

Abstract
Human African trypanosomiasis (HAT) is a deadly neglected tropical disease caused by the protozoan parasite Trypanosoma brucei. During the course of screening a collection of diverse nitrogenous heterocycles, we discovered two novel compounds that contain the tetracyclic core of the Yohimbine and Corynanthe alkaloids, were potent inhibitors of T. brucei proliferation and T. brucei methionyl-tRNA synthetase (TbMetRS) activity. Inspired by these key findings, we prepared several novel series of hydroxyalkyl δ-lactam, δ-lactam, and piperidine analogs and tested their anti-trypanosomal activity. A number of inhibitors are more potent against T. brucei than these initial hits with one hydroxyalkyl δ-lactam derivative being 25-fold more effective in our assay. Surprisingly, most of these active compounds failed to inhibit TbMetRS. This work underscores the importance of verifying, irrespective of close structural similarities, that new compounds designed from a lead with a known biological target engage the putative binding site.
Keywords: focused compound libraries, analog synthesis, structure-activity-relationships, tropical disease, growth inhibition
Graphical Abstract
Introduction
As part of a broad program directed toward the discovery of novel compounds that would serve as tools for chemical biology or as therapeutic leads, we recently developed a general platform for the rapid assembly of diverse molecular libraries of natural product-like compounds.1 The approach features multi-component assembly processes (MCAPs) to create versatile intermediates that can then be quickly transformed by selective ring-forming reactions into various heterocyclic scaffolds bearing functional groups suitable for further diversification. A variety of small molecular libraries derived from more than 40 different nitrogenous heterocycles were prepared using this approach, and compounds were submitted for screening as part of the NIH Molecular Libraries Initiative.2–7
One focused library contained the tetracyclic heterocyclic framework that is a structural subunit of the Yohimbine and Corynanthe alkaloids,7 and the representative compounds 1 and 2 (Figure 1)were found to inhibit the proliferation of the protozoan parasite Trypanosoma brucei (T. brucei), the infectious agent that causes human African trypanosomiasis (HAT). These compounds also inhibit T. brucei methionyl-tRNA synthetase (TbMetRS),8 which plays an essential role in protein synthesis. HAT is a deadly, yet sadly neglected, tropical disease with an at-risk population of more than 60 million people in sub-Saharan Africa.9 The progression of the disease is divided into two stages – haemolymphatic and encephalitic.During the first stage, the parasite resides in the bloodstream and lymph of the host and causes a variety of flu-like symptoms and organ abnormalities. Progression to the second stage occurs when the parasite crosses the blood-brain barrier, leading to a variety of central nervous system disturbances such as dementia, sleep-wake cycle alteration, motor/visual system impairment, meningoencephalitis, and ultimately death. Unfortunately, treatment options for HAT are limited. Suramin and pentamidine are used to treat the first stage, whereas melarsoprol, eflornithine, and eflornithine/nifurtimox combination therapy are treatments for the second stage.9,10 However, each of these treatments suffer serious shortcomings, including toxicity and drug resistance. Recently fexinidazole, which is the first drug candidate for treating late-stage HAT in the past 30 years, was approved for use in non-European markets.11
Figure 1.

Initial screening hits identified for T. brucei and TbMetRS.8
In view of the deficiencies associated with drugs currently used to treat HAT, the unmet need for new chemotherapeutic agents with fewer off-target effects is apparent. Accordingly, the promising preliminary findings that 1 and 2 both decrease proliferation of T. brucei and inhibit TbMetRS inspired us to initiate a campaign to design and synthesize simplified analogs with improved physicochemical and biological properties. Owing to the substantial differences in active site residues of the parasite MetRS enzyme compared to its human ortholog,12 we envisioned that such compounds for treating HAT would have minimal adverse side effects. Indeed, this approach was pioneered by Buckner and colleagues who developed and studied libraries of aminoquinoline and urea-based TbMetRS inhibitors.12,13 The lead compound 3 (Figure 2) that emerged from these studies significantly suppressed proliferation of T. brucei rhodesiense in mice, but the treatment was not curative because the mice succumbed to parasitemia.13a This finding led to studies of other analogs.14
Figure 2.

Flexible, urea-based inhibitor 3 of TbMetRS.13
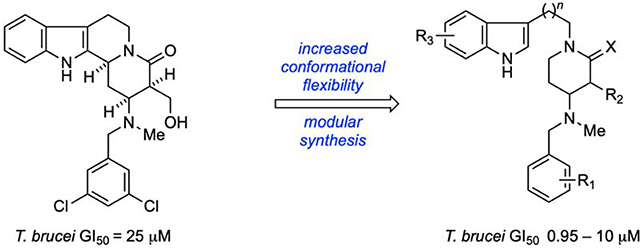
Comparing the structure of 3 with our initial hits 1 and 2 reveals some notable similarities and differences that provided a rationale for the design of analogs based on 1 and 2. For example, 3 and related compounds possess heteroaromatic and N-benzyl substituents that are separated by a flexible linker. Compounds 1 and 2 likewise feature heteroaromatic and N-benzyl substituents, but these subunits are connected by a rigid quinolizidine core. In the crystal structure of 3 bound to TbMetRS (PDB: 4MVW),13b the two aromatic substituents occupy separate pockets on the extended binding site of the enzyme. Specifically, the N-benzyl group occupies the methionine-binding pocket, and the heteroaromatic moiety resides in an auxiliary pocket; the flexible linker appears to allow these substituents to occupy their respective binding sites in favorable orientations. Based upon these observations and some preliminary modeling studies, we hypothesized that analogs of our initial hits 1 and 2 having the increased conformational flexibility of 3 would be superior inhibitors because the added flexibility allows the N-benzyl and indole moieties to interact more optimally with TbMetRS than is possible in the more rigid tetracyclic framework present in 1 and 2.
To test this hypothesis, we envisioned scission of the C(2)–C(3) bond (Figure 3) of the core structure 4 leading to the set of compounds having the general structures 5–7. In addition to increasing the conformational flexibility of the core framework using different linking subunits, novel inhibitors belonging to these new chemotypes may be accessed by a modular synthesis that enables facile late-stage diversification of the indole and the N-benzyl substituents. We also planned to examine the that enables facile late-stage diversification of the indole and the N-benzyl substituents. We also planned to examine the effects simplifying the core of 5 in an iterative fashion by removing the hydroxymethylene side-chain and reducing the lactam to give the three distinct series of hydroxyalkyl δ-lactam, lactam, and piperidine analogs as generally defined by 5–7, respectively.
Figure 3.

Design of more flexible scaffolds 5–7 from parent 4.
Results and Discussion
Synthesis
Synthesis of hydroxyalkyl δ-lactam analogs having the general form 5 was accomplished by first converting indole (8) into the amide 10 in a one-pot process involving sequential reaction of 8 with oxalyl chloride and the amine 9 (Scheme 1). Reduction of 10 furnished 12 in 62% overall yield from 8. Acid-catalyzed removal of the acetal protecting group in 10 using CF3CO2H (TFA) in aqueous trifluoroethanol (TFE) produced an unstable aldehyde that was treated directly with N-methylhydroxylamine, and the intermediate nitrone underwent an intramolecular dipolar cycloaddition upon heating to furnish 13 in 85% yield. Reductive opening of the isoxazolidine ring and subsequent reductive alkylation of the resultant secondary amine with a series of substituted benzaldehydes delivered the hydroxyalkyl δ-lactam analogs 14a–h in overall yields ranging from 24–46%.
Scheme 1.

Synthesis of hydroxyalkyl δ-lactam analogs 14a–h.
In addition to probing the effects of varying the substitution on the N-benzyl group, we prepared one series of hydroxyalkyl δ-lactams in which the length of the chain linking the indole and lactam rings was varied (Scheme 2) and another series in which the nature of the substituents on the indole ring was changed (Scheme 3). Toward preparing analogs with variable linker lengths, the indole carboxylic acids 15a,b were sequentially treated with thionyl chloride and then 9, and the intermediate amides were reduced with LiAlH4 to give amines that were acylated with 11 to furnish amides 16a,b in 52–76% overall yields (Scheme 2). Elaboration of 16a,b into 14i,j was then achieved in four steps (8–27%, Scheme 2) following the same steps previously used to convert 12 into 14a–h (Scheme 1).
Scheme 2.

Synthesis of hydroxyalkyl δ-lactam analogs 14i,j with varying linker lengths.
Scheme 3.

Synthesis of hydroxyalkyl δ-lactam analogs 14k–n with indolyl substituents.
