [DOI] [PubMed] [Google Scholar]
- 161.Marnef A, Cohen S & Legube G Transcription-coupled DNA double-strand break repair: active genes need special care. J. Mol. Biol 429, 1277–1288 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Groh M, Albulescu LO, Cristini A & Gromak N Senataxin: genome guardian at the interface of transcription and neurodegeneration. J. Mol. Biol 429, 3181–3195 (2017). [DOI] [PubMed] [Google Scholar]
- 163.Tarsounas M & Tijsterman M Genomes and G-quadruplexes: for better or for worse. J. Mol. Biol 425, 4782–4789 (2013). [DOI] [PubMed] [Google Scholar]
- 164.Zimmer J et al. Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol. Cell 61, 449–460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu H et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun 8, 14432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tacconi EM et al. BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol. Med 9, 1398–1414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ray Chaudhuri A & Nussenzweig A Thwarting endogenous stress: BRCA protects against aldehyde toxicity. EMBO Mol. Med 9, 1331–1333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Garcia MJ & Benitez J The Fanconi anaemia/BRCA pathway and cancer susceptibility. Searching for new therapeutic targets. Clin. Transl Oncol 10, 78–84(2008). [DOI] [PubMed] [Google Scholar]
- 169.Kim H & D’Andrea AD Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 26, 1393–1408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Clauson C, Scharer OD & Niedernhofer L Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol 5, a012732 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Michl J, Zimmer J & Tarsounas M Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 35, 909–923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sawyer SL et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 5, 135–142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Domchek SM et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 3, 399–405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 175.Bryant HE et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 176.Morales J et al. Review of poly(ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr 24, 15–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pommier Y, O’Connor MJ & de Bono J Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl Med 8, 362ps17 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Lord CJ & Ashworth A BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016). [DOI] [PubMed] [Google Scholar]
- 179.Ledermann J et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 15, 852–861 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Litton JK et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med 379, 753–763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Robson M et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med 377, 523–533 (2017). [DOI] [PubMed] [Google Scholar]
- 182.Mirza MR et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl.J. Med 375, 2154–2164 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Swisher EM et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 18, 75–87 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Turner N, Tutt A & Ashworth A Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 4, 814–819 (2004). [DOI] [PubMed] [Google Scholar]
- 185.Mateo J et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med 373, 1697–1708 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jaspers JE et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tomida J et al. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 37, e99543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dev H et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol 20, 954–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Barazas M et al. Radiosensitivity is an acquired vulnerability of PARPi-resistant BRCA1-deficient tumors. Cancer Res. 79, 452–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tacconi EM et al. Chlorambucil targets BRCA1/2-deficient tumours and counteracts PARP inhibitor resistance. EMBO Mol. Med 11, e9982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cruz C et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol 29, 1203–1210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ang JE et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin. Cancer Res 19, 5485–5493 (2013). [DOI] [PubMed] [Google Scholar]
- 193.Rottenberg S et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc. Natl Acad. Sci. USA 104, 12117–12122 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rottenberg S et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl Acad. Sci. USA 105, 17079–17084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gogola E et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 33, 1078–1093 (2018). [DOI] [PubMed] [Google Scholar]
- 196.Michelena J et al. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun 9, 2678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang J et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 28, 2693–2698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Callen E et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153, 1266–1280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Swisher EM et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 68, 2581–2586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Norquist B et al. Secondary somatic mutations predict restoring BRCA1/2 chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol 29, 3008–3015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Castroviejo-Bermejo M et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med 10, e9172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ter Brugge P et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J. Natl Cancer Inst 108, djw148 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Zoppoli G et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl Acad. Sci. USA 109, 15030–15035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zimmermann M et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tomimatsu N et al. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun 5, 3561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Densham RM & Morris JR Moving mountains-the BRCA1 promotion of DNA resection. Front. Mol. Biosci 6, 79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Minchom A, Aversa C & Lopez J Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol 10, 1758835918786658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Brandsma I, Fleuren EDG, Williamson CT & Lord CJ Directing the use of DDR kinase inhibitors in cancer treatment. Expert. Opin. Investig. Drugs 26, 1341–1355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Drean A et al. Modeling therapy resistance in BRCA1/2-mutant cancers. Mol. Cancer Ther 16, 2022–2034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Blackford AN & Jackson SP ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817 (2017). [DOI] [PubMed] [Google Scholar]