Analogs bearing different oxygen substituents at the 4- and 5-positions of the indole ring were likewise prepared by the three-step transformation of indoles 17a–c into the crotonamides 18a–c in 54–63% overall yields following the procedures previously outlined in Scheme 1 to convert indole (8) into 12 (Scheme 3). These amides were then elaborated to their respective analogs 14k–m in four steps (10–64% overall yield) as described for the syntheses of 14a–h (Scheme 1). The 5-hydroxy analog 14n was also prepared from 18b in four steps (16% overall yield) involving nitrone formation and cycloaddition followed by hydrogenolysis to simultaneously remove the O-benzyl protecting group and cleave the isoxazolidine ring. The 5-hydroxy intermediate 19d thus formed was then converted to 14n via reductive N-alkylation.
The 4-amino- δ-lactams 25a–h were then prepared from tryptamine (21) via the sequence of reactions summarized in Scheme 4. In the event, reaction of 21 with one equivalent of ethyl acrylate provided a secondary amine that was coupled with malonic acid monoethyl ester using N-ethyl N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) to furnish 22 in 79% overall yield. Dieckmann cyclization of 22 followed by hydrolysis and decarboxylation provided the β-keto-lactam 23 in 64% yield. Reductive amination of 23 with methylamine then delivered the amine 24 (71% yield), which was reductively alkylated with a set of substituted benzaldehydes to deliver the 25a–h in 58–81% yield.
Scheme 4.

Synthesis of δ-lactam analogs 25a–h.
Toward preparing the 4-aminopiperidines 29a–l, the key intermediates 27a–e were first synthesized from the Bocprotected piperidine 26, itself prepared from N-Boc-4-piperidinone (see SI) by sequential reductive alkylation with various substituted benzaldehydes and acid-catalyzed removal of the Boc-protecting group (Equation 1).
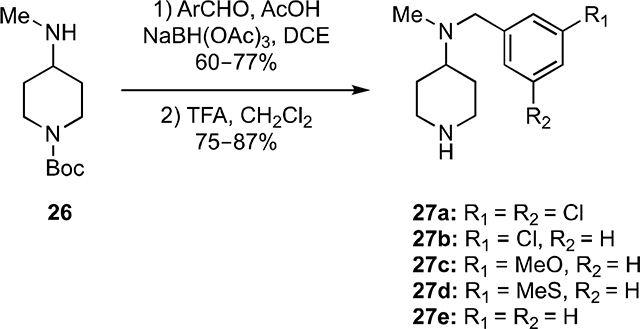 |
(1) |
Compounds 27a–e were then elaborated via divergent synthetic sequences to deliver the desired analogs. For example, coupling of 27a or 27b with various heteroaromatic alkyl carboxylic acids using EDCI gave the N-acyl piperidines 28a–c (70–87% yields) that were selectively reduced with either alane or borane to give 29a–c in 62–82% overall yields (Scheme 5). Use of a Lewis-acidic hydride reducing agent was crucial to avoid reduction of the aryl chloride substituents, as was observed when performing the reduction with lithium aluminum hydride (LiAlH4). The related fluoro-indole analog 29d was prepared from 27a in a similar fashion except the acid chloride 30a was used as an acylating agent, and CsF/Na2CO3 was used in an additional step after borane-mediated ketoamide reduction to decompose the amine-borane adducts. This step was required rather than the alternative aqueous acid workup because the addition of aqueous HCl following the ketoamide reduction led to reduction of the indole ring.15 The phenylurea and 7-azaindole derivatives 29e and 29f were prepared by treating 27a with phenyl isocyanate and 31, respectively (Scheme 5).16
Scheme 5.

Synthesis of piperidine analogs 29a–f.
Finally, analogs 29g–i were prepared in 62–96% overall yields from 27c–e via acylation with 30b followed by reduction with LiAlH4 (Equation 2).
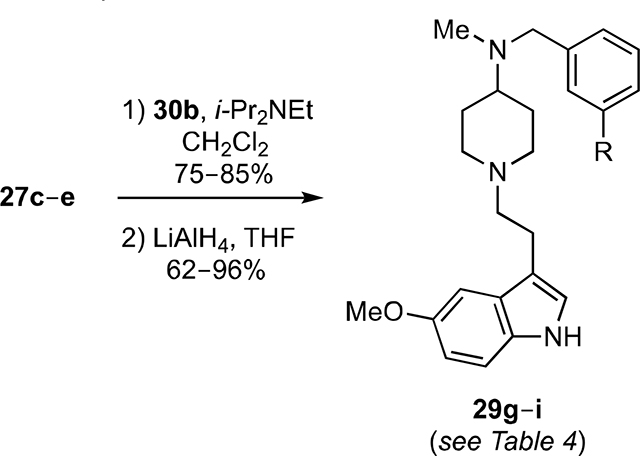 |
(2) |
Biological Analysis
Having prepared a suite of different analogs of our initial leads 1 and 2, we turned to the evaluation of their efficacy using a phenotypic screen to determine their effects on the growth of T. brucei in vitro. In order to ensure meaningful comparisons of structure activity relationships (SAR) relative to the more potent lead 1 (Figure 1), we assayed 1 using the same growth inhibition assay that we would employ to evaluate analogs of 1 and found that it displayed a GI50 valueof 25 μM. Owing perhaps to variations in experimental conditions, this value is substantially different from the 2 μM value originally determined (see Figure 1).8 Analysis of the GI50values of the hydroxyalkyl δ-lactams 14a–h and the δ-lactams 25a–h reveals how increasing the flexibility of the central core, modifying the substitution on the δ-lactam ring, and varying the nature of the N-benzyl group affects potencies (Table 1). Indeed, both 14a and 25a are roughly five-fold more active against T. brucei than 1,thus supportingour central hypothesis that increased flexibility would improve activity. As evidenced by the observation that 25a–h are less active than their counterparts 14a–h, removing the hydroxyethyl side chain on the δ-lactam ring consistently reduces the ability of compounds to inhibit growth. The SAR associated with the N-benzyl substituent on the various scaffolds roughly tracks trends observed by Buckner and coworkers. For example, the dimethoxy analogs 14d and 25d and the unsubstituted benzyl analogs 14h and 25h do not exhibit any activity,12,13 whereas compounds having 3-chlorobenzyl groups appear to outperform other substituted analogs.
Table 1.
T. brucei GI50 values hydroxyalkyl δ-lactams 14a–h and δ-lactams 25a–h.
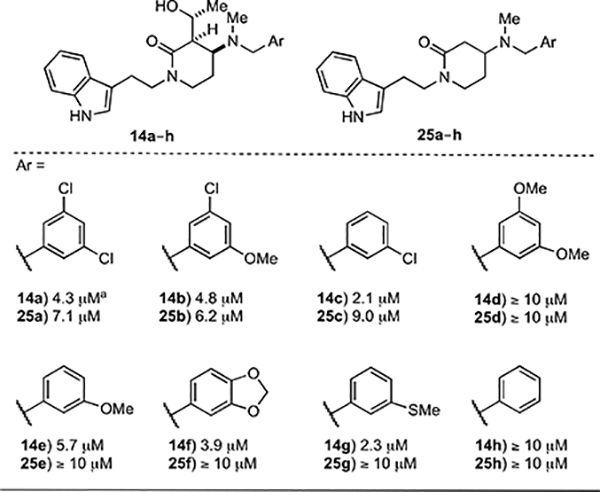 |
error values were consistently <10% with exceptions of 14f (18%) and 14g (26%)
The GI50 values for the set of hydroxyalkyl δ-lactam analogs 14i–n enable us to assess the effects of varying the length of the chain linking the indole ring and the δ-lactam core and of modifying the nature of substituents on the indole ring (Table 2). With regard to the linker length, compounds having one to three carbon atoms in the chain (e.g., 14a, 14i, and 14j, respectively) are approximately equipotent. Based upon our original design rationale, this result is somewhat surprising. Specifically, we hypothesized that the length of the linking unit would modulate potency because the increased flexibility that accompanies longer chains could affect how the two aromatic moieties on the δ-lactam ring interact with their respective binding pockets. The presence of alkoxy groups on the indole ring as found in 14k–m offers a 2–5-fold increase in growth inhibitory activity relative to the unsubstituted parent 14a, whereas the 5’-hydroxy analog 14n is equipotent with 14a.
Table 2.
T. brucei GI50 values of hydroxyalkyl δ-lactams 14i–n.
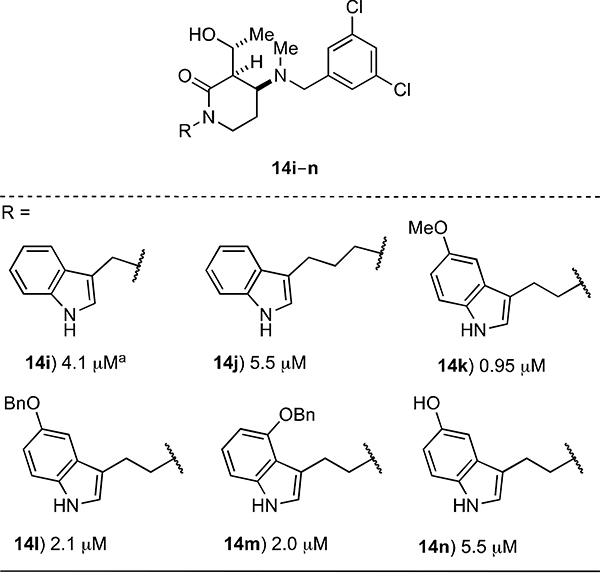 |
error values were consistently <10% with exceptions of 14i (15%), 14l (19%) and 14n (11%)
The GI50 values for the N-(3,5-dichlorobenzyl)piperidines 28a and 29a,c–f (Table 3) provide additional SAR data for analogs of 1. For example, 28a, which has an amide linker between the piperidine ring and the indole groups, displays significantly diminished activity relative to its alkyl counterpart 29a. We surmise that this difference likely arises from a conformational effect, because the potencies of the lactams 25a–h (Table 1) show that a basic nitrogen atom in the piperidine ring is not essential for activity. Having a fluorine substituent on the indole ring as in 29d or replacing the indole moiety with a benzimidazole ring as in 29c has no notable effect on potency, whereas replacing the indole with the 7-azaindole ring in 29f leads to a four-fold loss in growth inhibitory activity. Finally, the phenyl-urea analog 29e, which we prepared for a direct comparison of our piperidine scaffold with Buckner’s phenyl-urea diaminopropane inhibitors, is inactive, perhaps because of conformational effects.14
Table 3.
T. brucei GI50 values of N-(3,5-dichlorobenzyl)piperidine analogs 28a and 29a,c–f.
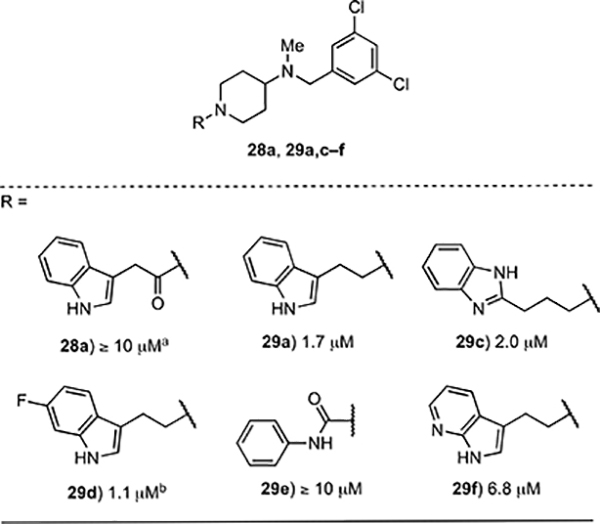 |
error values were consistently <10% with exception of 29c (30%).
error not determined.
The set of compounds 29b,g–i was designed to explore the SAR of a set of analogs of 1 having a piperidine core in the spacer linking the 5-methoxyindole, which appears to be an optimal indole substituent (see 14k, Table 2), and the N-benzyl groups (Table 4). Of these compounds, the thiomethyl compound 29h (GI50 = 1.6 μM) is the most potent, while the potencies of the other N-benzyl analogs 29b,g,i are 2–4-fold less active.
Table 4.
T. brucei GI50 values of piperidine analogs 29b,g–i.
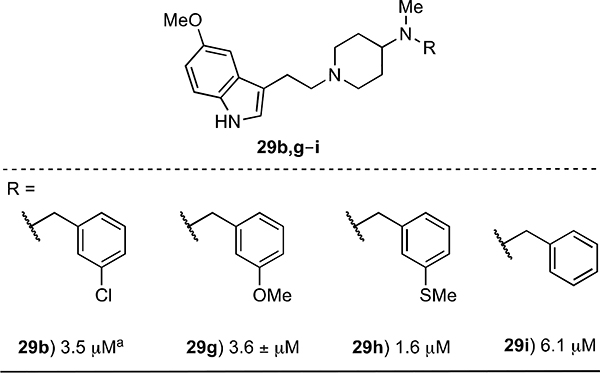 |
error values were consistently <10% with exception of 29l (11%)
Examination of the growth inhibition data for our analogs of 1 (see Tables 1–4)reveals a relatively flat SAR in which a fairly large number of structural modifications seem to have little impact on the observed GI50 values. We initially focused upon GI50 values because we lacked the capability of performing the established radiolabeled methionine assay for TbMetRS activity that was employed in the initial screening.13 However, we also wanted to show that the new analogs of 1 inhibited TbMetRS, so we assessed the inhibitory activity of our compounds towards recombinant TbMetRS using a colorimetric pyrophosphatase-coupled enzyme inhibition assay.17 We first tested the original hit 1 and found that it exhibited an IC50 value of 4.9 ± 1.5 μM in this assay. The discrepancy between this IC50 value and the 0.6 μM value (Figure 1) is expected, because the concentration of TbMetRS and other reagents in the colorimetric assay is much higher than those used in the radiolabeled methionine assay. Indeed, variations in IC50 values arising from concentration differences are common, so it is important to ensure concentrations are the same before making any correlations of IC50 values. Surprisingly, however, although a few compounds having the hydroxyalkyl δ-lactam scaffoldfound in 1 (i.e., 14a,b,h, and n) display 15–30 % inhibition of TbMetRS at 200 μM, most analogs are inactive against TbMetRS at concentrations up to 200 μM. Even for those compounds that inhibit TbMetRS, growth inhibition values and enzyme inhibition data correlate poorly, suggesting that the mechanism of action for the growth inhibition of more flexible analogs of 1 likely differs from that of 1.
Summary and Conclusion
In summary, following the discovery of 1 and 2 as novel leads against HAT, we designed several sets of analogs as molecular hybrids of 1 and 3, a more flexible inhibitor of TbMetRS, that we hypothesized would bind with higher affinity to TbMetRS. The rationale for these experiments was based upon preliminary modeling studies suggesting that the terminal aromatic rings, which comprise the pharmacophore of TbMetRS inhibitors, in more flexible analogs of 1 would interact better with the enzyme. Small collections of hydroxyalkyl δ-lactams, δ-lactams and piperidines were thus prepared and evaluated in growth inhibition assays of T. brucei. Although a number of compounds were discovered that are up to 25-fold more potent than 1 in our assay, there is little variation overall in the SAR. Moreover, each of the new compounds is less active against TbMetRS than 1. Accordingly, the majority of analogs in this study appear to inhibit proliferation of T. brucei by an alternate, as yet unknown, mechanism that does not involve inhibition of TbMetRS. This work underscores the importance of ensuring that new compounds that are designed from an initial hit engage the same binding site; nothing should be assumed, irrespective of close structural similarities.
EXPERIMENTAL SECTION
General
Solvents and reagents were reagent grade and were used without purification unless otherwise noted. Methanol (MeOH) and N,N,-dimethylformamide (DMF) were dried by filtration through two columns of activated molecular sieves prior to use. Tetrahydrofuran (THF) was passed through two columns of activated neutral alumina prior to use. Benzene was distilled from sodium and benzophenone. Methylene chloride, diisopropylamine, triethylamine, and diisopropylethylamine were distilled from calcium hydride immediately prior to use. Pyridine was distilled from potassium hydroxide (KOH) and calcium hydride and stored over KOH. Dioxane was distilled from sodium metal and benzophenone prior to use. All solvents were determined to have less than 50 ppm H2O by Karl Fischer coulometric moisture analysis. All reactions were performed under an atmosphere of argon or nitrogen. Removal of solvent under reduced pressure was performed using a rotary evaporator. Flash chromatography was performed using SiliaFlash® F60 silica gel (Silicycle, 40–63 μM, 60 Å). Infrared (IR) spectra were obtained either neat on sodium chloride or as solutions in the solvent indicated and reported as wavenumbers (cm−1). 1H and 13C NMR spectra were obtained at the specified frequency as a solution in CDCl3 unless otherwise indicated. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) (δ 0.00) and referenced relative to deuterated solvent. Coupling constants (J) are reported in Hz, and the splitting abbreviations used are: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; ddd, doublet of doublet of doublets; m, multiplet; comp, overlapping multiplets of magnetically non-equivalent protons; br, broad. For HRMS with compounds containing chlorine, the calculated and measured masses are for the isotope 35Cl.