- 211.Matos J & West SC Holliday junction resolution: regulation in space and time. DNA Repair 19, 176–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Scully R, Panday A, Elango R & Willis NA DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol 20, 698–714 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Orthwein A et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 214.Her J & Bunting SF How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem 293, 10502–10511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A & de Lange T 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 339, 700–704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gupta R et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173, 972–988 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Noordermeer SM et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Mirman Z et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Barazas M et al. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep. 23, 2107–2118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ghezraoui H et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Stewart JA, Wang Y, Ackerson SM & Schuck PL Emerging roles of CST in maintaining genome stability and human disease. Front. Biosci 23, 1564–1586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.He YJ et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 563, 522–526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Becker JR et al. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun 9, 5406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zgheib O, Pataky K, Brugger J & Halazonetis TD An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell Biol 29, 1050–1058 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC & de Lange T Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl Acad. Sci. USA 110, 2146–2151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wilson MD et al. The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103 (2016). [DOI] [PubMed] [Google Scholar]
- 227.Cortez D Replication-coupled DNA repair. Mol. Cell 74, 866–876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ait Saada A, Lambert SAE & Carr AM Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 71, 135–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pasero P & Vindigni A Nucleases acting at stalled forks: how to reboot the replication program with a few shortcuts. Annu. Rev. Genet 51, 477–499 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Wong AK et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene 17, 2279–2285 (1998). [DOI] [PubMed] [Google Scholar]
- 231.Cantor SB et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160 (2001). [DOI] [PubMed] [Google Scholar]
- 232.Folias A et al. BRCA1 interacts directly with the Fanconi anemia protein FANCA. Hum. Mol. Genet 11, 2591–2597 (2002). [DOI] [PubMed] [Google Scholar]
- 233.Davis AJ et al. bRcA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Res. 42, 11487–11501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wang Q et al. Adenosine nucleotide modulates the physical interaction between hMSH2 and BRCA1. Oncogene 20, 4640–4649 (2001). [DOI] [PubMed] [Google Scholar]
- 235.Batenburg NL et al. CSB interacts with BRCA1 in late S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic Acids Res. 47, 10678–10692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Cortez D, Wang Y, Qin J & Elledge SJ Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 286, 1162–1166 (1999). [DOI] [PubMed] [Google Scholar]
- 237.Venere M, Snyder A, Zgheib O & Halazonetis TD Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 67, 6100–6105 (2007). [DOI] [PubMed] [Google Scholar]
- 238.Chen J Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 60, 5037–5039 (2000). [PubMed] [Google Scholar]
- 239.Gatei M et al. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J. Biol. Chem 276, 17276–17280 (2001). [DOI] [PubMed] [Google Scholar]
- 240.Verma S et al. BRCA1/BARD1-dependent ubiquitination of NF2 regulates Hippo-YAP1 signaling. Proc. Natl Acad. Sci. USA 116, 7363–7370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang B et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194–1198 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kim H, Huang J & Chen J CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol 14, 710–715 (2007). [DOI] [PubMed] [Google Scholar]
- 243.Ruffner H, Jiang W, Craig AG, Hunter T & Verma IM BRCA1 is phosphorylated at serine 1497 in vivo at a cyclin-dependent kinase 2 phosphorylation site. Mol. Cell Biol 19, 4843–4854 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wang H et al. BRCA1 proteins are transported to the nucleus in the absence of serum and splice variants BRCA1a, BRCA1b are tyrosine phosphoproteins that associate with E2F, cyclins and cyclin dependent kinases. Oncogene 15, 143–157 (1997). [DOI] [PubMed] [Google Scholar]
- 245.Liu Y, Virshup DM, White RL & Hsu LC Regulation of BRCA1 phosphorylation by interaction with protein phosphatase 1alpha. Cancer Res. 62, 6357–6361 (2002). [PubMed] [Google Scholar]
- 246.Dubrovska A et al. TGFbeta1/Smad3 counteracts BRCA1-dependent repair of DNA damage. Oncogene 24, 2289–2297 (2005). [DOI] [PubMed] [Google Scholar]
- 247.Scully R et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl Acad. Sci. USA 94, 5605–5610 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Schlegel BP, Green VJ, Ladias JA & Parvin JD BRCA1 interaction with RNA polymerase II reveals a role for hRPB2 and hRPB10alpha in activated transcription. Proc. Natl Acad. Sci. USA 97, 3148–3153 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chai YL et al. The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene 18, 263–268 (1999). [DOI] [PubMed] [Google Scholar]
- 250.Pao GM, Janknecht R, Ruffner H, Hunter T & Verma IM CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl Acad. Sci. USA 97, 1020–1025 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Nair SJ et al. Genetic suppression reveals DNA repair-independent antagonism between BRCA1 and COBRA1 in mammary gland development. Nat. Commun 7, 10913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Jensen DE et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 16, 1097–1112 (1998). [DOI] [PubMed] [Google Scholar]
- 253.Aprelikova ON et al. BRCA1-associated growth arrest is RB-dependent. Proc. Natl Acad. Sci. USA 96, 11866–11871 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kais Z et al. KIAA0101 interacts with BRCA1 and regulates centrosome number. Mol. Cancer Res 9, 1091–1099 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Matsuzawa A et al. The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Mol. Cell 53, 101–114 (2014). [DOI] [PubMed] [Google Scholar]
- 256.Lou Z, Minter-Dykhouse K & Chen J BRCA1 participates in DNA decatenation. Nat. Struct. Mol. Biol 12, 589–593 (2005). [DOI] [PubMed] [Google Scholar]
- 257.Bochar DA et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102, 257–265 (2000). [DOI] [PubMed] [Google Scholar]
- 258.Harte MT et al. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 70, 2538–2547 (2010). [DOI] [PubMed] [Google Scholar]
- 259.Yarden RI & Brody LC BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl Acad. Sci. USA 96, 4983–4988 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Magnard C et al. BRCA1 interacts with acetyl-CoA carboxylase through its tandem of BRCT domains. Oncogene 21, 6729–6739 (2002). [DOI] [PubMed] [Google Scholar]
- 261.Chen CF et al. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J. Biol. Chem 271, 32863–32868 (1996). [DOI] [PubMed] [Google Scholar]
- 262.Sato K et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr. Biol 22, 1659–1666 (2012). [DOI] [PubMed] [Google Scholar]
- 263.Kim BJ et al. The histone variant MacroH2A1 is a BRCA1 ubiquitin ligase substrate. Cell Rep. 19, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kleiman FE et al. BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev. 19, 1227–1237 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Starita LM et al. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J. Biol. Chem 280, 24498–24505 (2005). [DOI] [PubMed] [Google Scholar]
- 266.Velimezi G et al. Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48. Nat. Commun 9, 2280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Uckelmann M et al. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun 9, 229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]