N-(3,3-Dimethoxypropyl)-2-(1H-indol-3-yl)-2-oxoacetamide (10)
A solution of oxalyl chloride (1.52 g, 12.0 mmol) in ether (1 mL) was added dropwise over 5 min to a solution of indole (1.41 g, 12.0 mmol) in ether (24 mL) at 0 °C. The reaction was stirred at 0 °C for 1 h, then warmed to room temperature, and stirred for an additional 1 h. The yellow precipitate was collected via vacuum filtration and dried in vacuo to give 2.28 g (87%) of indole-3-glyoxal chloride. A suspension of indole-3-glyoxal chloride (1.91 g, 8.7 mmol) in CH2Cl2 (20 mL) was added dropwise over 30 min to a solution of 9 (1.47 g, 7.3 mmol) and triethylamine (3.70 g, 36.5 mmol) in CH2Cl2 (75 mL) at 0 °C. The reaction was stirred at 0 °C for 0.5 h, then warmed to room temperature and stirred for an additional 1 h. Saturated aqueous NaHCO3 (100 mL) was added to the reaction and stirred for 15 min. The mixture was extracted with CH2Cl2 (3 × 100 mL), and the combined organic extracts were washed with aqueous NaOH (1 M, 2 × 100 mL), water (100 mL), and brine (100 mL), dried (Na2SO4), filtered, and concentrated in vacuo to give 2.23 g (97%) of crude 10. The crude material was >90% purity, and was further purified by dissolving in CH2Cl2 (300 mL) and washing successively with saturated aqueous NH4Cl (200 mL), saturated aqueous NaHCO3 (200 mL), water (200 mL), and brine (200 mL). The organic fraction was dried (Na2SO4), and concentrated in vacuo to give 1.91 g (90%) of 10 as a pale yellow solid (>95% purity, 1H NMR). 1H NMR (400 MHz) δ 9.99 (s, 1 H), 9.01 (d, J = 3.2 Hz, 1 H), 8.41 (d, J = 7.2 Hz, 1 H), 7.97 (t, J = 6.0 Hz, 1 H), 7.40 – 7.38 (m, 1 H), 7.31 (ddd, J = 8.4, 7.2, 1.2 Hz, 1 H), 7.25 (ddd, J = 8.4, 7.2, 1.2 Hz, 1 H), 4.48 (t, J = 6.4 Hz, 1 H), 3.47 (q, J = 6.4 Hz, 2 H), 3.36 (s, 6 H), 1.91 (q, J = 6.4 Hz, 2 H). 13C NMR (100 MHz, CD3CN) δ 181.6, 163.0, 139.1, 136.5, 126.9, 124.0, 123.1, 121.9, 112.8, 112.5, 103.7, 53.1, 35.1, 32.3. HRMS (ESI) m/z calcd for C15H18N2O4(M+Na)+, 313.1159; found, 313.1166.
(E)-N-(2-(1H-Indol-3-yl)ethyl)-N-(3,3-dimethoxypropyl)but-2-enamide (12)
A solution of 10 (0.50 g, 1.70 mmol) in THF (5 mL) was added dropwise over 15 min to a stirred suspension of lithium aluminum hydride (0.65 g, 17.0 mmol) in THF (40 mL) at 0 °C. The reaction was heated at 65 °C for 14 h. The reaction was cooled to 0 °C, and the reaction was quenched via successive addition of water (0.6 mL), aqueous sodium hydroxide (15% w/v, 0.6 mL), and water (3 mL). The suspension was warmed to room temperature and MgSO4 was added. The solution was filtered through a fritted funnel, the solids were washed with excess CH2Cl2 and the filtrate was concentrated in vacuo. The crude material was taken up in aqueous HCl (0.2 M, 150 mL) and washed with ether (2 × 100 mL). The aqueous fraction was basified with aqueous NaOH (40% w/v) to pH 12–14, as judged by pH paper. The basic solution was extracted with CH2Cl2 (3 × 200 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to give 0.41 g (90%) of N-(2-(1H-indol-3-yl)ethyl)-3,3-dimethoxypropan-1-amine as a viscous oil (>95% purity, 1H NMR); 1H NMR (400 MHz) δ 8.61 (s, 1 H), 7.57 (d, J = 7.2 Hz, 1 H), 7.42 (dt, J = 8.0, 1.2 Hz, 1 H), 7.27 – 7.20 (m, 1 H), 7.17 (dd, J = 8.0, 1.2 Hz, 1 H), 7.14 (ddd, J = 8.0, 7.2, 1.2 Hz, 1 H), 4.25 (t, J = 4.0 Hz, 1 H), 3.20 – 3.18 (comp, 4 H), 3.03 (t, J = 5.6 Hz, 2 H), 3.00 (s, 6 H), 1.88 (tt, J = 7.2, 4.0 Hz, 2 H). 13C NMR (100 MHz) δ 134.0, 123.8, 121.5, 120.0, 117.4, 115.6, 109.2, 105.8, 101.3, 52.3, 45.6, 41.1, 25.8, 19.6; IR (neat) 3411, 2337, 2173, 1640, 1458 cm−1. HRMS (CI) m/z calcd for C15H23N2O2(M+H)+, 263.1760; found, 263.1760.
A solution of crotonoyl chloride (51 mg, 0.48 mmol) in CH2Cl2 (1 mL) was added dropwise to a solution of triethylamine (82 mg, 0.81 mmol) and N-(2-(1H-indol-3-yl)ethyl)-3,3-dimethoxypropan-1-amine (106 mg, 0.40 mmol) in CH2Cl2 (1 mL) at – 78 °C. The reaction was stirred for 2 h at –78 °C, whereupon saturated aqueous NaHCO3 (5 mL) was added. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic fractions were dried (Na2SO4), filtered, and the was solvent removed in vacuo. The resulting residue was purified via flash column chromatography eluting with CH2Cl2 : methanol (99 : 1 → 97 : 1 along a gradient) to give 94 mg (71%) of 12 as a clear oil. 1H NMR (400 MHz, CD3CN) (1 : 1 rotamer mixture) δ 9.24 (brs, 0.5 H), 9.20 (brs, 0.5 H), 7.65 (d, J = 8.0 Hz, 0.5 H), 7.58 (d, J = 8.0 Hz, 0.5 H), 7.42 (d, J = 3.6 Hz, 0.5 H), 7.40 (d, J = 3.6 Hz, 0.5 H), 7.17 – 7.14 (comp, 1 H), 7.10 – 7.06 (comp, 2 H), 6.87 (dq, J = 14.8, 7.2 Hz, 0.5 H), 6.64 (dq, J = 14.8, 7.2 Hz, 0.5 H), 6.40 (dd, J = 14.8, 1.6 Hz, 0.5 H), 6.16 (dd, J = 14.8, 1.6 Hz, 0.5 H), 4.38 – 4.34 (comp, 1 H), 3.63 (t, J = 6.4 Hz, 1 H), 3.61 (t, J = 6.4 Hz, 1 H), 3.39 – 3.35 (comp, 2 H), 3.27 (s, 6 H), 3.00 (t, J = 8.0 Hz, 1 H), 2.98 (t, J = 8.0 Hz, 1 H), 1.99 (dd, J = 6.8, 1.6 Hz, 1.5 H), 1.83 – 1.79 (comp, 2 H), 1.69 (dd, J = 6.8, 1.6 Hz, 1.5 H). 13C NMR (1:1 rotamer mixture) (100 MHz, CD3CN) δ 136.8, 127.8, 127.7, 123.4, 122.7, 122.6, 122.6, 121.8, 121.7, 119.1, 119.0, 118.9, 118.5, 112.9, 111.9, 111.7, 111.6, 103.1, 102.6, 52.8, 52.5, 48.8, 47.5, 44.0, 42.5, 32.7, 31.1, 25.3, 23.7, 17.5, 17.3. HRMS (ESI) m/z calcd for C19H26N2O3 (M+Na)+, 353.1836; found, 353.1836.
(3S,3aR,7aS)-5-(2-(1H-Indol-3-yl)ethyl)-1,3-dimethylhexahydroisoxazolo[4,3-c]pyridin-4(1H)-one (13)
A solution of trifluoroacetic acid (5 drops) and12 (110 mg, 0.33 mmol) in TFE/water (3 : 1, 7 mL) was stirred for 1 h at room temperature. Saturated aqueous NaHCO3 (20 mL) was added, and the aqueous layer extracted with CH2Cl2 (3 × 20 mL). The combined organic fractions were dried (Na2SO4), filtered, and concentrated in vacuo. The crude mixture was dissolved in toluene (5 mL), followed by addition of N-methylhydroxylamine hydrochloride (41 mg, 0.50 mmol) and triethylamine (83 mg, 0.83 mmol). The reaction was heated under reflux for 1 h, then cooled to room temperature, and partitioned between saturated aqueous NaHCO3 (20 mL), ethyl acetate (20 mL), and methanol (2 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with water (30 mL), brine (30 mL), dried (Na2SO4), filtered, and concentrated in vacuo to give 88 mg (85%) of 13 as an oil (>90% purity, 1H NMR). 1H NMR (500 MHz) δ 8.26 (brs, 1 H), 6.62 (d, J = 8.1 Hz, 1 H), 7.32 (d, J = 8.1 Hz, 1 H), 7.16 (t, J = 7.1 Hz, 1 H), 7.09 (t, J = 7.1 Hz, 1 H), 6.99 (d, J = 1.7 Hz, 1 H), 3.89 (p, J = 6.1 Hz, 1 H), 3.73 (m, 1 H), 3.64 (m, 1 H), 3.53 (td, J = 12.0, 2.9 Hz, 1 H), 2.99 – 2.95 (comp, 3 H), 2.81 – 2.76 (comp, 2 H), 2.63 (s, 3 H), 1.75 (ddt, J = 14.5, 11.2, 4.1 Hz, 1 H) 1.59 (dq, J = 14.5, 3.2 Hz, 1 H), 1.44 (d, J = 5.9 Hz, 3 H). 13C NMR (125 MHz) δ 169.0, 136.3, 127.4, 122.0, 121.9, 119.3, 118.7, 112.8, 111.2, 77.4, 66.1, 55.3, 48.5, 44.0, 43.1, 25.2, 23.4, 19.2. HRMS (CI) m/zcalcd for C18H23N3O2 (M+H)+, 314.1869; found, 314.1867.
(±)-(3R,4S)-1-(2-(1H-Indol-3-yl)ethyl)-3-((R)-1-hydroxyethyl)-4-(methylamino)piperidin-2-one
Zinc (powder, 530 mg, 8.14 mmol) was added in three portions over 1.5 h to a solution of 13 (85 mg, 0.27 mmol) in acetic acid (aq 80%, 20 mL) at 60 °C, and stirred at 60 °C for an additional 1 h. The reaction was cooled to room temperature, whereupon excess zinc was filtered and washed with ethyl acetate. Ethyl acetate (100 mL) was added to the filtrate and zinc acetate immediately precipitated out of solution as a fluffy white solid. The zinc acetate was filtered, and washed with excess ethyl acetate. The solvent was removed in vacuo, and the crude material was taken up in aqueous HCl (1 M, 30 mL) and washed with ether (2 × 30 mL). The aqueous fraction was basified with aqueous NaOH (40% w/v) to pH ~ 14. The basic solution was extracted with CH2Cl2 (4 × 50 mL). The combined organic fractions were washed with saturated aqueous NaHCO3 (100 mL), water (100 mL), brine (1 × 100 mL), dried (Na2SO4), filtered, and concentrated in vacuo to give 78 mg (91%) of (±)-(3R,4S)-1-(2-(1H-indol-3-yl)ethyl)-3-((R)-1-hydroxyethyl)-4-(methylamino)piperidin-2-one as an oil (>95% purity, 1H NMR). 1H NMR (500 MHz, CD3OD) δ 7.58 (dt, J = 7.8, 0.7 Hz, 1 H), 7.32 (dt, J = 7.8, 0.7 Hz, 1 H), 7.09 – 7.05 (comp, 2 H), 7.00 (td, J = 7.1, 1.0 Hz, 1 H), 4.37 (pent, J = 6.3 Hz, 1 H), 3.67 – 3.60 (m, 1 H), 3.56 – 3.49 (m, 1 H), 3.33 – 3.17 (comp, 1 H), 3.15 (pent, J = 3.9 Hz, 1 H), 3.11 – 3.04 (comp, 3 H), 2.40 (dd, J = 6.1, 4.2 Hz, 1 H), 2.34 (s, 3 H), 1.97 (app sex, J = 7.8 Hz, 1 H), 1.74 (dtd, J = 13.9, 7.1, 3.2 Hz, 1 H), 1.23 (d, J = 6.4 Hz, 3 H). 13C NMR (125 MHz, CD3OD) δ 171.0, 138.1, 128.9, 123.6, 122.4, 119.7, 119.3, 113.1, 112.3, 67.5, 56.1, 51.4, 45.7, 33.5, 30.8, 24.8, 23.7, 22.5. HRMS (ESI) m/z calcd for C18H25N3O2 (M+H)+, 316.2020; found, 316.2024.
(3R,4S)-1-(2-(1H-Indol-3-yl)ethyl)-4-((3,5-dichlorobenzyl)(methyl)amino)-3-((R)-1-hydroxyethyl)piperidin-2-one (14a)
A solution of 4.28 (50 mg, 0.16 mmol) and 3,5-dichlorobenzaldehyde (45 mg, 0.26 mmol) in acetonitrile (1 mL) was heated under reflux for 2 h. The reaction was cooled to room temperature, at which time NaBH3CN (22 mg, 0.35 mmol) and acetic acid (53 μL, 0.89 mmol) were added. The reaction was stirred at room temperature for 20 h. The mixture was partitioned between saturated aqueous NaHCO3 (6.5 mL), ethyl acetate (6.5 mL), and methanol (0.5 mL). The two phases were separated, and the aqueous phase was extracted with ethyl acetate (2 × 6.5 mL). The combined organic fractions were washed with brine (6.5 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The crude material was purified via flash column chromatography eluting with a gradient of hexanes : ethyl acetate (1 : 1) → ethyl acetate : methanol (20 : 1), to give 36 mg (48%) of 14a as a white solid; 1H NMR (600 MHz) δ 8.15 (d, J = 1.9 Hz, 1 H), 7.55 (d, J = 8.2 Hz, 1 H), 7.34 (dt, J = 8.1, 0.8 Hz, 1 H), 7.27 (t, J = 1.9 Hz, 1 H), 7.18 (ddd, J = 8.1, 7.1, 1.1 Hz, 1 H), 7.12 – 7.08 (comp, 3 H), 7.02 (d, J = 1.9 Hz, 1 H), 4.35 (dq, J = 6.5, 6.2 Hz, 1 H), 3.74 – 3.78 (m, 1 H), 3.64 – 3.69 (m, 1 H), 3.41 – 3.26 (comp, 3 H), 3.24 (dt, J = 13.4, 5.4 Hz, 1 H), 3.12 (dt, J = 16.9, 9.2 Hz, 1 H), 3.04 – 2.99 (m, 1 H), 2.97 – 2.92 (m, 1 H), 2.31 (t, J = 5.2 Hz, 1H), 2.03 (s, 3 H), 1.83 – 1.87 (comp, 3 H), 1.15 (d, J = 6.5 Hz, 3 H); 13C NMR (150 MHz) δ 171.7, 140.6, 136.3, 135.2, 128.1, 127.6, 127.3, 122.4, 122.1, 119.4, 118.4, 112.3, 111.5, 66.2, 58.9, 48.0, 47.4, 44.9, 37.3, 29.7, 23.2, 21.7, 20.1; HRMS (ESI) m/z calcd for C25H29Cl2N3O2 (M+H)+, 474.1710 and 476.1686; found, 474.1712 and 476.1690.
Ethyl 3-((2-(1H-indol-3-yl)ethyl)(3-ethoxy-3-oxopropyl)amino)-3-oxopropanoate (22)
Ethyl 3-((2-(1H-indol-3-yl)ethyl)amino)propanoate18 (3.00 g, 11.5 mmol) and 3-ethoxy-3-oxopropanoic acid (1.68 g, 12.6 mmol) were dissolved in CH2Cl2 (250 mL) and the solution was cooled to 0 °C, whereupon EDCI•HCl (2.43 g, 12.6 mmol) and iPr2NEt (3.30 g, 25.2 mmol, 4.38 mL) were added. The reaction was warmed to room temperature and stirred for 40 h, whereupon the reaction was diluted with CH2Cl2 (250 mL) and washed with 1 M HCl (500 mL). The aqueous layer was then extracted with an additional portion of CH2Cl2 (250 mL). The combined organic layers were washed with brine (500 mL), dried (Na2SO4) and concentrated under reduced pressure to yield 4.3g (99%) of crude 22 as an amber oil (4.3g 99%) which was judged to be >90% pure by 1H NMR and carried forward without further purification. For characterization, a portion of the crude product was purified via column chromatography, eluting with hexanes/EtOAc (3:1 → 1:1) to provide 22 as an off-white solid. 1H NMR (3:2 rotamer mixture) (400 MHz) δ 8.24 (s, 0.5 H), 8.15 (s, 0.3 H), 7.71 (d, J = 7.8 Hz, 0.3 H), 7.59 (d, J = 7.8 Hz, 0.6 H), 7.40 (t, J = 8.7 Hz, 0.9 H), 7.25 – 7.18 (m, 1 H), 7.18 – 7.11 (m, 0.9 H), 7.06 (d, J = 2.3 Hz, 0.3 H), 7.00 (d, J = 2.4 Hz, 0.6 H), 4.26 (q, J = 7.1 Hz, 0.7 H), 4.18 – 4.11 (comp, 3 H), 3.70 (t, J = 7.0 Hz, 1.2 H), 3.65 – 3.59 (comp, 1.9 H), 3.58 (s, 0.7 H), 3.55 (t, J = 7.1 Hz, 0.7 H), 3.16 (s, 1.2 H), 3.08 – 3.01 (comp, 1.9 H), 2.69 (t, J = 7.0 Hz, 1.2 H), 2.57 (t, J = 7.1 Hz, 0.7 H), 1.32 (t, J = 7.1 Hz, 1.2 H), 1.28 – 1.22 (comp, 4.7 H); 13C NMR (100 MHz) (3:2 rotamer mixture) δ 172.1, 170.1, 167.8, 167.6, 166.4, 166.1, 136.3, 127.3, 126.9, 122.4, 122.3, 122.1, 122.0, 119.7, 119.4, 118.8, 118.1, 112.9, 111.7, 111.4, 111.1, 61.4, 61.3, 61.0, 60.6, 49.9, 46,9, 44.5, 42.4, 41.2, 40.9, 36.6, 32.6, 29.7, 24.8, 23.2, 14.2, 14.1, 14.0; IR (NaCl, film) 3417, 2983, 2920, 2361, 2255, 2128, 1731, 1642, 1458, 1370, 1319, 1260, 1190, 1159, 1097, 1027, 1007, 824, 746, 618 cm−1; HRMS (ESI) m/z calcd for C20H26N2O5 (M+Na)+ 397.1734; found 397.1733.
1-(2-(1H-Indol-3-yl)ethyl)piperidine-2,4-dione (23)
A suspension of NaH (60% w/w in mineral oil, 135 mg, 3.80 mmol) in cyclohexane (20 mL) was heated under reflux, and a solution of 22 (700 mg, 1.90 mmol) in toluene (3 mL) was added dropwise. The reaction was stirred under reflux for 6 h during which time a tan precipitate formed. The reaction was then cooled to room temperature, and the solvent was removed under reduced pressure. The flask was then cooled to 0 °C, EtOH (20 mL) was added, and the reaction was stirred for an additional 2 h. The solution was then diluted with H2O (200 mL) and 1 M HCl was added until pH = 1, and the aqueous solution was extracted with CH2Cl2 (3× 100 mL). The combined organic layers were washed with brine (100 mL), dried (MgSO4) and concentrated under reduced pressure to afford a tan solid which was then resuspended in AcOH (2 mL) and H2O (18 mL). The solution was heated under reflux until all starting material was consumed, as judged by LC-MS. The solution was cooled to room temperature, neutralized with sat. NaHCO3 (100 mL), and extracted with CH2Cl2 (3× 100 mL). The combined organic extracts were then washed with brine (200 mL), dried (MgSO4) and concentrated under reduced pressure to afford crude 23. The crude material was purified via flash column chromatography, eluting with hexanes:EtOAc (75:25 → 40:60) to furnish 313 mg (64%) of 23 as an off-white solid. 1H NMR (400 MHz) δ 8.04 (bs, 0.5 H), 7.65 (d, J = 7.8 Hz, 0.99 H),7.39 (d, J = 8.0 Hz, 1.05 H), 7.23 (t, J = 7.2 Hz, 1.18 H), 7.16 (t, J = 7.2, 1.08 H), 7.07 (s, 0.96 H), 3.85 (t, J = 6.9 Hz, 2.00 H), 3.34 (t,s overlapping, 3.90 H), 3.11 (t, J = 7 Hz, 2.05 H), 2.34 (t, J = 6.1 Hz, 1.91 H); 13C NMR (100 MHz) δ 203.8, 168.1, 136.3, 127.3, 122.3, 122.1, 119.6, 118.5, 112.9, 111.4, 49.0, 44.4, 38.5, 23.7; IR (NaCl, film) 3283, 2924, 2854, 2362, 1726, 1644, 1489, 1644, 1489, 1457, 1350, 1227, 1095, 1011, 745 cm−1; HRMS (ESI) m/z calcd for C15H16N2O2 (M+Na)+ 279.1104; found 279.1100.
1-(2-(1H-Indol-3-yl)ethyl)-4-(methylamino)piperidin-2-one (24)
To a solution of ketone 23 (65 mg, 0.25 mmol) in methanol (2.5 mL) was added methylamine (78 mg, 2.5 mmol, 33% wt. soln. in EtOH), AcOH (150 mg, 2.5 mmol), and NaBH3CN (31 mg, 0.5 mmol), and the resulting mixture was stirred for 16 h at room temperature, whereupon an additional portion of NaBH3CN (31 mg, 0.5 mmol) was added. After stirring for an additional 3 h, the reaction was diluted in 1 M HCl (50 mL) and washed with Et2O (50 mL). The aqueous layer was treated with 1 M NaOH until the solution turned cloudy and pH > 10, and the resulting mixture was diluted with brine (20 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude material was purified via column chromatography, eluting with CH2Cl2:MeOH:Et3N (95 : 5 : 1) to yield 45 mg (66%) of 24 as a white foam.1H NMR (400 MHz) δ 8.02 (s, 1 H), 7.66 (d, J = 7.9 Hz, 1 H), 7.37 (dt, J = 8.1, 1.0 Hz, 1 H), 7.20 (ddd, J = 8.1, 7.0, 1.3 Hz, 1 H), 7.13 (ddd, J = 7.9, 7.0, 1.1 Hz, 1 H), 7.06 (d, J = 2.3 Hz, 1 H), 3.66 (td, J = 7.1, 2.9 Hz, 2 H), 3.25 – 3.18 (m, 1 H), 3.16 – 3.08 (m, 1 H), 3.07 – 3.02 (comp, 2 H), 2.86 – 2.78 (m, 1 H), 2.68 (ddd, J = 17.1, 5.3, 1.8 Hz, 1 H), 2.41 (s, 3 H), 2.22 (dd, J = 17.1, 8.6 Hz, 1 H), 1.90 (dt, J = 8.1, 5.0 Hz, 1 H), 1.58 – 1.46 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 168.3, 136.3, 127.5, 122.00, 121.97, 119.3, 118.7, 113.1, 111.2, 53.2, 48.12, 45.7, 43.42, 38.8, 33.3, 28.4, 22.9; IR (NaCl, film) 3252, 3053, 2926, 2856, 2799, 2361, 1624, 1498, 1456, 1344, 1302, 1264, 1233, 1158, 1102, 1011 cm−1; HRMS (ESI) m/z calcd for C16H21N3O (M+Na)+ 294.1577; found 294.1574.
1-(2-(1H-Indol-3-yl)ethyl)-4-((3,5-dichlorobenzyl)(methyl)amino)piperidin-2-one (25a)
To a stirred solution of 24 (16 mg, 0.11 mmol) and 3,5-dichlorobenzaldehyde (25 mg, 0.314 mmol) in 1,2-dichloroethane (1.4 mL) was added sodium triacetoxyborohydride (45 mg, 0.21 mmol). The reaction was stirred for 4 h, whereupon it was diluted with CH2Cl2 (50 mL) and washed with sat. NaHCO3 (50 mL) and brine (50 mL). The organic layer was dried (Na2SO4)and concentrated under reduced pressure to afford crude 25a. The crude material was purified via flash column chromatography, eluting with hexanes:EtOAc:TEA (40:60:1) to furnish 17 mg (68%) of 25a as a yellow solid. 1H NMR (400 MHz) δ 8.10 (s, 1 H), 7.66 (d, J = 7.9 Hz, 1 H), 7.37 (d, J = 8.1 Hz, 1 H), 7.24 (s, 1 H), 7.31 – 7.25 (comp, 3 H), 7.18 (td, J = 8.1, 7.0 Hz, 1 H), 7.10 (td, J = 7.9, 7.0 Hz, 1 H), 7.05 (d, J = 2.3 Hz, 1 H), 3.74 – 3.50 (comp, 4 H), 3.17 – 3.00 (comp, 4 H), 2.94 – 2.81 (m, 1 H), 2.65 (dd, J = 17.2, 5.2 Hz, 1 H), 2.51 – 2.41 (m, 1 H), 2.16 (s, 3 H), 2.05 – 1.91 (m, 1 H), 1.60 (qd, J = 11.5, 6.0 Hz, 1 H); 13C NMR (100 MHz) δ 144.0, 137.0, 135.6, 128.2, 127.9, 127.4, 122.7, 122.5, 120.0, 119.4, 113.9, 111.8, 57.31, 57.29, 48.4, 46.9, 37.6, 35.0, 26.6, 23.7; IR (NaCl, film) 3624, 2509, 2929, 2855, 2793, 2242, 1623, 1569, 1499, 1456, 1433, 1345, 1303, 1231, 1209, 1158, 1125, 1098, 1074, 1036 cm−1; HRMS (ESI) m/z calc’d for C23H25Cl2N3O (M+H)+ 430.1447; found 430.1448.
N-(3,5-Dichlorobenzyl)-N-methylpiperidin-4-amine (27a)
A solution of tert-butyl 4-(methylamino)piperidine-1-carboxylate (44 mg, 0.2 mmol), 3,5-dichlorobenzaldehyde (53 mg, 0.3 mmol), acetic acid (12 mg, 0.2 mmol), and sodium triacetoxyborohydride (85 mg, 0.4 mmol) in 1,2-dichloroethane (2 mL) was stirred at room temperature for 18 h, whereupon the solution was diluted with 1 M NaOH (15 mL) and extracted with Et2O (3 × 15 mL). The combined organic extracts were dried (Na2SO4) and concentrated via rotary evaporation. The crude material was purified by flash chromatography eluting with hexane:EtOAc:Et3N (95:5:1) to give 48 mg (64%) of tert-butyl 4-((3,5-dichlorobenzyl)(methyl)amino)piperidine-1-carboxylate as an opaque white oil. 1H NMR (400 MHz) δ 7.23 – 7.19 (comp, 3 H), 4.16 (s, 2 H), 3.50 (s, 2 H), 2.75 – 2.61 (comp, 2 H), 2.55 (tt, J = 11.5, 3.6 Hz, 1 H), 2.17 (s, 3 H), 1.83 – 1.70 (comp, 2 H), 1.45 (comp, 11 H). 13C NMR (100 MHz) δ 154.7, 143.9, 134.7, 126.9, 126.7, 79.4, 61.1, 57.0, 43.1, 37.7, 28.4, 27.9; IR (NaCl, film) 2975, 2940, 2854, 2788, 1694, 1590, 1569, 1451, 1425, 1365, 1330, 1275, 1244, 1159, 1111, 1046, 1004 cm−1. HRMS (ESI) m/z calcd for C18 H26Cl2N2O2 (M+H)+ 373.1444; found 373.1454.
A solution of tert-butyl 4-((3,5-dichlorobenzyl)(methyl)amino)piperidine-1-carboxylate (52 mg, 0.14 mmol) and trifluoroacetic acid (160 mg, 1.4 mmol) in CH2Cl2 (2.8 mL) was stirred at room temperature for 24 h, whereupon the solvent was removed by rotary evaporation. The resulting solid was resuspended in 1 M HCl (20 mL) and washed with Et2O (20 mL). 3 M NaOH was added to the aqueous layer until the solution became cloudy, whereupon it was extracted with CH2Cl2 (3 × 20 mL), dried (Na2SO4) and concentrated via rotary evaporation to give 29 mg (76%) of 27a as a yellow paste. 1H NMR (400 MHz) δ 7.22 (t, J = 0.6 Hz, 3 H), 3.52 (s, 2 H), 3.18 (d, J = 12.2 Hz, 2 H), 2.66 – 2.46 (m, 4 H), 2.19 (s, 3 H), 1.51 (qd, J = 12.1, 4.1 Hz, 3 H); 13C NMR (100 MHz) δ 144.1, 134.7, 126.9, 126.8, 61.1, 56.9, 46.1, 37.7, 29.0; IR (NaCl, film) 2941, 2851, 2791, 1589, 1568, 1449, 1432, 1384, 1353, 1273, 1208, 1045 cm−1; HRMS (ESI) m/z calcd for C13H18Cl2N2 (M+H)+ 273.0920; found 273.0923.
1-(4-((3,5-Dichlorobenzyl)(methyl)amino)piperidin-1-yl)-2-(1H-indol-3-yl)ethan-1-one (28a)
A solution 27a (33 mg, 0.12 mmol), indole-3-acetic acid (32 mg, 0.18 mmol), EDCI·HCl (35 mg, 0.18 mmol), and iPr2NEt (0.8 mL, 0.48 mmol) in THF (1.2 mL) was stirred at rt for 2 h. The reaction was then diluted with 1 M aq. NaOH (20 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic extracts were dried (Na2SO4) and concentrated via rotary evaporation. The crude material was purified by flash chromatography, eluting with hexanes:EtOAc:Et3N (40:60:1) to give 28a (40 mg, 78%) as a white solid.1H NMR (400 MHz) δ 8.17 (s, 1 H), 7.65 (dq, J = 7.9, 0.9 Hz, 1 H), 7.35 (dt, J = 8.1, 1.0 Hz, 1 H), 7.22 (t, J = 2.0 Hz, 1 H), 7.19 (ddd, J = 8.2, 7.0, 1.2 Hz, 1 H), 7.16 – 7.14 (m, 2 H), 7.13 (ddd, J = 8.0, 7.1, 1.1 Hz, 1 H), 7.09 (dd, J = 2.3, 1.1 Hz, 1 H), 4.74 (d, J = 13.7 Hz, 1 H), 4.05 – 3.93 (m, 1 H), 3.86 (dd, J = 4.4, 1.0 Hz, 2 H), 3.37 (s, 2 H), 2.93 (td, J = 12.9, 2.7 Hz, 1 H), 2.55 (td, J = 11.2, 10.6, 3.0 Hz, 2 H), 2.07 (s, 3 H), 1.80 (d, J = 13.0 Hz, 1 H), 1.58 (d, J = 13.0 Hz, 1 H), 1.40 (qd, J = 12.3, 4.4 Hz, 1 H), 1.12 (qd, J = 12.3, 4.3 Hz, 1 H); 13C NMR (126 MHz) δ 170.1, 144.0, 136.4, 135.0, 127.3, 127.3, 126.9, 122.6, 122.5, 119.9, 119.1, 111.4, 109.8, 61.1, 57.1, 46.1, 41.8, 37.9, 31.9, 28.7, 27.7; IR (NaCl, film) 3058, 2926, 2857, 2790, 1624, 1568, 1454, 1434, 1353, 1268, 1227, 1149, 1125, 1098, 1047 cm−1; HRMS (ESI) m/zcalcd for C23H25Cl2N3O (M+Na)+ 452.1267; found, 452.1268.
1-(2-(1H-Indol-3-yl)ethyl)-N-(3,5-dichlorobenzyl)-N-methylpiperidin-4-amine (29a)
To a solution of 28a (21 mg, 0.05 mmol) in THF (1 mL) was added a 0.5 M solution of alane in toluene (0.15 mmol, 0.3 mL) and stirred for 3 h at room temperature. The reaction was then diluted with 1 M NaOH (20 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic extracts were dried (Na2SO4) and concentrated via rotary evaporation. The crude material purified by flash chromatography eluting with hexanes/EtOAc/Et3N (40:60:1) to give 15 mg (70%) of 29a as a yellow paste.1H NMR (400 MHz) δ 8.13 – 8.05 (s, 1 H), 7.61 (d, J = 7.8 Hz, 1 H), 7.34 (dt, J = 8.0, 0.9 Hz, 1 H), 7.23 (s, 3 H), 7.21 – 7.16 (m, 1 H), 7.15 – 7.09 (m, 1 H), 7.01 (d, J = 2.2 Hz, 1 H), 3.80 – 3.71 (m, 1 H), 3.53 (s, 2 H), 3.21 – 3.11 (comp, 2 H), 3.02 – 2.93 (comp, 2 H), 2.75 – 2.65 (comp, 2 H), 2.46 (tt, J = 11.5, 3.8 Hz, 1 H), 2.21 (s, 3 H), 2.05 (td, J = 11.8, 2.4 Hz, 2 H), 1.85 (comp, 3 H), 1.70 (qd, J = 12.0, 3.8 Hz, 2 H). 13C NMR (100 MHz) δ 144.2, 136.2, 134.7, 127.4, 126.9, 126.8, 122.0, 121.4, 119.2, 118.8, 114.5, 111.1, 61.3, 59.3, 57.0, 53.4, 37.9, 27.9, 23.2. IR (NaCl, film) 3412, 3179, 3057, 2943, 2853, 2808, 2360, 1620, 1589, 1568, 1455, 1433, 1376, 1353, 1303, 1228, 1145, 1097, 1049, 1014 cm−1. HRMS (ESI) m/zcalcd for C23H27Cl2N3 (M+H)+ 416.1655; found 416.1656.
Supplementary Material
ACKNOWLEDGMENT
We thank the Robert A. Welch Foundation (F-0652) for generous support of this research. We also thank the UT Austin Mass Spectrometry Facility for high-resolution mass spectral data, the National Institutes of Health (1 S10 OD021508-01) for funding a Bruker AVANCE III 500 NMR spectrometer, Prof. Wim G.J. Hol for generous donation of recombinant TbMetRS plasmids, and Prof. Frederick S. Buckner for assaying our initial hits and analogs. Work in the Mensa-Wilmot laboratory was supported by National Institutes of Health (R01AI126311).
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ASSOCIATED CONTENT
Supporting Information
Experimental procedures and analytical data for all new compounds (PDF), purification of recombinant TbMetRS and TbMetRS aminoacylation assay procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) For leading references, see: Sunderhaus JD; Dockendorff C; Martin SF. Synthesis of Diverse Heterocyclic Scaffolds via Tandem Additions to Imine Derivatives and Ring-Forming Reactions. Tetrahedron 2009, 65, 6454–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin SF Strategies for the Synthesis of Alkaloids and Novel Nitrogen Heterocycles. In Adv. Heterocyclic Chem 2013, 110, 73–117. [Google Scholar]; (c) Sahn JJ; Granger BA; Martin SF Evolution of a Strategy for Preparing Bioactive Small Molecules by Sequential Multicomponent Assembly Processes, Cyclizations, and Diversification. Org. Biomol. Chem. 2014, 12, 7659–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Donald JR; Martin SF; Synthesis and Diversification of 1,2,3-Triazole-Fused 1,4-Benzodiazepine Scaffolds. Org. Lett. 2011, 13, 852–855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Donald JR; Wood RR; Martin SF Application of a Sequential Multicomponent Assembly Process/Huisgen Cycloaddition Strategy to the Preparation of Libraries of 1,2,3-Triazole-Fused 1,4-Benzodiazepines. ACS Comb. Sci. 2012, 14, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Sahn JJ; Su JY; Martin SF Facile and Unified Approach to Skeletally Diverse, Privileged Scaffolds. Org. Lett. 2011, 13, 2590–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sahn JJ; Martin SF Expedient Synthesis of Norbenzomorphan Library via Multicomponent Assembly Process Coupled with Ring-Closing Reactions ACS Comb Sci. 2012, 14, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Hardy S; Martin SF Multicomponent Assembly and Diversification of Novel Heterocyclic Scaffolds Derived from 2-Arylpiperidines. Org. Lett. 2011, 13, 3102–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hardy S; Martin SF Multicomponent, Mannich-type assembly process for generating novel, biologically-active 2-arylpiperidines and derivatives. Tetrahedron 2014, 70, 7142–7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Granger BA; Kaneda K; Martin SF Multicomponent Assembly Strategies for the Synthesis of Diverse Tetrahydroisoquinoline Scaffolds. Org. Lett. 2011, 13, 4542–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Granger BA; Kaneda K; Martin SF Libraries of 2,3,4,6,7,11b-Hexahydro-1H-pyrido[2,1-a]isoquinolin-2-amine Derivatives via a Multicomponent Assembly Process/1,3-Dipolar Cycloaddition Strategy. ACS Comb. Sci. 2012, 14, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahn JJ; Martin SF Facile syntheses of substituted, conformationally-constrained benzoxazocines and benzazocines via sequential multicomponent assembly and cyclization Tetrahedron Lett. 2011, 52, 6855–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wang Z; Kaneda K; Fang Z; Martin SF Diversity Oriented Synthesis: Concise Entry to Novel Derivatives of the Yohimbine and Corynanthe Alkaloids. Tetrahedron Lett. 2012, 53, 477–479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Granger BA; Wang Z; Kaneda K; Fang Z; Martin SF Multicomponent assembly processes for the synthesis of diverse yohimbine and corynanthe alkaloid analogues. ACS Combi. Sci.2013, 15, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) National Center for Biotechnology Information. PubChem Database. YPSDOYLDQXJDKB-BVSLBCMMSA-N, CID=46902334, https://pubchem.ncbi.nlm.nih.gov/compound/46902334 (accessed on June 13, 2019); (b) National Center for Biotechnology Information. PubChem Database. CFNBSJDMYAEBMY-HOOUFKQNSA-N, CID=46902355, https://pubchem.ncbi.nlm.nih.gov/compound/46902355 (accessed on June 13, 2019).
- 9.Kennedy PG Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurology 2013, 12, 186–194. [DOI] [PubMed] [Google Scholar]
- 10.Barrett MP; Boykin DW; Brun R; Tidwell RR Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. British J. Pharm. 2007, 152, 1155–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesu VKBK; Kalonji WM; Bardonneau C; Mordt OV; Blesson S; Simon F; Delhomme S; Bernhard S; Kuziena W; Lubaki J-PF; et al. Oral Fexinidazole for Late-Stage African Trypanosoma Brucei Gambiense Trypanosomiasis: A Pivotal Multicentre, Randomised, Non-Inferiority Trial. The Lancet 2018, 391, 144–154. [DOI] [PubMed] [Google Scholar]
- 12.(a) Shibata S; Gillespie JR; Kelley AM; Napuli AJ; Zhang Z; Kovzun KV; Pefley RM; Lam J; Zucker FH; Van Voorhis WC; Merritt EA; Hol WGJ; Verlinde CLMJ; Fan E; Buckner FS Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei infection in mice. Antimicrob. Agents Chemother.2011, 51, 1982–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koh CY; Kim JE; Shibata S; Ranade RM; Yu M; Liu J; Gillespie JR; Buckner FS; Verlinde CLMJ; Fan E; Hol WJG; Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 2012, 20, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Shibata S; Gillespie JR; Ranade RM; Koh CY; Kim JE; Laydbak JU; Zucker FH; Hol WGJ; Verlinde CLMJ; Buckner FS; Fan E Urea-based inhibitors of trypanosoma brucei methionyl-trna synthetase: Selectivity and in vivo characterization. J. Med. Chem.2012, 55, 6342–6351; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koh CY; Kim JE; Wetzel AB; Van der Schueren WJ; Shibata S; Ranade RM; Liu J, Zhang Z; Gillespie JR; Buckner FS; Verlinde CLMJ; Fan E; Hol WJG Structures of Trypanosoma brucei Methionyl-tRNA Synthetase with Urea-Based Inhibitors Provide Guidance for Drug Design against Sleeping Sickness. PLoS NTD 2014, 8, 4, e2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W; Zhang Z; Ranade RM; Gillespie JR; Barros-Álvarez X; Creason SA; Shibata S; Verlinde CLMJ; Hol WGJ; Buckner FS; et al. Optimization of a Binding Fragment Targeting the “enlarged Methionine Pocket” Leads to Potent Trypanosoma Brucei Methionyl-tRNA Synthetase Inhibitors. Bioorg. Med. Chem. Lett.2017, 27, 2702–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macor JE; Fox CB; Johnson C; Koe BK; Lebel LA; Zorn SH 1-(2-Aminoethyl)-3-Methyl-8,9-dihydropyrano[3,2-e]indole: A Rotationally Restricted Phenolic Analog of the Neurotransmitter Serotonin and Agonist Selective for Serotonin (5-HT2-Type) Receptors. J. Med. Chem.1992, 35, 3625–3632. [DOI] [PubMed] [Google Scholar]
- 16.Gálvez C; Viladoms P Reactivity of 1H-Pyrrolo[2,3-b]pyridine. II. Synthesis of 3-(β-Haloethyl)-7-azaindole. J. Heterocyclic Chem 1984, 21, 421–423. [Google Scholar]
- 17.(a) Cestari I; Stuart K A Spectrophotometric Assay for Quantitative Measurement of Aminoacyl tRNA Synthetase Activity. J. Biomol. Screen. 2012, 20, 1–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baykov AA; Evtushenko OA; Avaeva SM A Malachite Green Procedure for Orthophosphate Determination and Its Use in Alkaline Phosphatase-Based Enzyme Immunoassay. Anal. Biochem. 1988, 171, 266–270. [DOI] [PubMed] [Google Scholar]
- 18.Huizenga RH; Pandit UK Models of Folate Cofactors - 22. Lewis Acid Catalyzed Cyclization of Carbon-Fragment Transfer Products of Folate Cofactor Models. Synthesis of Enantiomerically Pure Tetracyclic (ABCE) Ring System of Aspidosperma Alkaloids. Tetrahedron 1991, 47, 4155–4164. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.